

This review discusses the genomic revolution, its impact on our understanding of myelodysplastic syndrome (MDS) biology and risk stratification, and the current role and challenges of the application of genetic mutational data into daily clinical practice. This article also examines how genomic data could be used clinically to aid with the diagnosis, prognosis, prediction of response to specific therapies, and the development of novel and rationally targeted therapies for MDS.
Keywords: MDS, Mutations, Molecular testing, Myelodysplastic syndromes, Prognostication, Biomarkers
Abstract
Myelodysplastic syndromes (MDS) are heterogeneous hematopoietic neoplasms that are driven by somatically acquired genetic mutations and epigenetic alterations. Accurate risk stratification is essential for delivery of risk-adaptive therapeutic interventions. The current prognostic tools sum the impact of clinical, pathologic, and laboratory parameters. Newer technologies with next-generation targeted deep sequencing and whole-genome and -exome sequencing have identified several recurrent mutations that play a vital role in the pathophysiology of MDS and the impact of these genetic changes on disease phenotype. Equally important, well-annotated databases of MDS patients with paired clinicopathologic and genetic data have enabled better understanding of the independent prognostic impact of several molecular mutations on important clinical endpoints such as overall survival and probability of leukemic progression. Cumulative evidence suggests that genomic data can also be used clinically to aid with the diagnosis, prognosis, prediction of response to specific therapies, and the development of novel and rationally targeted therapies. However, the optimal use of this mutational profiling remains a work in progress and currently there is no standard set of genes or techniques that are recommended for routine use in the clinic. In this review, we discuss the genomic revolution and its impact on our understanding of MDS biology and risk stratification. We also discuss the current role and the challenges of the application of genetic mutational data into daily clinical practice and how future research could help improve the prognostication precision and specific therapy selection for patients with MDS.
Implications for Practice:
Heterogeneity in clinical outcomes of MDS is partly related to interpatient variability of recurrent somatic mutations that drive disease phenotype and progression. Although clinical risk stratification tools have functioned well in prognostication for patients with MDS, their ability to predict clinical benefits of specific MDS therapies is limited. Molecular testing shows promise in aiding diagnosis, risk stratification, and therapy-specific benefit prediction for MDS patients. Nonetheless, logistical issues related to assay performance standardization, validation, interpretation, and development of guidelines for how to use the results to inform clinical decisions are yet to be resolved.
Introduction
Myelodysplastic syndromes (MDS) comprise a group of hematopoietic neoplasms that result in dysplastic changes and ineffective hematopoiesis and manifest clinically with varying degrees of peripheral blood (PB) cytopenias and propensity to progress to acute myeloid leukemia (AML) [1–3]. Traditionally, the diagnosis of MDS is based on the presence of dysplasia in one or more hematopoietic lineages and/or the presence of characteristic chromosomal abnormalities. However, dysplasia is also seen in other myeloid malignancies, and approximately half of MDS patients have a normal karyotype, complicating the diagnosis [4–7].
After the completion of the human genome sequencing project in 2001, several studies that used high-throughput sequencing technologies have identified recurrent somatic mutations that are important in MDS pathophysiology and contribute to its eclectic phenotype [5–9]. This genomic information also plays a role in establishing the diagnosis and prognostication [5–7] and may lead to more effective therapies for MDS. Here we discuss the recent advances in the understating of the molecular underpinnings in MDS as they relate to the practice in the clinic. We also discuss current limitations of the applications of these findings in daily practice.
Genomic Revolution and MDS
Despite significant technological strides that have been achieved over the past 15 years, we are still far from fully understanding the function of the majority of the genome [10–13]. The human genome consists of approximately 3 billion base pairs (bp) of DNA, of which only ∼1.5% code for proteins [14–16].
Several methods have been developed to analyze the human genome. One of the oldest and most commonly used methods is conventional cytogenetics [17–20]. After the invention of chromosome banding techniques in the 1960s [21], several characteristic cytogenetic changes have been described in patients with MDS (Table 1). To date, cytogenetic analysis remains one of the most important diagnostic and prognostic tools in MDS patients. Although the detection of cytogenetic abnormalities that are associated with MDS may aid the diagnostic workup in patients with peripheral blood cytopenias without aberrant dysplasia in the bone marrow (BM) [22], cytogenetic abnormalities are present in only 50% of patients with known MDS, limiting their use as a negative predictive factor [18–20]. The most common cytogenetic abnormalities in MDS include: del(5q), trisomy 8, del(20q), monosomy 7, or del(7q) [17–20]. These abnormalities are likely secondary events in the setting of genomic instability caused by the founding mutation [23], with the notable exception of the isolated del(5q) abnormality. The revised International Prognostic Scoring System (IPSS-R) uses the most widely accepted five-group cytogenetic risk classification schema (Table 2) that replaced the three-group cytogenetic classification used in the original IPSS [24].
Table 1.
Common cytogenetic abnormalities in MDS
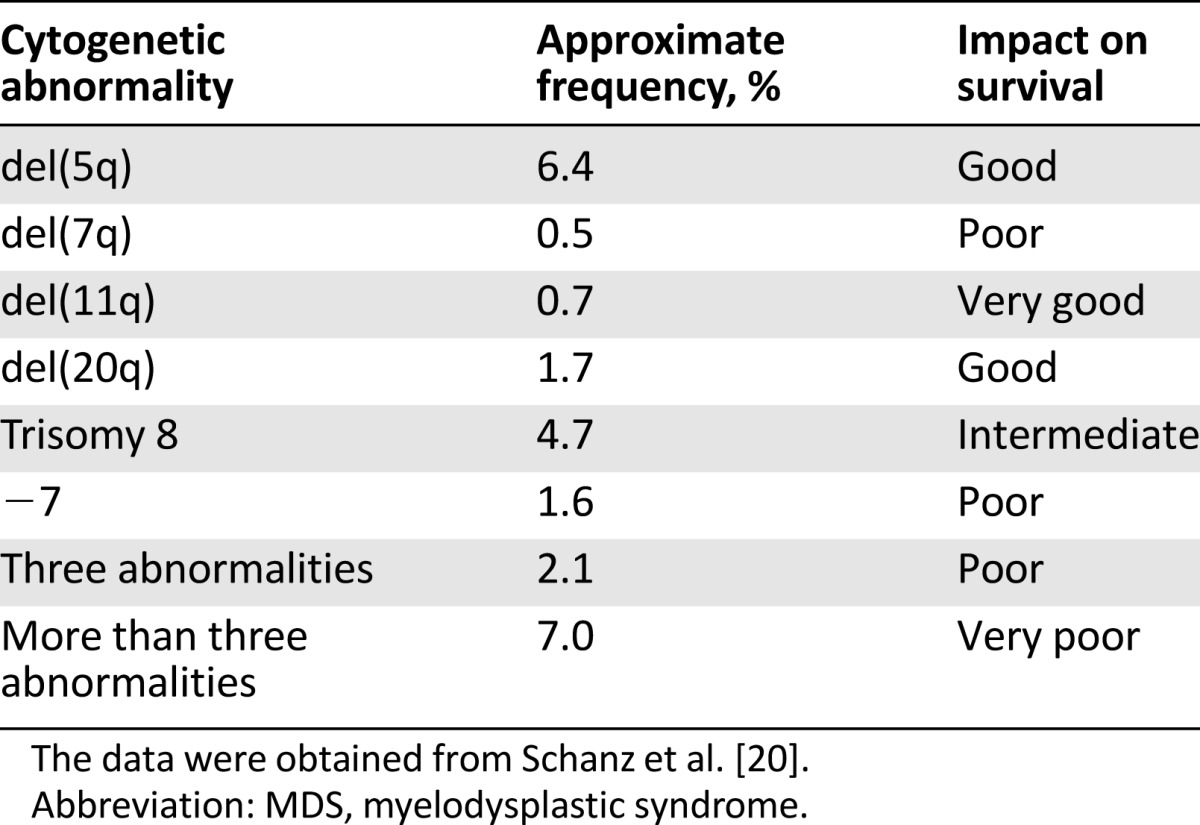
Table 2.
Cytogenetic abnormalities of prognostic importance in MDS

Notably, cytogenetic analysis is effectively a low-resolution view of the entire genome. To overcome this limitation, newer methods such as fluorescence-in-situ-hybridization (FISH) and comparative genomic analysis were developed [25, 26]. The utility of a FISH assay depends mainly on the size of the fluorescently labeled DNA probe. To further improve the genomic coverage of cytogenetic abnormalities, several array platforms have been developed [26–29]. The use of single nucleotide polymorphism array (SNP-A) technology by hybridization of tumor DNA to arrays containing probes that are specific for allelic variants allowed the identification of gene copy number and loss of heterozygosity [30].
More recently, DNA sequencing has become an important technology for identifying molecular changes. The first DNA sequencing effort was developed by Fred Sanger et al. [31] in the 1970s. A typical Sanger DNA sequencing can only read a sequence of approximately 1000 bp in length, which is less than one millionth of the human genome. Furthermore, the sequenced DNA fragments have to be cloned or polymerase chain reaction-amplified, a labor-intensive process [31, 32]. Although Sanger sequencing provides high resolution to analyze a known mutational hotspot, it represents an expensive method to sequence a large proportion of the genome. Using this method, the first human genome was sequenced but at a cost of more than $3 billion over a period of more than 10 years [10–13]. With the development of next-generation sequencing methods and the improvement of bioinformatics techniques, it is possible today to sequence the entire genome (whole-genome sequencing), part of the genome (whole-exome sequencing [WES]), or just the transcribed RNA (transcriptome sequencing) at a much lower cost [33–38].
Clinical Implications of Molecular Testing in MDS
The ultimate goal of genomic sequencing is to translate our knowledge of these recurrent gene mutations into clinical use so that this information can aid the physician in diagnosis, prognostication, and prediction of clinical benefit from specific therapies. It can also facilitate the development of rationally designed targeted therapies that have the potential of improving patient outcomes. Although there have been significant advances in identifying recurrent gene mutations that are relevant for these purposes, translating this huge amount of information into the clinic has lagged behind the discovery phase.
Diagnostic Utility of Genomic Information
Applying the 2008 World Health Organization standards, the diagnosis of MDS is made based on identification of dysplastic changes and/or detection of recurrent chromosomal abnormalities. This is challenging in patients with low bone-marrow cellularity (hypoplastic MDS [hypo-MDS]), and normal karyotype by metaphase cytogenetic (MC) analysis may hinder the ability to distinguish this subtype of MDS from other BM failure disorders such as aplastic anemia (AA) [39, 40]. Using SNP-A karyotyping, the detection of chromosomal lesions can be increased to 19% in patients with AA and 59% in patients with hypo-MDS [30]. More importantly, additional clonal lesions can be detected in 36% of patients with normal/noninformative MC. In a subset of AA patients studied at presentation, persistent chromosomal genomic lesions were found in 10 of 33 patients, suggesting that the initial diagnosis actually may have been hypo-MDS [30]. In a study of 150 AA patients without morphological evidence of MDS, several genes known to be associated with BM failure disorders and myeloid malignancies were screened [41]. Interestingly, 19% of AA patients had detectable somatic mutations [41]. AA patients with somatic mutations were more likely to evolve to MDS compared with those without mutations [41].
Emerging data suggest that recurrent somatic mutations occur in patients with pancytopenia without enough dysplastic features to meet the diagnosis of MDS. In a study of 250 patients with idiopathic cytopenia of unknown significance, somatic mutations obtained by next-generation sequencing were found in 33% [42], the most common being TET2 (38%), DNMT3A (20%), ASXL1 (18%), SRSF2 (15%), and ZRSR2 (11%). The mean allele frequency for mutations was 33%, with 14% of mutations having an allele frequency of <10% [42]. In another analysis of 82 patients with paired nondiagnostic and diagnostic samples, 69 had adequate molecular material at both time points for assessment [43]. A driver mutation, with or without a structural variant, was identified using targeted deep sequencing in 91% of nondiagnostic samples; this increased to 94% at the point of diagnosis [43]. These findings suggest that MDS patients with ambiguous morphology at the time of presentation often carry readily detectable driver mutations that may identify patients with early-stage disease [43].
In a recent study of WES of DNA obtained from 17,182 people who did not have known hematologic disorders, somatic mutations were rare in persons younger than 40 years but rose appreciably in frequency with advanced age [44]. Among persons aged 70–79, 80–89, and 90–108 years, clonal mutations were observed in 9.5%, 11.7%, and 18.4%, respectively. The most common mutations were DNMT3A, TET2, and ASXL1. More importantly, the presence of a somatic mutation was associated with a significant increase in the risk of hematologic cancers (hazard ratio [HR], 11.1), all-cause mortality (HR, 1.4), incident coronary heart disease (HR, 2.0), and ischemic stroke (HR, 2.6) [44]. In similar study, investigators in Sweden analyzed WES of DNA in peripheral blood cells from 12,380 people who were unselected for cancer or hematologic phenotype [45]. Somatic mutations were found in 10% of people older than 65 years but only in 1% of those younger than 50 years. Similar to the previous study, mutations in DNMT3A, ASXL1, and TET2 were among the most commonly find mutations [45]. Furthermore, the presence of these mutations predispose these persons to develop hematologic malignancies. Interestingly, analysis of two patients at the time of diagnosis of AML showed that their leukemia arose from prior clones [45]. Other studies have also found similar findings [46, 47]. Given these findings of ambient somatic mutations in an unselected population, findings of genomic abnormalities in patients who do not have MDS must be interpreted with caution, and in and of themselves should not render an MDS diagnosis. The term “clonal hematopoiesis of indeterminate potential” has recently been proposed to describe individuals with hematologic malignancy-associated somatic mutation in blood or bone marrow but without other diagnostic criteria for hematologic malignancy [48].
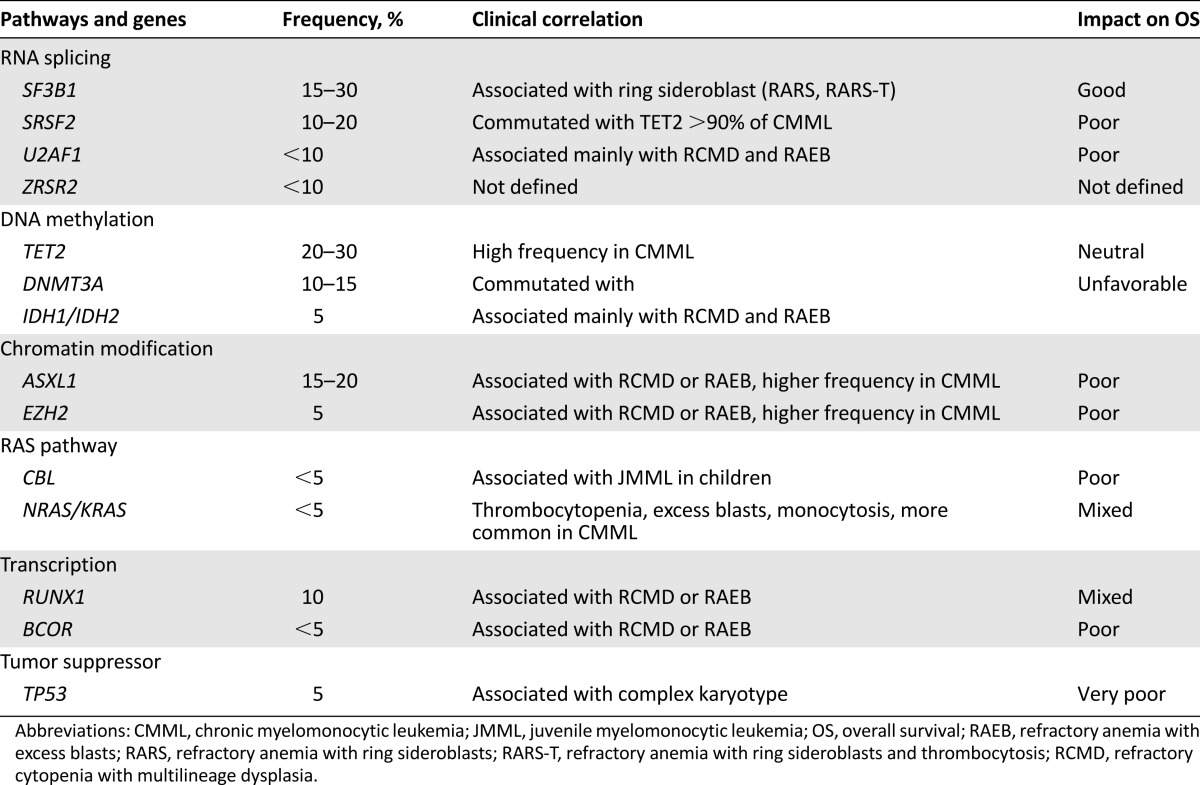
Genomic data can also be used to correlate genotype with disease phenotype. A recent study evaluated 308 patients with MDS, MDS/myeloproliferative neoplasm overlap, or AML evolving from MDS [8]. Unsupervised statistical analyses showed that MDS associated with SF3B1 mutation (20.8%) is a distinct entity irrespective of current morphologic classification criteria [8] (Table 3). Additionally, patients with concurrent mutations of TET2 and SRSF2 or ZRSR2 showed significantly higher hemoglobin levels (12.1 vs. 9.6 g/dL; p = .003) and higher monocyte counts (2.95 vs. 2.05 × 109/L; p = .017) compared with those without comutations [8]. This study highlights the importance of somatic gene mutations in the morphological classification of MDS and sets the stage for molecular abnormalities being incorporated into disease definitions.
Table 3.
Mutations and their clinical and prognostic relevance
Risk Stratification and Prognostic Utility of Genomic Data in MDS
Because MDS are associated with high interpatient variability in survival and risk of progression to AML, several prognostic models have been developed and validated to predict outcomes for individual patients. The most commonly used prognostic tools are the International Prognostic Scoring System (IPSS) [49], the World Health Organization (WHO) classification-based Prognostic Scoring System (WPSS) [50], the MD Anderson Prognostic Scoring System (MDAPSS) [51], and the revised IPSS (IPSS-R) [24]. All use clinicopathologic variables including BM blast percentage and cytopenias, in addition to cytogenetic analysis. The WPSS also includes transfusion dependence and WHO classification, whereas the MDAPSS adds transfusion dependence and performance status.
Existing Clinicopathologic Prognostic Tools in MDS Have Significant Limitations
A full discussion of the advantages and disadvantages of each prognostic model is beyond the scope of this review, and the interested reader is referred to recent comprehensive reviews [52–55]. The practical use of these risk stratification models is hampered by multiple challenges. With the exception of the MDAPSS, all have been developed and validated in patients with newly diagnosed MDS who were generally not treated with disease-modifying therapies, although some models have been subsequently validated in patients who received active therapies such as hypomethylating agents (HMAs) [56] and lenalidomide [57]. The application of these models in patients with HMA failure has not been validated. Importantly, several of these tools excluded important subsets of patients from their derivation process (e.g., therapy-related MDS). Furthermore, there is significant interpatient variability in outcomes within any risk group of each prognostic tool, and thus the degree to which the reported median outcome prediction applies to any individual patient is less reliable. Importantly, a significant subset of patients with IPSS low and intermediate-1 have been shown to exhibit significantly worse outcomes than predicted by the IPSS [58]. The WPSS also depends on an accurate WHO classification, which requires an experienced hematopathologist, and is further hampered by difficulties in reproducibility among hematopathologists [59].
Some Recurrent Molecular Mutations in MDS Carry Independent Prognostic Significance
In a landmark study, Bejar et al. [5] evaluated the presence of 18 gene mutations in bone marrow (BM) samples from 439 patients with MDS. At least one mutation was present in 50.1% of patients, and some mutations were associated with characteristic clinicopathologic features. For example, TET2 mutations were over-represented in samples with normal cytogenetic features, TP53 mutations were strongly associated with a complex karyotype, and mutations in RUNX1, TP53, and NRAS were each strongly associated with severe thrombocytopenia [5]. After adjusting for clinical variables, only six genes have negative impact on overall survival (OS): ASXL1, RUNX1, TP53, EZH2, CBL, and ETV6 [5].
In a larger study, samples from 944 MDS patients were evaluated for 104 gene mutations [7]. In total, 89.5% harbored at least one mutation, with each bone-marrow sample having a median of 3 (range, 0–12), including the 69.7% of cases who had normal karyotype. The six most frequently mutated genes were TET2, SF3B1, ASXL1, SRSF2, DNMT3A, and RUNX1 [7]. After grouping the mutations into several biological pathways, mutations in RNA splicing (SF3B1, SRSF2, ZRSR2, and UA2F1) and mutations in DNA methylation pathway (DNMT3A, TET2, ISH1, and IDH2) were among the most affected pathways. The majority of mutations occurred in patients with higher-risk MDS and, similar to prior reports, mutations in SF3B1 were found in 82.2% of patients with ring sideroblasts (RS) [7]. In univariate analysis, 25 of 48 genes affected survival (p < .05), and only SF3B1 mutations were associated with improved outcome [7].
In another important study [9], 111 genes were sequenced in 738 patients with MDS and closely related neoplasms. In total, 78% of patients had one or more oncogenic mutations identified in 43 genes. The most frequently mutated genes were: SF3B1 (24%), TET2 (22%), and SRSF2 (14%) [6]. Interestingly, only 4 mutations were present in >10% of patients. Mutational data again correlated with disease phenotype. SF3B1 mutations were associated with refractory anemia with RS, whereas SRSF2 was more frequent in chronic monocytic leukemia (CMML). Clonal architecture analyses showed that 62% of patients had only clonal driver mutations, and 34% had clonal and subclonal mutations. Furthermore, the data suggested that mutations in RNA splicing machinery and DNA methylation occurred early in the disease process, whereas mutations in chromatin modification and signaling occurred later [6]. Additionally, the number of driver mutations correlated with leukemia-free survival (LFS). Patients with 1 driver mutation or cytogenetic lesion had a LFS of 49 months compared with 42, 27, 18, and 4 months for patients with 2, 3, 4–5, and ≥6 mutations, respectively [6].
Efforts at Integrating Molecular Mutations in the Existing Prognostic Tools
Because several somatic mutations have been shown to have an impact on survival and correlate with specific clinicopathologic characteristics (Table 3), questions have arisen regarding the best way to combine the independent prognostic data derived from molecular mutations with the information derived from the traditional clinicopathologic parameters. Although these mutations enhance the prognostic utility of current clinicopathologic models, the optimal ways to incorporate the prognostic information derived from oncogenic mutations to the currently used risk stratification schemes in MDS have yet to be defined.
Many of the described mutations in MDS correlate with other clinicopathologic features such as WHO class, BM blast percentage, cytogenetics, and blood counts; incorporating these parameters into prognostic schemas may change the independent prognostic impact of these variables. For example, NRAS mutations are associated with increased BM blasts and thrombocytopenia; however, when these clinical characteristics are controlled for, the presence of this mutation does not appear to impact survival [5]. Thus, the mutations most likely to enhance the prognostic performance of existing tools are likely to be those without clear clinicopathologic phenotypic associations.
Many of the described mutations in MDS correlate with other clinicopathologic features such as WHO class, BM blast percentage, cytogenetics, and blood counts; incorporating these parameters into prognostic schemas may change the independent prognostic impact of these variables.
In one study, patients who had at least one of the mutations that were independently associated with OS had a worse OS compared with a patient in the same IPSS risk group without a mutation and were upstaged to the next highest IPSS prognostic group [5]. For example, one third of the patients with low- and intermediate-1-risk groups by IPSS who had a significant mutation had an OS similar to patient with intermediate-2 and high-risk group [5]. In another study, a novel prognostic model was developed by combining 14 prognostic mutated genes with age, gender, and IPSS-R variables [7]. Using this model, four risk groups were identified (low, intermediate, high, and very high risk), with significantly different 3-year OS rates of 95.2%, 69.3%, 32.8%, and 5.3%, respectively, p < .001 [7]. The authors then used genetic data only to build another model. The risk stratification of the combined model (genes and clinical variables) was only slightly better than the gene-only models, further suggesting the prognostic overlap between the genomic and clinical data.
Similarly, Papaemmanuil et al. [6] compared three data sets: the IPSS; a data set derived from all standard clinicopathologic variables (including peripheral blood [PB] counts, BM morphology, cytogenetics, and demographic data); and a data set that combines both sets of variables together. Incorporating genomic data achieved a marginal but not significant increase in the area under the curve of the combined model, suggesting a significant overlap between the two data sets. Furthermore, somatic mutations can play a role in risk stratification of subgroup of patients with MDS.
The impact of somatic mutations on prognosis among patients with low risk MDS stratified by the MD Anderson Lower-Risk Prognostic Scoring System (LR-PSS) was evaluated [60]. Mutations of EZH2, RUNX1, TP53, and ASXL1 were associated with shorter OS independent of the LR-PSS. Only EZH2 mutations retained prognostic significance in a multivariable analysis [60]. Similarly, researchers studied the impact of genetic mutations on outcome of patients with CMML. In a study of 312 patients with CMML, several mutations including ASXL1 SRSF2, CBL, and IDH2 predicted for inferior OS in univariable analyses [61]. In multivariate analyses, ASXL1 status, age, hemoglobin, white blood cell (WBC) count, and platelet counts defined three groups of CMML patients with median OS not reached, 38.5 months, and 14.4 months, respectively [61]. In another study, the prognostic impact of ASXL1 and SETBP1 mutations in 466 patients with CMML was evaluated [62]. In multivariable analyses, ASXL1 mutations, absolute monocyte count >10 × 109/L, hemoglobin < 10 g/dL, platelet count <100 × 109/L, and circulating immature myeloid cells identified four risk groups with median OS of 16, 31, 59, and 97 months, respectively [62]. These findings highlight the potential utility of combining the molecular and clinicopathologic parameters in prognostic models, and also the complexity of identifying molecular data not already accounted for by clinical parameters.
Prediction of Clinical Benefit From Specific MDS Therapies Using Genomic Data
The HMAs (azacitidine and decitabine) and lenalidomide are the only Food and Drug Administration-approved therapy for patients with MDS in the United States [1, 63, 64]. Treatment with HMAs improves cytopenias and can prolong survival for patients treated with azacitidine; however, only 40%–50% of patients respond to treatment with HMAs [65–68]. Additionally, six cycles of continuous treatment might be needed before a response is observed [69–71]. Thus, identification of biomarkers that can predict which patients are likely to respond to therapies can prevent prolonged exposure to ineffective therapy, decrease cost, and prevent unnecessary side effects.
Because the proposed mechanisms of action of HMAs involve reduction in methylation of cytosine residues in CpG islands in gene promoters by inhibiting DNA methyltransferese, correlating the presence of somatic mutations that affect DNA methylation pathways in MDS patients with clinical benefit from HMAs has been attempted by several groups [72]. In a small study of 82 patients with MDS and oligoblastic AML (20%–30% BM blasts) treated with azacitidine, TET2 mutations were found in 15% of the patients [73]. Patients with TET2 mutations had a higher response to azacitidine compared with wild-type patients (82% vs. 45%, p = .04, respectively); however, response duration and OS were similar between the two groups [73]. Furthermore, TET2 mutations predicted response to azacitidine independently from karyotype analysis [73].
In a larger retrospective study, samples from 213 patients with MDS were sequenced prior to their treatment with HMAs for the presence of 40 recurrent gene mutations [74]. The most frequently mutated genes were ASXL1 (46%), TET2 (27%), RUNX1 (20%), TP53 (18%), and DNMT3A (16%) followed by the splicing factor genes SRSF2 (16%), SF3B1 (15%), and U2AF1 (14%). TET2 mutant patients showed only a trend toward increased overall response rate (ORR) compared with WT TET2 (55% vs. 44%; odds ratio [OR], 1.58; 95% CI, 0.86–2.89, p = .14). No other mutated genes were associated with a significantly improved ORR in univariate analyses. However, in a revised analysis considering only TET2 mutations with variant allelic frequency (VAF) >10% (>20% for heterozygous mutations), TET2 mutations were associated with a significantly higher ORR compared with WT (60% vs. 43%; odds ratio [OR], 1.99; 95% confidence interval [CI], 1.05–3.80, p = .036; adjusted OR, 1.98; 95% CI, 1.02–3.85, p = .044) [74]. Furthermore, mutated TET2 and unmutated ASXL1 demonstrated an increased ORR compared with other mutational profiles (65% vs. 44%; OR, 2.37; 95% CI, 1.00–5.58, p = .049). This effect was more pronounced when mutations were required to have a VAF of ≥10% (74% vs. 44%; OR, 3.65; 95% CI, 1.38–9.67, p = .009), representing >10% of patients in this cohort. However, TET2 mutation status did not impact OS [74]. It should be noted that the frequency of ASXL1, TET2, and TP53 mutations in this study was higher compared with what was seen in previous studies that included larger number of patients [5–7]. In addition, the higher complete remission rate for this patient cohort (31%) compared with other studies, the lack of OS advantage with TET2 mutations, and the lack of other approved therapies that prolong OS for patients with higher-risk MDS aside from HMAs make the practical application of these results in the clinic uncertain.
Mutational analysis was performed on samples from 92 patients with MDS and related disorders who received azacitidine, decitabine, or both. In univariate analysis, the presence of TET2MUT, DNMT3AMUT, and/or IDH1/2MUT were associated with higher ORR compared with WT patients, and the ORR for patients with 2 versus 1 versus 0 gene mutations were 75% versus 25% versus 20%. In multivariate analysis and after accounting for clinical variables, TET2MUT and/or DNMT3AMUT, platelet count, and white blood cell count were found to be independent predictors of ORR [75]. The authors also developed a scoring system based on the number of poor features present (TET2WT and DNMT3AWT, platelets <100 × 109/L, and WBC ≥ 3.0 × 109/L). Based on this scoring system, three groups were identified with ORRs of 43%, 23%, and 0% for patients with 0/1 versus 2 versus 3 poor features, respectively [75].
One of the most difficult decision points in treating a patient with MDS is when to recommend allogeneic hematopoietic cell transplantation (alloHCT). Massive parallel sequencing was used to evaluate samples from 87 patients with MDS prior to alloHCT in an attempt to correlate pre-alloHCT genetic mutation status with post-transplantation outcomes [76]. In univariable analyses, only TP53 mutations were associated with shorter OS (HR, 3.74; p < .001) and progression-free survival (HR, 3.97; p < .001). After adjustment for clinical variables associated with these end points, mutations in TP53 (HR, 2.30; p = .027), TET2 (HR, 2.40; p = .033), and DNMT3A (HR, 2.08; p = .049) were associated with decreased OS [76]. More importantly, patients with TP53 mutation had very poor outcomes with alloHCT, raising the question of whether these patients should be offered alloHCT at all. Equally important, patients with MDS and complex karyotype have been well known to have very poor outcomes. However, recent evidence suggests that TP53 mutation, which is present in approximately 50% of patients with complex karyotype MDS, may in fact drive the poor outcome in those patients, because patients with complex karyotype who do not carry TP53 mutations have better outcomes approaching patients without complex karyotype, suggesting that the multiple cytogenetic abnormalities in patients with TP53 is a secondary phenomenon related to genomic instability [77]. Furthermore, the negative impact of TP53 mutations is also seen in patients with isolated del(5q) (present in approximately 20% of patients) [78], in whom TP53 mutations confer lenalidomide resistance and worse survival [79]. The evidence regarding very poor outcomes of MDS patients with TP53 mutations treated with HMAs or alloHCT suggests these therapies do not abrogate the negative prognostic effect of these mutations, and therefore early referral of these patients for clinical trials should be considered when feasible.
The evidence regarding very poor outcomes of MDS patients with TP53 mutations treated with HMAs or alloHCT suggests these therapies do not abrogate the negative prognostic effect of these mutations, and therefore early referral of these patients for clinical trials should be considered when feasible.
Conclusion
Heterogeneity in clinical outcomes of MDS is partly related to interpatient variability of recurrent somatic mutations that drive disease phenotype and progression. Although clinical risk stratification tools have functioned well in prognostication for patients with MDS, their ability to predict clinical benefits of specific MDS therapies is limited. Molecular testing shows promise in aiding diagnosis, risk stratification, and therapy-specific benefit prediction for MDS patients. In addition, testing for molecular mutations and alterations can guide clinical trial referrals, especially after failure of HMAs [80]. For example, patients with MLL rearrangements, which are rare but confer a poor prognosis, could be considered for clinical trials using bromodomain inhibitors or Dot1L inhibitors, because preclinical evidence suggests particular sensitivity of myeloid malignancies carrying these mutations to these experimental agents [81, 82]. Similarly, although the prognostic impact of IDH mutations in MDS is less clear, testing for these mutations should be considered as evidence suggesting they are important in pathogenesis of IDH-mutant myeloid malignancies, and their inhibitors have also entered early clinical trials [83–85]. In addition to molecular mutations, other predictive biomarkers (e.g., methylation signatures, UCK1 enzyme expression) are also under investigation [86–89]. Nonetheless, logistical issues related to assay performance standardization, validation, interpretation, and development of guidelines for how to use the results to inform clinical decisions are yet to be resolved [88–91]. Success at these efforts will likely require the use of very large, international, well-annotated data sets with comprehensive paired clinicopathologic and mutational data [92–94].
This article is available for continuing medical education credit at CME.TheOncologist.com.
Author Contributions
Manuscript writing: Aziz Nazha, Mikkael A. Sekeres, Steven D. Gore, Amer M. Zeidan
Disclosures
Steven D. Gore: Celgene, Kyowa, Sunesis, Boehringer Ingelheim (C/A), Celgene (RF). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Garcia-Manero G. Myelodysplastic syndromes: 2014 update on diagnosis, risk-stratification, and management. Am J Hematol. 2014;89:97–108. doi: 10.1002/ajh.23642. [DOI] [PubMed] [Google Scholar]
- 2.Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med. 2009;361:1872–1885. doi: 10.1056/NEJMra0902908. [DOI] [PubMed] [Google Scholar]
- 3.Cazzola M, Della Porta MG, Travaglino E, et al. Classification and prognostic evaluation of myelodysplastic syndromes. Semin Oncol. 2011;38:627–634. doi: 10.1053/j.seminoncol.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 4.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood. 2009;114:937–951. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- 5.Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–2506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papaemmanuil E, Gerstung M, Malcovati L, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–3627; quiz 3699. doi: 10.1182/blood-2013-08-518886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–247. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malcovati L, Papaemmanuil E, Ambaglio I, et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood. 2014;124:1513–1521. doi: 10.1182/blood-2014-03-560227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cazzola M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and its clinical relevance. Blood. 2013;122:4021–4034. doi: 10.1182/blood-2013-09-381665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 11.Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 12.International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 13.Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–197. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- 14.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 16.Bohlander SK. ABCs of genomics. Hematology. American Society of Hematology Education Program. 2013;2013:316–323. doi: 10.1182/asheducation-2013.1.316. [DOI] [PubMed] [Google Scholar]
- 17.Deeg HJ, Scott BL, Fang M, et al. Five-group cytogenetic risk classification, monosomal karyotype, and outcome after hematopoietic cell transplantation for MDS or acute leukemia evolving from MDS. Blood. 2012;120:1398–1408. doi: 10.1182/blood-2012-04-423046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haase D, Germing U, Schanz J, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: Evidence from a core dataset of 2124 patients. Blood. 2007;110:4385–4395. doi: 10.1182/blood-2007-03-082404. [DOI] [PubMed] [Google Scholar]
- 19.Schanz J, Steidl C, Fonatsch C, et al. Coalesced multicentric analysis of 2,351 patients with myelodysplastic syndromes indicates an underestimation of poor-risk cytogenetics of myelodysplastic syndromes in the international prognostic scoring system. J Clin Oncol. 2011;29:1963–1970. doi: 10.1200/JCO.2010.28.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schanz J, Tüchler H, Solé F, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30:820–829. doi: 10.1200/JCO.2011.35.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caspersson T, Farber S, Foley GE, et al. Chemical differentiation along metaphase chromosomes. Exp Cell Res. 1968;49:219–222. doi: 10.1016/0014-4827(68)90538-7. [DOI] [PubMed] [Google Scholar]
- 22.Cazzola M, Malcovati L. Myelodysplastic syndromes: Coping with ineffective hematopoiesis. N Engl J Med. 2005;352:536–538. doi: 10.1056/NEJMp048266. [DOI] [PubMed] [Google Scholar]
- 23.Lindsley RC, Ebert BL. The biology and clinical impact of genetic lesions in myeloid malignancies. Blood. 2013;122:3741–3748. doi: 10.1182/blood-2013-06-460295. [DOI] [PubMed] [Google Scholar]
- 24.Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–2465. doi: 10.1182/blood-2012-03-420489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coleman JF, Theil KS, Tubbs RR, et al. Diagnostic yield of bone marrow and peripheral blood FISH panel testing in clinically suspected myelodysplastic syndromes and/or acute myeloid leukemia: A prospective analysis of 433 cases. Am J Clin Pathol. 2011;135:915–920. doi: 10.1309/AJCPW10YBRMWSWYE. [DOI] [PubMed] [Google Scholar]
- 26.Maciejewski JP, Tiu RV, O’Keefe C. Application of array-based whole genome scanning technologies as a cytogenetic tool in haematological malignancies. Br J Haematol. 2009;146:479–488. doi: 10.1111/j.1365-2141.2009.07757.x. [DOI] [PubMed] [Google Scholar]
- 27.Maciejewski JP, Mufti GJ. Whole genome scanning as a cytogenetic tool in hematologic malignancies. Blood. 2008;112:965–974. doi: 10.1182/blood-2008-02-130435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Keefe CL, Tiu R, Gondek LP, et al. High-resolution genomic arrays facilitate detection of novel cryptic chromosomal lesions in myelodysplastic syndromes. Exp Hematol. 2007;35:240–251. doi: 10.1016/j.exphem.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmad A, Iqbal MA. Significance of genome-wide analysis of copy number alterations and UPD in myelodysplastic syndromes using combined CGH-SNP arrays. Curr Med Chem. 2012;19:3739–3747. doi: 10.2174/092986712801661121. [DOI] [PubMed] [Google Scholar]
- 30.Afable MG, 2nd, Wlodarski M, Makishima H, et al. SNP array-based karyotyping: Differences and similarities between aplastic anemia and hypocellular myelodysplastic syndromes. Blood. 2011;117:6876–6884. doi: 10.1182/blood-2010-11-314393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zagursky RJ, Berman ML. Cloning vectors that yield high levels of single-stranded DNA for rapid DNA sequencing. Gene. 1984;27:183–191. doi: 10.1016/0378-1119(84)90139-2. [DOI] [PubMed] [Google Scholar]
- 33.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu W, O’Connor TD, Jun G, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coffey AJ, Kokocinski F, Calafato MS, et al. The GENCODE exome: Sequencing the complete human exome. Eur J Hum Genet. 2011;19:827–831. doi: 10.1038/ejhg.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steijger T, Abril JF, Engström PG, et al. Assessment of transcript reconstruction methods for RNA-seq. Nat Methods. 2013;10:1177–1184. doi: 10.1038/nmeth.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 39.Young NS, Maciejewski J. The pathophysiology of acquired aplastic anemia. N Engl J Med. 1997;336:1365–1372. doi: 10.1056/NEJM199705083361906. [DOI] [PubMed] [Google Scholar]
- 40.Gondek LP, DeZern AE. I walk the line: How to tell MDS from other bone marrow failure conditions. Curr Hematol Malig Rep. 2014;9:389–399. doi: 10.1007/s11899-014-0224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kulasekararaj AG, Jiang J, Smith AE, et al. Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplastic syndrome. Blood. 2014;124:2698–2704. doi: 10.1182/blood-2014-05-574889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brian Kwok PR, Lin K, Flamholz R et al. Next-Generation Sequencing (NGS)-Based Profiling of Idiopathic Cytopenia of Undetermined Significance (ICUS) Identifies a Subset of Patients with Genomic Similarities to Lower-Risk Myelodysplastic Syndrome (MDS). Paper presented at: American Society of Hematology 56th Annual Meeting; December 6–9, 2014; San Francisco, CA.
- 43.Cargo CNR, Evans P, Barrans S et al. Early Diagnosis of Myelodysplastic Syndromes Can be Improved By Deep Sequencing and Array Based Cytogenetics. Paper presented at: American Society of Hematology 56th Annual Meeting; December 6–9, 2014; San Francisco, CA.
- 44.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McKerrell T, Park N, Moreno T, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Reports. 2015;10:1239–1245. doi: 10.1016/j.celrep.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015:pii. doi: 10.1182/blood-2015-03-631747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079–2088. [PubMed] [Google Scholar]
- 50.Malcovati L, Germing U, Kuendgen A, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol. 2007;25:3503–3510. doi: 10.1200/JCO.2006.08.5696. [DOI] [PubMed] [Google Scholar]
- 51.Kantarjian H, O’Brien S, Ravandi F, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer. 2008;113:1351–1361. doi: 10.1002/cncr.23697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zeidan AM, Gore SD, Padron E, et al. Current state of prognostication and risk stratification in myelodysplastic syndromes. Curr Opin Hematol. 2015;22:146–154. doi: 10.1097/MOH.0000000000000110. [DOI] [PubMed] [Google Scholar]
- 53.Faltas B, Zeidan A, Gergis U. Myelodysplastic syndromes: Toward a risk-adapted treatment approach. Expert Rev Hematol. 2013;6:611–624. doi: 10.1586/17474086.2013.840997. [DOI] [PubMed] [Google Scholar]
- 54.Zeidan AM, Smith BD, Komrokji RS, et al. Prognostication in myelodysplastic syndromes: Beyond the International Prognostic Scoring System (IPSS) Am J Med. 2013;126:e25. doi: 10.1016/j.amjmed.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bejar R. Prognostic models in myelodysplastic syndromes. Hematology. American Society of Hematology Education Program. 2013;2013:504–510. doi: 10.1182/asheducation-2013.1.504. [DOI] [PubMed] [Google Scholar]
- 56.Lamarque M, Raynaud S, Itzykson R, et al. The revised IPSS is a powerful tool to evaluate the outcome of MDS patients treated with azacitidine: The GFM experience. Blood. 2012;120:5084–5085. doi: 10.1182/blood-2012-09-453555. [DOI] [PubMed] [Google Scholar]
- 57.Sekeres MA, Swern AS, Fenaux P, et al. Validation of the IPSS-R in lenalidomide-treated, lower-risk myelodysplastic syndrome patients with del(5q) Blood Cancer J. 2014;4:e242. doi: 10.1038/bcj.2014.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garcia-Manero G, Shan J, Faderl S, et al. A prognostic score for patients with lower risk myelodysplastic syndrome. Leukemia. 2008;22:538–543. doi: 10.1038/sj.leu.2405070. [DOI] [PubMed] [Google Scholar]
- 59.Naqvi K, Jabbour E, Bueso-Ramos C, et al. Implications of discrepancy in morphologic diagnosis of myelodysplastic syndrome between referral and tertiary care centers. Blood. 2011;118:4690–4693. doi: 10.1182/blood-2011-03-342642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bejar R, Stevenson KE, Caughey BA, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol. 2012;30:3376–3382. doi: 10.1200/JCO.2011.40.7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Itzykson R, Kosmider O, Renneville A, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31:2428–2436. doi: 10.1200/JCO.2012.47.3314. [DOI] [PubMed] [Google Scholar]
- 62.Patnaik MM, Itzykson R, Lasho TL, et al. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: A two-center study of 466 patients. Leukemia. 2014;28:2206–2212. doi: 10.1038/leu.2014.125. [DOI] [PubMed] [Google Scholar]
- 63.Sekeres MA, Cutler C. How we treat higher-risk myelodysplastic syndromes. Blood. 2014;123:829–836. doi: 10.1182/blood-2013-08-496935. [DOI] [PubMed] [Google Scholar]
- 64.Fenaux P, Ades L. Review of azacitidine trials in Intermediate-2-and High-risk myelodysplastic syndromes. Leuk Res. 2009;33(suppl 2):S7–S11. doi: 10.1016/S0145-2126(09)70227-9. [DOI] [PubMed] [Google Scholar]
- 65.List AF, Fenaux P, Mufti GJ, et al. Effect of azacitidine (AZA) on overall survival in higher-risk myelodysplastic syndromes (MDS) without complete remission. J Clin Oncol. 2008;26:7006a. [Google Scholar]
- 66.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–232. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: Results of a phase III randomized study. Cancer. 2006;106:1794–1803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 68.Kantarjian HM, O’Brien S, Huang X, et al. Survival advantage with decitabine versus intensive chemotherapy in patients with higher risk myelodysplastic syndrome: Comparison with historical experience. Cancer. 2007;109:1133–1137. doi: 10.1002/cncr.22508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Silverman LR, Fenaux P, Mufti GJ, et al. Continued azacitidine therapy beyond time of first response improves quality of response in patients with higher-risk myelodysplastic syndromes. Cancer. 2011;117:2697–2702. doi: 10.1002/cncr.25774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gore SD, Hermes-DeSantis ER. Enhancing survival outcomes in the management of patients with higher-risk myelodysplastic syndromes. Cancer Control. 2009;16(suppl):2–10. doi: 10.1177/107327480901604s03. [DOI] [PubMed] [Google Scholar]
- 71.Gore SD, Fenaux P, Santini V, et al. A multivariate analysis of the relationship between response and survival among patients with higher-risk myelodysplastic syndromes treated within azacitidine or conventional care regimens in the randomized AZA-001 trial. Haematologica. 2013;98:1067–1072. doi: 10.3324/haematol.2012.074831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123:8–13. doi: 10.1002/ijc.23607. [DOI] [PubMed] [Google Scholar]
- 73.Itzykson R, Kosmider O, Cluzeau T, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25:1147–1152. doi: 10.1038/leu.2011.71. [DOI] [PubMed] [Google Scholar]
- 74.Bejar R, Lord A, Stevenson K, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014;124:2705–2712. doi: 10.1182/blood-2014-06-582809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Traina F, Visconte V, Elson P, et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia. 2014;28:78–87. doi: 10.1038/leu.2013.269. [DOI] [PubMed] [Google Scholar]
- 76.Bejar R, Stevenson KE, Caughey B, et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J Clin Oncol. 2014;32:2691–2698. doi: 10.1200/JCO.2013.52.3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bejar RPE, Haferlach T. TP53 mutation status divides MDS patients with complex karyotypes into distinct prognostic risk groups: Analysis of combined datasets from the International Working Group for MDS-Molecular Prognosis Committee. Blood. 2014;124:532a. [Google Scholar]
- 78.Kulasekararaj AG, Smith AE, Mian SA, et al. TP53 mutations in myelodysplastic syndrome are strongly correlated with aberrations of chromosome 5, and correlate with adverse prognosis. Br J Haematol. 2013;160:660–672. doi: 10.1111/bjh.12203. [DOI] [PubMed] [Google Scholar]
- 79.Saft L, Li JS, Greenberg PL, et al. p53 mutant independently impacts risk: Analysis of deletion 5q, lower-risk myelodysplastic syndromes (MDS) patients treated with lenalidomide (LEN) in the MDS-004 Study. Blood. 2014;124:414a. [Google Scholar]
- 80.Zeidan AM, Komrokji RS. There’s risk, and then there’s risk: The latest clinical prognostic risk stratification models in myelodysplastic syndromes. Curr Hematol Malig Rep. 2013;8:351–360. doi: 10.1007/s11899-013-0172-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dilsaver SC, DelMedico VJ, Quadri AB. Lithium-induced worsening of wintertime depression in a bipolar patient. J Clin Psychiatry. 1990;51:347–348. [PubMed] [Google Scholar]
- 82.Deshpande AJ, Bradner J, Armstrong SA. Chromatin modifications as therapeutic targets in MLL-rearranged leukemia. Trends Immunol. 2012;33:563–570. doi: 10.1016/j.it.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stein EMAJ, Collins R, et al. AG-221, an oral, selective, first-in-class, potent inhibitor of the IDH2 mutant metabolic enzyme, induces durable remissions in a phase I study in patients with IDH2 mutation positive advanced hematologic malignancies. Blood. 2014;124:437. [abstract 115] [Google Scholar]
- 84.Chaturvedi A, Araujo Cruz MM, Goparaju R, et al. A novel inhibitor of mutant IDH1 induces differentiation in vivo and prolongs survival in a mouse model of leukemia. Blood. 2014;124:3598a. [Google Scholar]
- 85.Kon Kim T, Gore SD, Zeidan AM. Epigenetic therapy in acute myeloid leukemia: Current and future directions. Semin Hematol. 2015;52:172–183. doi: 10.1053/j.seminhematol.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Valencia A, Masala E, Rossi A, et al. Expression of nucleoside-metabolizing enzymes in myelodysplastic syndromes and modulation of response to azacitidine. Leukemia. 2014;28:621–628. doi: 10.1038/leu.2013.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Voso MT, Santini V, Fabiani E, et al. Why methylation is not a marker predictive of response to hypomethylating agents. Haematologica. 2014;99:613–619. doi: 10.3324/haematol.2013.099549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zeidan AM, Lee JW, Prebet T, et al. Platelet count doubling after the first cycle of azacitidine therapy predicts eventual response and survival in patients with myelodysplastic syndromes and oligoblastic acute myeloid leukaemia but does not add to prognostic utility of the revised IPSS. Brit J Haematol. 2014;167:62–68. doi: 10.1111/bjh.13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meldi K, Qin T, Buchi F, et al. Specific molecular signatures predict decitabine response in chronic myelomonocytic leukemia. J Clin Invest. 2015;125:1857–1872. doi: 10.1172/JCI78752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zeidan AM, Lee JW, Prebet T, et al. Comparison of the prognostic utility of the revised International Prognostic Scoring System and the French Prognostic Scoring System in azacitidine-treated patients with myelodysplastic syndromes. Brit J Haematol. 2014;166:352–359. doi: 10.1111/bjh.12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zeidan AM, Linhares Y, Gore SD. Current therapy of myelodysplastic syndromes. Blood Rev. 2013;27:243–259. doi: 10.1016/j.blre.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zeidan AM, Prebet T, Saad Aldin E, et al. Risk stratification in myelodysplastic syndromes: Is there a role for gene expression profiling? Expert Rev Hematol. 2014;7:191–194. doi: 10.1586/17474086.2014.891437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee EJ, Zeidan AM. Genome sequencing in myelodysplastic syndromes: can molecular mutations predict benefit from hypomethylating agent therapy? Expert Rev Hematol. 2015;8:155–158. doi: 10.1586/17474086.2015.1016905. [DOI] [PubMed] [Google Scholar]
- 94.Lee EJ, Podoltsev N, Gore SD, et al. The evolving field of prognostication and risk stratification in MDS: Recent developments and future directions. Blood Rev. 2015 doi: 10.1016/j.blre.2015.06.004. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]