

Abstract
Gene therapy is a promising modality for the treatment of inherited and acquired cardiovascular diseases. The identification of the molecular pathways involved in the pathophysiology of heart failure and other associated cardiac diseases led to encouraging preclinical gene therapy studies in small and large animal models. However, the initial clinical results yielded only modest or no improvement in clinical endpoints. The presence of neutralizing antibodies and cellular immune responses directed against the viral vector and/or the gene-modified cells, the insufficient gene expression levels, and the limited gene transduction efficiencies accounted for the overall limited clinical improvements. Nevertheless, further improvements of the gene delivery technology and a better understanding of the underlying biology fostered renewed interest in gene therapy for heart failure. In particular, improved vectors based on emerging cardiotropic serotypes of the adeno-associated viral vector (AAV) are particularly well suited to coax expression of therapeutic genes in the heart. This led to new clinical trials based on the delivery of the sarcoplasmic reticulum Ca2+-ATPase protein (SERCA2a). Though the first clinical results were encouraging, a recent Phase IIb trial did not confirm the beneficial clinical outcomes that were initially reported. New approaches based on S100A1 and adenylate cyclase 6 are also being considered for clinical applications. Emerging paradigms based on the use of miRNA regulation or CRISPR/Cas9-based genome engineering open new therapeutic perspectives for treating cardiovascular diseases by gene therapy. Nevertheless, the continuous improvement of cardiac gene delivery is needed to allow the use of safer and more effective vector doses, ultimately bringing gene therapy for heart failure one step closer to reality.
Keywords: Gene therapy, Cardiovascular disease, Heart failure, Clinical trials, Adeno-associated viral vector, SERCA2a, S100A1, Adenylate cyclase, miRNA, CRISPR
1. Introduction
Cardiovascular disease (CVD) remains a major public health problem with increasing prevalence, poor clinical outcomes, and large health care costs. Approximately 1–2% of the adult population in developed countries suffer from heart failure (HF), with prevalence rising to ≥10% among individuals aged 70 years or older.1 While pharmacological2 and invasive therapies for CVD achieve symptom reduction and slow disease progression, there is still an urgent need for alternative therapeutic approaches to effectively treat or even cure CVD and HF. The molecular pathways and causative genes involved in the induction and progression of CVD have been elucidated through advances in molecular cardiology.3 These emerging insights pave the way towards the use of gene therapy as a novel treatment modality for CVD and HF,3 fueled by the emerging clinical successes in the field at large.4
Depending on the underlying CVD, short-term or sustained expression of a given therapeutic gene may be required that in turn determines the type of gene delivery vector needed to achieve the desired therapeutic effect5 (Table 1). For instance, short-term expression may suffice to induce vasculogenenesis for the treatment of myocardial infarction (MI), whereas more sustained expression may be required to achieve left ventricular remodelling in HF. The obvious target cells for CVD gene therapy include cardiomyocytes and endothelial cells/smooth muscle cells. However, distal cell types can also be considered (i.e. skeletal muscle, liver) that could be used for the systemic release of paracrine factors.6–8
Table 1.
Plasmids and viral vectors for CVD
| Delivery method | Plasmid | AAV | Lentivirus | Ad |
|---|---|---|---|---|
| Schematic diagram |  |
 |
 |
 |
| Diameter (nM) | N/A | 20 | 80–100 | 70–90 |
| Genome/Size (Kb) | DNA/N/A | (ds)ssDNA/±4,8 | RNA/±10 | dsDNA/±36 |
| Cardiac gene transfer | Low cardiac transfection | Cardiotropic AAV serotypes | Low cardiac transduction | High cardiac transduction |
| Duration of expression | Expression up to 2 months | Long-term cardiac expression | Long-term cardiac expression | Expression up to 2 weeks |
| Major disadvantage | Low transfection efficiency | Risk of neutralizing antibodies and T-cell responses | Risk of insertional mutagenesis | High antibody and inflammatory response |
| Used in clinical trials for CVD | + | + | − | + |
In this review, we will focus on gene therapy approaches that have been explored in clinical trials or are approaching clinical translation. Since the use of miRNA for modulating heart function has been extensively reviewed elsewhere,9 this falls out of the scope of this review.
2. Gene therapy and CVDs
2.1. Non-viral vectors for CVD
Despite the initial promising results based on non-viral vectors (mainly plasmid transfections) and the hype that surrounded the initial trials, most recent studies have shown that this approach is generally not very efficient to treat CVD by gene therapy.10,11 Different strategies have been developed to improve its overall efficiency such as the use of liposome-DNA complexes that increase plasmid stability, though the plasmid is rapidly cleared from the systemic circulation.12 Polymer-based DNA complexes based on poly-l-lysine (PLL) and polyethyleneimine (PEI) products protect plasmids from nuclease digestion and facilitate cellular uptake;10 however, when used in vivo, these complexes tend to aggregate and accumulate in different tissues. Electroporation and the use of micro-bubbles as echo contrast agents have also been explored but with variable results.13 The ultrasound-targeted micro-bubbles (UTM) strategy has gained interest as an alternative delivery strategy for CVD due to its intrinsic low levels of toxicity and immunogenicity, which is compounded by the potential for re-administration and organ-specific delivery of the genes of interest.14 Recent reports have shown benefits in MI and HF animal models, using UTM for targeted delivery of DNA or microRNA. In particular, lipid micro-bubbles carrying VEGF and stem cell factor (SCF) genes significantly improved myocardial perfusion and ventricular function after coronary artery ligation in rodent models.15 Similarly, repeated UTM-based delivery of SCF and stromal cell-derived factor (SDF)-1α genes in a rat model of MI resulted in an increased vascular density, improved myocardial function, and reduced infarct size.16 UTM was also well suited to enhance delivery of microRNAs to cardiomyocytes without discernable toxicity. In particular, UTM-mediated delivery of miR-133 in cardiomyocytes in vitro led to a reversal of hypertrophy.17 One of the challenges consists of translating these findings to large animal models and ultimately to the clinic, which is compounded by the relative low efficiency and/or short-term gene expression.
2.2. Viral vectors for CVD
Viral vectors consist of genetic material surrounded by a protein-based capsid or a lipidic envelope that interacts with specific cell surface receptors to aid binding, internalization, and delivery of the therapeutic gene into the target cell.18 The capsid or envelope protein directs trafficking of the therapeutic gene towards the nucleus and protects it from degradation in the lysosomes.4 In general, viral vectors are more efficient than non-viral vectors and have the potential for long-term gene expression (Table 1). Viral vectors resemble the parental viruses from which they have been derived except for the lack of viral genes in the vector backbone. With the exception of early-generation adenoviral vectors (see below), that retain some residual viral genes in their backbone, all other vector types are devoid of viral genes. Consequently, vector manufacturing is more challenging than when non-viral vectors are employed and requires packaging cells that complement the missing viral genes in trans. The clinical use of viral vectors also raises important regulatory challenges.19 Depending on the type of viral vector used, the immune response to the vector and/or the gene-modified cells may be a limiting factor.20 Vector-specific immune responses can preclude gene transfer after vector re-administration and/or limit the duration of gene expression or result in immune clearance of gene-modified cells.
2.2.1. Adenoviral vectors
Adenoviral (Ad) vectors are non-enveloped, non-integrating double-stranded (ds)DNA vectors that enter the cells, predominantly via clathrin-mediated endocytosis upon binding with coxsackie-adenovirus receptor (CAR). The dsDNA is subsequently transported to the nucleus allowing efficient transduction of a wide range of dividing and non-dividing cells, including cardiomyocytes, skeletal muscle, or smooth muscle cells.21–23 In the heart, transgene expression after Ad vectors transduction is robust but transient (1–2 weeks),24,25 which limits its applications in CVD for HF. Nevertheless, it is a useful system for short-term pro-angiogenic therapies in ischaemic heart disease,26 peripheral arterial occlusive disease, and limb ischaemia.27 One major disadvantage of Ad vectors relates to their ability to induce inflammation, which compromise their efficacy and safety in clinical trials.28,29 In particular, the early-generation Ad vectors contain residual adenoviral genes in the vector backbone that can be induced in vivo and trigger T-cell-mediated immune responses that eliminate the gene-modified cells. The latest generation Ad vectors exhibit decreased T-cell immune responses by eliminating all of the residual viral genes (i.e. gutless or helper-dependent Ad vectors) expanding the cargo capacity to 30 kb.30 Nevertheless, both early- and late-generation Ad vector particles can rapidly activate the innate immune system contributing to significant dose-limiting toxicity.31 Though catheter-mediated localized delivery in the myocardium may minimize this risk,32 the intrinsic risks associated with immune system activation remain. This risk is compounded by the broad tropism of Ad vectors resulting in ectopic transduction of non-target cells (e.g. hepatocytes, antigen-presenting cells).33 Consequently, the utility of Ad vectors in cardiovascular gene therapy trials in humans must be carefully evaluated.
Recombinant vectors derived from the serotype 5 adenovirus (Ad5) have been predominantly used in preclinical and clinical trials in gene therapy for CVD.34 The CAR is the primary cell surface receptor for Ad5, though other cellular co-receptors are also implicated in vector entry (i.e. integrins). CAR is highly expressed on cardiomyocytes, whereas its expression is reduced in vascular smooth muscle and endothelial cells. This impacts on the transduction efficiency in these different cell types after systemic administration.35 Although Ad vectors cannot easily cross the endothelial barrier after systemic administration, it has been reported that Ad vectors can selectively transduce endothelial cells after local administration.36 Additionally, Ad vectors also achieve high levels of myocardial transduction after local delivery, either by intracoronary infusion or by direct intramyocardial injection.37 The transduction efficiency varies depending on the Ad serotype. In particular, Ad serotype 49 (Ad49) showed increased transduction of endothelial cells and smooth muscle cells in vitro and in vascular graft ex vivo.38
2.2.2. Adeno-associated viral vectors
Adeno-associated viral vectors (AAV) are single-stranded (ss)DNA vectors with a favourable safety profile and capable of achieving persistent transgene expression in a wide range of target tissues, including the heart.39 Since they provoke much less inflammation compared with Ad vectors, they have garnered a lot of interest for cardiac gene therapy applications. Long-term expression is predominantly mediated by episomally retained, non-integrated AAV genomes, mostly organized as high molecular weight concatemers.40 More than 100 serotypes of the wild-type AAV have been reported.41 Many exhibit a distinct tissue tropism that is determined by the capsid protein structure.3 AAV1, AAV6, AAV8, and AAV9 have been identified as the most cardiotropic serotypes after systemic delivery.42 These distinct serotypes predominantly transduce cardiomyocytes. However, the extent of AAV transduction in other cardiac cells (e.g. fibroblasts) has not been carefully examined. We and others demonstrated that AAV9 was the most efficient serotype for cardiac gene delivery, at least in mice.40,43,44 Recent studies using mouse models have reported a >200-fold increase in myocardial transduction efficiency after only a single intravenous dose of AAV9 compared with AAV1, confirming the superiority of AAV9 for cardiac gene transfer.44 The ability of certain AAV serotypes to efficiently transduce cardiomyocytes following intravenous vector administration suggests that these AAV vectors are capable of transcytosis, depending on the serotype.45 Though direct intramyocardial AAV injection may also result in transduction of cardiomyocytes, its effect is typically more localized and restricted to the injection site itself.46 However, the superior cardiotropic properties of AAV9 in mouse models may not necessarily translate to larger animal models and ultimately to human subjects and may depend—at least in part—on the delivery method (e.g. anteriograde vs. retrograde coronary artery delivery) warranting further preclinical studies (Table 2).47,48 Overall, transduction of smooth muscle cells and endothelial cells in blood vessels using AAV has been relatively modest, even after using retargeted, peptide-modified AAV.49–51
Table 2.
Gene therapy delivery strategies for CVD targeting the heart
| Delivery method | Procedures | Indications | Advantages | Disadvantages |
|---|---|---|---|---|
| Catheter-based delivery methods | ||||
| Anterograde arterial infusion | a. Intracoronary perfusion | Patients with unstable and advanced heart failure |
|
|
| b. Intracoronary perfusion + Balloon occlusion | ||||
| c. Intracoronary perfusion + Balloon occlusion + Venous occlusion | ||||
| Retrograde intravenous | a. Intravenous perfusion + Venous occlusion | Patients with impaired coronary artery circulation and limited potential for revascularization |
|
|
| Direct intramyocardial injection | a. Percutaneous for endocardial delivery | Therapeutic angiogenesis and focal arrhythmia therapy when restricted area is needed |
|
|
| b. Surgically invasive intramyocardial administration | ||||
| c. NOGA system | ||||
| Pericardial delivery | a. Percutaneous approach |
|
|
|
Despite their increased cardiotropism, AAV9 transduction is not restricted to the myocardium, since other tissues, including liver and skeletal muscle can also be transduced.40,52 To improve the cardiac specificity of AAV, AAV2 variants were selected that exhibit different receptor-binding properties, with improved transduction profiles and the ability to at least partially evade neutralizing antibodies53 (Figure 1). One particular approach consisted of constructing a random viral display peptide AAV library allowing subsequent in vivo selection of cardiotropic AAV variants.54 Alternatively, using an AAV cap gene library produced by DNA shuffling of different AAV serotype capsid genes, Yang et al. obtained a myocardium-tropic AAV strain, AAVM41, through direct evolution strategies and DNA shuffling. This variant exhibited enhanced transduction to cardiac muscle and diminished tropism to the liver after systemic administration.55 Finally, Samulski et al. replaced a hexapeptide in a previously identified heparan sulfate receptor footprint sequence from an AAV2 vector with corresponding residues from other AAV strains. Consequently, such an AAV2/AAV8 chimera designated AAV2i8 selectively transduced cardiac and whole-body skeletal muscle tissues, while exhibiting significantly reduced hepatic tropism.56 Such liver-detargeted AAV vectors could also be obtained by random mutagenesis of residues within a surface-exposed region of the major AAV9 capsid protein.57 Using a combination of sequence analysis, structural models, and in vivo screening, several functionally diverse AAV9 variants were identified, notably, variants AAV9.45 and AAV9.61 that displayed a 10- to 25-fold lower gene transfer efficiency in liver, while transducing the myocardium as efficiently as AAV9. In conclusion, these emerging strategies can be used to enhance tissue tropism and can also be utilized to produce AAV that are able to evade neutralizing antibodies, at least partially.
Figure 1.
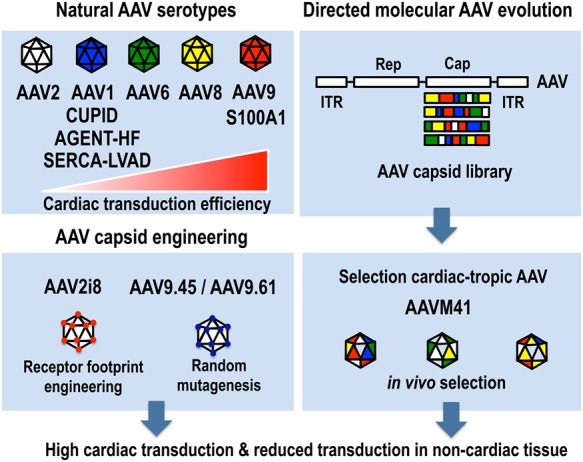
Strategies to increase cardiac-specific gene transfer using AAV vectors using naturally occurring AAV serotypes, AAV capsid engineering, or directed molecular evolution and in vivo selection of cardiotropic AAV variants (see text for details). The relative cardiac transduction efficiency of the naturally occurring AAV serotypes (AAV2, AAV1, AAV6, AAV8, and AAV9) based on preclinical murine studies is shown schematically.
Cardiac-specific promoters could be employed to restrict expression to the heart. Typically these promoters are quite large, consequently restricting the size of the therapeutic gene that can readily be accommodated in an AAV vector.58 Moreover, gene expression levels are often reduced compared with when more robust (viral) promoters were used, such as the CMV promoter.44,58 The alpha myosin heavy chain (αMHC) promoter has been among the most commonly used myocardial-specific promoters. It drives transgene expression throughout the entire heart, including the ventricles and atria.59,60 The myosin light-chain (MLC) 2v promoter has also been used in various cardiac gene therapy applications, by virtue of its cardiac-specific expression pattern.61–65 Comparative analysis after intra-vascular gene delivery in newborn mice using AAV vectors revealed that the αMHC promoter is among the most cardiac-specific promoters, whereas the desmin promoter resulted in robust expression in both heart and skeletal muscle.44 Nevertheless, both promoters were not as potent compared with the CMV promoter. We have recently explored novel computational approaches, to identify potent, yet small evolutionary conserved cardiac-specific cis-regulatory modules (CRMs) that boost the performance of cardiac-specific promoters, as in the case of the αMHC promoter.66 We have also validated this in silico approach to identify and validate tissue-specific CRMs that improve gene expression in other tissues, such as liver.48,67 This underscores the validity of computational vector design as a novel strategy to improve vector performance in gene therapy. An alternative approach to achieve cardiac-specific expression consists of incorporating a miRNA-regulated cassette that selectively represses gene expression in non-cardiac tissues.68,69
One major drawback of AAV is the limited packaging capacity of the vector particles (i.e. 4.7 kb), which constrains the size of the transgene expression cassette that can be used.70 Dual vector strategies have been developed to overcome the packaging constraints that rely on the concatemerization of AAV genomes to generate functional expression cassettes in situ in the transduced cells.21 In particular, the use of trans-splicing AAV enables the in situ reconstitution a functional expression cassette of up to 10 kb.71 Such trans-splicing AAV9 vectors were able to transduce the hearts of neonatal and adult dystrophic mdx mice at the same efficiency as conventional non-split ssAAV9.72
Though the risk of inflammatory immune responses following AAV transduction is significantly reduced compared with when Ad vectors are employed, there are still some important immune issues to address. In particular, the high prevalence of pre-existing antibodies to wild-type AAV in the population can result in rapid neutralization of AAV precluding gene transfer.73 Similarly, the induction of neutralizing antibodies after AAV-based gene therapy will prevent gene transfer after AAV vector re-administration.39,74 However, the use of alternative AAV serotypes or capsid variants, AAV capsid decoys, pharmacological immune modulation, and/or plasmapheresis treatment may allow AAV transduction in the face of pre-existing antibodies.39
In addition, T-cell immune responses against the AAV capsid antigens that are presented in association with MHC class I (MHC-I) antigens on the surface of transduced target cells may curtail long-term gene expression after immune rejection of the gene-modified target cells.20 The use of transient immune suppression and/or AAV capsid variants that results in reduced MHC-I presentation of AAV capsid-derived antigenic peptides may reduce this risk.75–77
2.2.3. Lentiviral vectors
The quintessential lentiviral vectors (LV) are derived from the HIV type 1 (HIV-1).78 LV are enveloped single-stranded (ss)RNA vectors that have the ability to stably integrate their genome as cDNA into the chromosomes of both dividing and non-dividing target cells. They are therefore well suited to achieve long-term expression of the therapeutic gene.79 LV have been used successfully for the treatment of monogenetic haematopoietic disorders, resulting in long-term therapeutic effects.80,81 However, their use in gene therapy applications for CVD/HF is more limited given their relatively poor transduction of myocardium after in vivo LV gene delivery. This may depend, at least on part, on the species, age, and/or mode of LV delivery.82 Indeed, recently it was shown that rat myocardium could be transduced by LV.83,84 These results are very promising in that the LV platform could be tailored towards an effective treatment option for CVD that require long-term expression of the therapeutic gene, without the adverse effects of adenoviral vectors obviating the need for repeated viral vector administration.
Since LV could also transduce endothelial cells or endothelial progenitors, these properties have potential implications for the treatment of peripheral vascular diseases by LV gene therapy or as a means to induce therapeutic angiogenesis/vasculogenesis.85,86 The endothelial tropism of the LV can be enhanced either by using single-chain Fv retargeting moieties or by pseudotyping the LV with naturally occurring endotheliotropic envelopes (e.g. Nipah virus).87,88 It would be important to now validate the therapeutic potential of such endothelial-specific LV in an experimental model of CVD. Despite their promise, it is important to also consider some of the safety issues of using LV. Since they can integrate randomly into the target cell genome, with a preference for genes, their use carries an intrinsic risk of triggering insertional oncogenesis.89 However, this risk can be reduced by optimizing the vector design and depends on the target cell type. Consequently, the risk of insertional oncogenesis may be significantly lower in post-mitotic terminally differentiated cardiomyocyte than in lentivirally transduced gene-modified haematopoietic stem cells (HSC) in the context of myeloablative haematopoietic reconstitution.
2.3. Enhancing uptake of viral vectors
Several strategies have been developed to improve the delivery of therapeutic genes and increase the vector uptake by cardiomyocytes. Initially, improvements in vector delivery techniques were evaluated, from intracoronary delivery of the viral particles by percutaneous administration to direct intramyocardial injections and image-guided injection strategies based on NOGA® electromechanical mapping of the heart.11 Though these methods have increased the overall vector uptake, they still yielded relatively low transduction efficiencies in cardiomyocytes. Consequently, this requires high doses of vector particles, increasing the risk of immune responses against the vector and/or the gene-modified cells. Moreover, the need for higher vector doses also resulted in an increased risk of ectopic transduction in undesired tissues. Therefore, the necessity to achieve higher cardiac transduction efficiencies by using minimally invasive techniques required the development of adjuvants that enhanced vector uptake into the myocardium. Sasano et al.90 reported an increase of up to 80% in cardiac transduction efficiencies in a porcine model by using a combination of VEGF, adenosine, calcium, and nitroglycerin (NTG) infusion prior to the viral vector administration. However, this adversely impacted on the haemodynamic parameters, which impedes the possible clinical translation of this particular strategy. Prior to the CUPID trial, Hajjar and co-workers91 reported a modest yet significant increase in mRNA and protein concentration of SERCA2a in the left ventricle of treated pigs compared with the control groups by simultaneous intravenous infusion of NTG and the vector of interest (i.e. AAV1/SERCA2a). This strategy was posteriorly implemented in the CUPID trial (see below).
3. Therapeutic genes for gene therapy for CVD
To ultimately achieve the greatest therapeutic impact by gene therapy, it is necessary to identify the appropriate therapeutic gene. Promising results have been obtained with gene delivery systems that induce angiogenesis/vasculogenesis or that target proteins involved in the handling of cardiomyocytic calcium (Ca2+) (e.g. sarcoplasmic reticulum Ca2+-ATPase, S100A1, and phospholamban) and the β-adrenergic system (the β1- and β2-adrenergic receptors, or the G-protein-coupled receptor (GPCR) kinase-2 (GRK2) (Table 3).
Table 3.
Preclinical gene therapy studies for heart failure and other cardiovascular diseases
| Molecular target | Model | Vector | Findings | Refs |
|---|---|---|---|---|
| VEGF-A165 | Coronary artery occlusion model in adult sheep | Plasmid DNA CMVenh/p-VEGF-A165 (3.8 mg) |
|
92 |
| Occlusion of the LAD coronary artery in dogs | Adeno-associated virus AAV6-CMVp- VEGF-A165 (5 × 1012 vp) |
|
93 | |
| Occlusion of the LAD coronary artery in pigs | Adeno-associated virus AAV1-MLCp- VEGF-A165 (1 × 1012 vp) |
|
94 | |
| FGF4 | Pig model of chronic myocardial ischaemia | Adenovirus Ad5-CMVp-PR39 (3 × 109 vp) |
|
95 |
| Stress-induced myocardial ischaemia in pigs | Adenovirus Ad5-CMVp-FGF4 (up to 1.6 × 1012 vp) |
|
34 | |
| Sarcoplasmic reticulum Ca2+-ATPase | Human ventricular myocytes from patients with end-stage heart failure | Adenovirus Ad5-CMVp-SERCA2a |
|
96 |
| Ascending aortic constriction in rats | Adenovirus Ad5-CMVp-SERCA2a |
|
97 | |
| Heart failure post-MI model in sheep | Adeno-associated virus AAV6-CMVp-SERCA2a (5 × 1012 vp) |
|
98 | |
| Rats with ligation of the left anterior descending artery | Adenovirus Ad-CMVp-SERCA2a (2 × 1011 vp) |
|
99 | |
| Pressure-overload model of heart failure in guinea pigs | Adeno-associated virus AAV1-CMVp-SERCA2a |
|
100 | |
| S100A1 | Rat model of heart failure | Adeno-associated virus AAV6-actin-S100A1 (2.5 × 1011 vp) |
|
101 |
| Heart failure due to balloon occlusion of the left circumflex coronary artery in domestic pigs | Adeno-associated virus AAV9-CMVenh/MLCp-S100A1 (1.5 × 1013 vp) |
|
102 | |
| β-Adrenergic receptor | Catheterization of coronary artery in rabbits | Adenovirus Ad-CMVp-β2-AR (5 × 1011 vp) |
|
103 |
| Rat model of hypertrophied heart failure | Plasmid DNA |
|
104 | |
| Rat model of post-MI heart failure | Adenovirus Ad-β2-AR |
|
105 | |
| Adenylyl-cyclase 6 | Wild-type C57/B6 mice | Adenovirus Ad.AC6 (2.5 × 1010 vp) |
|
106 |
| Transgenic mice overexpressing adenylyl cyclase (AC) type 6 | n/a |
|
107 | |
| Transgenic mice overexpressing adenylyl cyclase (AC) type 6 | n/a |
|
108 | |
| Porcine model of heart failure | Adenovirus Ad-CMVp-AC6 (1.4 × 1012 vp) |
|
109 |
3.1. Angiogenic factors for CVD: ischaemic heart disease
Angiogenic factors induce the formation of new vascular networks, which make them suitable therapeutic options for treating acute coronary syndromes and peripheral vascular diseases. It is beyond the scope of this review to provide an exhaustive overview and discussion of the underlying molecular biological aspects of angiogenesis and vasculogenesis. Instead, only the most salient features and controversies relevant to the subsequent clinical trials (see below) will be highlighted.
Several types of angiogenic factors that exhibit different properties have been explored in gene therapy for CVD. These include the different subtypes of VEGF, such as VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PlGF).110 Some of these factors also yield distinct isoforms (e.g. VEGF-A165). The intracellular signals of these VEGF subtypes are mediated mainly by three different tyrosine kinase receptors: VEGFR1, VEGFR2, and VEGFR3. Specific interaction between these VEGF subtypes and their cognate cellular receptors evokes a differential cellular response in endothelial cells and cardiomyocytes. In particular, an isoform of VEGF-A (VEGF-A165) reportedly has high angiogenic activity and plays a significant role in ischaemic diseases.111 VEGF-A165 interacts principally with VEGFR1 and VEGFR2. Interaction of VEGF-A165 with VEGFR1 on endothelial cells contributes to vascular stability of newly formed vessels (Figure 2). Its interaction with VEGFR2 on endothelial cells induces angiogenesis, vasculogenesis, and arteriogenesis, vasodilation, cell survival, and increase of cell permeability (Figure 2). Simultaneously, activation of VEGFR2 in newly formed cardiomyocytes increased expression of anti-apoptotic proteins and reduced expression of pro-apoptotic proteins,111 suggesting a direct effect on cardiomyocytes. Additionally, VEGFR2 activation induces recruitment of local cardiac stem cells in ischaemic areas.112 Importantly, the angiogenic response to VEGF is depending on the blood flow. In normal flow conditions, VEGF response is characterized by dilatation of the existing capillaries. In contrast, in an ischaemic situation, a more robust angiogenic response is observed. PlGF also binds on VEGFR1 and synergizes with VEGF.113 PlGF stimulates angiogenesis and collateral growth in ischaemic heart and limb with at least a comparable efficiency to VEGF through its action on different cell types (i.e. endothelial, smooth muscle, and inflammatory cells and their precursors) that play a cardinal role in blood vessel formation. However, PlGF did not cause side effects associated with VEGF, such as oedema or hypotension.
Figure 2.
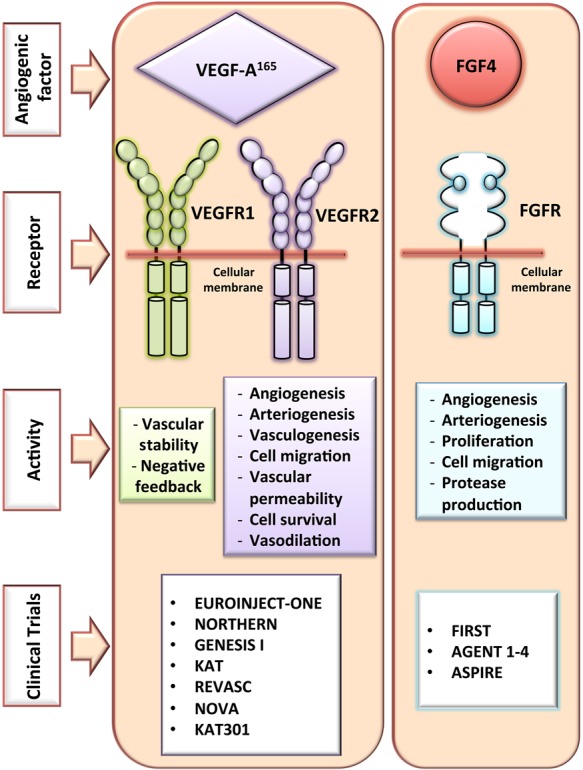
Angiogenic factors in gene therapy for CVD. Description of the membrane receptor involved in the angiogenic factor pathways and the physiological effects. VEGF-A165, vascular endothelial growth factor; FGF-4, fibroblast growth factor 4; VEGFR, vascular endothelial growth factor receptor; FGFR, fibroblast growth factor receptor.
Studies in small and large animal ischaemic heart disease models have reported that myocardial overexpression of VEGF following naked DNA transfection, Ad vectors, or AAV transduction induce angiogenesis, improving myocardial blood flow and overall left ventricular function.114 Preclinical animal studies demonstrated that VEGF-A165 gene therapy resulted in the induction of angiogenesis, arteriogenesis, and vasculogenesis,92,93 recovery of ventricular function during ischaemia, reduction in apoptosis of the infarcted area.94 VEGF also plays a role in the recruitment and homing of cardiac and endothelial progenitor cells115 and maintenance of physiological homeostasis in the heart.116 Translational studies in large animal models confirmed some of the beneficial effects of VEGF, either by itself or in combination with other angiogeneic factors (e.g. Ang 1). For instance, direct intramyocardial injection of AAV expressing VEGF and Ang1 resulted in activation of pro-survival pathways and reduction of cell apoptosis, consistent with improvement in the perfusion and function of the heart in a porcine MI model.94
There are 22 identified members of the fibroblast growth factor (FGF) family in humans and they interact with four high-affinity specific co-receptor systems consisting of tyrosine kinase FGF receptors (FGFRs) and heparin-like GAGs (HLGAG).117 The FGFR2 is mainly present in cardiovascular cells and interacts with FGF1, 2, 4, and 5. FGF induces cell proliferation, migration, and production of proteases in endothelial cells and cardiomyocytes. Additionally, interaction of FGF5 with cardiomyocytes has been reported to stimulate angiogenesis, enhance collateral blood flow, and relieve stress-induced ischaemia.118 Previous studies reported that FGF2 can act on most cardiac cells, including cardiomyocytes, endothelial cells, smooth muscle cells, and fibroblasts inducing a cardio-protective effect in ischaemic events by inducing angiogenesis and increasing vascular remodelling.119 In pig models of MI, the injection of a plasmid encoding FGF2 resulted in increased vascular perfusion and cardiac contractility compared with control pigs.95 Lastly, FGF4 has additional paracrine effects compared with FGF1 and FGF2. Intracoronary injection of Ad vectors encoding human FGF4 in a pig model of stress-induced MI resulted in improved myocardial perfusion and regional myocardial function (Figure 2).34,120–122 Other angiogenic factors, including VEGF-B, PlGF, and hypoxia-inducible factor-1α (HIF1α), were also shown to increase angiogenesis and improve the therapeutic outcome in small and large animal models following treatment by either recombinant proteins and/or gene therapy (Table 3).
These preclinical studies justified the use of angiogenic gene therapy in clinical trials (see below). Nevertheless, despite their promise, angiogenic gene therapies suffered several limitations: (i) an increase of vascular permeability was often apparently raising important safety concerns (i.e. vascular leak syndrome);123 (ii) a single angiogenic factor was often insufficient to obtain fully functional, stable, and mature blood vessels, requiring the combination of multiple growth factors instead; (iii) unnatural large capillary structures were often formed124 requiring approaches to also recruit smooth muscle cells to obtain a more effective revascularization; and (iv) finally, expression of angiogenic factors may contribute to pathological angiogenesis and increase the risk of haemangioma formation125 and tumour progression and metastasis. Nevertheless, careful selection of the type of angiogenic factor used may mitigate some of these risks (i.e. PlGF).113
3.1.1. Clinical trials using angiogenic factors for CVD
The initial gene therapy trials for CVD focus on the administration of genes encoding angiogenic growth factors, such as VEGF-A165, angiopoietins, FGF,126 and HIF-1α26 (Table 4). The main objective of these trials is to promote the development of collateral blood vessels in ischaemia-related conditions, such as chronic critical limb ischaemia, myocardial ischaemia, angina, or peripheral arterial occlusive disease.11,115,127–129 EUROINJECT-ONE, a multicentre, double-blind, randomized trial, included 80 patients with severe stable ischaemic heart disease. VEGF-A165 cDNA plasmid (0.5 mg) was administered by intramyocardial catheter injection. The study reported functional and symptomatic improvements in patients, but without any significant difference in perfusion compared with placebo.11 In the NORTHERN trial, the dose of VEGF-A165 cDNA plasmid was increased to 2 mg. The plasmid was administered through an endocardial route using an electro-anatomical guidance catheter in patients with Class 3 or 4 angina. After 6 months of follow-up, no evidence of improvement was observed in myocardial perfusion assessed by single-photon emission tomography (SPECT).130
Table 4.
Gene therapy clinical trials for therapeutic angiogenesis
| Trial | Gene | Country | Vector | Delivery method | Dose | Clinical condition (no. of patients) | Findings |
|---|---|---|---|---|---|---|---|
|
FIRST (FGF Initiating RevaScularization Trial) 2002 |
FGF2 | USA | Plasmid DNA | Intracoronary infusion (single dose) | Dose escalation (0.3, 3, and 30 µg/kg) | Coronary artery disease (337) |
|
|
AGENT-I Angiogenic GENe Therapy (2002) |
FGF4 | USA | Adeno-virus | Intracoronary infusion (single dose) | Five doses: (3.3 × 108, 1.0 × 109, 3.3 × 109, 1.0 × 1010, 3.3 × 1010 viral particles) | Chronic stable angina (79) |
|
|
AGENT-Ii Angiogenic GENe Therapy (2003) |
FGF4 | USA | Adeno-virus | Intracoronary infusion (single dose) | 1010 adenoviral particles | Chronic stable angina (52) |
|
|
KAT Kuopio Angiogenesis Trial (2003) |
VEGF-A165 | Finland | Adeno-virus/plasmid liposome | Intracoronary infusion (single dose) | Two groups: VEGF-Adv, 2 × 1010pfu VEGF-P/L; 2000 µg of DNA with 2000 µL of DOTMA:DOPE |
Coronary artery disease (103) |
|
|
VIVA (Vascular endothelial growth factor in Ischaemia for Vascular Angiogenesis) (2003) |
VEGF-A165 | USA | Plasmid DNA | Intracoronary infusion (day 0) plus intravenous infusions (Days 3, 6, and 9) | Low dose: 17 ng kg−1 min−1 High dose: 50 ng kg−1 min−1 |
Stable exertional angina (178) |
|
| EUROINJECT—One (2005) | VEGF-A165 | Denmark, Poland, Sweden, and Austria | Plasmid DNA | Direct intramyocardial injection via NOGA-Myostar© | 0.5 mg of phVEGF-A165 | Severe stable ischaemic heart disease (80) |
|
| REVASC-II (2006) | VEGF-A121 | Canada | Adeno-virus | Direct intramyocardial injections | 4 × 1010 viral particles AdVEGF121 | Severe coronary artery disease (67) |
|
|
AGENT-III/IV Angiogenic GENe Therapy (2008) |
FGF4 | USA, Europe | Adeno-virus | Intracoronary infusion (single dose) | Low dose of 4, 1 × 109 viral particles (vp), and a high dose of 1 × 1010 vp | Recurrent stable angina (AGENT-III: 416; AGENT-IV: 116) |
|
| Phase I intracoronary administration of Ad-Hhgf (2009) | hHGF | China | Adeno-virus | Intracoronary infusion (single dose) | Three doses: 5.0 × 109, 1.0 × 1010, 2.0 × 1010 pfu | Severe and diffuse triple vessel coronary disease (18) |
|
|
NORTHERN (NOGA angiogenesis Revascularization THErapy: assessment by RadioNuclide imaging) (2009) |
VEGF-A165 | Canada | Plasmid DNA | Endocardial route using an electro-anatomical NOGA guidance catheter | Total dose, 2 mg | Refractory Canadian Cardiovascular Society (CCS) Class 3 or 4 angina symptoms (93) |
|
| Multicentre Phase I and Safety Study (2010) | HIF1α | Germany, UK | Adeno-virus | Intramyocardial injections during CABG | Three doses: 1.0 × 1010, 3.0 × 1010, and 1.0 × 1011 viral particles | Hypo-perfused area of viable ventricular muscle (13) |
|
| VIF-CAD (2011) | Bicistronic [VEGF/FGF] plasmid | Poland | Plasmid DNA | Percutaneous intramyocardial injection using NOGA guidance catheter | Total dose, 0.5 mg | Refractory coronary artery disease (52) |
|
| GENESIS I (2012) | VEGF-A165 | Argentina | Plasmid DNA | Intramyocardial injections | Total dose, 3.8 mg | Severe CAD not amenable for revascularization (10) |
|
| ASPIRE (2013) | FGF4 | Russia | Adeno-virus | intracoronary infusion during induced transient ischaemia | 6 × 109 viral particles Ad5FGF4 | Stable angina pectoris (100) |
|
| KAT301 (2013) | VEGF-D | Finland | Adeno-virus | Endocardial injection system (NOGATM) | Escalating dose of 1 × 109, 1 × 1010, and 1 × 1011 vpu injected into 10 sites of the myocardium | Severe coronary artery disease (30) |
|
In an effort to improve the gene delivery efficiency of the angiogenic factors, viral vectors were used instead. The Phase II REVASC trial131 was therefore conducted to evaluate the efficacy of an adenoviral vector-containing VEGF (Ad. VEGF-A121) in patients with severely symptomatic coronary artery disease who are not candidates for conventional revascularization. Though one of the primary endpoints of the trial, exercise treadmill evaluation was significantly improved after 6 months of follow-up, there was no evidence of improvement in myocardial perfusion, evaluated by SPECT. Different limitations inherent to the study such as the surgical vector delivery technique employed, lack of blindness for the treatment groups, and the advances in catheter systems for percutaneous intramyocardial delivery prompted new clinical studies. The KAT301 trial was based on the use of an adenoviral vector to administer VEGF-D cDNA. Using a catheter-mediated trans-endocardial injection strategy, patients with coronary heart disease (CHD) with no other therapeutic options will be recruited in an escalating dose protocol. Doses of 1 × 109, 1 × 1010, and 1 × 1011 vp of Ad.VEGF-D will be injected into 10 myocardial sites. The main goals of the study are to evaluate safety and efficacy (http://clinicaltrials.gov/show/NCT01002430). These trials constitute the major advances in VEGF-based gene therapy.
The AGENT trial (Angiogenic GENe Therapy)118 evaluated the safety and anti-ischaemic effects of five ascending doses of adenoviral vectors carrying the FGF4 cDNA (Ad5-FGF4). Selected patients with chronic stable angina exhibited symptomatic improvement in exercise time at 4 weeks after injection, and the overall safety profile of the viral vector system was ascertained. The same strategy was followed in the AGENT-2 trial.132 Intracoronary administration of Ad5-FGF4 was evaluated in patients with coronary artery disease. After 8 weeks of follow-up, a trend towards improvement in stress-induced myocardial perfusion was observed compared with baseline. Nonetheless, there was no significant difference compared with the placebo group. In the AGENT-3 and AGENT-4 trials, low and high doses of Ad5-FGF4 for chronic angina were evaluated. The results demonstrated a sex-specific beneficial effect on the exercise treadmill test; however, this effect was mainly due to a poor placebo response among women.133 Moreover, both studies were stopped after an interim analysis of the AGENT-3 trial indicated that there were no significant differences regarding the primary endpoint in the treatment arm compared with placebo.
Several factors could have influenced the negative results obtained in the clinical trials using angiogenic factors compared with the promising preclinical data or earlier clinical trials.134,135 The clinical data were only conclusive when the later studies were randomized and blinded compared with the initial ones. This suggested a strong placebo effect, in part due to the strict selection of patients, the delivery method used (intramuscular injection can increase the production of growth factors), or a possible bias by lack of a blinded protocol. The lack of efficacy could also be explained by the fact that the patients selected for clinical trials were in end stages of the disease, with the more severe presentation, despite previous pharmacological interventions. It cannot be excluded that these angiogenic gene therapies may be more effective in the less severe patients, which constitutes the higher percentage of patients in the population. Finally, especially for naked plasmid strategies, the poor DNA uptake of cardiac and muscle cells is low, the short transient expression limited up to 2 weeks can be insufficient for achieve an effective angiogenic stimulus with substantial quantifiable changes in the heart parameter in comparison to the results obtained in preclinical models. Given the intrinsic complexity of angiogenesis, it is unlikely that administration of a single gene would suffice to obtain sustainable effects in patients with CVD. Indeed, preclinical studies have shown that additional factors are needed to lead to optimal endothelial and smooth cell proliferation and integration into sustainable and functional blood vessel development.136,137
3.2. Myocyte Ca2+ handling/contractility target genes: HF
In healthy cardiomyocytes, the norepinephrine (NE) released by the sympathetic system stimulates the β-adrenergic receptor, thereby inducing the entry of Ca2+ through the L-channels into the cells. The increase of calcium concentration activates the ryanodine receptor (RyR) to release stored Ca2+ from the sarcoplasmic reticulum into the cytoplasm, which in turn leads to contraction of the myofilaments. The NE stimulus also activates adenylyl-cyclase 6 (AC6), which induces the conversion of ATP in cyclic-AMP (cAMP), which in turn causes the phosphorylation (P) of phospholamban (PLN). This phosphorylation step, which is mediated by phosphokinase-A (PKA), releases PLN's inhibitory effect on the sarcoplasmic reticulum Ca2+–ATPase pump (SERCA2a). Next, SERCA2a binds Ca2+ ions from the cytoplasm and pumps them back into the sarcoplasmic reticulum, reducing the cytoplasmic concentration of calcium and permitting the relaxation of the myofilaments. The alteration of Ca2+-handling proteins during the process of excitation–contraction coupling plays an important role in the development and evolution of HF. In failing cardiomyocytes, a reduced Ca2+ cycling activity by a reduced expression of SERCA2a, an imbalance in phosphorylation of the RyR and abnormal expression of regulatory proteins such as S100A1 and AC6 have been demonstrated, suggesting these proteins as possible therapeutic targets for gene therapy (Table 3).
3.2.1. Sarcoplasmic reticulum Ca2+ATPase up-regulation improves cardiac function in HF
Initial studies demonstrated that HF is partly caused by decreased sarcoplasmic/endoplasmic reticulum Ca2+ATPase2a (SERCA2a) levels or activity, independent of the aetiology of the HF.138 The calcium pump SERCA2a causes muscle relaxation by lowering the cytosolic calcium and restores the calcium reserves in the sarcoplasmic reticulum, which are necessary for muscle contraction.139 Subsequent studies demonstrated an association between decreased SERCA2a mRNA level and low SERCA2a protein concentration and Ca2+-ATPase activity, especially during the transition from compensated hypertrophy to decompensated HF, leading to faster and more severe HF.140 Overexpression of SERCA2a in human ventricular cardiomyocytes obtained from patients with ischaemic and dilated cardiomyopathy resulted in increased SERCA2a pump activity consistent with improved contraction and relaxation velocity.96 In a rat model of pressure-overload hypertrophy HF, Ad vector-mediated overexpression of SERCA2a raised left ventricular, systolic, and diastolic function to levels similar to those observed in control mice.97 Using a percutaneous delivery system for AAV6 encoding SERCA2a, Beeri et al.98 demonstrated long-term overexpression of SERCA2a in a sheep model of mitral regurgitation and MI, with improved contractility and inhibition of the remodelling process.
Most importantly, SERCA2a gene transfer improves contractile function survival rates and the energy potential in failing hearts without increasing mortality or worsening metabolism.99 In addition, SERCA2a overexpression in a rat model of ischaemia/reperfusion injury significantly decreased ventricular arrhythmias and (more importantly) led to reduced infarct size and improved wall thickening in the anterior wall.99 It is also encouraging that long-term in vivo overexpression of SERCA2a after AAV1-mediated gene transfer in cardiomyocytes in a preclinical volume-overload model of HF can preserve cardiac function by increasing eNOS expression in coronary arteries.100 Furthermore, Ad SERCA2a transduction in an animal model of pressure-overload hypertrophy with transition to HF resulted in decreased pro-apoptotic (e.g. Bax) and apoptotic and inflammatory (e.g. TNF-α) cytokines, suggesting a SERCA2a regulatory role in controlling inflammation and apoptotic events during HF and MI.
Before clinical translation, the effects of SERCA2a were evaluated in a Yorkshire-Landrace swine model of volume-overload HF. An AAV1 vector was used to deliver human SERCA2a cDNA. The CMV immediate-early promoter/enhancer was incorporated into the transgene cassette. Two months after HF induction, pigs received AAV1-SERCA2a vectors, resulting in normalization of SERCA2a expression levels consistent with improvements in left ventricular ionotropic activity and remodelling.141 Nevertheless, the results obtained in these preclinical models must be carefully and critically evaluated. In particular, it was shown that human cardiomyocytes rely on SERCA2a pump function (around 71%) to a lesser extent than rat and mouse cardiomyocytes (around 90%).142 Ultimately, only more clinical trials will provide the necessary data to corroborate the therapeutic potential of SERCA2a gene therapy in patients with HF.
Apart from Ad vectors and AAV vectors, LV have also been used to deliver SERCA2 to the myocardium. LV containing the SERCA2 gene were delivered by a hypothermic intracoronary delivery method in rat myocardium, 2 weeks after MI, Significantly, this LV-SERCA2 gene therapy resulted in long-term improvement of systolic and diastolic function, prevention of left ventricular remodelling even up to 6 months after gene therapy, consistent with significant improvement of the survival rate.84 Though these findings offer a potentially translatable therapy, the results would still need to be confirmed in a preclinical large animal model.
3.2.2. S100A1 is a key protein in cycling Ca2+ capacity of myofilaments
S100A1 is preferentially expressed in myocardial tissue though low levels have also been reported in other tissues.143 It exerts profound ionotropic actions through the modulation of cardiomyocyte Ca2+ homeostasis and myofilament function independent of β-adrenergic stimulation.144 S100A1 interacts in a Ca2+-dependent manner with the RyR and stabilizes the SERCA2a-PLN complex.145 S100A1 diminishes the diastolic leak of Ca2+ and influences cardiac titin and mitochondrial F1-ATPase. Cardiomyocytes that overexpress S100A1 present a higher ATP content than control cells, suggesting a role for S100A1 in energy metabolism.146 Considering the potential therapeutic effects of S100A1 protein, gene therapy studies have evaluated its performance in HF models. Pleger et al. evaluated the response of an AAV6-S100A1 vector in a rat model of HF. The results demonstrated long-term improvement in cardiac dysfunction and attenuated left ventricular remodelling. Even non-transduced cardiomyocytes showed a trend towards functional improvement, suggesting that S100A1-overexpressing cardiomyocytes have an indirect bystander effect on neighbouring cardiomyocytes. The therapeutic effects of S100A1 were preserved during β-adrenergic antagonist treatment with metoprolol, improving its cardiac reverse remodelling, ionotropic action, and anti-arrhythmic effects.101 Intracoronary adenovirus-mediated S100A1 gene delivery in a rat model of post-MI improved myocardial contractility and Ca2+ handling.147 Additionally, S100A1 gene transfer improved force generation in engineered cardiac grafts without interfering with the mechanical structure of the engrafted heart.148 In a large animal model of HF, Pleger et al.102 evaluated the long-term effectiveness of an AAV9 vector encoding the S100A1 gene. Retrograde coronary venous delivery strategy was used in post-ischaemic pig model of HF after 2 weeks of occlusion of the left circumflex coronary artery. The follow-up resulted in long-term benefits in cardiac performance by improving mitochondrial function in failing cardiomyocytes and reverse remodelling by improving systolic and diastolic left ventricular performance at 12 weeks post intervention. The use of S100A1 gene therapy for the treatment of HF was further supported by Brinks et al.149 using an in vitro evaluation of human failing ventricular cardiomyocytes strategy. An Ad vector encoding S100A1 was used to transduce cardiomyocytes isolated from the heart of patients undergoing transplant surgery. S100A1 levels returned to normal in transduced cardiomyocytes, with clear improvement in contractibility performance and restoring of sarcoplasmic reticulum functions. These preclinical data support the use of S100A1 as a gene therapy modality in patients with HF.
3.2.3. Adenylyl-cyclase type 6 is a central regulator of calcium cycling in cardiomyocytes and SERCA2a activity.
Adenylyl-cyclase type 6 (AC6) catalyzes the conversion of ATP to 3′,5′-cyclic-AMP (cAMP) and pyrophosphate. It improves the affinity of SERCA2a for calcium by activating a cAMP-dependent protein kinase of PLN. Upon phosphorylation, PLN loses its inhibitory effect on SERCA2a. This AC6 pathway also results in the transcription factor-3-dependent suppression of PLN promoter activity. The overall effect of PLN activation is a decrease in contractility and the rate of muscle relaxation, thereby decreasing stroke volume and heart rate.150 Conversely, inhibiting PLN expression of function has the reverse effect and is therapeutically beneficial. Indeed, studies in transgenic mice overexpressing AC6 demonstrated increased cAMP generation in cardiomyocytes, which in turn triggered an improvement in cardiac function, particularly left ventricular contractile function.106 A decrease in ventricular hypertrophy and increased survival were reported in cardiomyopathy after AC6 activation.151 This finding is consistent with improved left ventricle function, reduced ventricular dilation, and reduced mortality rates in transgenic mice overexpressing AC6 compared with control mice.107 In contrast to conventional sympathomimetic interventions, increasing AC6 expression in a post-MI mouse model did not increase susceptibility to ventricular arrhythmias. Nevertheless, AC6 overexpression in a long-term murine model of pressure-overload HF was correlated with a worse outcome, possibly because of increased systolic ventricular wall stress. These results suggest a different molecular imbalance depending on the cause of the HF.108 Intracoronary delivery of an Ad vector encoding AC6 into a large-animal model of HF improved left ventricle function and attenuated deleterious left ventricular remodelling. These results were associated with increased cAMP-generating capacity.109 In summary, the pivotal effects of AC6 on the β-AR signalling pathway and calcium handling, consistent with its impact on improving left ventricular function and survival rates, reaffirm the use of AC6 as a rational approach to HF treatment. Moreover, intracoronary delivery of AC6 in small and large preclinical models confirms its favourable safety profile.
3.2.4. Alterations of the β-adrenergic system modulate cardiomyocyte contractile function in HF.
It was demonstrated >20 years ago that the β-adrenergic receptor (β-AR) system is centrally involved in HF.152 Activation of the sympathetic system in patients with HF correlated with morbidity and mortality levels. Reduction and de-sensitization of the β-AR receptors caused by the up-regulation of β-AR kinases and increased function of the inhibitory guanine nucleotide-binding protein (Gi) during HF have led to the employment of this system as a treatment option for HF.153 Results from transgenic mice revealed differential roles for different components of the β-AR system. Up-regulation of β1-AR is related to cardiomyocyte hypertrophy, followed by fibrosis and HF induced by activation of the β1-AR-Gs pathway.154 In contrast, moderate up-regulation of β2-AR improved basal contractility and rescued left ventricular contractility after MI.155 This difference has been explained by the ability of β2-AR to couple both Gi and the stimulatory guanine nucleotide-binding protein (Gs). In contrast, β1-AR couples only to Gs. The effects of the β2-AR on Gi proteins antagonize the contractile response controlled by the Gs proteins. Thus, the beneficial and pro-survival signalling derived from the β2-AR effect is mediated primarily by cAMP-PKA signalling activation and preferential activation of Gi during HF.156
Percutaneous-mediated intracoronary delivery of Ad vectors encoding β2-AR in rabbit models improved global left ventricular systolic and contractility performance.103 In rat models of pressure-overloaded HF, β2-AR cDNA was transfected by intracoronary infusion using a liposomal delivery method. The results revealed an enhanced response to isoproterenol (β-adrenergic agonist) in failing hearts.104 Recent evidence suggested that Ad vector-mediated β2-AR overexpression leads to enhanced endothelial cell proliferation and migration compounded by VEGF production, which subsequently led to improved ischaemia-induced angiogenesis in an ischaemic hind limb model.157 These results were extended to a rat model of post-MI via direct intramyocardial injection. Four weeks post-injection, rats exhibited improved left ventricular remodelling and cardiac function, increased capillary density, increased arteriolar length density, and enhanced in vivo myocardial blood flow and coronary reserve were observed, strengthening the role of β2-AR in the regulation of cardiac angiogenesis in the context of HF.105
Furthermore, homologous desensitization, a process by which kinases decrease the interaction between activated β receptors and their G proteins, is mediated by GRKs. Homologous desensitization reduces the neurohormonal response in the heart, which leads to worsening heart function during HF.158
Animal studies have shown that expression of a peptide inhibitor of GRK2 (βARKct) can improve the contractility of failing myocardium, promote reverse remodelling of the left ventricle, and improve outcome post-MI.159–162 Translational studies in human cardiomyocytes showed that the delivery of βARKct using Ad vector-mediated gene transfer in ventricular cardiomyocytes from patients with end-stage HF improved contractile function and β-adrenergic responsiveness.163 The long-term therapeutic impact and feasibility of the βARKct gene transfer was evaluated in a clinically relevant pig model of ischaemic cardiomyopathy using AAV6-mediated βARKct delivery.164 Retrograde injection into the coronary veins resulted in efficient and long-term βARKct expression, with significant systolic performance improvement overall at 6 weeks post-treatment. The results also suggest a correction of the catecholaminergic overdrive in post-MI HF. Finally, long-term βARKct expression in this porcine FH model exhibited a positive effect on the adverse cardiac remodelling process after MI. The favourable efficacy and safety data in preclinical models support the use of βARKct gene therapy for future clinical trials.
3.2.5. Clinical trials for gene therapy to treat HF by targeting the myocyte Ca2+ handling/contractility pathway
The first-in-human Phase I/II clinical trial for the treatment of HF (Table 4), the ‘calcium up-regulation by percutaneous administration of gene therapy in cardiac disease trial’, (CUPID Trial) prompted renewed interest in gene therapy.165 During Phase I, 9 patients with severe HF received an intracoronary delivery of AAV1 vector encoding SERCA2a cDNA in an open-label dose-escalation protocol (doses ranged from 1.4 × 1011 to 3 × 1012 vector particles per patient). After 6 months of follow-up, a tendency towards improvement in the functional, symptomatic, biomarker, and left ventricular/remodelling parameters was reported.166 The Phase II trial is based on a randomized, double-blind, placebo-controlled, dose-escalation protocol. Thirty-nine patients with Class III/IV HF received intracoronary infusion of placebo (n = 14) or low-dose (6 × 1011 vector particles; n = 8), mid-dose (3 × 1012 vector particles; n = 8), or high-dose (1 × 1013 vector particles; n = 9) AAV1-SERCA2a vector via percutaneous intracoronary artery infusion. After 12 months of follow-up, improvement or stabilization was reported based on the Heart Failure Questionnaire, a 6-min walk test, peak maximum oxygen consumption, N-terminal pro-hormone brain natriuretic peptide levels, and left ventricular end-systolic volume.167 Importantly, no adverse events could be attributed to the SERCA2a gene therapy and the AAV1-SERCA2a emerged as a viable therapeutic option for patients with severe HF with poor response to traditional pharmacologic treatments140 with a general consensus that the CUPID results could constitute a basis for larger pivotal trials.168 Recently, a 36-month follow-up analysis of the patients enrolled in the CUPID1 Phase IIa trial reported a lower number of cardiovascular events, including death in the treated groups compared with placebo.169 Additionally, no adverse immune events related to the AAV1-SERCA2a treatment were apparent in a long-term assessment.170
Two new clinical trials are based on SERCA2a. The Phase II study ‘Investigation of the safety and feasibility of AAV1-SERCA2a gene transfer in patients with chronic HF and a left ventricular assist device’ (SERCA-LVAD; NCT00534703) evaluates the effects of SERCA2a in patients with chronic HF that have received previously a left ventricular assist device (LVAD). Up to 24 patients will be randomized to receive a unique dose of 1 × 1013 DRP (DNase resistant particles) of AAV1-SERCA2a or placebo by a percutaneous method. The first results are expected in mid-2016. The second Phase II study, ‘AAV1-CMV-Serca2a GENe Therapy Trial in Heart Failure’ (AGENT-HF; NCT01966887), is a double-blind, randomized, placebo-controlled, parallel study that aims to evaluate the effects of the AAV1-CMV-SERCA2a vector in the cardiac remodelling parameters of 44 patients with symptomatic HF NYHA IIb/IV and EF of 35% or less, after intracoronary infusion of a single dose of 1 × 1013 DRP of the vector. The first results are expected at the beginning of 2016.
Given the intrinsic limitations of the low number of patients in a Phase IIa study, a Phase IIb, double-blind, placebo-controlled, multinational, multicentre, randomized event-driven study was needed (CUPID2) to confirm the initial CUPID1 results in a large number of patients (n = 250).171 CUPID2 is a randomized, double-blind, placebo-controlled, multinational trial evaluating a single, one-time, intracoronary infusion of the AAV1/SERCA2a vector vs. placebo added to a maximal, optimized HF drug and device regimen (NCT01643330). However, recently it was announced that this Phase 2b CUPID2 trial did not meet its primary and secondary endpoints (http://ir.celladon.com/releasedetail.cfm?releaseid=908592). In this study, the primary endpoint comparison of the AAV1/SERCA2a vector to placebo defined as HF-related hospitalizations or ambulatory treatment for worsening heart failure did not show a significant treatment effect. The secondary endpoint comparison of the AAV1-SERCA2a vector to placebo, defined as all-cause death, heart transplant or need for a mechanical circulatory support device, likewise failed to show a significant treatment effect. All other exploratory efficacy endpoints (improvement in NYHA classification, 6-min Walk Test, and Quality of Life) were also inconsistent with a treatment effect. No safety issues were noted, however. The exact reason for this outcome is not fully understood, and further studies are required to assess the efficiency of cardiac gene delivery using qPCR analysis on available patient biopsies. Intracoronary delivery of the AAV1 vector may not have resulted in efficient transduction of cardiomyocytes. This may reflect the relatively low transduction efficiencies in cardiomyocytes after anterograde delivery with AAV1-SERCA2a in porcine models,91,172 consistent with the relatively low vector copy number determined by qPCR. Unfortunately, in these preclinical studies,91 a placebo control group without AAV1-SERCA2a treatment was lacking. Consequently, since SERCA2a is normally also expressed in the heart, the contribution of the de novo expressed SERCA2a protein encoded by the AAV1-SERCA2a vector to the total SERCA2a protein levels could not be determined. Moreover, the presence of vector DNA in the myocardium does not necessarily imply bona fide gene transduction. In the CUPID2 trial, the vectors were administered by anterograde coronary delivery without any vessel balloon occlusion. The main advantage of this delivery strategy is that it does not increase the risk of ischaemic events in an already functionally compromised heart. However, under those circumstances the vector rapidly disseminates via the circulation into distal tissues, diminishing the overall efficacy of cardiac transduction. Consequently, vector dissemination may result in the inadvertent transduction of non-target tissues, particularly the liver. The use of retrograde delivery methods may potentially increase cardiac transduction efficiencies.172 Furthermore, the development of alternative AAV capsids by directed molecular evolution, permitting efficient transcytosis, and/or the use of reagents that enhance vector uptake may ultimately also overcome the limitations observed in the CUPID2 trial.
An alternative Phase I/II clinical gene therapy strategy for HF that is also based on Ca2+ handling has been initiated (NCT00787059). The objective of the AC6 trial for HF is to evaluate the safety and performance of human AC6 as a therapeutic option for patients HF. To achieve this goal, it is planned to include ∼56 patients and deliver an Ad5 vector carrying the AC6 gene. In this dose-escalation study, patients with an ejection fraction <40% will receive the vector by intracoronary delivery. Five different doses (ranging from 3.2 × 109 to 3.2 × 1011) will be compared with placebo. After 12 weeks of follow-up, patients will undergo exercise treadmill testing, and left ventricular functional parameters will be examined by echocardiography before and during the isoproterenol test.
4. Conclusions and future perspectives
The progress of unravelling the molecular pathways involved in cardiac function in normal and pathological states has increased efforts to develop co-adjuvant therapies as pharmacological and interventional options. Gene therapy is emerging as a suitable alternative, with substantial progress in preclinical models of CVD. Advances in therapies using angiogenic factors such as VEGF or FGF, the modification of β-adrenergic pathways, and molecules involved in cardiomyocyte Ca2+ cycling constitute clear examples of promising therapeutic alternatives. Additionally, an antagonist to the LDL receptor (LDLR), the proprotein convertase subtilisin/kexin type 9 (PCSK9) has been described as a promising therapeutic target for the prevention of CHD due to its role in the lipids metabolism,8 a major risk factor for CVD. Mutations of the PCSK9 have been associated with significant reduction of LDL-C and a subsequent decrease in CVD risk. Ding et al.8 demonstrated that using CRISP/Cas9 system was possible to induce a disruption in the PCSK9 gene and decrease blood cholesterol levels in mice up to 40% compared with control mice.
The advantages of the AAV vector-based therapeutic strategies over the other available recombinant vectors have positioned this delivery system as the preferred option for many of the gene therapy approaches for HF. However, the ability to obtain sustained expression of the gene of interest may not always be warranted and sometimes transient expression may be preferred based on safety considerations (e.g. angiogenic gene therapy). Though Ad vectors may at first glance seem ideally suited to achieve robust yet transient expression of a given therapeutic gene, this short-term expression results from immune complications intrinsic to this type of vectors. This inflammatory risk undermines the safety of adenoviral vector for CVD gene therapy and needs to be carefully assessed, even in the context of loco-regional catheter-mediated vector delivery into the myocardium.
The physiological and structural differences between animal models and humans and the development of immune response against the transgene products, the gene-modified cells, or the vectors themselves pose important challenges for clinical translation. The use of safer and more efficient gene delivery methods is warranted by improving the tissue specificity of the vectors using cardiac-specific enhancer/promoter to achieve better ‘transcriptional targeting’ and by using modified capsids to restrict vector entry into the desired target cells (in casu cardiomyocytes). For some gene therapy applications, it would be desirable to be able to control the duration and strength of the expression of the therapeutic gene, using clinically relevant approaches, which are now being developed.
Gene therapy clinical trials in ischaemic heart disease yielded limited results compared with preclinical models, with some improvement in secondary endpoints but no improvement in perfusion or myocardial function. The first clinical trial for HF reported substantial benefits in patients with severe angina, which justified the progression to larger clinical trials. In the future, consensus clinical and functional endpoints as well as the most appropriated measurement methods should be established to allow a clear and comparable understanding of the results between the different clinical trials. As gene therapy is currently also being used to treat non-lethal diseases in children, the hope is that the majority of patients who suffer from less severe heart disease may ultimately benefit from the advances in gene therapy.
Funding
M.Y.R. received funding from the ‘Patrimonio Autónomo, Fondo Nacional de Financiamiento para la Ciencia, la Tecnología y la Innovación Francisco José de Caldas’, Free University of Brussels Strategic Research Project and Association Française contre les Myopathies (AFM). T.V. and M.K.C. are supported by grants from the Flanders Fund for Scientific Research (FWO), AFM and VUB Strategic Research Projects (GENEFIX) and Industrial Research (IOF, VUB) GENECURE.
Acknowledgements
We thank the members of the Department of Gene Therapy and Regenerative Medicine and our collaborators for their various contributions to some of the work presented in this review.
Conflict of interest: none declared.
References
- 1.Bleumink GS, Knetsch AM, Sturkenboom MCJM, Straus SMJM, Hofman A, Deckers JW, Witteman JCM, Stricker BHC. Quantifying the heart failure epidemic: prevalence, incidence rate, lifetime risk and prognosis of heart failure The Rotterdam Study. Eur Heart J 2004;25:1614–1619. [DOI] [PubMed] [Google Scholar]
- 2.Gheorghiade M, Pang PS. Acute heart failure syndromes. J Am Coll Cardiol 2009;53:557–573. [DOI] [PubMed] [Google Scholar]
- 3.Tilemann L, Ishikawa K, Weber T, Hajjar RJ. Gene therapy for heart failure. Circ Res 2012;110:777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kay MA. State-of-the-art gene-based therapies: the road ahead. Nat Rev Genet 2011;12:316–328. [DOI] [PubMed] [Google Scholar]
- 5.Yerevanian A, Yerevanian A, Hajjar RJ. Progress in gene therapy for heart failure. J Cardiovasc Pharmacol 2014;63:95–106. [DOI] [PubMed] [Google Scholar]
- 6.Gao MH, Lai NC, Miyanohara A, Schilling JM, Suarez J, Tang T, Guo T, Tang R, Parikh J, Giamouridis D, Dillmann WH, Patel HH, Roth DM, Dalton ND, Hammond HK. Intravenous adeno-associated virus serotype 8 encoding urocortin-2 provides sustained augmentation of left ventricular function in mice. Hum Gene Ther 2013;24:777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai NC, Tang T, Gao MH, Saito M, Miyanohara A, Hammond HK. Improved function of the failing rat heart by regulated expression of insulin-like growth factor I via intramuscular gene transfer. Hum Gene Ther 2012;23:255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding Q, Strong A, Patel KM, Ng S-L, Gosis BS, Regan SN, Cowan CA, Rader DJ, Musunuru K. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res 2014;115:488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olson EN. MicroRNAs as Therapeutic Targets and Biomarkers of Cardiovascular Disease. Sci Transl Med 2014;6:239ps3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su C-H, Wu Y-J, Wang H-H, Yeh H-I. Nonviral gene therapy targeting cardiovascular system. Am J Physiol Heart Circ Physiol 2012;303:H629–H638. [DOI] [PubMed] [Google Scholar]
- 11.Gyöngyösi M, Khorsand A, Zamini S, Sperker W, Strehblow C, Kastrup J, Jorgensen E, Hesse B, Tägil K, Bøtker HE, Ruzyllo W, Teresiñska A, Dudek D, Hubalewska A, Rück A, Nielsen SS, Graf S, Mundigler G, Novak J, Sochor H, Maurer G, Glogar D, Sylven C. NOGA-guided analysis of regional myocardial perfusion abnormalities treated with intramyocardial injections of plasmid encoding vascular endothelial growth factor A-165 in patients with chronic myocardial ischemia: subanalysis of the EUROINJECT-ONE multicenter double-blind randomized study. Circulation 2005;112:I157–I165. [DOI] [PubMed] [Google Scholar]
- 12.Scimia MC, Cannavo A, Koch WJ. Gene therapy for heart disease: molecular targets, vectors and modes of delivery to myocardium. Expert Rev Cardiovasc Ther 2013;11:999–1013. [DOI] [PubMed] [Google Scholar]
- 13.Mali B, Jarm T, Corovic S, Paulin-Kosir MS, Cemazar M, Sersa G, Miklavcic D. The effect of electroporation pulses on functioning of the heart. Med Biol Eng Comput 2008;46:745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrara K, Pollard R, Borden M. Ultrasound microbubble contrast agents: fundamentals and application to gene and drug delivery. Annu Rev Biomed Eng 2007;9:415–447. [DOI] [PubMed] [Google Scholar]
- 15.Fujii H, Sun Z, Li S-H, Wu J, Fazel S, Weisel RD, Rakowski H, Lindner J, Li R-K. Ultrasound-targeted gene delivery induces angiogenesis after a myocardial infarction in mice. JACC Cardiovasc Imaging 2009;2:869–879. [DOI] [PubMed] [Google Scholar]
- 16.Fujii H, Li S-H, Wu J, Miyagi Y, Yau TM, Rakowski H, Egashira K, Guo J, Weisel RD, Li R-K. Repeated and targeted transfer of angiogenic plasmids into the infarcted rat heart via ultrasound targeted microbubble destruction enhances cardiac repair. Eur Heart J 2011;32:2075–2084. [DOI] [PubMed] [Google Scholar]
- 17.Gill S-L, O'Neill H, McCoy RJ, Logeswaran S, O'Brien F, Stanton A, Kelly H, Duffy GP. Enhanced delivery of microRNA mimics to cardiomyocytes using ultrasound responsive microbubbles reverses hypertrophy in an in-vitro model. Technol Health Care Off J Eur Soc Eng Med 2014;22:37–51. [DOI] [PubMed] [Google Scholar]
- 18.Petrus I, Chuah M, VandenDriessche T. Gene therapy strategies for hemophilia: benefits versus risks. J Gene Med 2010;12:797–809. [DOI] [PubMed] [Google Scholar]
- 19.Schneider CK, Salmikangas P, Jilma B, Flamion B, Todorova LR, Paphitou A, Haunerova I, Maimets T, Trouvin JH, Flory E, Tsiftsoglou A, Sarkadi B, Gudmundsson K, O'Donovan M, Migliaccio G, Ancāns J, Maciulaitis R, Robert JL, Samuel A, Ovelgönne JH, Hystad M, Fal AM, Lima BS, Moraru AS, Turcáni P, Zorec R, Ruiz S, Akerblom L, Narayanan G, Kent A, Bignami F, Dickson JG, Niederwieser D, Figuerola-Santos MA, Reischl IG, Beuneu C, Georgiev R, Vassiliou M, Pychova A, Clausen M, Methuen T, Lucas S, Schüssler-Lenz M, Kokkas V, Buzás Z, MacAleenan N, Galli MC, Linē A, Gulbinovic J, Berchem G, Fraczek M, Menezes-Ferreira M, Vilceanu N, Hrubisko M, Marinko P, Timón M, Cheng W, Crosbie GA, Meade N, di Paola ML, VandenDriessche T, Ljungman P, D'Apote L, Oliver-Diaz O, Büttel I, Celis P. Challenges with advanced therapy medicinal products and how to meet them. Nat Rev Drug Discov 2010;9:195–201. [DOI] [PubMed] [Google Scholar]
- 20.Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood 2013;122:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wasala NB, Shin J-H, Duan D. The evolution of heart gene delivery vectors. J Gene Med 2011;13:557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parker AL, Nicklin SA, Baker AH. Interactions of adenovirus vectors with blood: implications for intravascular gene therapy applications. Curr Opin Mol Ther 2008;10:439–448. [PubMed] [Google Scholar]
- 23.Du L, Dronadula N, Tanaka S, Dichek DA. Helper-dependent adenoviral vector achieves prolonged, stable expression of interleukin-10 in rabbit carotid arteries but does not limit early atherogenesis. Hum Gene Ther 2011;22:959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hajjar RJ. Potential of gene therapy as a treatment for heart failure. J Clin Invest 2013;123:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wright MJ, Wightman LM, Lilley C, de Alwis M, Hart SL, Miller A, Coffin RS, Thrasher A, Latchman DS, Marber MS. In vivo myocardial gene transfer: optimization, evaluation and direct comparison of gene transfer vectors. Basic Res Cardiol 2001;96:227–236. [DOI] [PubMed] [Google Scholar]
- 26.Kilian EG, Sadoni S, Vicol C, Kelly R, van Hulst K, Schwaiger M, Kupatt C, Boekstegers P, Pillai R, Channon K, Hetzer R, Reichart B. Myocardial transfection of hypoxia inducible factor-1alpha via an adenoviral vector during coronary artery bypass grafting. A multicenter phase I and safety study. Circ J Off J Jpn Circ Soc 2010;74:916–924. [DOI] [PubMed] [Google Scholar]
- 27.Muona K, Mäkinen K, Hedman M, Manninen H, Ylä-Herttuala S. 10-year safety follow-up in patients with local VEGF gene transfer to ischemic lower limb. Gene Ther 2012;19:392–395. [DOI] [PubMed] [Google Scholar]
- 28.Gahéry-Ségard H, Farace F, Godfrin D, Gaston J, Lengagne R, Tursz T, Boulanger P, Guillet JG. Immune response to recombinant capsid proteins of adenovirus in humans: antifiber and anti-penton base antibodies have a synergistic effect on neutralizing activity. J Virol 1998;72:2388–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krause A, Joh JH, Hackett NR, Roelvink PW, Bruder JT, Wickham TJ, Kovesdi I, Crystal RG, Worgall S. Epitopes expressed in different adenovirus capsid proteins induce different levels of epitope-specific immunity. J Virol 2006;80:5523–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schiedner G, Morral N, Parks RJ, Wu Y, Koopmans SC, Langston C, Graham FL, Beaudet AL, Kochanek S. Genomic DNA transfer with a high-capacity adenovirus vector results in improved in vivo gene expression and decreased toxicity. Nat Genet 1998;18:180–183. [DOI] [PubMed] [Google Scholar]
- 31.Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao G, Wilson JM, Batshaw ML. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab 2003;80:148–158. [DOI] [PubMed] [Google Scholar]
- 32.Kaski JC, Consuegra-Sanchez L. Evaluation of ASPIRE trial: a Phase III pivotal registration trial, using intracoronary administration of Generx (Ad5FGF4) to treat patients with recurrent angina pectoris. Expert Opin Biol Ther 2013;13:1749–1753. [DOI] [PubMed] [Google Scholar]
- 33.Chuah MKL, Collen D, VandenDriessche T. Biosafety of adenoviral vectors. Curr Gene Ther 2003;3:527–543. [DOI] [PubMed] [Google Scholar]
- 34.Gao MH, Lai NC, McKirnan MD, Roth DA, Rubanyi GM, Dalton N, Roth DM, Hammond HK. Increased regional function and perfusion after intracoronary delivery of adenovirus encoding fibroblast growth factor 4: report of preclinical data. Hum Gene Ther 2004;15:574–587. [DOI] [PubMed] [Google Scholar]
- 35.Ylä-Herttuala S, Alitalo K. Gene transfer as a tool to induce therapeutic vascular growth. Nat Med 2003;9:694–701. [DOI] [PubMed] [Google Scholar]
- 36.Vassalli G, Büeler H, Dudler J, von Segesser LK, Kappenberger L. Adeno-associated virus (AAV) vectors achieve prolonged transgene expression in mouse myocardium and arteries in vivo: a comparative study with adenovirus vectors. Int J Cardiol 2003;90:229–238. [DOI] [PubMed] [Google Scholar]
- 37.Williams PD, Ranjzad P, Kakar SJ, Kingston PA. Development of viral vectors for use in cardiovascular gene therapy. Viruses 2010;2:334–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dakin RS, Parker AL, Delles C, Nicklin SA, Baker AH. Efficient transduction of primary vascular cells by the rare adenovirus serotype 49 vector. Hum Gene Ther 2015;26:312–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet 2011;12:341–355. [DOI] [PubMed] [Google Scholar]
- 40.Vandendriessche T, Thorrez L, Acosta-Sanchez A, Petrus I, Wang L, Ma L, DE Waele L, Iwasaki Y, Gillijns V, Wilson JM, Collen D, Chuah MKL. Efficacy and safety of adeno-associated viral vectors based on serotype 8 and 9 vs. lentiviral vectors for hemophilia B gene therapy. J Thromb Haemost JTH 2007;5:16–24. [DOI] [PubMed] [Google Scholar]
- 41.Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, Wilson JM. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol 2004;78:6381–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zincarelli C, Soltys S, Rengo G, Koch WJ, Rabinowitz JE. Comparative cardiac gene delivery of adeno-associated virus serotypes 1–9 reveals that AAV6 mediates the most efficient transduction in mouse heart. Clin Transl Sci 2010;3:81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inagaki K, Fuess S, Storm TA, Gibson GA, Mctiernan CF, Kay MA, Nakai H. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther 2006;14:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DE, Zolotukhin I, Tarantal AF, Byrne BJ. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ Res 2006;99:e3–e9. [DOI] [PubMed] [Google Scholar]
- 45.Di Pasquale G, Chiorini JA. AAV transcytosis through barrier epithelia and endothelium. Mol Ther 2006;13:506–516. [DOI] [PubMed] [Google Scholar]
- 46.Prasad K-MR, Smith RS, Xu Y, French BA. A single direct injection into the left ventricular wall of an adeno-associated virus 9 (AAV9) vector expressing extracellular superoxide dismutase from the cardiac troponin-T promoter protects mice against myocardial infarction. J Gene Med 2011;13:333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao G, Bish LT, Sleeper MM, Mu X, Sun L, Lou Y, Duan J, Hu C, Wang L, Sweeney HL. Transendocardial delivery of AAV6 results in highly efficient and global cardiac gene transfer in rhesus macaques. Hum Gene Ther 2011;22:979–984. [DOI] [PubMed] [Google Scholar]
- 48.Chuah MK, Petrus I, De Bleser P, Le Guiner C, Gernoux G, Adjali O, Nair N, Willems J, Evens H, Rincon MY, Matrai J, Di Matteo M, Samara-Kuko E, Yan B, Acosta-Sanchez A, Meliani A, Cherel G, Blouin V, Christophe O, Moullier P, Mingozzi F, VandenDriessche T. Liver-specific transcriptional modules identified by genome-wide in silico analysis enable efficient gene therapy in mice and non-human primates. Mol Ther 2014;22:1605–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nicklin SA, Buening H, Dishart KL, de Alwis M, Girod A, Hacker U, Thrasher AJ, Ali RR, Hallek M, Baker AH. Efficient and selective AAV2-mediated gene transfer directed to human vascular endothelial cells. Mol Ther 2001;4:174–181. [DOI] [PubMed] [Google Scholar]
- 50.White SJ, Nicklin SA, Büning H, Brosnan MJ, Leike K, Papadakis ED, Hallek M, Baker AH. Targeted gene delivery to vascular tissue in vivo by tropism-modified adeno-associated virus vectors. Circulation 2004;109:513–519. [DOI] [PubMed] [Google Scholar]
- 51.Work LM, Büning H, Hunt E, Nicklin SA, Denby L, Britton N, Leike K, Odenthal M, Drebber U, Hallek M, Baker AH. Vascular bed-targeted in vivo gene delivery using tropism-modified adeno-associated viruses. Mol Ther 2006;13:683–693. [DOI] [PubMed] [Google Scholar]
- 52.Yue Y, Ghosh A, Long C, Bostick B, Smith BF, Kornegay JN, Duan D. A single intravenous injection of adeno-associated virus serotype-9 leads to whole body skeletal muscle transduction in dogs. Mol Ther 2008;16:1944–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J, Faust SM, Rabinowitz JE. The next step in gene delivery: molecular engineering of adeno-associated virus serotypes. J Mol Cell Cardiol 2011;50:793–802. [DOI] [PubMed] [Google Scholar]
- 54.Ying Y, Müller OJ, Goehringer C, Leuchs B, Trepel M, Katus HA, Kleinschmidt JA. Heart-targeted adeno-associated viral vectors selected by in vivo biopanning of a random viral display peptide library. Gene Ther 2010;17:980–990. [DOI] [PubMed] [Google Scholar]
- 55.Yang L, Jiang J, Drouin LM, Agbandje-McKenna M, Chen C, Qiao C, Pu D, Hu X, Wang D-Z, Li J, Xiao X. A myocardium tropic adeno-associated virus (AAV) evolved by DNA shuffling and in vivo selection. Proc Natl Acad Sci USA 2009;106:3946–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Asokan A, Conway JC, Phillips JL, Li C, Hegge J, Sinnott R, Yadav S, DiPrimio N, Nam H-J, Agbandje-McKenna M, McPhee S, Wolff J, Samulski RJ. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat Biotechnol 2010;28:79–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pulicherla N, Shen S, Yadav S, Debbink K, Govindasamy L, Agbandje-McKenna M, Asokan A. Engineering liver-detargeted AAV9 vectors for cardiac and musculoskeletal gene transfer. Mol Ther 2011;19:1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang B, Li J, Fu FH, Chen C, Zhu X, Zhou L, Jiang X, Xiao X. Construction and analysis of compact muscle-specific promoters for AAV vectors. Gene Ther 2008;15:1489–1499. [DOI] [PubMed] [Google Scholar]
- 59.Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J. Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res 2003;92:609–616. [DOI] [PubMed] [Google Scholar]
- 60.Bostick B, Yue Y, Long C, Marschalk N, Fine DM, Chen J, Duan D. Cardiac expression of a mini-dystrophin that normalizes skeletal muscle force only partially restores heart function in aged Mdx mice. Mol Ther 2009;17:253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Phillips MI, Tang Y, Schmidt-Ott K, Qian K, Kagiyama S. Vigilant vector: heart-specific promoter in an adeno-associated virus vector for cardioprotection. Hypertension 2002;39:651–655. [DOI] [PubMed] [Google Scholar]
- 62.Su H, Joho S, Huang Y, Barcena A, Arakawa-Hoyt J, Grossman W, Kan YW. Adeno-associated viral vector delivers cardiac-specific and hypoxia-inducible VEGF expression in ischemic mouse hearts. Proc Natl Acad Sci USA 2004;101:16280–16285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Choi S-C, Shim W-J, Lim D-S. Specific monitoring of cardiomyogenic and endothelial differentiation by dual promoter-driven reporter systems in bone marrow mesenchymal stem cells. Biotechnol Lett 2008;30:835–843. [DOI] [PubMed] [Google Scholar]
- 64.Bizy A, Guerrero-Serna G, Hu B, Ponce-Balbuena D, Willis BC, Zarzoso M, Ramirez RJ, Sener MF, Mundada LV, Klos M, Devaney EJ, Vikstrom KL, Herron TJ, Jalife J. Myosin light chain 2-based selection of human iPSC-derived early ventricular cardiac myocytes. Stem Cell Res 2013;11:1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boecker W, Bernecker OY, Wu JC, Zhu X, Sawa T, Grazette L, Rosenzweig A, del Monte F, Schmidt U, Hajjar RJ. Cardiac-specific gene expression facilitated by an enhanced myosin light chain promoter. Mol Imaging 2004;3:69–75. [DOI] [PubMed] [Google Scholar]
- 66.Rincon MY, Sarcar S, Danso-Abeam D, Keyaerts M, Matrai J, Samara-Kuko E, Acosta-Sanchez A, Athanasopoulos T, Dickson G, Lahoutte T, De Bleser P, VandenDriessche T, Chuah MK. Genome-wide computational analysis reveals cardiomyocyte-specific transcriptional cis-regulatory motifs that enable efficient cardiac gene therapy. Mol Ther 2015;23:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nair N, Rincon MY, Evens H, Sarcar S, Dastidar S, Samara-Kuko E, Ghandeharian O, Man Viecelli H, Thöny B, De Bleser P, VandenDriessche T, Chuah MK. Computationally designed liver-specific transcriptional modules and hyperactive factor IX improve hepatic gene therapy. Blood 2014;123:3195–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Geisler A, Schön C, Größl T, Pinkert S, Stein EA, Kurreck J, Vetter R, Fechner H. Application of mutated miR-206 target sites enables skeletal muscle-specific silencing of transgene expression of cardiotropic AAV9 vectors. Mol Ther 2013;21:924–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Geisler A, Jungmann A, Kurreck J, Poller W, Katus HA, Vetter R, Fechner H, Müller OJ. microRNA122-regulated transgene expression increases specificity of cardiac gene transfer upon intravenous delivery of AAV9 vectors. Gene Ther 2011;18:199–209. [DOI] [PubMed] [Google Scholar]
- 70.Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther 2010;18:80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lai Y, Yue Y, Liu M, Ghosh A, Engelhardt JF, Chamberlain JS, Duan D. Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat Biotechnol 2005;23:1435–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ghosh A, Yue Y, Shin J-H, Duan D. Systemic Trans-splicing adeno-associated viral delivery efficiently transduces the heart of adult mdx mouse, a model for duchenne muscular dystrophy. Hum Gene Ther 2009;20:1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, Rasko J, Ozelo MC, Hoots K, Blatt P, Konkle B, Dake M, Kaye R, Razavi M, Zajko A, Zehnder J, Rustagi PK, Nakai H, Chew A, Leonard D, Wright JF, Lessard RR, Sommer JM, Tigges M, Sabatino D, Luk A, Jiang H, Mingozzi F, Couto L, Ertl HC et al. . Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med 2006;12:342–347. [DOI] [PubMed] [Google Scholar]
- 74.Kwon I, Schaffer DV. Designer gene delivery vectors: molecular engineering and evolution of adeno-associated viral vectors for enhanced gene transfer. Pharm Res 2008;25:489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nathwani AC, Tuddenham EGD, Rangarajan S, Rosales C, McIntosh J, Linch DC, Chowdary P, Riddell A, Pie AJ, Harrington C, O'Beirne J, Smith K, Pasi J, Glader B, Rustagi P, Ng CYC, Kay MA, Zhou J, Spence Y, Morton CL, Allay J, Coleman J, Sleep S, Cunningham JM, Srivastava D, Basner-Tschakarjan E, Mingozzi F, High KA, Gray JT, Reiss UM et al. . Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 2011;365:2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martino AT, Basner-Tschakarjan E, Markusic DM, Finn JD, Hinderer C, Zhou S, Ostrov DA, Srivastava A, Ertl HCJ, Terhorst C, High KA, Mingozzi F, Herzog RW. Engineered AAV vector minimizes in vivo targeting of transduced hepatocytes by capsid-specific CD8+ T cells. Blood 2013;121:2224–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.European Society of Gene and Cell Therapy. French Society of Cell and Gene Therapy Collaborative Congress 2012 October 25–29, 2012 Palais des Congrès de Versailles Versailles, France. Hum Gene Ther 2012;23:A1–A173. [Google Scholar]
- 78.Cooray S, Howe SJ, Thrasher AJ. Retrovirus and lentivirus vector design and methods of cell conditioning. Methods Enzymol 2012;507:29–57. [DOI] [PubMed] [Google Scholar]
- 79.Mátrai J, Chuah MKL, VandenDriessche T. Recent advances in lentiviral vector development and applications. Mol Ther 2010;18:477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, Dionisio F, Calabria A, Giannelli S, Castiello MC, Bosticardo M, Evangelio C, Assanelli A, Casiraghi M, Di Nunzio S, Callegaro L, Benati C, Rizzardi P, Pellin D, Di Serio C, Schmidt M, Von Kalle C, Gardner J, Mehta N, Neduva V, Dow DJ, Galy A, Miniero R, Finocchi A, Metin A et al. . Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 2013;341:1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, Baldoli C, Martino S, Calabria A, Canale S, Benedicenti F, Vallanti G, Biasco L, Leo S, Kabbara N, Zanetti G, Rizzo WB, Mehta NAL, Cicalese MP, Casiraghi M, Boelens JJ, Del Carro U, Dow DJ, Schmidt M, Assanelli A, Neduva V, Di Serio C, Stupka E, Gardner J, von Kalle C et al. . Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013;341:1233158. [DOI] [PubMed] [Google Scholar]
- 82.VandenDriessche T, Thorrez L, Naldini L, Follenzi A, Moons L, Berneman Z, Collen D, Chuah MKL. Lentiviral vectors containing the human immunodeficiency virus type-1 central polypurine tract can efficiently transduce nondividing hepatocytes and antigen-presenting cells in vivo. Blood 2002;100:813–822. [DOI] [PubMed] [Google Scholar]
- 83.Bonci D, Cittadini A, Latronico MVG, Borello U, Aycock JK, Drusco A, Innocenzi A, Follenzi A, Lavitrano M, Monti MG, Ross J, Naldini L, Peschle C, Cossu G, Condorelli G. ‘Advanced’ generation lentiviruses as efficient vectors for cardiomyocyte gene transduction in vitro and in vivo. Gene Ther 2003;10:630–636. [DOI] [PubMed] [Google Scholar]
- 84.Niwano K, Arai M, Koitabashi N, Watanabe A, Ikeda Y, Miyoshi H, Kurabayashi M. Lentiviral vector–mediated SERCA2 gene transfer protects against heart failure and left ventricular remodeling after myocardial infarction in rats. Mol Ther 2008;16:1026–1032. [DOI] [PubMed] [Google Scholar]
- 85.Cherqui S, Kingdon KM, Thorpe C, Kurian SM, Salomon DR. Lentiviral gene delivery of vMIP-II to transplanted endothelial cells and endothelial progenitors is proangiogenic in vivo. Mol Ther 2007;15:1264–1272. [DOI] [PubMed] [Google Scholar]
- 86.VandenDriessche T, Chuah MK. Targeting endothelial cells by gene therapy. Blood 2013;122:1993–1994. [DOI] [PubMed] [Google Scholar]
- 87.Abel T, El Filali E, Waern J, Schneider IC, Yuan Q, Münch RC, Hick M, Warnecke G, Madrahimov N, Kontermann RE, Schüttrumpf J, Müller UC, Seppen J, Ott M, Buchholz CJ. Specific gene delivery to liver sinusoidal and artery endothelial cells. Blood 2013;122:2030–2038. [DOI] [PubMed] [Google Scholar]
- 88.Witting SR, Vallanda P, Gamble AL. Characterization of a third generation lentiviral vector pseudotyped with Nipah virus envelope proteins for endothelial cell transduction. Gene Ther 2013;20:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Papayannakos C, Daniel R. Understanding lentiviral vector chromatin targeting: working to reduce insertional mutagenic potential for gene therapy. Gene Ther 2013;20:581–588. [DOI] [PubMed] [Google Scholar]
- 90.Sasano T, Kikuchi K, McDonald AD, Lai S, Donahue JK. Targeted high-efficiency, homogeneous myocardial gene transfer. J Mol Cell Cardiol 2007;42:954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Karakikes I, Hadri L, Rapti K, Ladage D, Ishikawa K, Tilemann L, Yi G-H, Morel C, Gwathmey JK, Zsebo K, Weber T, Kawase Y, Hajjar RJ. Concomitant intravenous nitroglycerin with intracoronary delivery of AAV1.SERCA2a enhances gene transfer in porcine hearts. Mol Ther 2012;20:565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vera Janavel GL, De Lorenzi A, Cortés C, Olea FD, Cabeza Meckert P, Bercovich A, Criscuolo M, Laguens R, Crottogini A. Effect of vascular endothelial growth factor gene transfer on infarct size, left ventricular function and myocardial perfusion in sheep after 2 months of coronary artery occlusion. J Gene Med 2012;14:279–287. [DOI] [PubMed] [Google Scholar]
- 93.Ferrarini M, Arsic N, Recchia FA, Zentilin L, Zacchigna S, Xu X, Linke A, Giacca M, Hintze TH. Adeno-Associated Virus-mediated transduction of VEGF165 improves cardiac tissue viability and functional recovery after permanent coronary occlusion in conscious dogs. Circ Res 2006;98:954–961. [DOI] [PubMed] [Google Scholar]
- 94.Tao Z, Chen B, Tan X, Zhao Y, Wang L, Zhu T, Cao K, Yang Z, Kan YW, Su H. Coexpression of VEGF and angiopoietin-1 promotes angiogenesis and cardiomyocyte proliferation reduces apoptosis in porcine myocardial infarction (MI) heart. Proc Natl Acad Sci 2011;108:2064–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Post MJ, Sato K, Murakami M, Bao J, Tirziu D, Pearlman JD, Simons M. Adenoviral PR39 improves blood flow and myocardial function in a pig model of chronic myocardial ischemia by enhancing collateral formation. Am J Physiol Regul Integr Comp Physiol 2006;290:R494–R500. [DOI] [PubMed] [Google Scholar]
- 96.del Monte F, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW, Gwathmey JK, Rosenzweig A, Hajjar RJ. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation 1999;100:2308–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Miyamoto MI, del Monte F, Schmidt U, DiSalvo TS, Kang ZB, Matsui T, Guerrero JL, Gwathmey JK, Rosenzweig A, Hajjar RJ. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sci USA 2000;97:793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Beeri R, Chaput M, Guerrero JL, Kawase Y, Yosefy C, Abedat S, Karakikes I, Morel C, Tisosky A, Sullivan S, Handschumacher MD, Gilon D, Vlahakes GJ, Hajjar RJ, Levine RA. Gene delivery of sarcoplasmic reticulum calcium ATPase inhibits ventricular remodeling in ischemic mitral regurgitation. Circ Heart Fail 2010;3:627–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.del Monte F, Lebeche D, Guerrero JL, Tsuji T, Doye AA, Gwathmey JK, Hajjar RJ. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc Natl Acad Sci USA 2004;101:5622–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hadri L, Bobe R, Kawase Y, Ladage D, Ishikawa K, Atassi F, Lebeche D, Kranias EG, Leopold JA, Lompré A-M, Lipskaia L, Hajjar RJ. SERCA2a gene transfer enhances eNOS expression and activity in endothelial cells. Mol Ther 2010;18:1284–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pleger ST, Most P, Boucher M, Soltys S, Chuprun JK, Pleger W, Gao E, Dasgupta A, Rengo G, Remppis A, Katus HA, Eckhart AD, Rabinowitz JE, Koch WJ. Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation 2007;115:2506–2515. [DOI] [PubMed] [Google Scholar]
- 102.Pleger ST, Shan C, Ksienzyk J, Bekeredjian R, Boekstegers P, Hinkel R, Schinkel S, Leuchs B, Ludwig J, Qiu G, Weber C, Raake P, Koch WJ, Katus HA, Müller OJ, Most P. Cardiac AAV9-S100A1 gene therapy rescues post-ischemic heart failure in a preclinical large animal model. Sci Transl Med 2011;3:92ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shah AS, Lilly RE, Kypson AP, Tai O, Hata JA, Pippen A, Silvestry SC, Lefkowitz RJ, Glower DD, Koch WJ. Intracoronary adenovirus-mediated delivery and overexpression of the beta(2)-adrenergic receptor in the heart : prospects for molecular ventricular assistance. Circulation 2000;101:408–414. [DOI] [PubMed] [Google Scholar]
- 104.Kawahira Y, Sawa Y, Nishimura M, Sakakida S, Ueda H, Kaneda Y, Matsuda H. In vivo transfer of a beta 2-adrenergic receptor gene into the pressure-overloaded rat heart enhances cardiac response to beta-adrenergic agonist. Circulation 1998;98:II262–II267; discussion II267–II268. [PubMed] [Google Scholar]
- 105.Rengo G, Zincarelli C, Femminella GD, Liccardo D, Pagano G, de Lucia C, Altobelli GG, Cimini V, Ruggiero D, Perrone-Filardi P, Gao E, Ferrara N, Lymperopoulos A, Koch WJ, Leosco D. Myocardial β(2)-adrenoceptor gene delivery promotes coordinated cardiac adaptive remodelling and angiogenesis in heart failure. Br J Pharmacol 2012;166:2348–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Roth DM, Lai NC, Gao MH, Drumm JD, Jimenez J, Feramisco JR, Hammond HK. Indirect intracoronary delivery of adenovirus encoding adenylyl cyclase increases left ventricular contractile function in mice. Am J Physiol Heart Circ Physiol 2004;287:H172–H177. [DOI] [PubMed] [Google Scholar]
- 107.Takahashi T, Tang T, Lai NC, Roth DM, Rebolledo B, Saito M, Lew WYW, Clopton P, Hammond HK. Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation 2006;114:388–396. [DOI] [PubMed] [Google Scholar]
- 108.Guellich A, Gao S, Hong C, Yan L, Wagner TE, Dhar SK, Ghaleh B, Hittinger L, Iwatsubo K, Ishikawa Y, Vatner SF, Vatner DE. Effects of cardiac overexpression of type 6 adenylyl cyclase affects on the response to chronic pressure overload. Am J Physiol Heart Circ Physiol 2010;299:H707–H712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lai NC, Roth DM, Gao MH, Tang T, Dalton N, Lai YY, Spellman M, Clopton P, Hammond HK. Intracoronary adenovirus encoding adenylyl cyclase VI increases left ventricular function in heart failure. Circulation 2004;110:330–336. [DOI] [PubMed] [Google Scholar]
- 110.Rissanen TT, Ylä-Herttuala S. Current status of cardiovascular gene therapy. Mol Ther 2007;15:1233–1247. [DOI] [PubMed] [Google Scholar]
- 111.Taimeh Z, Loughran J, Birks EJ, Bolli R. Vascular endothelial growth factor in heart failure. Nat Rev Cardiol 2013;10:519–530. [DOI] [PubMed] [Google Scholar]
- 112.Urbanek K, Rota M, Cascapera S, Bearzi C, Nascimbene A, De Angelis A, Hosoda T, Chimenti S, Baker M, Limana F, Nurzynska D, Torella D, Rotatori F, Rastaldo R, Musso E, Quaini F, Leri A, Kajstura J, Anversa P. Cardiac stem cells possess growth factor-receptor systems that after activation regenerate the infarcted myocardium, improving ventricular function and long-term survival. Circ Res 2005;97:663–673. [DOI] [PubMed] [Google Scholar]
- 113.Autiero M, Luttun A, Tjwa M, Carmeliet P. Placental growth factor and its receptor, vascular endothelial growth factor receptor-1: novel targets for stimulation of ischemic tissue revascularization and inhibition of angiogenic and inflammatory disorders. J Thromb Haemost 2003;1:1356–1370. [DOI] [PubMed] [Google Scholar]
- 114.Zachary I, Morgan RD. Therapeutic angiogenesis for cardiovascular disease: biological context, challenges, prospects. Heart Br Card Soc 2011;97:181–189. [DOI] [PubMed] [Google Scholar]
- 115.Xiao N, Qi X-Y, Tang L-N, Tan L-L, Chen Y-Q, Zhao H-M. VEGF promotes cardiac stem cells differentiation into vascular endothelial cells via the PI3K/Akt signaling pathway. Artif Cells Nanomedicine Biotechnol 2014;42:400–405. [DOI] [PubMed] [Google Scholar]
- 116.Giacca M, Zacchigna S. VEGF gene therapy: therapeutic angiogenesis in the clinic and beyond. Gene Ther 2012;19:622–629. [DOI] [PubMed] [Google Scholar]
- 117.Itoh N, Ohta H. Pathophysiological roles of FGF signaling in the heart. Front Physiol 2013;4 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3764331/ (30 April 2015, date last accessed). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Grines CL, Watkins MW, Helmer G, Penny W, Brinker J, Marmur JD, West A, Rade JJ, Marrott P, Hammond HK, Engler RL. Angiogenic Gene Therapy (AGENT) trial in patients with stable angina pectoris. Circulation 2002;105:1291–1297. [DOI] [PubMed] [Google Scholar]
- 119.Detillieux KA, Sheikh F, Kardami E, Cattini PA. Biological activities of fibroblast growth factor-2 in the adult myocardium. Cardiovasc Res 2003;57:8–19. [DOI] [PubMed] [Google Scholar]
- 120.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011;473:298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 2011;10:417–427. [DOI] [PubMed] [Google Scholar]
- 122.Ylä-Herttuala S. Cardiovascular gene therapy with vascular endothelial growth factors. Gene 2013;525:217–219. [DOI] [PubMed] [Google Scholar]
- 123.Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol 2006;8:1223–1234. [DOI] [PubMed] [Google Scholar]
- 124.Rissanen TT, Korpisalo P, Markkanen JE, Liimatainen T, Ordén M-R, Kholová I, de Goede A, Heikura T, Gröhn OH, Ylä-Herttuala S. Blood flow remodels growing vasculature during vascular endothelial growth factor gene therapy and determines between capillary arterialization and sprouting angiogenesis. Circulation 2005;112:3937–3946. [DOI] [PubMed] [Google Scholar]
- 125.Lee RJ, Springer ML, Blanco-Bose WE, Shaw R, Ursell PC, Blau HM. VEGF gene delivery to myocardium: deleterious effects of unregulated expression. Circulation 2000;102:898–901. [DOI] [PubMed] [Google Scholar]
- 126.Kapur NK, Rade JJ. Fibroblast growth factor 4 gene therapy for chronic ischemic heart disease. Trends Cardiovasc Med 2008;18:133–141. [DOI] [PubMed] [Google Scholar]
- 127.Sarkar N, Rück A, Källner G, Y-Hassan S, Blomberg P, Islam KB, van der Linden J, Lindblom D, Nygren AT, Lind B, Brodin LA, Drvota V, Sylvén C. Effects of intramyocardial injection of phVEGF-A165 as sole therapy in patients with refractory coronary artery disease—12-month follow-up: angiogenic gene therapy. J Intern Med 2001;250:373–381. [DOI] [PubMed] [Google Scholar]
- 128.Losordo DW, Vale PR, Hendel RC, Milliken CE, Fortuin FD, Cummings N, Schatz RA, Asahara T, Isner JM, Kuntz RE. Phase 1/2 placebo-controlled, double-blind, dose-escalating trial of myocardial vascular endothelial growth factor 2 gene transfer by catheter delivery in patients with chronic myocardial ischemia. Circulation 2002;105:2012–2018. [DOI] [PubMed] [Google Scholar]
- 129.Favaloro L, Diez M, Mendiz O, Janavel GV, Valdivieso L, Ratto R, Garelli G, Salmo F, Criscuolo M, Bercovich A, Crottogini A. High-dose plasmid-mediated VEGF gene transfer is safe in patients with severe ischemic heart disease (Genesis-I). A phase I, open-label, two-year follow-up trial. Catheter Cardiovasc Interv Off J Soc Card Angiogr Interv 2013;82:899–906. [DOI] [PubMed] [Google Scholar]
- 130.Stewart DJ, Kutryk MJB, Fitchett D, Freeman M, Camack N, Su Y, Della Siega A, Bilodeau L, Burton JR, Proulx G, Radhakrishnan S, NORTHERN Trial Investigators. VEGF gene therapy fails to improve perfusion of ischemic myocardium in patients with advanced coronary disease: results of the NORTHERN trial. Mol Ther 2009;17:1109–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Stewart DJ, Hilton JD, Arnold JMO, Gregoire J, Rivard A, Archer SL, Charbonneau F, Cohen E, Curtis M, Buller CE, Mendelsohn FO, Dib N, Page P, Ducas J, Plante S, Sullivan J, Macko J, Rasmussen C, Kessler PD, Rasmussen HS. Angiogenic gene therapy in patients with nonrevascularizable ischemic heart disease: a phase 2 randomized, controlled trial of AdVEGF(121) (AdVEGF121) versus maximum medical treatment. Gene Ther 2006;13:1503–1511. [DOI] [PubMed] [Google Scholar]
- 132.Grines CL, Watkins MW, Mahmarian JJ, Iskandrian AE, Rade JJ, Marrott P, Pratt C, Kleiman N, Angiogene GENe Therapy (AGENT-2) Study Group. A randomized, double-blind, placebo-controlled trial of Ad5FGF-4 gene therapy and its effect on myocardial perfusion in patients with stable angina. J Am Coll Cardiol 2003;42:1339–1347. [DOI] [PubMed] [Google Scholar]
- 133.Henry TD, Grines CL, Watkins MW, Dib N, Barbeau G, Moreadith R, Andrasfay T, Engler RL. Effects of Ad5FGF-4 in patients with angina: an analysis of pooled data from the AGENT-3 and AGENT-4 trials. J Am Coll Cardiol 2007;50:1038–1046. [DOI] [PubMed] [Google Scholar]
- 134.Hedman M, Hartikainen J, Ylä-Herttuala S. Progress and prospects: hurdles to cardiovascular gene therapy clinical trials. Gene Ther 2011;18:743–749. [DOI] [PubMed] [Google Scholar]
- 135.Katz MG, Fargnoli AS, Williams RD, Bridges CR. The road ahead: working towards effective clinical translation of myocardial gene therapies. Ther Deliv 2014;5:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dewerchin M, Carmeliet P. PlGF: a multitasking cytokine with disease-restricted activity. Cold Spring Harb Perspect Med 2012;2:a011056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Iwasaki H, Kawamoto A, Tjwa M, Horii M, Hayashi S, Oyamada A, Matsumoto T, Suehiro S, Carmeliet P, Asahara T. PlGF repairs myocardial ischemia through mechanisms of angiogenesis, cardioprotection and recruitment of myo-angiogenic competent marrow progenitors. PLoS ONE 2011;6:e24872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H, Drexler H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circ Res 1994;75:434–442. [DOI] [PubMed] [Google Scholar]
- 139.Shareef MA, Anwer LA, Poizat C. Cardiac SERCA2A/B: therapeutic targets for heart failure. Eur J Pharmacol 2014;724:1–8. [DOI] [PubMed] [Google Scholar]
- 140.Gwathmey JK, Yerevanian A, Hajjar RJ. Targeting sarcoplasmic reticulum calcium ATPase by gene therapy. Hum Gene Ther 2013;24:937–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kawase Y. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J Am Coll Cardiol 2008;51:1112–1119. [DOI] [PubMed] [Google Scholar]
- 142.Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan G-C, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW II, MacLennan DH, Kremastinos DT, Kranias EG. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest 2003;111:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kato K, Kimura S. S100ao (alpha alpha) protein is mainly located in the heart and striated muscles. Biochim Biophys Acta 1985;842:146–150. [DOI] [PubMed] [Google Scholar]
- 144.Most P, Seifert H, Gao E, Funakoshi H, Völkers M, Heierhorst J, Remppis A, Pleger ST, DeGeorge BR, Eckhart AD, Feldman AM, Koch WJ. Cardiac S100A1 protein levels determine contractile performance and propensity toward heart failure after myocardial infarction. Circulation 2006;114:1258–1268. [DOI] [PubMed] [Google Scholar]
- 145.Kettlewell S, Most P, Currie S, Koch WJ, Smith GL. S100A1 increases the gain of excitation-contraction coupling in isolated rabbit ventricular cardiomyocytes. J Mol Cell Cardiol 2005;39:900–910. [DOI] [PubMed] [Google Scholar]
- 146.Boerries M, Most P, Gledhill JR, Walker JE, Katus HA, Koch WJ, Aebi U, Schoenenberger C-A. Ca2+ -dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol Cell Biol 2007;27:4365–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Most P, Pleger ST, Völkers M, Heidt B, Boerries M, Weichenhan D, Löffler E, Janssen PML, Eckhart AD, Martini J, Williams ML, Katus HA, Remppis A, Koch WJ. Cardiac adenoviral S100A1 gene delivery rescues failing myocardium. J Clin Invest 2004;114:1550–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Remppis A, Pleger ST, Most P, Lindenkamp J, Ehlermann P, Schweda C, Löffler E, Weichenhan D, Zimmermann W, Eschenhagen T, Koch WJ, Katus HA. S100A1 gene transfer: a strategy to strengthen engineered cardiac grafts. J Gene Med 2004;6:387–394. [DOI] [PubMed] [Google Scholar]
- 149.Brinks H, Rohde D, Voelkers M, Qiu G, Pleger ST, Herzog N, Rabinowitz J, Ruhparwar A, Silvestry S, Lerchenmüller C, Mather PJ, Eckhart AD, Katus HA, Carrel T, Koch WJ, Most P. S100A1 genetically targeted therapy reverses dysfunction of human failing cardiomyocytes. J Am Coll Cardiol 2011;58:966–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Marks AR. Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Invest 2013;123:46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Roth DM, Bayat H, Drumm JD, Gao MH, Swaney JS, Ander A, Hammond HK. Adenylyl cyclase increases survival in cardiomyopathy. Circulation 2002;105:1989–1994. [DOI] [PubMed] [Google Scholar]
- 152.Brodde OE. Beta-adrenoceptors in cardiac disease. Pharmacol Ther 1993;60:405–430. [DOI] [PubMed] [Google Scholar]
- 153.Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol 2009;54:1747–1762. [DOI] [PubMed] [Google Scholar]
- 154.Milano CA, Allen LF, Rockman HA, Dolber PC, McMinn TR, Chien KR, Johnson TD, Bond RA, Lefkowitz RJ. Enhanced myocardial function in transgenic mice overexpressing the beta 2-adrenergic receptor. Science 1994;264:582–586. [DOI] [PubMed] [Google Scholar]
- 155.Zhu W, Petrashevskaya N, Ren S, Zhao A, Chakir K, Gao E, Chuprun JK, Wang Y, Talan M, Dorn GW, Lakatta EG, Koch WJ, Feldman AM, Xiao R-P. Gi-biased β2AR signaling links GRK2 upregulation to heart failure. Circ Res 2012;110:265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gong H, Adamson DL, Ranu HK, Koch WJ, Heubach JF, Ravens U, Zolk O, Harding SE. The effect of Gi-protein inactivation on basal, and beta(1)- and beta(2)AR-stimulated contraction of myocytes from transgenic mice overexpressing the beta(2)-adrenoceptor. Br J Pharmacol 2000;131:594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Iaccarino G, Ciccarelli M, Sorriento D, Galasso G, Campanile A, Santulli G, Cipolletta E, Cerullo V, Cimini V, Altobelli GG, Piscione F, Priante O, Pastore L, Chiariello M, Salvatore F, Koch WJ, Trimarco B. Ischemic neoangiogenesis enhanced by beta2-adrenergic receptor overexpression: a novel role for the endothelial adrenergic system. Circ Res 2005;97:1182–1189. [DOI] [PubMed] [Google Scholar]
- 158.Lohse MJ. G-protein-coupled receptor kinases and the heart. Trends Cardiovasc Med 1995;5:63–68. [DOI] [PubMed] [Google Scholar]
- 159.Koch WJ, Rockman HA, Samama P, Hamilton RA, Bond RA, Milano CA, Lefkowitz RJ. Cardiac function in mice overexpressing the beta-adrenergic receptor kinase or a beta ARK inhibitor. Science 1995;268:1350–1353. [DOI] [PubMed] [Google Scholar]
- 160.Ungerer M, Kessebohm K, Kronsbein K, Lohse MJ, Richardt G. Activation of beta-adrenergic receptor kinase during myocardial ischemia. Circ Res 1996;79:455–460. [DOI] [PubMed] [Google Scholar]
- 161.Harding VB, Jones LR, Lefkowitz RJ, Koch WJ, Rockman HA. Cardiac beta ARK1 inhibition prolongs survival and augments beta blocker therapy in a mouse model of severe heart failure. Proc Natl Acad Sci USA 2001;98:5809–5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Raake PW, Vinge LE, Gao E, Boucher M, Rengo G, Chen X, DeGeorge BR Jr, Matkovich S, Houser SR, Most P, Eckhart AD, Dorn GW II, Koch WJ. G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ Res 2008;103:413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Williams ML, Hata JA, Schroder J, Rampersaud E, Petrofski J, Jakoi A, Milano CA, Koch WJ. Targeted beta-adrenergic receptor kinase (betaARK1) inhibition by gene transfer in failing human hearts. Circulation 2004;109:1590–1593. [DOI] [PubMed] [Google Scholar]
- 164.Raake PWJ, Schlegel P, Ksienzyk J, Reinkober J, Barthelmes J, Schinkel S, Pleger S, Mier W, Haberkorn U, Koch WJ, Katus HA, Most P, Müller OJ. AAV6.βARKct cardiac gene therapy ameliorates cardiac function and normalizes the catecholaminergic axis in a clinically relevant large animal heart failure model. Eur Heart J 2013;34:1437–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hajjar RJ, Zsebo K, Deckelbaum L, Thompson C, Rudy J, Yaroshinsky A, Ly H, Kawase Y, Wagner K, Borow K, Jaski B, London B, Greenberg B, Pauly DF, Patten R, Starling R, Mancini D, Jessup M. Design of a phase 1/2 trial of intracoronary administration of AAV1/SERCA2a in patients with heart failure. J Card Fail 2008;14:355–367. [DOI] [PubMed] [Google Scholar]
- 166.Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ, Calcium Up-Regulation by Percutaneous Administration of Gene Therapy In Cardiac Disease (CUPID) Trial Investigators. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail 2009;15:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ, Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) Investigators. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 2011;124:304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mearns BM. Gene therapy: can CUPID rescue the broken hearted? Nat Rev Cardiol 2011;8:481. [DOI] [PubMed] [Google Scholar]
- 169.Zsebo K, Yaroshinsky A, Rudy JJ, Wagner K, Greenberg B, Jessup M, Hajjar RJ. Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ Res 2014;114:101–108. [DOI] [PubMed] [Google Scholar]
- 170.Pleger ST, Raake P, Katus HA, Most P. Cardiac calcium handling on trial targeting the failing cardiomyocyte signalosome. Circ Res 2014;114:12–14. [DOI] [PubMed] [Google Scholar]
- 171.Greenberg B, Yaroshinsky A, Zsebo KM, Butler J, Felker GM, Voors AA, Rudy JJ, Wagner K, Hajjar RJ. Design of a phase 2b trial of intracoronary administration of AAV1/SERCA2a in patients with advanced heart failure: the CUPID 2 trial (calcium up-regulation by percutaneous administration of gene therapy in cardiac disease phase 2b). JACC Heart Fail 2014;2:84–92. [DOI] [PubMed] [Google Scholar]
- 172.Weber C, Neacsu I, Krautz B, Schlegel P, Sauer S, Raake P, Ritterhoff J, Jungmann A, Remppis AB, Stangassinger M, Koch WJ, Katus HA, Müller OJ, Most P, Pleger ST. Therapeutic safety of high myocardial expression levels of the molecular inotrope S100A1 in a preclinical heart failure model. Gene Ther 2014;21:131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]