

Abstract
Interstitial lung disease (ILD) classification requires a multidisciplinary review that includes input from an ILD clinician, chest radiologist, and lung pathologist. We report a case of ILD that remained unclassifiable due to discordant clinical, radiological, and pathological findings despite a thorough evaluation that included examination of explanted lung tissue. This case demonstrates that ILD can remain unclassifiable even with a complete evaluation and illustrates one approach to the management of such patients.
Keywords: Diffuse lung disease, idiopathic interstitial pneumonia, interstitial lung disease, unclassifiable interstitial lung disease
Introduction
Interstitial lung disease (ILD) includes a large number of disorders that cause inflammation or fibrosis of the pulmonary parenchyma. Many fibrotic ILDs are attributed to well-defined underlying causes, commonly including connective tissue disease (CTD) and hypersensitivity pneumonitis (HP). Idiopathic interstitial pneumonias are other well-characterized ILDs that lack a clear etiology or pathobiology [1]; however, approximately 10% of fibrotic ILD patients cannot be confidently classified with a specific ILD subtype [2], [3]. We herein report a case of ILD that remained unclassifiable despite a thorough evaluation for underlying causes.
Case
A previously well 52-year-old man presented with 1 week of mild fatigue and exertional dyspnea. He had no medical history and was on no medication. He had a 30 pack-year smoking history and quit 2 years previously. Review of systems, family history, environmental history, and occupational history were unremarkable. Chest X-ray demonstrated basilar-predominant opacification suggestive of consolidation. He was treated for community-acquired pneumonia with moxifloxacin, but subsequently required admission to hospital with increasing dyspnea and hypoxemia.
On admission to hospital, his oxygen saturation was 90% on oxygen at 5 L/min via nasal prongs and his vital signs were normal. Physical examination revealed bilateral coarse crackles at the lung bases. He was not clubbed. There were no features of CTD or cardiac disease. Complete blood counts, liver function tests, and renal function tests were normal, with a borderline increased creatinine kinase and C-reactive peptide. Rheumatoid factor and anti-citrullinated C-peptide antibody were positive (33 IU/mL [upper limit of normal 15] and >250 EU [upper limit of normal 20], respectively). Other CTD serology was negative. Serum precipitins were negative. Spirometry showed severe restriction (forced vital capacity (FVC) 1.75 L [33% predicted]). Bronchoalveolar lavage revealed neutrophilic predominance at 29%, and lymphocytes at 13%. Bacterial cultures, viral polymerase chain reaction, and Pneumocystis stains were negative. A high-resolution computed tomography scan demonstrated consolidation and ground glass superimposed on a background of reticulation and architectural distortion (Fig. 1). A surgical lung biopsy was not performed due to ongoing hypoxemia.
Figure 1.
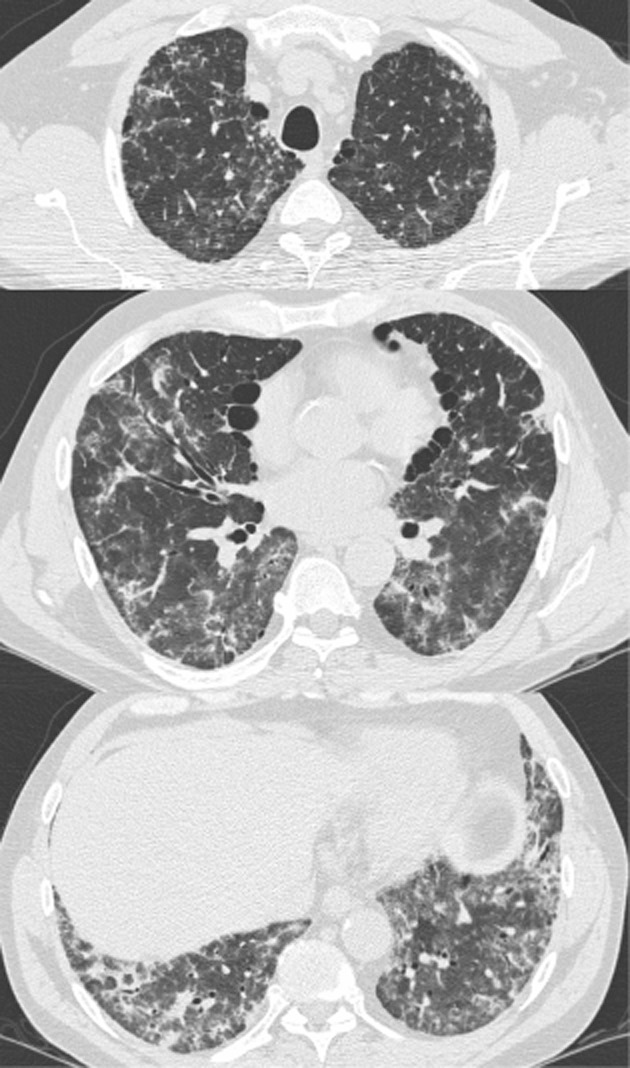
Selected transverse axial images from a high-resolution computed tomography displaying findings of architectural distortion, ground glass, and traction bronchiectasis. Importantly, there is no lobar predilection and there is an absence of honeycombing.
He did not improve with multiple courses of broad-spectrum intravenous antibiotics and high-dose corticosteroids. He was transferred to a tertiary hospital where multidisciplinary evaluation concluded the underlying pathological pattern was likely a combination of organizing pneumonia and nonspecific interstitial pneumonia (NSIP), however with an unclear etiology. He was treated with pulse corticosteroids and subsequently with intravenous cyclophosphamide. He had mild improvement while on this therapy and was discharged from hospital 3 weeks after admission, still requiring oxygen at 1 L/min at rest and 5–6 L/min on ambulation. He completed six cycles of cyclophosphamide and was then treated with mycophenolate mofetil and ongoing low-dose prednisone. He continued to have worsening dyspnea and hypoxemia and underwent a successful bilateral lung transplant 11 months after presentation.
The explanted lungs showed an NSIP pattern, with some areas that resembled usual interstitial pneumonia (UIP) (Fig. 2). There were occasional interstitial and airspace giant cells and rare interstitial non-necrotizing granulomas. The initial pathology report of the explanted lung suggested a diagnosis of chronic HP; however, no clear exposures were identified on another detailed review of the patient's occupational and environmental history. Re-evaluation of the explanted lungs concluded that the pathological findings could represent a CTD-ILD; however, HP was considered to have a similar probability. The patient continues to do well 1-year post-transplantation, without newly identified environmental exposures or CTD manifestations that would confirm an underlying etiology.
Figure 2.
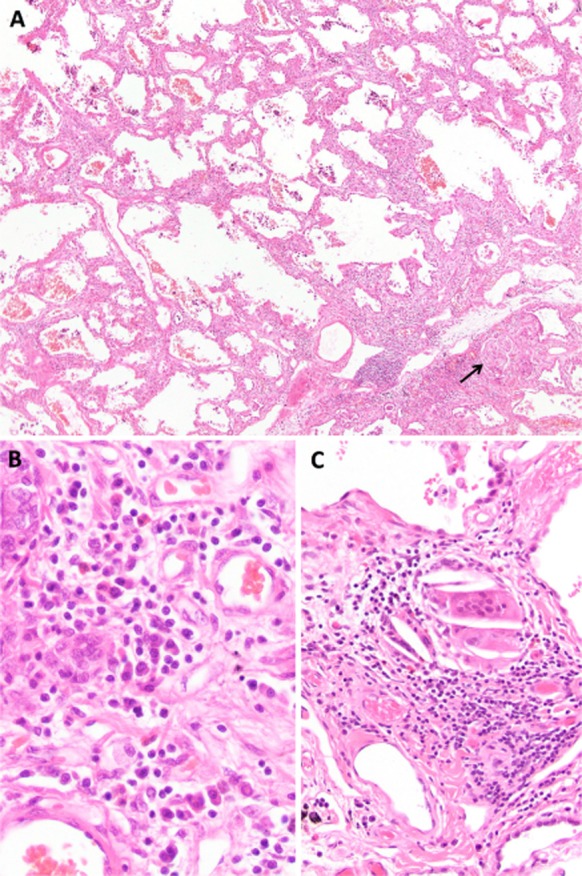
Explanted lung pathology. (A) Low power view showing a mixed cellular and fibrotic nonspecific interstitial pneumonia pattern. A small granuloma is present at the arrow (hematoxylin and eosin stain, ×80). (B) High power view demonstrating an inflammatory infiltrate consisted of lymphocytes as well as numerous plasma cells and a few eosinophils (hematoxylin and eosin stain, ×400). (C) High power view of a granuloma (hematoxylin and eosin stain, ×200).
Discussion
ILD classification requires a thorough review with input from a multidisciplinary panel that includes an experienced respirologist, radiologist, and pathologist [1]. As previously described [4], our case illustrates that some ILD patients cannot be classified with a specific ILD despite detailed review of a complete diagnostic evaluation. The presence of plasma cells and granulomas in the explanted lung raised a question of underlying CTD, but this could not be proved clinically and no evidence of CTD has developed since transplant.
ILD can remain unclassifiable if critical clinical, radiological, or pathological information is unavailable; if there is major discrepancy between clinical, radiological, and pathological findings; if interpretation of radiological or pathological findings is altered by prior treatment; or if there are overlapping or concurrent conditions [2]. All of these patients are grouped under the single category of “unclassifiable ILD”; however, there are important differences among these subgroups, and the definition and terminology of unclassifiable ILD is inconsistent. These issues highlight the need for greater consensus and a standardized approach to the evaluation and classification of this population.
Management of unclassifiable ILD is challenging as there is no direct evidence for this population. In this context, management of unclassifiable ILD can be guided by considering the most likely diagnosis and the anticipated disease behavior [1]. The radiological features in our case were consistent with an overlap of organizing pneumonia and NSIP. This suggested a possible role for aggressive immunosuppression, and provided justification for high-dose prednisone and steroid-sparing agents. Conversely, the absence of typical radiological UIP features argued against the use of antifibrotic agents. Similar to other ILD subtypes [5], the non-pharmacologic management of unclassifiable ILD should include smoking cessation, vaccinations, supplemental oxygen, pulmonary rehabilitation, and lung transplantation.
In conclusion, this case illustrates that unclassifiable ILD can exist despite multidisciplinary review of adequate clinical, radiological, and pathological findings. Additional research is required to improve the diagnostic approach to these patients, and specifically to identify non-invasive biomarkers that can distinguish idiopathic pulmonary fibrosis from other fibrotic ILD subtypes. Further studies are also required to clarify treatment strategies and validate the disease behavior approach to the management of unclassifiable ILD.
Disclosure Statements
CJR has received speaking honoraria and research funding from InterMune Inc. and Boehringer Ingelheim. The remaining authors report no conflicts of interest.
Appropriate written informed consent was obtained for publication of this case report and accompanying images.
References
- Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013;188:733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skolnik K. Ryerson CJ. Unclassifiable interstitial lung disease: a review. Respirology. 2015 doi: 10.1111/resp.12568. , and. doi: 10.1111/resp.12568. [DOI] [PubMed] [Google Scholar]
- Troy L, Glaspole I, Goh N, et al. Prevalence and prognosis of unclassifiable interstitial lung disease. Eur. Respir. J. 2014;43:1529–1530. doi: 10.1183/09031936.00003414. [DOI] [PubMed] [Google Scholar]
- Ryerson CJ, Urbania TH, Richeldi L, et al. Prevalence and prognosis of unclassifiable interstitial lung disease. Eur. Respir. J. 2013;42:750–757. doi: 10.1183/09031936.00131912. [DOI] [PubMed] [Google Scholar]
- Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]