

It is increasingly clear that there are many interconnected and compensatory pathways that can overcome vascular endothelial growth factor-targeted inhibition of angiogenesis. Maximizing the potential of antiangiogenic therapy is likely to require a broader therapeutic approach using a new generation of multitargeted antiangiogenic agents.
Keywords: Angiogenesis inhibitors; Antibodies, monoclonal, humanized; Molecular targeted therapy; Receptors; Fibroblast growth factor; Platelet-derived growth factor; Vascular endothelial growth factor
Abstract
Angiogenesis, or the formation of new capillary blood vessels, occurs primarily during human development and reproduction; however, aberrant regulation of angiogenesis is also a fundamental process found in several pathologic conditions, including cancer. As a process required for invasion and metastasis, tumor angiogenesis constitutes an important point of control of cancer progression. Although not yet completely understood, the complex process of tumor angiogenesis involves highly regulated orchestration of multiple signaling pathways. The proangiogenic signaling molecule vascular endothelial growth factor (VEGF) and its cognate receptor (VEGF receptor 2 [VEGFR-2]) play a central role in angiogenesis and often are highly expressed in human cancers, and initial clinical efforts to develop antiangiogenic treatments focused largely on inhibiting VEGF/VEGFR signaling. Such approaches, however, often lead to transient responses and further disease progression because angiogenesis is regulated by multiple pathways that are able to compensate for each other when single pathways are inhibited. The platelet-derived growth factor (PDGF) and PDGF receptor (PDGFR) and fibroblast growth factor (FGF) and FGF receptor (FGFR) pathways, for example, provide potential escape mechanisms from anti-VEGF/VEGFR therapy that could facilitate resumption of tumor growth. Accordingly, more recent treatments have focused on inhibiting multiple signaling pathways simultaneously. This comprehensive review discusses the limitations of inhibiting VEGF signaling alone as an antiangiogenic strategy, the importance of other angiogenic pathways including PDGF/PDGFR and FGF/FGFR, and the novel current and emerging agents that target multiple angiogenic pathways for the treatment of advanced solid tumors.
Implications for Practice:
Significant advances in cancer treatment have been achieved with the development of antiangiogenic agents, the majority of which have focused on inhibition of the vascular endothelial growth factor (VEGF) pathway. VEGF targeting alone, however, has not proven to be as efficacious as originally hoped, and it is increasingly clear that there are many interconnected and compensatory pathways that can overcome VEGF-targeted inhibition of angiogenesis. Maximizing the potential of antiangiogenic therapy is likely to require a broader therapeutic approach using a new generation of multitargeted antiangiogenic agents.
Introduction
Angiogenesis, a process that involves tight regulation of multiple signaling pathways, is the physiologic process by which new blood vessels form from pre-existing vessels. Although it is a homeostatic process that predominantly occurs during embryogenesis, angiogenesis also occurs in the adult during the ovarian cycle and in normal physiologic repair processes such as wound healing. Many cancers exploit angiogenic mechanisms to stimulate tumor growth and disease progression [1] (Fig. 1). Numerous proangiogenic and antiangiogenic factors, extracellular matrix components, and cell types act in concert to determine the type, location, and abundance of the angiogenic response [2]. However, there is universal agreement that vascular endothelial growth factor (VEGF) and its cognate receptor (VEGF receptor 2 [VEGFR-2]) are the most prominent regulators of angiogenesis. VEGF signaling stimulates cellular pathways that lead to the formation and branching of new tumor blood vessels, promotes rapid tumor growth, and facilitates metastatic potential [3]. Accordingly, there was a long-held perception that inhibiting the VEGF/VEGFR pathway alone would cause a rapid and sustained antiangiogenic/antitumor response [4]. Indeed, several VEGF/VEGFR targeted inhibitors have been approved after improving the prognosis of patients with cancer compared with chemotherapy alone across several indications [5–7]. However, because other mediators of angiogenesis, including the platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF) signaling pathways also regulate angiogenesis, tumor growth, and metastasis, compensatory mechanisms may come into play when VEGF signaling is blocked. Consequently, more recent antiangiogenic treatments aim to simultaneously block both VEGF/VEGFR signaling and other pathways that are critical to angiogenesis and tumor growth. The purpose of this review is to discuss the relevant signaling pathways involved in tumor angiogenesis, growth, and resistance to anti-VEGF therapy and to highlight the potential clinical benefits related to their pharmacologic inhibition.
Figure 1.

Tumor angiogenesis mechanisms. Soluble angiogenic factors (e.g., VEGF, PDGF, FGF) are secreted from the tumor and surrounding cells to induce and regulate key steps in angiogenesis. Reproduced with permission from [1].
Abbreviations: bFGF, basic fibroblast growth factor; bFGFR, basic fibroblast growth factor receptor; MMP, matrix metalloproteinase; PDGF, platelet-derived growth factor; PDGFR, platelet-derived growth factor receptor; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
Materials and Methods
To evaluate angiogenesis in cancer, a systematic review of the published literature during the period 2005–2014 was performed using PubMed. Following peer review, this paper was updated with any pertinent literature for the period 2014–2015. Articles were limited to the English language only, and the following key words were used: angiogenesis, vascular endothelial growth factor, platelet-derived growth factor, fibroblast growth factor, angiopoietins, TIE2, proto-oncogene protein RET, proto-oncogene protein MET, and hepatocyte growth factor. In addition, abstracts from annual meetings of the American Society of Clinical Oncology and the European Society for Medical Oncology, among others, were searched to identify recent presentations related to angiogenesis in cancer. The discussion of antiangiogenic agents was limited to agents that have progressed to phase III clinical trial status.
Angiogenesis and the VEGF/VEGFR Pathway
VEGF was initially identified as an endothelial cell-specific mitogen with the ability to induce physiologic and pathologic angiogenesis [8, 9]. Since this finding, much has been learned about the nature of VEGF signaling and its role in angiogenesis. VEGF comprises a family of ligands (VEGF-A to -D and placental growth factor [PlGF]) that bind to VEGFR tyrosine kinases [2, 10, 11]. VEGF-A, VEGF-B, and PlGF have decisive roles in angiogenesis. Although VEGF-A and -B have the greatest binding affinity for VEGFR-1 and -2, the majority of angiogenic effects are attributed to the interaction of VEGF-A with VEGFR-2 [11]. Less well understood, VEGFR-1 is thought to function predominantly as a decoy receptor by regulating the amount of free VEGF-A available to activate VEGFR-2 because VEGFR-1 negatively regulates VEGF-A/VEGFR-2 interaction [12]. The role of PlGF in angiogenesis remains controversial; however, gain- and loss-of-function experiments have shown that it may directly stimulate vessel growth and maturation and recruit proangiogenic bone marrow-derived progenitors and monocyte-macrophage lineage cells [13]. VEGF-C and -D appear to be the most important factors in lymphangiogenesis and have the greatest binding affinity for VEGFR-3 [14]. Not surprisingly, VEGFs are produced by several types of cells (Fig. 1), including fibroblasts, inflammatory cells, and many tumor cells, often in response to increasing tissue hypoxia [4].
The role of PlGF in angiogenesis remains controversial; however, gain- and loss-of-function experiments have shown that it may directly stimulate vessel growth and maturation and recruit proangiogenic bone marrow-derived progenitors and monocyte-macrophage lineage cells.
Inhibition of VEGF/VEGFR Signaling
Several agents, including bevacizumab, aflibercept, and, most recently, ramucirumab, that target the VEGF/VEGFR signaling pathway have been developed and are now approved across several indications.
Bevacizumab
Bevacizumab, a humanized monoclonal antibody (mAb) that targets VEGF-A to prevent its interaction with VEGFR-1 and -2, was the first targeted antiangiogenic approved for use in oncology [15]. Currently approved in the U.S. as combination therapy for first- and second-line treatment of metastatic colorectal cancer (mCRC) and metastatic renal cell carcinoma (mRCC) and for first-line therapy for unresectable, locally advanced, recurrent, or metastatic non-small cell lung cancer (NSCLC), bevacizumab is also approved as monotherapy for adults with progressive glioblastoma [5]. Most recently, bevacizumab has also been approved in combination with chemotherapy for the first-line treatment of persistent, recurrent, or metastatic cervical cancer and platinum-resistant recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer [5]. Notably, the U.S. Food and Drug Administration (FDA) revoked the product license in the U.S. for the treatment of breast cancer [16]. Although a positive impact on progression-free survival (PFS) and response rate had been demonstrated consistently, such an effect on overall survival (OS) had not. Coupled with an emerging unfavorable adverse events profile in this population, the use of bevacizumab in breast cancer was questioned [17], with the FDA concluding that the drug had not been shown to be safe and effective for that use [16].
Although an important advance in treatment, bevacizumab provides only a modest survival benefit, with inconsistent effects in different tumor types [18]. The addition of bevacizumab to first-line irinotecan/5-fluorouracil/leucovorin for CRC, for example, increases OS by 4.7 months, whereas its addition to first-line carboplatin/paclitaxel for unresectable, locally advanced, recurrent, and metastatic nonsquamous NSCLC increases OS by 2 months [18]. Responses to bevacizumab are often transient, and many patients experience disease progression as an adaptive response to ongoing therapy or following treatment withdrawal [19–21]. Furthermore, early studies with bevacizumab across a variety of cancer types established a set of adverse events (AEs) attributed to antiangiogenic therapy, with the most documented toxicity being hypertension, which was reported in up to 36% of patients [22]. Because clinical trials are conducted under varying conditions and patient types (e.g., different treatment regimens, cancer types, age groups), the frequency of AEs varies widely. The most common AEs observed in bevacizumab-treated patients at a rate of >10% and at least twice the control arm rate are epistaxis, headache, hypertension, rhinitis, proteinuria, taste alteration, dry skin, rectal hemorrhage, lacrimation disorder, back pain, and exfoliation [23, 24].
Aflibercept
Aflibercept is a fusion protein that consists of VEGF-binding portions from the extracellular domains of human VEGFR-1 and -2 fused to the Fc portion of human immunoglobulin G1 (IgG1) [25]. Aflibercept functions as a decoy receptor by neutralizing the available VEGF-A and -B and PlGF and making the ligands unavailable to bind and activate VEGFRs. The ability of aflibercept to bind multiple VEGF ligands may provide a more complete blockade of angiogenesis than bevacizumab, which targets only VEGF-A [26]. Preclinical studies with aflibercept showed antitumor and antiangiogenic activity in a variety of xenograft models, including human colon cancer [25–27]. Indicated for patients with mCRC that is resistant to or that has progressed following an oxaliplatin-containing regimen, approval of aflibercept was based on findings from a phase III trial that showed its addition to folinic acid, fluorouracil, and irinotecan (FOLFIRI) significantly improved OS relative to placebo plus FOLFIRI in patients with mCRC who had previously received oxaliplatin (median: 13.50 vs. 12.06 months, respectively; hazard ratio [HR]: 0.817; 95% confidence interval [CI]: 0.713–0.937; p = .0032) [22, 28]. AEs with aflibercept compared with placebo were very similar to, although less severe than, those seen with bevacizumab and other antiangiogenic agents. The most common AEs (≥20%) reported at a higher incidence (≥2%) than with placebo are leukopenia, diarrhea, neutropenia, proteinuria, increased aspartate aminotransferase, stomatitis, fatigue, thrombocytopenia, increased alanine aminotransferase, hypertension, decreased weight, decreased appetite, epistaxis, abdominal pain, dysphonia, increased serum creatinine, and headache [22].
Ramucirumab
Recently approved in the U.S. for advanced gastric cancer or gastro-esophageal junction (GEJ) adenocarcinoma after prior fluoropyrimidine- or platinum-containing chemotherapy, ramucirumab is a fully humanized IgG1 mAb targeting the extracellular domain of VEGFR-2 [7]. The phase III REGARD and RAINBOW trials, which were pivotal to FDA approval, evaluated ramucirumab as monotherapy and in combination with paclitaxel, respectively, in previously treated patients with advanced gastric cancer or GEJ adenocarcinoma [29]. The REGARD trial found that patients receiving ramucirumab with best supportive care (BSC) experienced median OS of 5.2 months compared with 3.8 months with placebo (HR: 0.776; 95% CI: 0.603–0.998; p = .047). In the phase III RAINBOW trial, the addition of ramucirumab significantly improved OS from 7.36 months to 9.63 months (HR: 0.807; 95% CI: 0.678–0.962; p = .0169) [30]. Ramucirumab has shown varying degrees of efficacy in renal, uterine, colorectal, and ovarian carcinoma [31, 32]. Most recently, results of ramucirumab trials in NSCLC and breast cancer have been reported. As a second-line treatment, ramucirumab plus docetaxel in the phase III REVEL study (NCT01168973) was reported to significantly increase OS among patients with stage IV NSCLC versus docetaxel alone (10.5 vs. 9.1 months; HR: 0.86; 95% CI: 0.75–0.98; p = .023) [33]. Furthermore, ramucirumab was well tolerated, with most treatment-emergent AEs occurring at a similar frequency in the ramucirumab and placebo arms. Ramucirumab is now approved in the U.S. in combination with docetaxel as a second-line therapy in advanced and/or metastatic NSCLC [7]. However, in metastatic, human epidermal growth factor receptor 2-negative (HER2-negative) breast cancer, results have been somewhat disappointing. In the ROSE/TRIO trial, ramucirumab in combination with docetaxel failed to demonstrate a meaningful improvement in important clinical outcomes versus docetaxel alone (OS: 27.3 vs. 27.2 months; HR: 1.01; 95% CI: 0.83–1.23; p = .915) [34]. Phase III trials of ramucirumab are also ongoing in mCRC, and results of its use in advanced hepatocellular carcinoma (HCC) as a second-line treatment (REACH trial) have been presented recently [35]. In that study, patients who had progressed during or following sorafenib or who were intolerant to it received ramucirumab plus BSC versus placebo plus BSC. A significant improvement in PFS (2.8 vs. 2.1 months; HR: 0.63; 95% CI: 0.52–0.75; p < .0001) was observed in the ramucirumab arm versus placebo, but this did not translate into a significant OS improvement (9.2 vs. 7.6 months; HR: 0.866; 95% CI: 0.717–1.046; p = .1391) [35].
Angiogenesis Beyond the VEGF/VEGFR Pathway
Although VEGF-mediated signaling can promote the growth, survival, migration, and invasion of cancer cells, a role for a number of signaling pathways working in combination with VEGF/VEGFR signaling is now appreciated. Studies of these proangiogenic signaling pathways have provided considerable insight into the molecular mechanisms that underlie tumor angiogenesis and provide a foundation for the development of antiangiogenic therapies that target these pathways (Fig. 2). Indeed, VEGF-independent signaling pathways have been shown to regulate tumor angiogenesis and serve as alternative inductors of tumor growth [36]. Several of these pathways have been well characterized, including the FGF/FGFR, PDGF/PDGFR, and hepatocyte growth factor (HGF)/MET signaling pathways.
Figure 2.

Angiogenesis signaling and targets of inhibition in approved antiangiogenic agents. Reproduced and adapted with permission from [74].
Abbreviations: Ang, angiopoietin; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; PDGF, platelet-derived growth factor; PDGFR, platelet-derived growth factor receptor; PlGF, placental growth factor; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
VEGF-independent signaling pathways have been shown to regulate tumor angiogenesis and serve as alternative inductors of tumor growth.
PDGF/PDGFR
The PDGF family consists of PDGF-A to -D polypeptide homodimers and the PDGF-AB heterodimer. These ligands exert their effects by binding to the PDGFR-α and -β tyrosine kinase receptors and activating pathways that are the same as and/or similar to those stimulated by VEGF [37, 38]. Accordingly, activation of PDGF signaling is implicated in growth, survival, and motility of a variety of cell types [39]. Overstimulation of PDGF signaling, either alone or in combination with FGF and VEGF, is associated with tumor vascularization in malignant disease, including but not limited to NSCLC, HCC, and ovarian cancer (OC) [39, 40]. Furthermore, direct activation of PDGF signaling has been observed in multiple tumor types, and coexpression of PDGF and its receptor suggests a role for autocrine and paracrine activation [41]. Roles for aberrant PDGF signaling in tumor angiogenesis include pericyte recruitment to vessels; secretion of proangiogenic factors; stimulation of endothelial cell proliferation, migration, sprouting, and tube formation in tumors; and promotion of lymphangiogenesis and subsequent lymphatic metastasis [42–45]. The importance of PDGF signaling in tumor angiogenesis is further supported by several studies demonstrating that PDGFR inhibitors improve the antitumor efficacy of VEGFR blocking agents [46]. Work is ongoing to clarify a role for PDGF in tumor angiogenesis, in the hope of developing more effective antiangiogenic treatments that reduce growth, maturation, and metastases of various tumor types.
FGF/FGFR
FGFs are heparin-binding growth factors that comprise a family of 23 members, 18 of which function as ligands for four receptor tyrosine kinases (RTKs), namely, FGFR-1 to -4 [47, 48]. FGFs and FGFRs are ubiquitously expressed and have numerous functions, including the regulation of normal cell growth and differentiation and of angiogenesis [49]. FGFR-1 is the primary FGFR expressed on endothelial cells, although FGFR-2 is also present in small amounts [50]. Among the FGFR ligands, FGF1 and FGF2 have been reported to have potent proangiogenic effects that induce the proliferation and migration of endothelial cells [51]. Overexpression of FGF and FGFR is reported in many cancers and is attributed to a number of mutations, including constitutive activation, gene amplification, translocations, gene fusions, and altered gene splicing, which may lead to enhanced angiogenesis through the stimulation and release of other proangiogenic factors [48, 49]. As discussed previously for PDGF, a collaborative interplay between FGF and VEGF signaling has also been demonstrated to be important for angiogenic and metastatic processes [52–54]. FGF can act synergistically with VEGF to amplify tumor angiogenesis; therefore, simultaneously targeting the FGF and VEGF pathways may more efficiently suppress angiogenesis and tumor growth than targeting either pathway alone. FGFs are implicated in the emerging phenomenon of resistance to VEGF inhibition. Resistance to VEGFR-2 blockade in late-stage tumors, for example, occurred in in vivo pancreatic cancer models, in which tumors regrew following an initial period of anti-VEGFR-2-mediated growth suppression [55].
ANG/TIE2
Angiopoietins play a critical role in the maintenance of vessel quiescence and comprise a family of four ligands (ANG1 to ANG4). ANG1 and ANG2 are the best-characterized members and bind to the TIE2 receptor. ANG1 binding enhances perivascular-endothelial cell interaction and endothelial cell survival, which, in turn, promotes the stabilization of blood vessels, whereas ANG2 is predominantly synthesized and secreted by endothelial cells at sites of vascular remodeling in response to proangiogenic signals (e.g., inflammation, cytokines, hypoxia) [56]. Of note, ANG2 overexpression in many cancers correlates with poor survival and more invasive cancer phenotypes [53, 57]; however, studies indicate that, depending on the context, ANG/TIE2-targeting therapy can promote either protumor or antitumor effects. ANG2/TIE2-stimulated tumor vascular destabilization, for example, also may render established vasculature more resistant to antiangiogenic therapy, whereas TIE2 inhibition is believed to promote vascular regression. Hampered in part by a limited understanding of the biological complexity that is generated by agonistic and antagonistic signaling, development of treatments targeting the ANG/TIE2 pathway has proved to be challenging [58].
HGF/MET
Produced as a single-chain inactive precursor protein, HGF is a pleiotropic growth factor that binds MET RTK. Not only does HGF/MET signaling regulate normal cell proliferation, motility, and survival, it also mediates tumor angiogenesis and growth in a variety of cell and tissue types, including various carcinomas, sarcomas, hematopoietic malignancies, melanomas, and central nervous system tumors [59, 60]. Proangiogenic effects of HGF/MET on tumors occur primarily by direct activation of endothelial cells to undergo motogenic or morphogenic changes and by indirect stimulation of the production of proangiogenic factors, including VEGF [61]. Comparisons of bevacizumab-resistant glioblastoma with pretreatment tumors from the same patients found increased MET expression in the former, suggesting that MET may play a role in antiangiogenic therapy resistance by compensating for the inhibition of VEGF and promoting an invasive tumor phenotype [62, 63]. The role of HGF/MET signaling in tumor angiogenesis continues to be a topic of intense investigation because better understanding could facilitate the development of MET-targeted therapies [59].
RET
The rearranged during transfection (RET) proto-oncogene encodes an RTK that is required for many biological processes, including normal development, maturation, and maintenance of several tissues and cell types [64]. When mutated, RET is associated with the growth, maintenance, and progression of several human cancers, including thyroid carcinoma, lung adenocarcinoma, chronic myelomonocytic leukemia, pancreatic cancer, breast cancer, acute myeloid leukemia, and colon carcinoma [64, 65]. Although a direct role in tumor angiogenesis and growth is not completely understood, RET appears to act in a tissue-specific manner by promoting tumor-associated inflammation and recruitment of proinflammatory mediators to stimulate tumor angiogenesis [64]. Furthermore, clinical studies of small-molecule tyrosine kinase inhibitors (TKIs) showed that inhibition of VEGFR-2 and epidermal growth factor receptor (EGFR) or MET also inhibits RET activity, suggesting that the effects of RET can occur, at least in part, through interaction among these pathways [66–68].
Overcoming Resistance to Antiangiogenic Agents: Targeting Multiple Angiogenic Signaling Pathways
Despite the efficacy that anti-VEGF/VEGFR targeted treatments can potentially provide, it is now apparent that many patients are intrinsically refractory or develop resistance to existing antiangiogenic agents that principally target VEGF-A or -B and VEGFR-2. Antiangiogenic resistance is most easily explained by the presence and utilization of various redundant and compensatory proangiogenic signaling pathways to recruit vasculature [69–71]. In support of this hypothesis, studies show that PlGF may mediate resistance by promoting proangiogenic signals when VEGF-A is blocked. In a phase II trial of FOLFIRI and bevacizumab in patients with previously treated mCRC, plasma levels of VEGF-C, VEGF-D, and PlGF were significantly elevated before or at the time of disease progression, suggesting that the increased levels of these proangiogenic factors may compensate for the anti-VEGF-A effects of bevacizumab [72]. Similar findings were reported in a study of bevacizumab-treated patients with CRC that demonstrated that those patients eventually developed increased levels of PlGF and VEGF-D, which coincided with resumption of angiogenesis [73]. Studies also show that, in the absence of VEGF-A activity, binding of VEGF-C and -D to VEGFR-2 and -3 may be sufficient to promote angiogenesis and tumor progression, which highlights another potential compensatory angiogenic mechanism in bevacizumab-treated patients [74].
Similarly, the suggestion of a role for FGF and PDGF signaling in the development of anti-VEGF resistance is borne out by clinical observations showing that increased plasma levels of FGF and PDGF precede disease progression in patients receiving bevacizumab chemotherapy [72]. FGF and PDGF are among the better-characterized proangiogenic pathways implicated in anti-VEGF resistance [55, 75–78]. Indeed, the VEGF, FGF, and PDGF signaling pathways appear to be closely integrated, as shown by data suggesting their redundancy and/or synergy in angiogenesis. FGF-dependent revascularization, for example, has been reported in anti-VEGF-resistant patients who have pancreatic tumors or recurrent glioblastoma [55, 79]. Similar findings are reported for PDGF signaling, with PDGFR expression found to be increased in a pancreatic cancer model that is resistant to VEGFR inhibition; combined targeting of VEGF and PDGF signaling induced regression of established tumor blood supply and inhibited tumor growth [77, 78].
Although data highlight the importance of VEGF signaling, it seems likely that, given the many intracellular pathways that influence tumorigenesis, treatments targeting this pathway alone may be less effective compared with multitargeting agents. A multitargeted approach to treatment is believed to limit the development of resistance and maximize antitumor efficacy [70].
Current and Emerging Multitargeting Antiangiogenic Agents
Antiangiogenic treatments that target multiple signaling pathways simultaneously have been and continue to be developed in the hope of increasing antitumor efficacy (Fig. 2). Several currently available cancer treatments aim to inhibit VEGF, FGF, PDGF, and/or other angiogenesis signaling pathways (Table 1) and have been approved across different cancer indications for a number of years. These agents include sorafenib, sunitinib, axitinib, and pazopanib. Sorafenib, sunitinib, and axitinib are small-molecule TKIs that simultaneously inhibit the VEGF and PDGF pathways and target other signaling pathways, whereas pazopanib inhibits the VEGF, PDGF, FGF, and other pathways [6, 83, 85, 86]. Preclinical studies suggest that the antitumor and antiangiogenic effects of these agents occur in part through activation of endothelial cell apoptosis, decreased vessel permeability, and reduced blood flow [80, 104–110].
Table 1.
Currently available multitargeting antiangiogenics
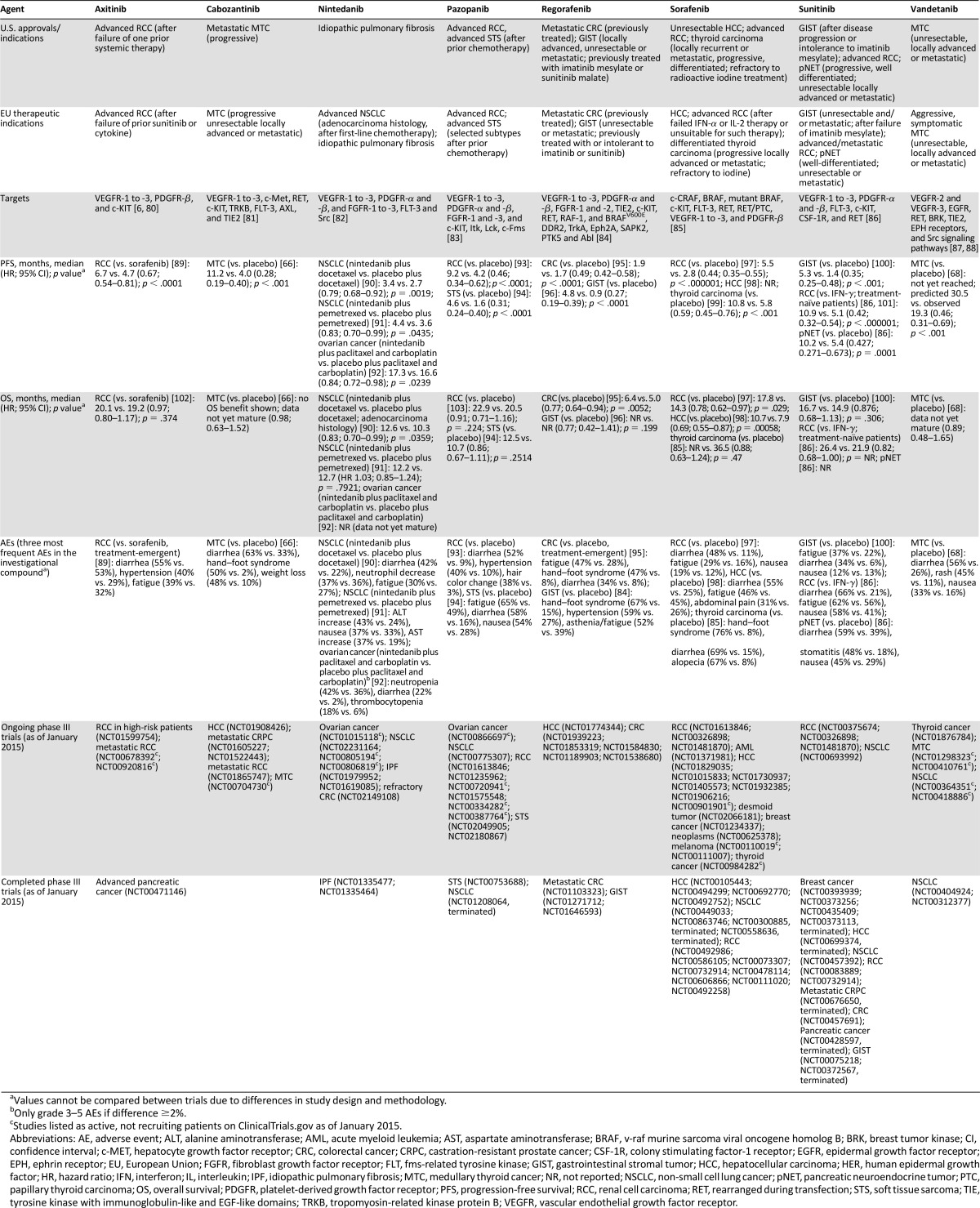
For some of the more recently approved agents, such as cabozanitinib and vandetanib, their efficacy may also be attributed in part to their effects on other tumor growth signaling pathways and mechanisms. Cabozantinib targets the VEGFR family as well as c-Met, RET, c-KIT, TRKB, FLT-3, AXL, and TIE2 [66, 67], whereas vandetanib targets the VEGFR family and EGFR in addition to RET, BRK, TIE2, and EPH receptors and Src signaling [87]. Cabozantinib and vandetanib are both approved for advanced/metastatic medullary thyroid cancer, a relatively rare malignancy [66]. OS data are not yet available, but both compounds have demonstrated significant improvements in PFS versus placebo in this population [66, 68].
Regorafenib is a potent TKI with activity against VEGFR-1 to -3, PDGFR-α and -β, FGFR-1 and -2, TIE2, c-KIT, RET, RAF-1, and BRAF. Regorafenib was approved by the FDA in 2012 for the second-line treatment of patients with mCRC who have been previously treated with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy, with anti-VEGF therapy and, if KRAS wild type, with anti-EGFR therapy [83]. Clinical studies show that regorafenib significantly reduces tumor vascularity, delays tumor growth, and prevents metastasis in colon cancer models, supporting its use as an antiangiogenic treatment for CRC [111, 112].
In November 2014, the multitargeting antiangiogenic agent nintedanib was granted marketing authorization in the European Union for use in combination with docetaxel to treat locally advanced, metastatic or locally recurrent NSCLC of adenocarcinoma histology after first-line chemotherapy [113]. This approval was based on the LUME-Lung 1 trial, which demonstrated a significant improvement in OS to more than 1 year in patients with lung adenocarcinoma treated with nintedanib plus docetaxel versus docetaxel alone [90]. Phase I and II clinical studies initially demonstrated beneficial clinical effects with nintedanib monotherapy in advanced HCC, RCC, and CRC, and in addition to standard chemotherapy combination regimens in various tumor types, including prostate cancer and gynecologic malignancies [114–122]. Encouraging PFS data in OC have been reported (Table 2), and OS data for this indication are awaited [92]. Nintedanib is an orally available angiokinase inhibitor of VEGFR-1 to -3, PDGFR-α and -β, and FGFR-1 to -3, in addition to FLT-3 and Src [82, 114, 138]. Human tumor model studies show that nintedanib can reduce vessel density, vessel integrity, and tumor growth via effects on endothelial and smooth muscle cells, pericytes, and tumor cells [82].
Table 2.
Investigational antiangiogenic agents in phase III development for any indicationa
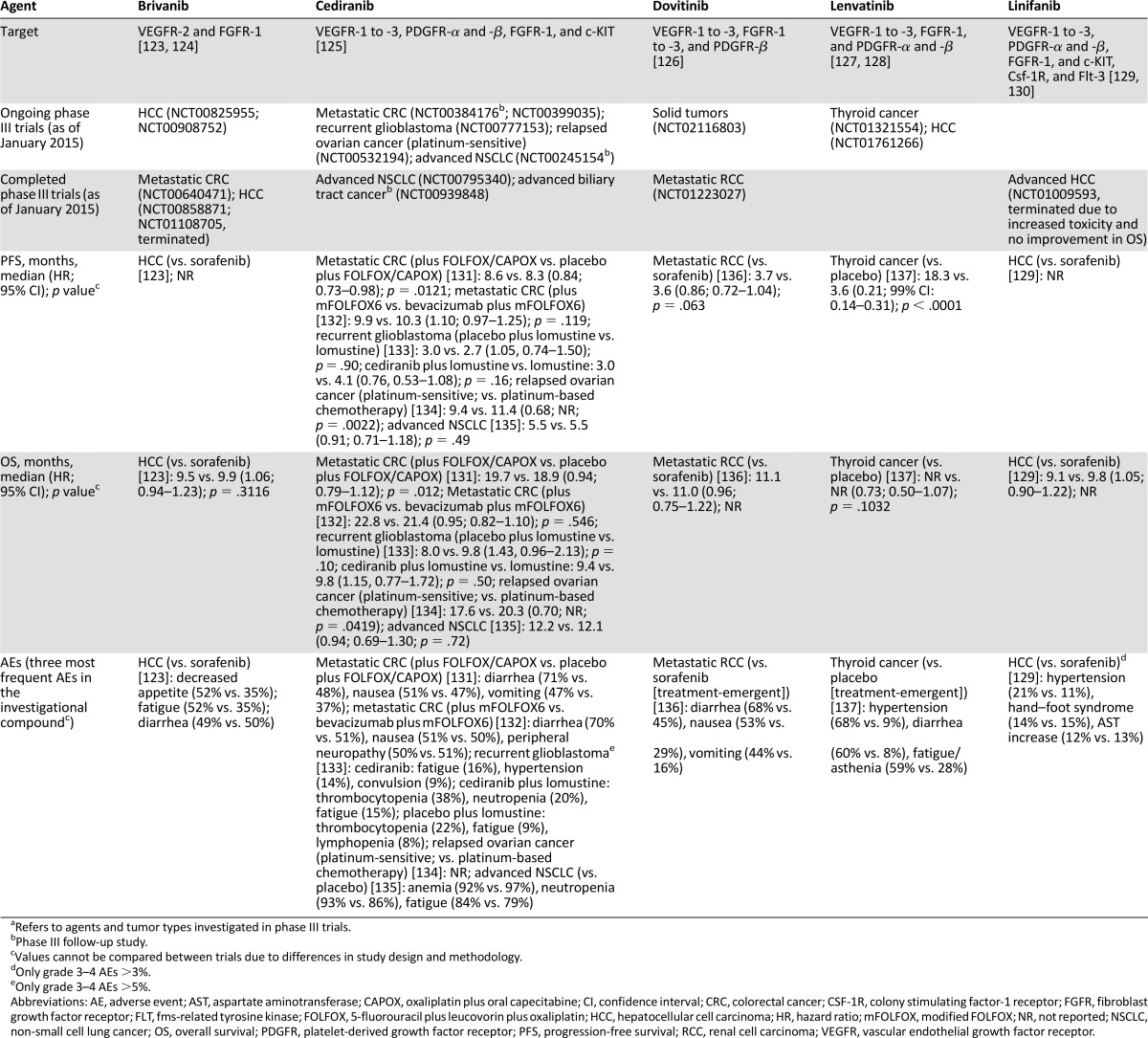
Several other multitargeting antiangiogenic agents are currently in late-stage clinical trials or are under review for approval. These include cediranib, dovitinib, linifanib, brivanib, and lenvatinib (Table 2), which are all investigational antiangiogenic agents that inhibit various VEGFR, FGFR, and PDGFR family members and associated aspects of angiogenesis. Of these agents, cediranib is perhaps the furthest advanced in development, with completed or ongoing phase III trials for a number of indications, including CRC, NSCLC, ovarian cancer, glioblastoma, and biliary tract cancer. Most recently, in 2013, initial results from the ICON-6 trial of relapsed platinum-sensitive ovarian cancer showed significant improvement in PFS when cediranib was given concurrently with platinum-based chemotherapy versus chemotherapy alone and a positive effect on both PFS and OS when continued as a maintenance therapy [134]. Despite initial signs of activity as a monotherapy or in combination with chemotherapy in advanced tumors, cediranib has not yet been approved for use [139], and phase III clinical trials continue to evaluate the utility of this drug in combination with chemotherapy.
The investigational agent dovitinib, which targets FGFR as well as VEGFR and PDGFR, is being evaluated in RCC and has reached phase III development. In phase I trials, although dovitinib demonstrated activity in heavily pretreated patients [126], it was not shown to be superior to sorafenib in the third-line setting in patients with RCC who had progressed during treatment with previous VEGF-targeted therapies and mammalian target of rapamycin inhibitors [136]. RCC is a highly vascularized tumor, which is often due to von Hippel-Lindau gene mutations that drive proangiogenic signaling pathways [140]. The antiangiogenic agents axitinib and pazopanib have demonstrated significant antitumor activity and have been approved in advanced RCC as second-line therapy (Table 1).
Lenvatinib and linifanib, which target VEGFR, FGFR, and PDGFR, and brivanib, which targets VEGFR and FGFR, have reached phase III development in HCC. Hypervascularization is a key characteristic of HCC disease progression [141], making this an attractive indication for the investigation of antiangiogenic agents. Indeed, sorafenib has been approved for this indication for a number of years [85]; however, resistance to sorafenib has been observed [141, 142] and has led to the investigation of other antiangiogenic agents. The development of both linifanib and brivanib, however, appears to have faltered. Neither compound has demonstrated superiority or noninferiority to sorafenib (in terms of OS), and this is coupled with increased toxicity in first-line treatment [123, 129] and the termination of phase III HCC trials for both compounds (Table 2). The efficacy and safety of antiangiogenic agents in HCC continues to be debated [141].
Discussion
Angiogenesis is a complex mechanism that depends on the tumor type. Indications including RCC, HCC, NSCLC, and OC, which are considered to be highly vascularized tumors, have been the focus of development for antiangiogenic agents. Several antiangiogenic agents are approved for these indications. Because multiple therapeutic options are available now, most patients on clinical trials receive additional lines of therapy when their tumors progress, thus it has been felt that the classic survival endpoint for approving novel compounds by the FDA may not be ideal. There is a push to move to PFS as an endpoint for approval by the FDA. Although this endpoint has not been fully adopted, there is evidence that the agency is moving in this direction.
It is interesting that few agents are continuing to be studied in solid tumors such as breast cancer, which are considered to be less vascularized (Tables 1, 2). To date, the efficacy of antiangiogenic agents such as bevacizumab, sorafenib, and ramucirumab in breast cancer has been very variable [17]. The variability in response to antiangiogenic therapy in breast cancer is most likely explained by the extent of vascularization in this tumor type, the highly heterogeneous nature of the disease, the development of drug resistance, and the utilization of compensatory angiogenic mechanisms [17]. Recent results with ramucirumab plus docetaxel in the ROSE/TRIO trial in advanced HER2-negative breast cancer were disappointing [34]. No meaningful improvement in important clinical outcomes such as OS versus docetaxel alone were observed, and there was significantly more toxicity [34]. However, results from the TANIA and IMELDA trials in this indication demonstrated that continued second-line treatment with bevacizumab plus chemotherapy significantly improved PFS compared with bevacizumab alone [143, 144]. It is clear that the use of antiangiogenic therapy in breast cancer remains to be fully evaluated [17].
High variability in patient response to antiangiogenic therapy across different indications exists, and this is coupled with the development of therapy resistance [145]. As with other targeted compounds, a biomarker to identify patients with cancer who will benefit from antiangiogenic therapy is still needed. One of the main challenges in identifying potential biomarkers for antiangiogenic therapy is the complex nature of the angiogenic signaling process, which is characterized by multiple pathways that not only overlap but that continuously cross-talk, making it difficult to eliminate an angiogenic stimulus [146]. Several possible types of biomarkers are being investigated across different indications: circulating biomarkers (e.g., concentrations of soluble angiogenic receptor ligands), genetic biomarkers (e.g., single nucleotide polymorphisms), tissue biomarkers (e.g., immunohistochemical staining of angiogenic receptors); and physiologic biomarkers (e.g., hypertension) [145]. However, the reproducibility of candidate biomarkers across indications is limited, and there is a paucity of studies omparing the same biomarkers for the same indication. The use of genomic and proteomic technologies will be key in improving our ability to match a target pathology with antiangiogenic therapy [17].
Conclusion
The focus of new and emerging antiangiogenic therapies is the simultaneous disruption of multiple signaling pathways. It is hoped that by using multitargeting, tumors will be less able to overcome the antiangiogenic and antitumor effects. Indeed, results from various clinical trials have already demonstrated the benefits of some multitargeting antiangiogenic agents in different tumor types.
Acknowledgments
Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Duncan Campbell of GeoMed and Christopher Ontiveros of inVentiv Medical Communications during the preparation of the manuscript.
Author Contributions
Conception/Design: Yujie Zhao, Alex A. Adjei
Data analysis and interpretation: Yujie Zhao, Alex A. Adjei
Manuscript writing: Yujie Zhao, Alex A. Adjei
Final approval of manuscript: Yujie Zhao, Alex A. Adjei
Disclosures
The authors indicated no financial relationships.
References
- 1.Folkman J. Angiogenesis: An organizing principle for drug discovery? Nature Rev Drug Discov . 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 2.Kieran MW, Kalluri R, Cho YJ. The VEGF pathway in cancer and disease: Responses, resistance, and the path forward. Cold Spring Harb Perspect Med. 2012;2:a006593. doi: 10.1101/cshperspect.a006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23:1011–1027. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 4.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avastin [prescribing information]. South San Francisco, CA: Genentech; 2014.
- 6.Inlyta [package insert]. New York, NY: Pfizer, Inc.; 2013.
- 7.Cyramza [package insert]. Indianapolis, IN: Eli Lilly and Company; 2014.
- 8.Leung DW, Cachianes G, Kuang WJ, et al. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 9.Tischer E, Gospodarowicz D, Mitchell R, et al. Vascular endothelial growth factor: A new member of the platelet-derived growth factor gene family. Biochem Biophys Res Commun. 1989;165:1198–1206. doi: 10.1016/0006-291x(89)92729-0. [DOI] [PubMed] [Google Scholar]
- 10.Cao Y. Positive and negative modulation of angiogenesis by VEGFR1 ligands. Sci Signal. 2009;2:re1. doi: 10.1126/scisignal.259re1. [DOI] [PubMed] [Google Scholar]
- 11.Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: A critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368–4380. doi: 10.1200/JCO.2002.10.088. [DOI] [PubMed] [Google Scholar]
- 12.Fischer C, Mazzone M, Jonckx B, et al. FLT1 and its ligands VEGFB and PlGF: Drug targets for anti-angiogenic therapy? Nat Rev Cancer. 2008;8:942–956. doi: 10.1038/nrc2524. [DOI] [PubMed] [Google Scholar]
- 13.De Falco S. The discovery of placenta growth factor and its biological activity. Exp Mol Med. 2012;44:1–9. doi: 10.3858/emm.2012.44.1.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tammela T, Alitalo K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. 2010;140:460–476. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 15.Ferrara N, Hillan KJ, Gerber HP, et al. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 16.FDA commissioner announces Avastin decision. Available at http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm280536.htm. Accessed May 5, 2015.
- 17.Kristensen TB, Knutsson ML, Wehland M, et al. Anti-vascular endothelial growth factor therapy in breast cancer. Int J Mol Sci. 2014;15:23024–23041. doi: 10.3390/ijms151223024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mortimer J, Zonder HB, Pal SK. Lessons learned from the bevacizumab experience. Cancer Contr. 2012;19:309–316. doi: 10.1177/107327481201900407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shojaei F. Anti-angiogenesis therapy in cancer: Current challenges and future perspectives. Cancer Lett. 2012;320:130–137. doi: 10.1016/j.canlet.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 20.Mancuso MR, Davis R, Norberg SM, et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest. 2006;116:2610–2621. doi: 10.1172/JCI24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pàez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen HX, Cleck JN. Adverse effects of anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol. 2009;6:465–477. doi: 10.1038/nrclinonc.2009.94. [DOI] [PubMed] [Google Scholar]
- 23.Loupakis F, Cremolini C, Fioravanti A, et al. Pharmacodynamic and pharmacogenetic angiogenesis-related markers of first-line FOLFOXIRI plus bevacizumab schedule in metastatic colorectal cancer. Br J Cancer. 2011;104:1262–1269. doi: 10.1038/bjc.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gressett SM, Shah SR. Intricacies of bevacizumab-induced toxicities and their management. Ann Pharmacother. 2009;43:490–501. doi: 10.1345/aph.1L426. [DOI] [PubMed] [Google Scholar]
- 25.Holash J, Davis S, Papadopoulos N, et al. VEGF-Trap: A VEGF blocker with potent antitumor effects. Proc Natl Acad Sci USA. 2002;99:11393–11398. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomez-Manzano C, Holash J, Fueyo J, et al. VEGF Trap induces antiglioma effect at different stages of disease. Neuro Oncol. 2008;10:940–945. doi: 10.1215/15228517-2008-061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim ES, Serur A, Huang J, et al. Potent VEGF blockade causes regression of coopted vessels in a model of neuroblastoma. Proc Natl Acad Sci USA. 2002;99:11399–11404. doi: 10.1073/pnas.172398399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Cutsem E, Tabernero J, Lakomy R, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol. 2012;30:3499–3506. doi: 10.1200/JCO.2012.42.8201. [DOI] [PubMed] [Google Scholar]
- 29.Fuchs CS, Tomasek J, Yong CJ, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2014;383:31–39. doi: 10.1016/S0140-6736(13)61719-5. [DOI] [PubMed] [Google Scholar]
- 30.Wilke H, Muro K, Van Cutsem E, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014;15:1224–1235. doi: 10.1016/S1470-2045(14)70420-6. [DOI] [PubMed] [Google Scholar]
- 31.Spratlin J. Ramucirumab (IMC-1121B): Monoclonal antibody inhibition of vascular endothelial growth factor receptor-2. Curr Oncol Rep. 2011;13:97–102. doi: 10.1007/s11912-010-0149-5. [DOI] [PubMed] [Google Scholar]
- 32.Aprile G, Bonotto M, Ongaro E, et al. Critical appraisal of ramucirumab (IMC-1121B) for cancer treatment: From benchside to clinical use. Drugs. 2013;73:2003–2015. doi: 10.1007/s40265-013-0154-8. [DOI] [PubMed] [Google Scholar]
- 33.Garon EB, Ciuleanu T-E, Arrieta O, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): A multicentre, double-blind, randomised phase 3 trial. Lancet. 2014;384:665–673. doi: 10.1016/S0140-6736(14)60845-X. [DOI] [PubMed] [Google Scholar]
- 34.Mackey JR, Ramos-Vazquez M, Lipatov O, et al. Primary results of ROSE/TRIO-12, a randomized placebo-controlled phase III trial evaluating the addition of ramucirumab to first-line docetaxel chemotherapy in metastatic breast cancer. J Clin Oncol. 2015;33:141–148. doi: 10.1200/JCO.2014.57.1513. [DOI] [PubMed] [Google Scholar]
- 35.Zhu AX, Ryoo BY, Yen CJ, et al. Ramucirumab (RAM) as a second-line treatment in patients (PTS) with advanced hepatocellular carcinoma (HCC) following first-line therapy with sorafenib: Results from the randomized phase III REACH study. Ann Oncol. 2014;25(suppl 5):LBA16a. [Google Scholar]
- 36.Ferrara N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 2010;21:21–26. doi: 10.1016/j.cytogfr.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 37.Heldin CH. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun Signal. 2013;11:97. doi: 10.1186/1478-811X-11-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu E, Palmer N, Tian Z, et al. Comprehensive dissection of PDGF-PDGFR signaling pathways in PDGFR genetically defined cells. PLoS One. 2008;3:e3794. doi: 10.1371/journal.pone.0003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol Med. 2013;19:460–473. doi: 10.1016/j.molmed.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 40.Levitzki A. PDGF receptor kinase inhibitors for the treatment of PDGF driven diseases. Cytokine Growth Factor Rev. 2004;15:229–235. doi: 10.1016/j.cytogfr.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 41.Tejada ML, Yu L, Dong J, et al. Tumor-driven paracrine platelet-derived growth factor receptor alpha signaling is a key determinant of stromal cell recruitment in a model of human lung carcinoma. Clin Cancer Res. 2006;12:2676–2688. doi: 10.1158/1078-0432.CCR-05-1770. [DOI] [PubMed] [Google Scholar]
- 42.Ding W, Knox TR, Tschumper RC, et al. Platelet-derived growth factor (PDGF)-PDGF receptor interaction activates bone marrow-derived mesenchymal stromal cells derived from chronic lymphocytic leukemia: Implications for an angiogenic switch. Blood. 2010;116:2984–2993. doi: 10.1182/blood-2010-02-269894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cao R, Björndahl MA, Religa P, et al. PDGF-BB induces intratumoral lymphangiogenesis and promotes lymphatic metastasis. Cancer Cell. 2004;6:333–345. doi: 10.1016/j.ccr.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 44.Kodama M, Kitadai Y, Sumida T, et al. Expression of platelet-derived growth factor (PDGF)-B and PDGF-receptor β is associated with lymphatic metastasis in human gastric carcinoma. Cancer Sci. 2010;101:1984–1989. doi: 10.1111/j.1349-7006.2010.01639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xue Y, Lim S, Yang Y, et al. PDGF-BB modulates hematopoiesis and tumor angiogenesis by inducing erythropoietin production in stromal cells. Nat Med. 2012;18:100–110. doi: 10.1038/nm.2575. [DOI] [PubMed] [Google Scholar]
- 46.Lu C, Shahzad MM, Moreno-Smith M, et al. Targeting pericytes with a PDGF-B aptamer in human ovarian carcinoma models. Cancer Biol Ther. 2010;9:176–182. doi: 10.4161/cbt.9.3.10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beenken A, Mohammadi M. The FGF family: Biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8:235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turner N, Pearson A, Sharpe R, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70:2085–2094. doi: 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao Y, Cao R, Hedlund EM. R Regulation of tumor angiogenesis and metastasis by FGF and PDGF signaling pathways. J Mol Med (Berl) 2008;86:785–789. doi: 10.1007/s00109-008-0337-z. [DOI] [PubMed] [Google Scholar]
- 50.Javerzat S, Auguste P, Bikfalvi A. The role of fibroblast growth factors in vascular development. Trends Mol Med. 2002;8:483–489. doi: 10.1016/s1471-4914(02)02394-8. [DOI] [PubMed] [Google Scholar]
- 51.Presta M, Dell’Era P, Mitola S, et al. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–178. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 52.Cao R, Ji H, Feng N, et al. Collaborative interplay between FGF-2 and VEGF-C promotes lymphangiogenesis and metastasis. Proc Natl Acad Sci USA. 2012;109:15894–15899. doi: 10.1073/pnas.1208324109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang H, Bhat A, Woodnutt G, et al. Targeting the ANGPT-TIE2 pathway in malignancy. Nat Rev Cancer. 2010;10:575–585. doi: 10.1038/nrc2894. [DOI] [PubMed] [Google Scholar]
- 54.Giavazzi R, Sennino B, Coltrini D, et al. Distinct role of fibroblast growth factor-2 and vascular endothelial growth factor on tumor growth and angiogenesis. Am J Pathol. 2003;162:1913–1926. doi: 10.1016/S0002-9440(10)64325-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Casanovas O, Hicklin DJ, Bergers G, et al. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 56.Cascone T, Heymach JV. Targeting the angiopoietin/Tie2 pathway: Cutting tumor vessels with a double-edged sword? J Clin Oncol. 2012;30:441–444. doi: 10.1200/JCO.2011.38.7621. [DOI] [PubMed] [Google Scholar]
- 57.Tait CR, Jones PF. Angiopoietins in tumours: The angiogenic switch. J Pathol. 2004;204:1–10. doi: 10.1002/path.1618. [DOI] [PubMed] [Google Scholar]
- 58.Gerald D, Chintharlapalli S, Augustin HG, et al. Angiopoietin-2: An attractive target for improved antiangiogenic tumor therapy. Cancer Res. 2013;73:1649–1657. doi: 10.1158/0008-5472.CAN-12-4697. [DOI] [PubMed] [Google Scholar]
- 59.Graveel CR, Tolbert D, Vande Woude GF. MET: A critical player in tumorigenesis and therapeutic target. Cold Spring Harb Perspect Biol. 2013;5:a009209. doi: 10.1101/cshperspect.a009209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.You WK, McDonald DM. The hepatocyte growth factor/c-Met signaling pathway as a therapeutic target to inhibit angiogenesis. BMB Rep. 2008;41:833–839. doi: 10.5483/bmbrep.2008.41.12.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wojta J, Kaun C, Breuss JM, et al. Hepatocyte growth factor increases expression of vascular endothelial growth factor and plasminogen activator inhibitor-1 in human keratinocytes and the vascular endothelial growth factor receptor flk-1 in human endothelial cells. Lab Invest. 1999;79:427–438. [PubMed] [Google Scholar]
- 62.Lu KV, Chang JP, Parachoniak CA, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. 2012;22:21–35. doi: 10.1016/j.ccr.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jahangiri A, De Lay M, Miller LM, et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin Cancer Res. 2013;19:1773–1783. doi: 10.1158/1078-0432.CCR-12-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mulligan LM. RET revisited: Expanding the oncogenic portfolio. Nat Rev Cancer. 2014;14:173–186. doi: 10.1038/nrc3680. [DOI] [PubMed] [Google Scholar]
- 65.Ibáñez CF. Structure and physiology of the RET receptor tyrosine kinase. Cold Spring Harb Perspect Biol. 2013;5:a009134. doi: 10.1101/cshperspect.a009134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Elisei R, Schlumberger MJ, Müller SP, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31:3639–3646. doi: 10.1200/JCO.2012.48.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kurzrock R, Sherman SI, Ball DW, et al. Activity of XL184 (cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol. 2011;29:2660–2666. doi: 10.1200/JCO.2010.32.4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wells SA, Jr, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134–141. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crinò L, Metro G. Therapeutic options targeting angiogenesis in nonsmall cell lung cancer. Eur Respir Rev. 2014;23:79–91. doi: 10.1183/09059180.00008913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ballas MS, Chachoua A. Rationale for targeting VEGF, FGF, and PDGF for the treatment of NSCLC. Onco Targets Ther. 2011;4:43–58. doi: 10.2147/OTT.S18155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Welti J, Loges S, Dimmeler S, et al. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest. 2013;123:3190–3200. doi: 10.1172/JCI70212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kopetz S, Hoff PM, Morris JS, et al. Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: Efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J Clin Oncol. 2010;28:453–459. doi: 10.1200/JCO.2009.24.8252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lieu CH, Tran H, Jiang ZQ, et al. The association of alternate VEGF ligands with resistance to anti-VEGF therapy in metastatic colorectal cancer. PLoS One. 2013;8:e77117. doi: 10.1371/journal.pone.0077117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Clarke JM, Hurwitz HI. Understanding and targeting resistance to anti-angiogenic therapies. J Gastrointest Oncol. 2013;4:253–263. doi: 10.3978/j.issn.2078-6891.2013.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ebos JM, Kerbel RS. Antiangiogenic therapy: Impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8:210–221. doi: 10.1038/nrclinonc.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Loges S, Mazzone M, Hohensinner P, et al. Silencing or fueling metastasis with VEGF inhibitors: Antiangiogenesis revisited. Cancer Cell. 2009;15:167–170. doi: 10.1016/j.ccr.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 77.Bergers G, Song S, Meyer-Morse N, et al. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Erber R, Thurnher A, Katsen AD, et al. Combined inhibition of VEGF and PDGF signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. FASEB J. 2004;18:338–340. doi: 10.1096/fj.03-0271fje. [DOI] [PubMed] [Google Scholar]
- 79.Batchelor TT, Sorensen AG, di Tomaso E, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilmes LJ, Pallavicini MG, Fleming LM, et al. AG-013736, a novel inhibitor of VEGF receptor tyrosine kinases, inhibits breast cancer growth and decreases vascular permeability as detected by dynamic contrast-enhanced magnetic resonance imaging. Magn Reson Imaging. 2007;25:319–327. doi: 10.1016/j.mri.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 81.Cometriq [package insert]. South San Francisco, CA; Exelixis, Inc. 2012.
- 82.Hilberg F, Roth GJ, Krssak M, et al. BIBF 1120: Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68:4774–4782. doi: 10.1158/0008-5472.CAN-07-6307. [DOI] [PubMed] [Google Scholar]
- 83.Votrient [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2013.
- 84.Stivarga [package insert]. Wayne, NJ: Bayer HealthCare Pharmaceuticals, Inc.; 2013.
- 85.Nexavar [package insert]. South San Francisco, CA: Bayer HealthCare Pharmaceuticals, Inc.; 2013.
- 86.Sutent [package insert]. New York, NY: Pfizer; 2013.
- 87.Caprelsa [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2013.
- 88.Chau NG, Haddad RI. Vandetanib for the treatment of medullary thyroid cancer. Clin Cancer Res. 2013;19:524–529. doi: 10.1158/1078-0432.CCR-12-2353. [DOI] [PubMed] [Google Scholar]
- 89.Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): A randomised phase 3 trial. Lancet. 2011;378:1931–1939. doi: 10.1016/S0140-6736(11)61613-9. [DOI] [PubMed] [Google Scholar]
- 90.Reck M, Kaiser R, Mellemgaard A, et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): A phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014;15:143–155. doi: 10.1016/S1470-2045(13)70586-2. [DOI] [PubMed] [Google Scholar]
- 91.Hanna NH, Kaiser R, Sullivan RN, et al. LUME-lung 2: A multicenter, randomized, double-blind, phase III study of nintedanib plus pemetrexed versus placebo plus pemetrexed in patients with advanced nonsquamous non-small cell lung cancer (NSCLC) after failure of first-line chemotherapy. J Clin Oncol. 2013;31:8034a. [Google Scholar]
- 92.Du Bois A, Kristensen G, Ray-Coquard I, et al. AGO-OVAR 12: A randomized placebo-controlled GCIG/ENGOT-intergroup phase III trial of standard frontline chemotherapy +/− nintedanib for advanced ovarian cancer. Int J Gynecol Cancer. 2013;23(suppl 1):7. [Google Scholar]
- 93.Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 94.van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379:1879–1886. doi: 10.1016/S0140-6736(12)60651-5. [DOI] [PubMed] [Google Scholar]
- 95.Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:303–312. doi: 10.1016/S0140-6736(12)61900-X. [DOI] [PubMed] [Google Scholar]
- 96.Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295–302. doi: 10.1016/S0140-6736(12)61857-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Escudier B, Eisen T, Stadler WM, et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol. 2009;27:3312–3318. doi: 10.1200/JCO.2008.19.5511. [DOI] [PubMed] [Google Scholar]
- 98.Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 99.Brose MS, Nutting CM, Jarzab B, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet. 2014;384:319–328. doi: 10.1016/S0140-6736(14)60421-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Demetri GD, Garrett CR, Schöffski P, et al. Complete longitudinal analyses of the randomized, placebo-controlled, phase III trial of sunitinib in patients with gastrointestinal stromal tumor following imatinib failure. Clin Cancer Res. 2012;18:3170–3179. doi: 10.1158/1078-0432.CCR-11-3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 102.Motzer RJ, Escudier B, Tomczak P, et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: Overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013;14:552–562. doi: 10.1016/S1470-2045(13)70093-7. [DOI] [PubMed] [Google Scholar]
- 103.Sternberg CN, Hawkins RE, Wagstaff J, et al. A randomised, double-blind phase III study of pazopanib in patients with advanced and/or metastatic renal cell carcinoma: Final overall survival results and safety update. Eur J Cancer. 2013;49:1287–1296. doi: 10.1016/j.ejca.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 104.Griffioen AW, Mans LA, de Graaf AM, et al. Rapid angiogenesis onset after discontinuation of sunitinib treatment of renal cell carcinoma patients. Clin Cancer Res. 2012;18:3961–3971. doi: 10.1158/1078-0432.CCR-12-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Majumder S, Piguet AC, Dufour JF, et al. Study of the cellular mechanism of sunitinib mediated inactivation of activated hepatic stellate cells and its implications in angiogenesis. Eur J Pharmacol. 2013;705:86–95. doi: 10.1016/j.ejphar.2013.02.026. [DOI] [PubMed] [Google Scholar]
- 106.Olson P, Chu GC, Perry SR, et al. Imaging guided trials of the angiogenesis inhibitor sunitinib in mouse models predict efficacy in pancreatic neuroendocrine but not ductal carcinoma. Proc Natl Acad Sci USA. 2011;108:E1275–E1284. doi: 10.1073/pnas.1111079108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chang YS, Adnane J, Trail PA, et al. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother Pharmacol. 2007;59:561–574. doi: 10.1007/s00280-006-0393-4. [DOI] [PubMed] [Google Scholar]
- 108.Kim S, Yazici YD, Calzada G, et al. Sorafenib inhibits the angiogenesis and growth of orthotopic anaplastic thyroid carcinoma xenografts in nude mice. Mol Cancer Ther. 2007;6:1785–1792. doi: 10.1158/1535-7163.MCT-06-0595. [DOI] [PubMed] [Google Scholar]
- 109.Liu L, Cao Y, Chen C, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–11858. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- 110.Hamberg P, Verweij J, Sleijfer S. (Pre-)clinical pharmacology and activity of pazopanib, a novel multikinase angiogenesis inhibitor. The Oncologist. 2010;15:539–547. doi: 10.1634/theoncologist.2009-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Abou-Elkacem L, Arns S, Brix G, et al. Regorafenib inhibits growth, angiogenesis, and metastasis in a highly aggressive, orthotopic colon cancer model. Mol Cancer Ther. 2013;12:1322–1331. doi: 10.1158/1535-7163.MCT-12-1162. [DOI] [PubMed] [Google Scholar]
- 112.Cyran CC, Kazmierczak PM, Hirner H, et al. Regorafenib effects on human colon carcinoma xenografts monitored by dynamic contrast-enhanced computed tomography with immunohistochemical validation. PLoS One. 2013;8:e76009. doi: 10.1371/journal.pone.0076009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.CHMP summary of positive opinion for Vargatef. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion_-_Initial_authorisation/human/002569/WC500173607.pdf. Accessed May 5, 2015.
- 114.Mross K, Stefanic M, Gmehling D, et al. Phase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumors. Clin Cancer Res. 2010;16:311–319. doi: 10.1158/1078-0432.CCR-09-0694. [DOI] [PubMed] [Google Scholar]
- 115.Reck M. BIBF 1120 for the treatment of non-small cell lung cancer. Expert Opin Investig Drugs. 2010;19:789–794. doi: 10.1517/13543784.2010.488220. [DOI] [PubMed] [Google Scholar]
- 116.Okamoto I, Kaneda H, Satoh T, et al. Phase I safety, pharmacokinetic, and biomarker study of BIBF 1120, an oral triple tyrosine kinase inhibitor in patients with advanced solid tumors. Mol Cancer Ther. 2010;9:2825–2833. doi: 10.1158/1535-7163.MCT-10-0379. [DOI] [PubMed] [Google Scholar]
- 117.Kudo K, Arao T, Tanaka K, et al. Antitumor activity of BIBF 1120, a triple angiokinase inhibitor, and use of VEGFR2+pTyr+ peripheral blood leukocytes as a pharmacodynamic biomarker in vivo. Clin Cancer Res. 2011;17:1373–1381. doi: 10.1158/1078-0432.CCR-09-2755. [DOI] [PubMed] [Google Scholar]
- 118.Eisen T, Shparyk Y, Macleod N, et al. Effect of small angiokinase inhibitor nintedanib (BIBF 1120) on QT interval in patients with previously untreated, advanced renal cell cancer in an open-label, phase II study. Invest New Drugs. 2013;31:1283–1293. doi: 10.1007/s10637-013-9962-7. [DOI] [PubMed] [Google Scholar]
- 119.Doebele RC, Conkling P, Traynor AM, et al. A phase I, open-label dose-escalation study of continuous treatment with BIBF 1120 in combination with paclitaxel and carboplatin as first-line treatment in patients with advanced non-small-cell lung cancer. Ann Oncol. 2012;23:2094–2102. doi: 10.1093/annonc/mdr596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bousquet G, Alexandre J, Le Tourneau C, et al. Phase I study of BIBF 1120 with docetaxel and prednisone in metastatic chemo-naive hormone-refractory prostate cancer patients. Br J Cancer. 2011;105:1640–1645. doi: 10.1038/bjc.2011.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ellis PM, Kaiser R, Zhao Y, et al. Phase I open-label study of continuous treatment with BIBF 1120, a triple angiokinase inhibitor, and pemetrexed in pretreated non-small cell lung cancer patients. Clin Cancer Res. 2010;16:2881–2889. doi: 10.1158/1078-0432.CCR-09-2944. [DOI] [PubMed] [Google Scholar]
- 122.du Bois A, Huober J, Stopfer P, et al. A phase I open-label dose-escalation study of oral BIBF 1120 combined with standard paclitaxel and carboplatin in patients with advanced gynecological malignancies. Ann Oncol. 2010;21:370–375. doi: 10.1093/annonc/mdp506. [DOI] [PubMed] [Google Scholar]
- 123.Johnson PJ, Qin S, Park JW, et al. Brivanib versus sorafenib as first-line therapy in patients with unresectable, advanced hepatocellular carcinoma: Results from the randomized phase III BRISK-FL study. J Clin Oncol. 2013;31:3517–3524. doi: 10.1200/JCO.2012.48.4410. [DOI] [PubMed] [Google Scholar]
- 124.Cai ZW, Zhang Y, Borzilleri RM, et al. Discovery of brivanib alaninate ((S)-((R)-1-(4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-5-methylpyrrolo[2,1-f][1,2,4]triazin-6-yloxy)propan-2-yl)2-aminopropanoate), a novel prodrug of dual vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1 kinase inhibitor (BMS-540215) J Med Chem. 2008;51:1976–1980. doi: 10.1021/jm7013309. [DOI] [PubMed] [Google Scholar]
- 125.Wedge SR, Kendrew J, Hennequin LF, et al. AZD2171: A highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65:4389–4400. doi: 10.1158/0008-5472.CAN-04-4409. [DOI] [PubMed] [Google Scholar]
- 126.Angevin E, Lopez-Martin JA, Lin CC, et al. Phase I study of dovitinib (TKI258), an oral FGFR, VEGFR, and PDGFR inhibitor, in advanced or metastatic renal cell carcinoma. Clin Cancer Res. 2013;19:1257–1268. doi: 10.1158/1078-0432.CCR-12-2885. [DOI] [PubMed] [Google Scholar]
- 127.Matsui J, Funahashi Y, Uenaka T, et al. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res. 2008;14:5459–5465. doi: 10.1158/1078-0432.CCR-07-5270. [DOI] [PubMed] [Google Scholar]
- 128.Matsui J, Yamamoto Y, Funahashi Y, et al. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer. 2008;122:664–671. doi: 10.1002/ijc.23131. [DOI] [PubMed] [Google Scholar]
- 129.Cainap C, Qin S, Huang WT, et al. Linifanib versus sorafenib in patients with advanced hepatocellular carcinoma: Results of a randomized phase III trial. J Clin Oncol. 2015;33:172–179. doi: 10.1200/JCO.2013.54.3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhou J, Goh BC, Albert DH, et al. ABT-869, a promising multi-targeted tyrosine kinase inhibitor: From bench to bedside. J Hematol Oncol. 2009;2:33. doi: 10.1186/1756-8722-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hoff PM, Hochhaus A, Pestalozzi BC, et al. Cediranib plus FOLFOX/CAPOX versus placebo plus FOLFOX/CAPOX in patients with previously untreated metastatic colorectal cancer: A randomized, double-blind, phase III study (HORIZON II) J Clin Oncol. 2012;30:3596–3603. doi: 10.1200/JCO.2012.42.6031. [DOI] [PubMed] [Google Scholar]
- 132.Schmoll HJ, Cunningham D, Sobrero A, et al. Cediranib with mFOLFOX6 versus bevacizumab with mFOLFOX6 as first-line treatment for patients with advanced colorectal cancer: A double-blind, randomized phase III study (HORIZON III) J Clin Oncol. 2012;30:3588–3595. doi: 10.1200/JCO.2012.42.5355. [DOI] [PubMed] [Google Scholar]
- 133.Batchelor TT, Mulholland P, Neyns B, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol. 2013;31:3212–3218. doi: 10.1200/JCO.2012.47.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ledermann JA, Perren TJ, Raja FA et al. Randomised double-blind phase III trial of cediranib (AZD 2171) in relapsed platinum sensitive ovarian cancer: Results of the ICON6 trial [abstract E17-7020]. Presented at: ECCO-ESMO-ESTRO European Cancer Congress; September 27–October 1, 2013; Amsterdam, The Netherlands. [Google Scholar]
- 135.Laurie SA, Solomon BJ, Seymour L, et al. Randomised, double-blind trial of carboplatin and paclitaxel with daily oral cediranib or placebo in patients with advanced non-small cell lung cancer: NCIC Clinical Trials Group study BR29. Eur J Cancer. 2014;50:706–712. doi: 10.1016/j.ejca.2013.11.032. [DOI] [PubMed] [Google Scholar]
- 136.Motzer RJ, Porta C, Vogelzang NJ, et al. Dovitinib versus sorafenib for third-line targeted treatment of patients with metastatic renal cell carcinoma: An open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:286–296. doi: 10.1016/S1470-2045(14)70030-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schlumberger MJ, Tahara M, Wirth LJ, et al. A phase 3, multicenter, double-blind, placebo-controlled trial of lenvatinib (E7080) in patients with 131I-refractory differentiated thyroid cancer (SELECT) J Clin Oncol. 2014;32:LBA6008a. [Google Scholar]
- 138.Stopfer P, Rathgen K, Bischoff D, et al. Pharmacokinetics and metabolism of BIBF 1120 after oral dosing to healthy male volunteers. Xenobiotica. 2011;41:297–311. doi: 10.3109/00498254.2010.545452. [DOI] [PubMed] [Google Scholar]
- 139.Sahade M, Caparelli F, Hoff PM. Cediranib: A VEGF receptor tyrosine kinase inhibitor. Future Oncol. 2012;8:775–781. doi: 10.2217/fon.12.73. [DOI] [PubMed] [Google Scholar]
- 140.Sonpavde G, Willey CD, Sudarshan S. Fibroblast growth factor receptors as therapeutic targets in clear-cell renal cell carcinoma. Expert Opin Investig Drugs. 2014;23:305–315. doi: 10.1517/13543784.2014.871259. [DOI] [PubMed] [Google Scholar]
- 141.Sun H, Zhu MS, Wu WR, et al. Role of anti-angiogenesis therapy in the management of hepatocellular carcinoma: The jury is still out. World J Hepatol. 2014;6:830–835. doi: 10.4254/wjh.v6.i12.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhu AX. New agents on the horizon in hepatocellular carcinoma. Ther Adv Med Oncol. 2013;5:41–50. doi: 10.1177/1758834012458480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gligorov J, Doval D, Bines J, et al. Maintenance capecitabine and bevacizumab versus bevacizumab alone after initial first-line bevacizumab and docetaxel for patients with HER2-negative metastatic breast cancer (IMELDA): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1351–1360. doi: 10.1016/S1470-2045(14)70444-9. [DOI] [PubMed] [Google Scholar]
- 144.von Minckwitz G, Puglisi F, Cortes J, et al. Bevacizumab plus chemotherapy versus chemotherapy alone as second-line treatment for patients with HER2-negative locally recurrent or metastatic breast cancer after first-line treatment with bevacizumab plus chemotherapy (TANIA): An open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:1269–1278. doi: 10.1016/S1470-2045(14)70439-5. [DOI] [PubMed] [Google Scholar]
- 145.Wehland M, Bauer J, Magnusson NE, et al. Biomarkers for anti-angiogenic therapy in cancer. Int J Mol Sci. 2013;14:9338–9364. doi: 10.3390/ijms14059338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pilotto S, Bonomi M, Massari F, et al. Anti-angiogenic drugs and biomarkers in non-small-cell lung cancer: A ‘hard days night’. Curr Pharm Des. 2014;20:3958–3972. doi: 10.2174/13816128113196660757. [DOI] [PubMed] [Google Scholar]