

All biotechnology-derived products (BDPs) with sufficient nonclinical and clinical data were identified to assess the safety of one-sixth highest nonseverely toxic dose (HNSTD) in advanced cancer first-in-human (FIH) trials. Using one-sixth HNSTD might reduce the number of dose escalations needed to reach the recommended dose in FIH studies and thus the number of patients treated at subtherapeutic doses and limit unnecessary exposure to high drug levels.
Keywords: Starting dose, Biotechnology derived products, Animal toxicology studies, Phase I clinical trials, Highest nonseverely toxic dose
Abstract
Background.
First-in-human (FIH) trials of low-molecular-weight anticancer agents conventionally derive a safe start dose (SD) from one-tenth the severely toxic dose in 10% of rodents or one-sixth the highest nonseverely toxic dose (HNSTD) in nonrodent species. No consensus has been reached on whether this paradigm can be safely applied to biotechnology-derived products (BDPs).
Materials and Methods.
A comprehensive search was conducted to identify all BDPs (excluding immune checkpoint inhibitors and antibody drug conjugates) with sufficient nonclinical and clinical data to assess the safety of hypothetical use of one-sixth HNSTD in an advanced cancer FIH trial.
Results.
The search identified 23 BDPs, of which 21 were monoclonal antibodies. The median ratio of the maximum tolerated or maximum administered dose (MTD or MAD) to the actual FIH SD was 36 (range, 8–500). Only 2 BDPs reached the MTD. Hypothetical use of one-sixth HNSTD (allometrically scaled to humans) would not have exceeded the MTD or MAD for all 23 BDPs and would have reduced the median ratio of the MTD or MAD to a SD to 6.1 (range, 3.5–55.3). Pharmacodynamic (PD) markers were included in some animal toxicology studies and were useful to confirm the hypothetical SD of one-sixth HNSTD.
Conclusion.
One-sixth HNSTD would not have resulted in unacceptable toxicities in the data available. Supporting its use could reduce the number of dose escalations needed to reach the recommended dose. A low incidence of toxicities in animals and humans underscores the need to identify the pharmacokinetic and PD parameters to guide SD selection of BDPs for FIH cancer trials.
Implications for Practice:
Start dose (SD) for biotechnology-derived products (BDPs) can be safely derived from one-sixth the highest nonseverely toxic dose in nonrodent species and may reduce the number of dose escalations needed to reach the recommended dose in first-in-human studies while limiting unnecessary exposure to high drug levels in humans. The use of this type of SD could improve the design of phase I studies of BDPs by making them more efficient. The role of preclinical pharmacodynamic markers was useful in confirming the hypothetical SD, and attempts should be explored in future animal studies to identify such parameters.
Introduction
An important objective of preclinical evaluation of new anticancer agents is to identify a safe starting dose for the first-in-human (FIH) clinical trial. These data are typically acquired from in vivo pharmacology and toxicology studies [1]. The ideal safe starting dose of a drug for a FIH trial should have no significant risk of toxicities but be sufficiently high to allow rapid attainment of the recommended phase 2 dose (RP2D).
Recommendations for determining the safe starting dose for investigational antitumor agents in FIH trials for patients with advanced cancer are described in the S9 guidance document developed by an expert working group of the International Conference on Harmonization [2]. Although the regulatory authorities in the United States, European Union, and Japan have adopted these guidelines, they are nonbinding, and the starting dose can be determined via various methods. The conventional paradigm in starting dose determination for low-molecular-weight anticancer drugs is one-tenth the severely toxic dose in ≤10% (STD10) of rodents, unless the nonrodent is the most appropriate species [3]. If so, one-sixth of the highest nonseverely toxic dose (HNSTD), the highest dose level that does not produce lethal, life-threatening, or irreversible toxicities, is considered an appropriate starting dose [4]. An appropriate or relevant species is one that demonstrates the pharmacologic effects of the drug.
Biotechnology-derived products (BDPs) are complex protein structures formulated via processes that use genetically modified organisms to produce peptide or protein pharmaceuticals such as fusion proteins and monoclonal antibodies [5]. Owing to the high specificity of BDPs for the human target, the cross-reactivity with the target in rodent species used in toxicology studies is often inadequate [6]. This results in most toxicology studies being conducted in nonhuman primates, preventing the use of STD10 to set the start dose. The suitability of using one-sixth HNSTD to select the start dose for BDPs has not been formally evaluated.
The most common approach to starting dose determination for BDPs has been to use a more conservative approach based on the “no observed adverse effect level” (NOAEL) [7], which is the highest dose in animals that does not produce a significant increase in biologically important adverse events compared with a control group. More recently, the use of pharmacokinetic (PK) and pharmacodynamic (PD) modeling to derive biologically optimal dose levels from data from preclinical toxicology studies and in vivo pharmacology studies has increased [8]. The ideal process to determine the starting dose for BDPs is not defined [9] and is usually a balance between the highest safe dose of a drug and the lowest potentially active dose.
The objective of the present systematic review was to evaluate the safety of using one-sixth HNSTD, a toxicity-based preclinical parameter, to guide the starting dose for anticancer BDPs.
Materials and Methods
Data Sources and Searches
Two search strategies were used to capture the clinical and preclinical data accurately. These are detailed in the supplemental online Appendix.
Study Selection
We defined the inclusion and exclusion criteria a priori. Preclinical studies were included if sufficient data were available to define the one-sixth HNSTD in a relevant species by the reviewing toxicologists (M.J.H. and M.S.R.); had followed good laboratory practice standards; was at least 4 weeks in duration; and had adequate histopathologic data. All studies of small molecules, immune checkpoint inhibitors, and antibody-drug conjugates were excluded. Immune checkpoint inhibitors were excluded because the toxicology data are often not predictive of fatal cytokine release because of species differences in the immune response. Antibody-drug conjugates were excluded because their structure and mechanism of action are different from those of BDPs, and toxicities could ensue from the antibody or toxin payload. Two reviewers (M.J.H. and M.S.R.) evaluated the titles and abstracts of the publications identified by the preclinical search strategy in duplicate, and any publication thought to be potentially relevant was retrieved in full.
Two reviewers (A.H. and N.C.) evaluated the titles and abstracts of the publications identified by the clinical search strategy in duplicate, and any publication thought to be potentially relevant was retrieved in full. The same two reviewers then assessed the full publications for eligibility. Single-agent phase I studies of BDPs were included. The following studies were excluded: nonclinical studies, nonphase I studies, studies conducted of patients without cancer or children, studies not testing an anticancer therapy, combination studies such as those combining drug with radiation, stem cell or bone marrow transplantation, supportive care studies, and studies published as a conference proceeding only.
The reviewers were not blinded of the study authors or outcomes. The studies were only included for the final review if sufficient preclinical, toxicology, and clinical data were available to assess the safety of the hypothetical use of one-sixth HNSTD in FIH trials. The decision to include a study for review was made by consensus among the authors (M.J.H., M.S.R., A.H., N.C., A.R., and C.L.T).
Data Extraction
Two pairs of reviewers extracted the data from the preclinical (M.J.H. and M.S.R.) and clinical (A.H. and N.C.) studies. No discrepancies were present among the preclinical reviewers. The discrepancies in the data extraction for the clinical review were resolved by consensus between the clinical reviewers (A.H. and N.C.), in addition to the other authors (C.L.T., A.R., and L.L.S.). The data extracted from clinical studies included drug type, actual starting dose, maximum tolerated dose (MTD) or maximum administered dose (MAD), RP2D and schedule, PK and PD data, number of cohorts, study sample size, dose-escalation method, and dose-dependent clinical toxicities. Data extracted from the preclinical studies included the drug type, species examined, NOAEL, MTD, schedule, maximum PD dose, PK and PD data, and dose-dependent toxicities. The maximum PD dose is the lowest level of drug that produces the greatest effect on a predetermined PD marker. One-sixth HNSTD was converted to human equivalent doses using allometric scaling factors as per standard regulatory guidance to normalize exposure between species. All human doses were converted to mg/kg using an average body weight of 65 kg for fixed doses and a scaling factor of 37 for doses reported in mg/m2 [10]. Additional information was collected from the U.S. Food and Drug Administration (FDA) website (available at: Drugs@FDA) for an approved drug’s prescribing information and label dose. In the present review, the RP2D or recommended dose of a BDP was the dose defined in the phase I study. The label dose was the dose listed in the prescribing information. Additionally, the dose used in an available phase II study was collected if the phase I trial did not state a recommended dose.
Statistical Analysis
The primary objective was to evaluate the safety of using one-sixth HNSTD based on the body mass or body surface area (allometric) to determine the starting dose in FIH trials of BDPs. A safe starting dose was defined as a dose that did not exceed the MTD or RP2D. The secondary aims included the impact on the number of dose levels if one-sixth HNSTD (allometric) was used as the starting dose and a correlation between dose-dependent toxicities in animals and humans. The role of a preclinical PD marker was examined in an exploratory manner to determine the relationship between one-sixth HNSTD and the maximum PD dose level. The data were tabulated, graphed, and summarized using descriptive statistics. No statistical comparisons were planned because of the small size and retrospective nature of our analysis.
Results
Search Results
Twenty-three agents were identified between January 2000 and November 2013 from the preclinical and clinical searches. These are detailed in supplemental online Figure 1.
Preclinical Study Characteristics
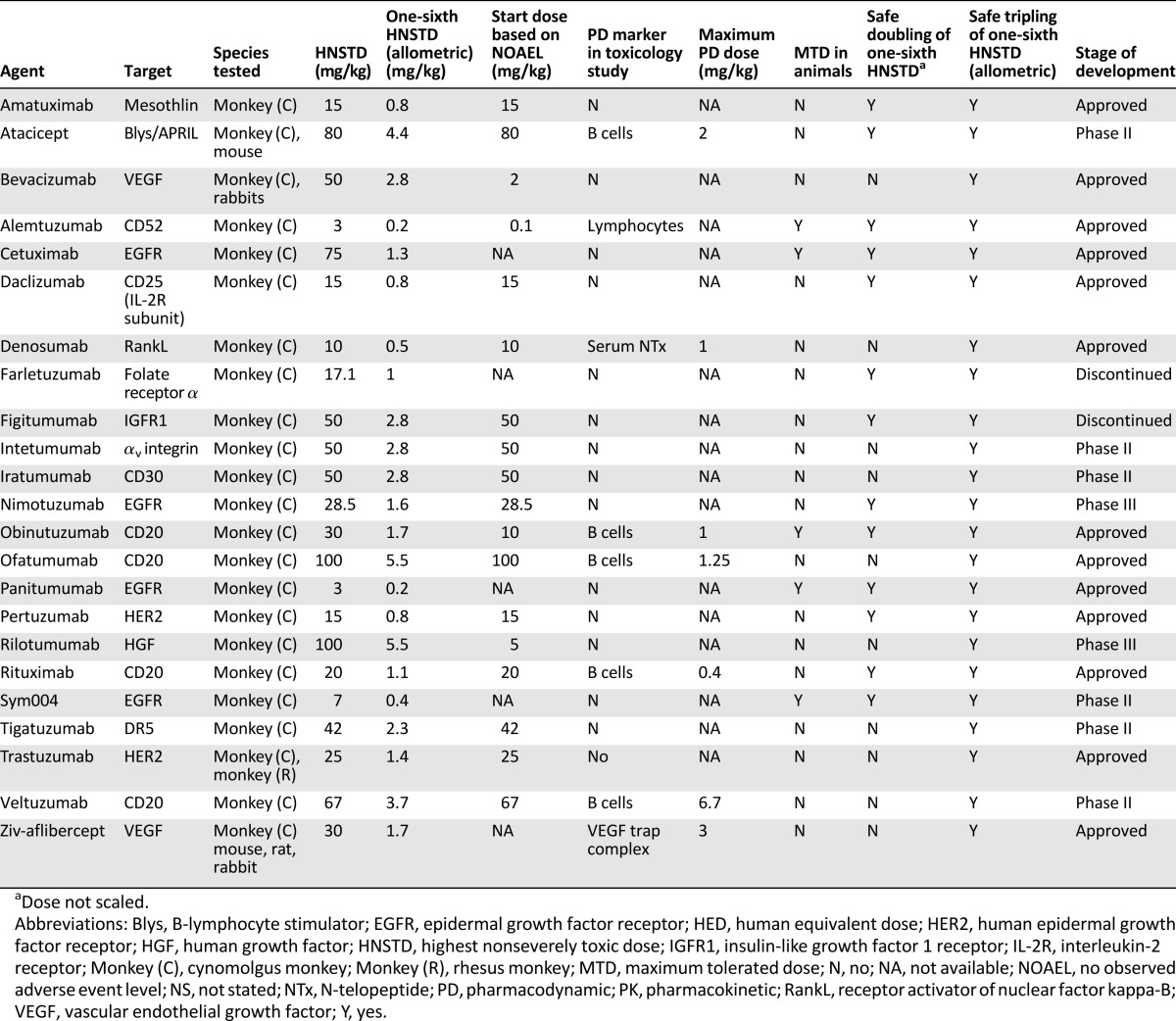
Most BDPs (n = 19) were tested in only 1 species. All BDPs were tested in cynomolgus monkeys, with only 2 agents (atacicept and aflibercept) tested in both rodent and nonrodent species. Bevacizumab and trastuzumab were both tested in two nonhuman primate species. For each agent, one-sixth HNSTD (allometric) was determined, but the NOAEL could only be calculated for 18 BDPs (78%). Eight toxicology studies presented PD data, and 75% (n = 6) of the PD markers reported were hematologic parameters (Table 1). N-telopeptide, a bone resorption biomarker, and a vascular endothelial growth factor trap complex were the nonhematologic PD markers used in the remaining two studies.
Table 1.
Preclinical information
Clinical Study Characteristics
The median study size of the FIH trials was 24 patients (range, 7–82), and the median number of dose levels was 5.5 (range, 2–12). The dose-escalation method was provided by 19 studies (83%), and all but 1 used the standard 3+3 design (78%). The MTD was declared for only 2 BDPs (Table 2). At the time of our review, 11 drugs (48%) had a label dose, 14 (61%) had declared a RP2D, and 7 BDPs (30%) had entered phase II testing. Atacicept and veltuzumab were the only agents without a label dose or RP2D. The label dose was the same as the RP2D for all drugs. The type and number of endpoints used to determine RP2D are charted in Figure 1 and supplemental online Figure 2, respectively. RP2D was determined by the toxicity combined with another measure (e.g., PK, PD, or efficacy) in only 5 agents. Amatuximab was the only BDP that used toxicity as a sole endpoint for RP2D. PK and PD data were presented in 19 (83%) and 18 (78%) phase I studies, respectively.
Table 2.
Clinical information
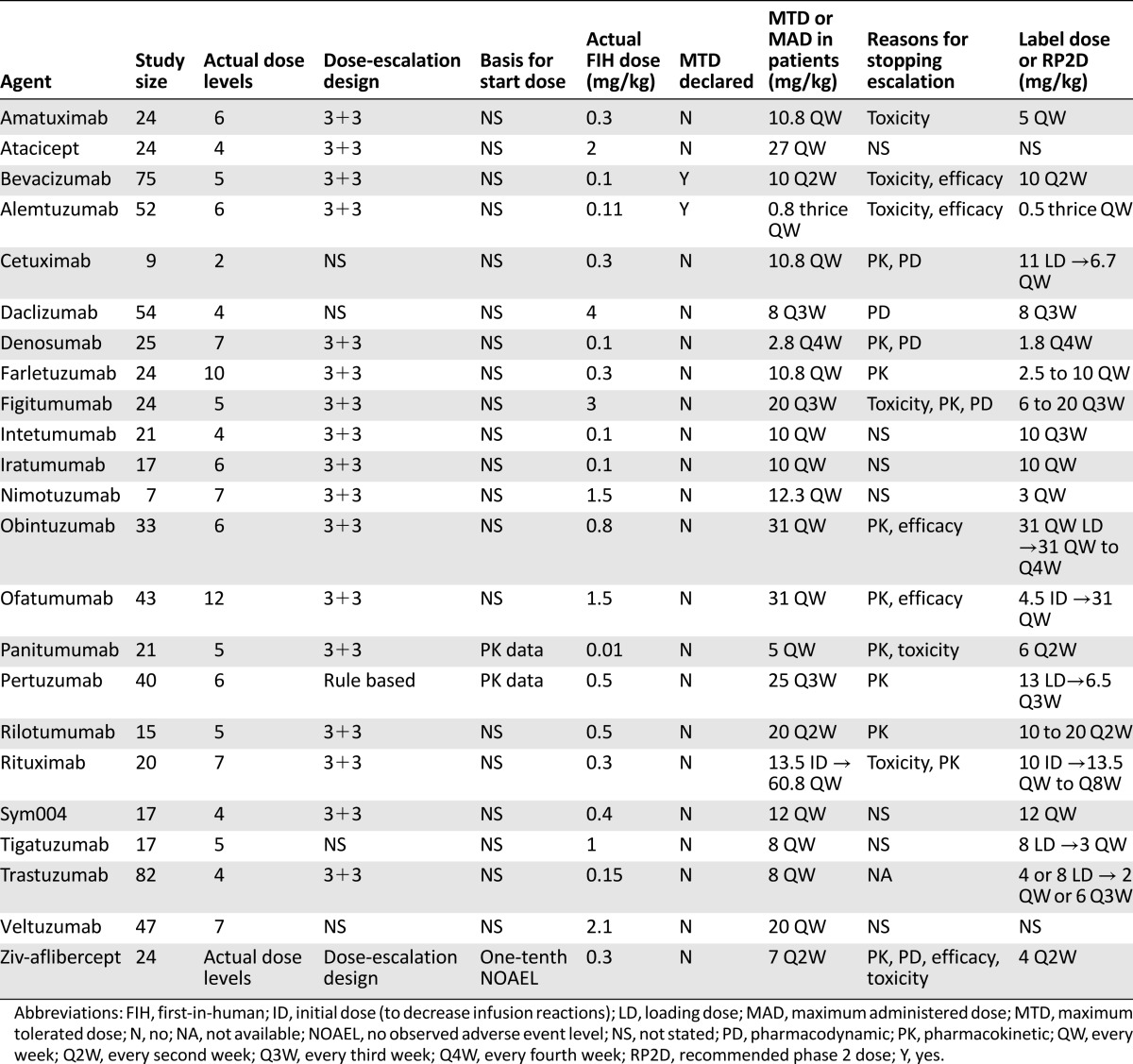
Figure 1.
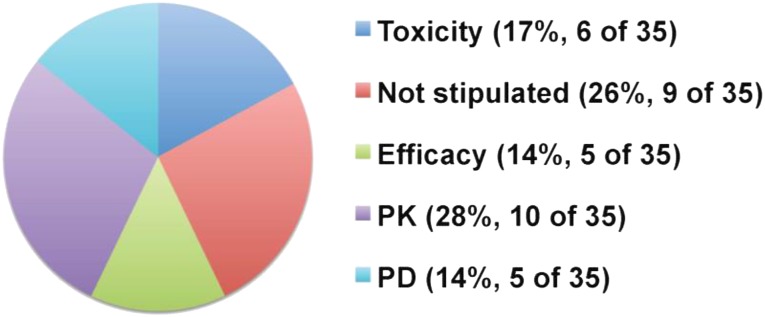
Endpoint used to determine RP2D. Cumulative measure because multiple endpoints could be used for one agent. The total number of endpoints used for all 23 agents was 35.
Abbreviations: PD, pharmacodynamic; PK, pharmacokinetic; RP2D, recommended phase 2 dose.
Use of One-Sixth HNSTD (Allometric) as Start Dose Is Unlikely to Lead to Unacceptable Toxicities
Only 3 clinical trials provided a reason for the actual BDP start dose. The use of one-sixth HNSTD (allometric and unscaled) would not have exceeded the highest dose tested (MTD, MAD) for all 23 agents. Furthermore, one-sixth HNSTD (allometric) did not did not exceed RP2D for all 23 agents; however, if one-sixth HNSTD was not scaled, only 1 agent (4%) would have exceeded RP2D. Most BDPs (21 of 23) were administered without reaching the MTD in phase I trials. Hypothetically, one-sixth HNSTD (allometric) would have safely allowed dose tripling for all agents (Table 1). However, if one-sixth HNSTD was not scaled for body surface area, hypothetical dose doubling would have exceeded the highest dose tested in 10 of the 23 BDPs (43%). Hence, one-sixth HNSTD (allometric) presumably would not have resulted in unacceptable toxicities, but this might not have been true if scaling for body surface area had not occurred.
Use of One-Sixth HNSTD Could Reduce the Number of Dose-Escalation Cohorts
The median ratio of MTD or MAD to the actual start dose was 36 (range, 8–500) (Fig. 2). One-sixth HNSTD would have reduced the median ratio of MTD or MAD to start dose to 2.0 (range, 1.1–10) and 6.1 (range, 3.5–55.3) if allometrically scaled. The one-sixth HNSTD (allometric) was always higher than the actual FIH start dose, except for daclizumab. The FIH start dose for daclizumab was 4 mg/kg, but the reason for selecting this dose level was not stated in the publication.
Figure 2.

Comparison of MTD or MAD ratios with one-sixth HNSTD (allometric) and MTD or MAD with FIH SD. The MAD was used if no MTD was declared. Red bars indicate the ratio of MTD or MAD with one-sixth HNSTD; yellow or blue bars, the ratio of MTD or MAD with the actual FIH SD.
Abbreviations: FIH, first-in-human; HNSTD, highest nonseverely toxic dose; MAD, maximum administered dose; MTD, maximum tolerated dose; SD, start dose.
The median ratio of one-sixth HNSTD (allometric) to the actual start dose for the 23 BDPs was 3.33 (range, 0.2–28). The hypothetical use of one-sixth HNSTD (allometric) would have decreased the number of cohorts needed to reach the MTD or MAD. Furthermore, one-sixth HNSTD (allometric) would have been safe, because even if the dose were tripled, it would not have exceeded the MTD or RP2D at the first dose escalation.
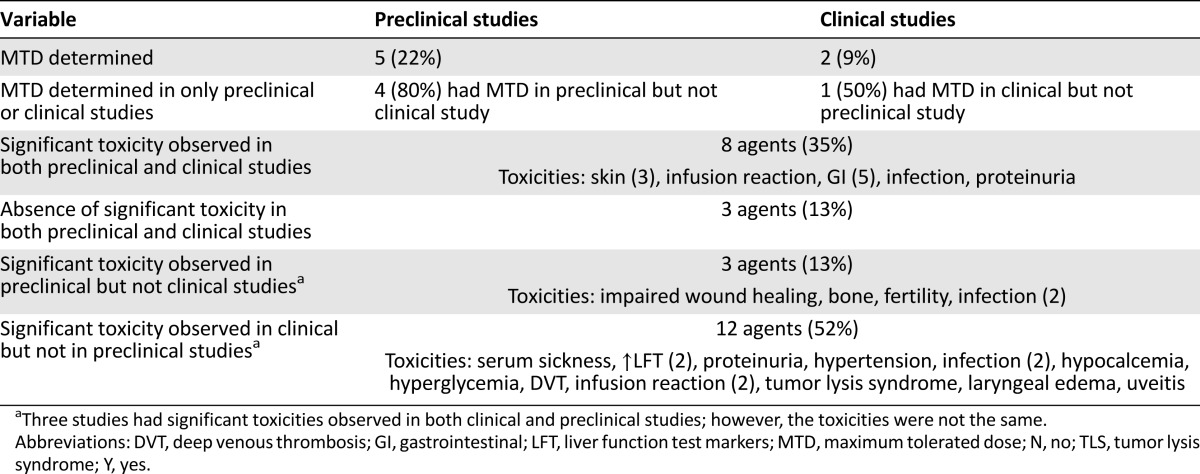
Dose-Dependent Toxicities Varied in FIH Studies Compared With Animal Studies
Alemtuzumab was the only drug to have had an MTD in both preclinical and clinical studies. Eleven BDPs had dose-dependent toxicities reported in the FIH study (Table 3). The typical adverse events observed in both preclinical and clinical studies for a BDP were skin and gastrointestinal (Table 3). The lack of adverse events in the preclinical study was replicated in the clinical trial for only 3 BDPs (13%). More than one half (52%) of the reviewed agents had significant toxicities in the FIH trial that were not observed in the animal toxicology study, and 3 drugs (13%) had toxicities in the preclinical phase that were not seen in the human clinical trials. These toxicities are listed in Table 3. Toxicities related to the mechanism of action such as dermatologic or gastrointestinal events were seen in both animals and humans. Harms that might be idiosyncratic or immune mediated in humans, such as infusion reactions, serum sickness, or uveitis, did not correlate well between the preclinical and clinical studies.
Table 3.
Overview of significant toxicities from both preclinical and clinical studies
Use of PD Markers Could Provide Increased Confidence to Set Start Dose Using HNSTD
PD markers can be used to assess the dynamic range of pharmacologic activity of the drug in preclinical studies. Determination of the maximum PD dose could affirm that one-sixth HNSTD (allometric) does not exceed the maximal pharmacologically active dose. Seven agents defined a maximum PD dose based on a PD biomarker used in a toxicology study (Table 1). Alemtuzumab did have a hematologic PD marker, but a maximum PD dose was not stated in the preclinical study. The one-sixth HNSTD (allometric) was in fact higher than the maximum PD dose for 4 drugs. All 4 agents used B cells as a hematologic PD marker. For 4 of 7 BDPs (71%), the maximum PD dose would have supported one-sixth HNSTD (allometric) to select the starting dose within the range of dynamic pharmacologic activity. In the cases in which one-sixth HNSTD (allometric) exceeded the maximum PD dose, consideration of the PD activity could have been used to adjust the starting dose to be closer to the optimal biologic dose.
Discussion
Predicting human toxicities of BDPs from preclinical studies is often confined to a single species because of the lack of target homology between species. Although the availability of a pharmacologically relevant species generally allows detection of target-mediated toxicities of BDPs, preclinical evaluations of these agents are limited in terms of identifying idiosyncratic or immune-mediated adverse events that can occur in humans. Therefore, sponsors and regulatory authorities typically adopt a cautious approach to setting the start dose of anticancer BDPs, such as the NOAEL [11, 12]. However, this conservative strategy has not been rigorously evaluated for anticancer BDPs [13]. Certainly, high-risk investigational products should have their start dose carefully selected to avoid unnecessary risks to patient safety [14]. Agents that should be considered high risk for causing harm include those that lack a relevant species for toxicity testing and immunostimulatory molecules or immune checkpoint inhibitors with the potential to produce a cytokine storm. For these drug classes and BDPs with a steep dose-response curve, the HNSTD can potentially underestimate the toxicity. When there is little confidence with preclinical safety models to predict human toxicity, a more conservative PD effects-based starting dose selection is recommended.
Assessing the safety of actual start doses for anticancer drugs has been the subject of previous evaluations. A European Organization for Research and Treatment of Cancer workshop analysis of 71 chemotherapy agents found that the murine-derived start dose was higher than the human MTD in approximately 2% of trials [15, 16]. After 2 reviews comparing animal toxicology studies with phase I trials, the National Cancer Institute concluded that determining the start dose from the most sensitive species for cytotoxic agents was safe in 98% of cases [17, 18]. In a review of 81 FIH trials of molecularly targeted agents, the start dose was defined by a variety of preclinical toxicology factors such as one tenth the lethal dose in rodents and various fractions of MTD, NOAEL, STD, and minimum effective dose in nonrodent species [19]. In addition, Le Tourneau et al. [19] reported that 3.7% of trials of molecularly targeted agents adopted a start dose that was unsafe, because it exceeded the MTD. The present study is the first to evaluate the start dose of BDPs.
The present review has demonstrated that using one-sixth HNSTD (allometric) would have improved the efficiency of a phase I trial by reducing the number of dose levels without compromising patient safety. Using the data for 23 BDPs that came from public sources, none of the hypothetical start doses would have exceeded the MTD. Despite most BDPs having a wide therapeutic window, as evidenced by the high MTD to start dose ratio, most studies (78%) used a more conservative dose-escalation strategy with the conventional 3+3 design. The toxicity parameters in the phase I trials of BDPs are less influential on MTD or RP2D determination compared with cytotoxic agents, as demonstrated in the present review, with only 2 MTDs (8.6%) declared and 6 agents (26%) using toxicity to define RP2D. The most common factor used in determining the recommended dose was a PK endpoint.
Given the minimal degree of toxicities seen in preclinical testing of many BDPs, the PK and PD measures derived from preclinical toxicology and pharmacology studies should be evaluated in the selection of the FIH starting dose. If the one-sixth HNSTD exceeds the maximum PD dose, the one-sixth HNSTD could place the starting dose far on the plateau of the dose-response curve and substantially higher than the optimal biologic dose. In such cases, in vitro and in vivo pharmacology data can guide PK/PD modeling to derive pharmacologically active starting doses that allow clinical determination of the recommended dose and schedule while still reducing the number of dose escalations in phase I studies [20]. The present review found that several agents had maximum PD doses identified in toxicology studies using translational PD markers. In some instances, this parameter was higher or well approximated with the one-sixth HNSTD, affirming that PD markers can be useful in the absence of toxicity. Characterizing PD biomarkers in preclinical studies could inform the start doses for BDPs, and our findings suggest that this approach is feasible.
The number of BDPs included in the present study was small; thus, all conclusions should be interpreted in this context. The present review was limited to BDPs that had their preclinical studies publicly available, which might have inadvertently biased the results toward agents with favorable pharmacologic and toxicologic profiles. Antibody-drug conjugates and immune checkpoint inhibitors were not included; therefore, these results cannot be applied to such agents. An opportunity to expand this analysis through collaboration with industry stakeholders may confirm the results of our review and strengthen the conclusions.
Conclusion
The use of one-sixth HNSTD (allometric) would not have resulted in unacceptable toxicities for the 23 BDPs evaluated. The absence of MTD in animals and humans underscores the need to identify and use alternative measures (PK or PD) to guide start dose selection for FIH trials. Using one-sixth HNSTD (allometric) might reduce the number of dose escalations needed to reach the recommended dose in FIH studies and, thus, the number of patients treated at subtherapeutic doses while limiting unnecessary exposure to high drug levels. Future preclinical and clinical studies using these important approaches are warranted.
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgments
This publication reflects the views of the authors and should not be construed to represent the U.S. Food and Drug Administration’s views or policies. We thank Nancy Muir for conducting the preclinical literature search and Junhui Zhang for conducting the clinical literature search.
Author Contributions
Conception/Design: Aaron R. Hansen, Natalie Cook, M. Stacey Ricci, Albiruni Razak, Christophe Le Tourneau, Kathleen McKeever, Lorin Roskos, Rakesh Dixit, Lillian L. Siu, Mary Jane Hinrichs
Provision of study material or patients: Aaron R. Hansen, Natalie Cook, M. Stacey Ricci, Albiruni Razak, Christophe Le Tourneau, Lillian L. Siu, Mary Jane Hinrichs
Collection and/or assembly of data: Aaron R. Hansen, Natalie Cook, M. Stacey Ricci, Albiruni Razak, Christophe Le Tourneau, Lillian L. Siu, Mary Jane Hinrichs
Data analysis and interpretation: Aaron R. Hansen, Natalie Cook, M. Stacey Ricci, Albiruni Razak, Christophe Le Tourneau, Kathleen McKeever, Lorin Roskos, Rakesh Dixit, Lillian L. Siu, Mary Jane Hinrichs
Manuscript writing: Aaron R. Hansen, Natalie Cook, M. Stacey Ricci, Albiruni Razak, Christophe Le Tourneau, Kathleen McKeever, Lorin Roskos, Rakesh Dixit, Lillian L. Siu, Mary Jane Hinrichs
Final approval of manuscript: Aaron R. Hansen, Natalie Cook, M. Stacey Ricci, Albiruni Razak, Christophe Le Tourneau, Kathleen McKeever, Lorin Roskos, Rakesh Dixit, Lillian L. Siu, Mary Jane Hinrichs
Disclosures
Albiruni Razak: Pfizer, Novartis, Karyopahrm, Bristol-Myers Squibb (RF); Kathleen McKeever: Ultragenyx (January 2015 to present), MedImmune/AstraZeneca (formerly employed) (E), AstraZeneca, Ultragenyx (OI); Lorin Roskos: AstraZeneca (E, OI); Lillian L. Siu: Novartis, Roche/Genentech, GlaxoSmithKline, Pfizer, Bayer, Karyopharm, Boehringer Ingelheim, AstraZeneca/MedImmune, Merck, Bristol-Myers Squibb, Regeneron (RF); Mary Jane Hinrichs: MedImmune (E, OI). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Schein PS. Preclinical toxicology of anticancer agents. Cancer Res. 1977;37:1934–1937. [PubMed] [Google Scholar]
- 2.International Conference for Harmonisation. S9 Nonclinical Evaluation for Anticancer Pharmaceuticals 2010. Available at http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm085389.pdf. Accessed January 5, 2015.
- 3.Newell DR, Burtles SS, Fox BW, et al. Evaluation of rodent-only toxicology for early clinical trials with novel cancer therapeutics. Br J Cancer. 1999;81:760–768. doi: 10.1038/sj.bjc.6690761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rozencweig M, Von Hoff DD, Staquet MJ, et al. Animal toxicology for early clinical trials with anticancer agents. Cancer Clin Trials. 1981;4:21–28. [PubMed] [Google Scholar]
- 5.Prugnaud JL. Similarity of biotechnology-derived medicinal products: specific problems and new regulatory framework. Br J Clin Pharmacol. 2008;65:619–620. doi: 10.1111/j.1365-2125.2007.03062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cavagnaro JA. Preclinical safety evaluation of biotechnology-derived pharmaceuticals. Nat Rev Drug Discov. 2002;1:469–475. doi: 10.1038/nrd822. [DOI] [PubMed] [Google Scholar]
- 7.Dorato MA, Engelhardt JA. The no-observed-adverse-effect-level in drug safety evaluations: Use, issues, and definition(s) Regul Toxicol Pharmacol. 2005;42:265–274. doi: 10.1016/j.yrtph.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 8.Muller PY, Milton M, Lloyd P, et al. The minimum anticipated biological effect level (MABEL) for selection of first human dose in clinical trials with monoclonal antibodies. Curr Opin Biotechnol. 2009;20:722–729. doi: 10.1016/j.copbio.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 9.International Conference for Harmonisation. S6 (R1) Preclinical safety evaluation of biotechnology-derived pharmaceuticals 1997. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM074957.pdf. Accessed January 5, 2015.
- 10.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 11.Milton MN, Horvath CJ. The EMEA guideline on first-in-human clinical trials and its impact on pharmaceutical development. Toxicol Pathol. 2009;37:363–371. doi: 10.1177/0192623309332997. [DOI] [PubMed] [Google Scholar]
- 12.Food and Drug Administration. Guidance for Industry: Estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers, 2005. Available at http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm078932.pdf. Accessed January 5, 2015.
- 13.Hansel TT, Kropshofer H, Singer T, et al. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov. 2010;9:325–338. doi: 10.1038/nrd3003. [DOI] [PubMed] [Google Scholar]
- 14.Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 15.Arbuck SG. Workshop on phase I study design. Ninth NCI/EORTC New Drug Development Symposium, Amsterdam, March 12, 1996. Ann Oncol. 1996;7:567–573. doi: 10.1093/oxfordjournals.annonc.a010672. [DOI] [PubMed] [Google Scholar]
- 16.Verweij J. Starting dose levels for phase I studies. Proceedings of the Ninth NCI-EORTC Symposium on New Drugs in Cancer Therapy. Amsterdam, 1996; March 12–15, 13 (Abstr). [Google Scholar]
- 17.Smith A, Tomaszewski J. Correlation of human toxicity and MTDs with preclinical animal data. Preclinical Pharmacology and Toxicology Workshop at the EORTC/NCI/AACR Meeting. Frankfurt, Germany, 2002. [Google Scholar]
- 18.Tomaszewski JE, Smith AC, Covey JM, et al. Relevance of preclinical pharmacology and toxicology to phase I trial extrapolation techniques: Relevance of animal toxicology. In: Baguely BC, editor. Anti-Cancer Drug Design. San Diego, CA: Academic Press; 2001. [Google Scholar]
- 19.Le Tourneau C, Stathis A, Vidal L, et al. Choice of starting dose for molecularly targeted agents evaluated in first-in-human phase I cancer clinical trials. J Clin Oncol. 2010;28:1401–1407. doi: 10.1200/JCO.2009.25.9606. [DOI] [PubMed] [Google Scholar]
- 20.Jones PS, Jones D. New regulatory framework for cancer drug development. Drug Discov Today. 2012;17:227–231. doi: 10.1016/j.drudis.2011.12.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.