

Background: E-selectin interactions with glycoprotein ligands mediate the initial capturing of cells out of flow.
Results: Adopting a Biacore-based immunoprecipitation binding assay unraveled differential binding kinetics of monomeric (m) versus dimeric (d) E-selectin to endogenous ligands.
Conclusion: Although mE-selectin binds transiently, dE-selectin binds with remarkably slow on- and off-rates.
Significance: Transitioning from monomeric to dimeric E-selectin could enable fast but firm capturing of cells out of flow.
Keywords: CD44, cell adhesion, cell migration, glycobiology, glycoprotein, surface plasmon resonance (SPR), PSGL-1, selectin, selectin ligand, sialyl Lewis x
Abstract
Selectins (E-, P-, and L-selectins) interact with glycoprotein ligands to mediate the essential tethering/rolling step in cell transport and delivery that captures migrating cells from the circulating flow. In this work, we developed a real time immunoprecipitation assay on a surface plasmon resonance chip that captures native glycoforms of two well known E-selectin ligands (CD44/hematopoietic cell E-/L-selectin ligand and P-selectin glycoprotein ligand-1) from hematopoietic cell extracts. Here we present a comprehensive characterization of their binding to E-selectin. We show that both ligands bind recombinant monomeric E-selectin transiently with fast on- and fast off-rates, whereas they bind dimeric E-selectin with remarkably slow on- and off-rates. This binding requires the sialyl Lewis x sugar moiety to be placed on both O- and N-glycans, and its association, but not dissociation, is sensitive to the salt concentration. Our results suggest a mechanism through which monomeric selectins mediate initial fast on and fast off kinetics to help capture cells out of the circulating shear flow; subsequently, tight binding by dimeric/oligomeric selectins is enabled to significantly slow rolling.
Introduction
The emigration of cells out of the circulating flow is a sophisticated process controlled by various adhesion molecules, particularly selectins (E-, P-, and L-selectins), chemokines, and integrins, functioning in a coordinated multistep cascade (1, 2). Selectins and their respective ligands mediate the first essential step in this process, which initially tethers the flowing cell to the vessel wall and, in the context of vascular shear stress, causes the cell to then roll along the endothelium. These interactions decrease the cellular velocity below that of the prevailing hemodynamic stream and bring the cell into close physical proximity to the vessel wall to initiate the subsequent cell adhesion steps. The mechanisms and post-translational modifications involved in the interaction of the identified ligands with selectins and their subsequent contribution to shear-resistant adhesion are crucial to understanding how cell migration is influenced by disease and how it can be manipulated in therapy.
One well defined example of migration is that of circulating hematopoietic stem/progenitor cells to the bone marrow. E-selectin is constitutively expressed on the bone marrow endothelium (3, 4), and it has been suggested to interact with several ligands on hematopoietic stem/progenitor cells to mediate the first step in the process of recruitment to the bone marrow. Like its siblings P- and L-selectin, E-selectin is a Ca2+-dependent lectin that binds to specialized carbohydrate determinants, sialyl Lewis X (sLex)2 (5, 6), displayed on either a protein scaffold as an N- or O-glycan (i.e. a glycoprotein) or a lipid scaffold (i.e. a glycolipid) (7–9).
Researchers have identified many E-selectin ligands on human and mouse cells (for a review, see Ref. 10). To be functionally classified as a selectin ligand, the glycoprotein should support rolling (through various in vitro assays). In addition, gene deletion/silencing studies and/or blocking monoclonal antibodies (mAbs) against specific ligands should impair selectin-mediated functions on intact cells. To date, no mAbs against glycoproteins that block binding to E-selectin have been identified. This has significantly hindered the ability of researchers to test the physiologic functions of candidate E-selectin ligands on human cells, such as hematopoietic stem/progenitor or leukemic cells, where gene knock-out and gene silencing methods are less feasible. The importance of each ligand to interact with E-selectin in human cells is thus debatable and requires additional attention and methodologies to determine and better understand their involvement.
Ideally, the in vitro characterization of direct E-selectin interactions with its ligands would comprise a pulldown of proteins in their native post-translationally modified form (often achieved through immunoprecipitation (IP) using mAbs) with recombinant proteins using Western blotting. However, IP from cell lysates has several limitations. First, long incubation times of the cell lysate with the antibody are required to enhance the capturing efficiency of the ligand, which assumes that protein stability in the lysate is maintained although this may not be the case. Second, extensive washing steps with buffer to remove nonspecifically bound proteins bias the IP to detect bimolecular interactions among stably bound proteins, resulting in the potential loss of important mechanistic information provided by weak or transient interactions that may exist. Third, the affinity and capturing efficiency of the mAb might be influenced by different post-translational modifications, posing a challenge to IP-based comparative approaches. Lastly, Western blotting-based IP binding studies do not provide quantitative measurements of binding kinetics, making it difficult to provide head-to-head comparisons of different ligands binding with a specific protein receptor.
In this study, we describe a powerful assay that is complementary to previous approaches in which we perform real time IP on a surface plasmon resonance (SPR) chip and directly measure the interaction of E-selectin with its ligands in a quantitative and rapid manner following cell lysis. In this assay, endogenous E-selectin ligands in their native post-translationally modified form are captured with high specificity from whole cell lysates prepared from a human leukemic progenitor (hematopoietic stem/progenitor cell model) cell line, KG1a, via a surface-immobilized mAb. Subsequently, their direct interaction with recombinant E-selectin protein in either monomeric (m) or dimeric (d) form is characterized. We demonstrate through several examples the quantitative nature of our SPR-based IP approach, including the ability to 1) capture residual and transient interactions, 2) directly characterize the contribution of different post-translational modifications on ligands (that contribute particular isoforms of proteins) to the unique antigenic efficiencies of the antibody used for IP, and 3) determine binding constants of antibody-isolated ligands with the adhesion molecule of interest, E-selectin. This assay enabled a comparative and comprehensive binding analysis of CD44/hematopoietic cell E-/L-selectin ligand (HCELL) and P-selectin glycoprotein ligand-1 (PSGL-1) (11–15, 53) in their native forms from KG1a cells with E-selectin. In addition, we include a comprehensive analysis of CD44/HCELL binding with E-selectin of two additional leukemic cell lines, THP-1 and HL-60. This work will help advance our understanding of the more detailed mechanisms involved in cell adhesion and migration.
Experimental Procedures
Cells
The human cell line KG1a (human acute myelogenous leukemia; serves as hematopoietic stem/progenitor cell-like (CD34+) model cell line), THP-1 (acute monocytic leukemia), and HL-60 (acute promyelocytic leukemia) cell lines were purchased from ATCC and cultured in RPMI 1640 medium supplemented with 10% FBS (Gibco) and 100 units/ml penicillin/streptomycin (Invitrogen). A transgenic Chinese hamster ovary (CHO) cell line stably expressing full-length mouse E-selectin (CHO-E) (or the plasmid alone (CHO-Mock)) was established in our laboratory by transfection of pEFdest51-based expression plasmid followed by blasticidin selection and isolation and maintained as described previously (11, 14).
Antibodies, Proteins, and Enzymes
Anti-human CD44 (clone 515, MsIgG1), anti-human/mouse CD44 (clone IM7, rat IgG2b), anti-human PSGL-1 (KPL1, MsIgG1), FITC-labeled anti-mouse IgGs (IgG1, IgG2a, and IgG2b), and HRP-labeled anti-mouse IgG antibodies were from BD Pharmingen. Anti-human CD44 antibody (Hermes-3, mouse IgG2a) was from Abgene, anti-CD34 antibody (EP373Y, rabbit IgG) that recognizes the C terminus of CD34 protein regardless of the glycosylation status and MsIgG isotype antibodies were from Abcam, and HRP-labeled anti-human IgG was from Southern Biotech. The glycoprotease-sensitive CD34 mAb QBend-10 (Novus Biologicals) is sensitive to removal of O- or N-linked glycans on CD34 and is routinely used to detect the efficiency of glycan removal (14, 16). Recombinant mouse E-selectin/human IgG Fc chimera (dE-selectin), recombinant human monomeric E-selectin derived from CHO cells (mE-selectin), recombinant human CD44 (rH-CD44; purified from NSO cells, a mouse myeloma line that lacks the necessary glycosylation machinery to create sLex) (17, 18), and anti-CD44 (2C5, MsIgG2a) were from R&D Systems. O Sialoglycoprotease was from Cedarlane Laboratories. Vibrio cholerae neuraminidase (Newcastle) and peptide: N-glycosidase F (PNGaseF) were from New England Biolabs.
Immunofluorescence Analysis
KG1a cells were cultured overnight on poly-l-ornithine-coated coverslips (Invitrogen). Attached cells were washed three times with washing buffer (HBSS containing 5% normal goat serum), blocked with blocking buffer (HBSS containing 10% normal goat serum), and fixed with 4% paraformaldehyde in HBSS for 15 min at room temperature. KG1a cells were washed three times in washing buffer and double immunostained with recombinant dE-selectin (10 μg/ml) and anti-CD44 mAb (5 μg/ml) in blocking buffer for 2 h at room temperature. The cells were washed and incubated with appropriate Alexa Fluor 488 anti-human IgG (green)- and Cy-5 anti-mouse IgG (red)-conjugated secondary antibody (Ab) at 10 μg/ml for 30 min at 4 °C. The cells were mounted on glass slides in ProLong Gold antifade reagent with DAPI (Life Technologies). Images were analyzed using a Zeiss LSM 710 Axio confocal microscope with a 100× oil immersion lens. The images shown are representative of at least three experiments with multiple images taken per slide. Cell surface labeling with secondary Ab or isotype control served as a background control.
Flow Cytometry Analysis
Aliquots of cells (2 × 105 cells) were washed with PBS containing 2% FBS and incubated with primary mAbs or with isotype control (either unconjugated or fluorochrome-conjugated). The cells were washed in washing buffer (PBS containing 2% FBS) and for indirect immunofluorescence incubated with appropriate secondary fluorochrome-conjugated Ab to isotype Abs. The cells were washed with the same washing buffer, and the fluorescence intensity was determined using a FACSCanto (BD Biosciences).
Cell Lysis, IP, and Western Blotting
Whole cell lysate was prepared by membrane disruption using Triton X-100 lysis buffer (1% Triton X-100, 50 mm Tris base (pH 8.0), 250 mm NaCl, 1 mm PMSF, and protease inhibitor mixture tablet (Roche Applied Science)) at a density of 1 × 107 cells/100 μl for 1 h at 4 °C. The insoluble materials were cleared by centrifugation at high speed for 30 min at 4 °C. The lysate was precleared three times (20 min each) with Protein G-agarose and then incubated with 3 μg of CD44 Ab (1:1 mixture of 515 and 2C5 unless otherwise stated) for 2 h at 4 °C. The lysate and Ab mixture was added to 50 μl of clean Protein G-agarose beads (the beads were prewashed three times with lysis buffer containing 2% BSA and three times with lysis buffer). The mixture was then incubated for 4 h at 4 °C under rotation (1400 rpm); washed three times in lysis buffer containing 2% BSA and three times in lysis buffer diluted with NuPAGE® lithium dodecyl sulfate sample buffer (2×) (Invitrogen) and β-mercaptoethanol (10 mm; Fisher); and heated for 10 min at 70 °C. For Western blot analysis, resultant proteins were run on a 4–20% SDS-polyacrylamide gradient gel and transferred to an immunoblot PVDF membrane (Bio-Rad) blocked with 5% nonfat milk in buffer containing TBS, Tween 20, 10 mm HEPES, and 2 mm CaCl2. Membranes were immunostained with 1 μg/ml recombinant dE-selectin chimera or the primary Ab of interest followed by incubation with the appropriate HRP-conjugated secondary Ab. Blots were visualized with chemiluminescence using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
CD44 Purification (for Reciprocal SPR Assay)
IP of CD44 and subsequent washing steps were performed as described under “Cell Lysis, IP, and Western Blotting” using Hermes-3 mAb. CD44 was eluted from the mAb and Protein G beads by incubating overnight at 4 °C in 500 μl of elution buffer (8 m urea, 4% Triton X-100, and 130 mm DTT). The supernatant was cleared by centrifugation (1500 rpm) and dialyzed overnight in dialysis buffer (50 mm NaCl, 50 mm Tris base (pH 8.0), 1 mm CaCl2, and 1% Triton X-100) using 10-kDa molecular mass-cutoff tubing. The concentration was determined using PierceTM BCA Protein Assay kit. The purified CD44 was confirmed by Western blot analysis.
Enzyme Treatment
The cell lysates were prepared as described above at a density of 1 × 107 cell/100 μl. N-Linked glycoprotein was removed by treating the lysate with commercially available PNGase F (8 units/ml) diluted with G7 buffer (50 mm sodium phosphate (pH 7.5)) and 1% Nonidet P-40 for 24 h at 37 °C. O-Glycans were removed by O-sialoglycoprotein endopeptidase (OSGE) (240 μg/ml) treatment for 4 h at 37 °C. OSGE was lyophilized in HEPES buffer (pH 7.4) and replenished in the same buffer containing 1% BSA. Sialic acid was removed by incubating V. cholerae neuraminidase (0.2 unit/ml) in 50 mm sodium acetate buffer (pH 5.5) containing 5 mm CaCl2 and 150 mm NaCl with lysate for 3 h at 37 °C. As a control for the treatment conditions, the lysates for each enzyme digestion were treated with the same buffer and incubated for the same duration and temperature but without the enzymes. These lysates were protected from degradation by protease inhibitor mixture from Roche Applied Science and PMSF (1 mm).
SPR
SPR binding experiments were performed using a Biacore T-100 system (GE Healthcare) at 25 °C. The system and flow cells were washed with the corresponding running buffer (filtered with a 0.2-μm filter and degassed) by conducting two priming steps. Immobilization of mAbs on a carboxymethylated (CM5) dextran sensor chip was performed using an amine coupling kit (Biacore) in accordance with the manufacturer's instructions. Briefly, the immobilization steps were performed in HBS-EP buffer (20 mm HEPES (pH 7.4), 150 mm NaCl, and 0.005% surfactant P20). A CM5 chip was activated with a 7-min injection of a 1:1 ratio mixture of N-hydroxysuccinimide (0.1 m) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (0.4 m) at a flow rate of 5 μl/min. The mAb was then injected at a concentration of 20 μg/ml in 10 mm sodium acetate buffer (pH 5.0) at a flow rate of 10 μl/min to reach an immobilization level between 4000 and 8000 response units (RU). The surface was then deactivated using a 7-min injection of 1 m ethanolamine hydrochloride at a flow rate of 5 μl/min. The exact number of RU for each immobilized mAb is specified in the text and/or corresponding figure legends. A control flow cell that was immobilized with an equal amount of the mAb corresponding isotype control was used to correct for the bulk refractive index of the buffer and nonspecific interactions with the surface and with the mAb. In the case of HL-60 and THP-1 cell lysates, the lysates were incubated with Fc receptor blocking reagent (15 μl/3 × 107 cells) for 1 h at 4 °C to minimize nonspecifically interacting materials in the lysate with anti-MsIgG2a isotype control. Analysis of nonspecific interactions with the CM5 surface was performed with a flow cell that underwent the same immobilization procedure but without the Ab. Further binding experiments were performed at a standard flow rate of 20 μl/min unless otherwise specified in running buffer (1% Triton X-100, 50 mm NaCl, 50 mm Tris base (pH 8.0), and 1 mm CaCl2). Whole cell lysates were prepared as described above, cleared by centrifugation, and injected over the immobilized Abs for a standard time of 500 s unless otherwise specified. The flow cells were washed with running buffer for a standard time of 3 min unless otherwise specified to remove nonspecifically mAb-bound proteins.
Reciprocal SPR binding experiments were performed similarly to that described above except that dE-selectin (13,000 RU) was immobilized on the CM5 chip, whereas purified CD44 (from KG1a lysate; 1150 nm) was introduced in the flow. A blank flow cell was used as a control to correct for the bulk refractive index of the buffer and nonspecific interactions with the surface.
To determine the purity of captured ligands, i.e. CD44, a CD44 recovery experiment was performed on a CM5 chip with three flow cells immobilized with 515 mAb and the fourth immobilized with the MsIgG1 isotype control. The experiment was conducted in accordance with the recovery method in the Biacore T-100 software. Briefly, KG1a lysate was injected at 20 μl/min for 600 s followed by a buffer washing step for either 1 or 5 min at 20 μl/min (the same flow rate as that used in the binding experiment). The captured proteins were incubated for 30 min with buffer containing 1% Triton X-100, 50 mm NaCl, 50 mm Tris base (pH 8.0), and 5 mm EDTA and subsequently eluted in a total volume of 10 μl. The cycle of CD44 binding and elution was repeated five times, and the total sample was analyzed by Western blotting as described above (without the Protein G beads and the associated washing steps).
dE-selectin was injected at a standard concentration of 177 nm (diluted in the running buffer) for a standard time of 130 s unless otherwise specified. The standard regeneration step for removing the bound dE-selectin was a 130-s injection of running buffer containing 1 m NaCl at a flow rate of 45 μl/min. The regeneration condition in the PNGase F, OSGE, and sialidase treatment study included, in addition to the standard regeneration step, an extra step to remove the bound CD44 from the CD44 mAb by washing with running buffer for 1 h. This regeneration procedure was effective in the case of 515 mAb, which tended to have lower affinity to CD44 in our experiments. However, in the case of Hermes-3 mAb, which bound with higher affinity to specific CD44 glycoforms, increased washing times were required. As a result, experiments were performed beginning with the enzyme-treated lysates because only minimal (to no) binding of CD44 to dE-selectin was observed.
Data analysis was performed using Biacore evaluation software. When sensorgram normalization was applied to even out the difference in the level of mAb-captured CD44 among different flow cells and/or different lysate injections, the entire sensorgram was multiplied by a normalization factor that was defined based on its relative RU just prior to dE-selectin injection to the RU in the flow cell with the highest accumulated CD44. The relationship among the amount of immobilized proteins (RUligand), the bound protein (RUanalyte), and their stoichiometry was calculated based on Equation 1.
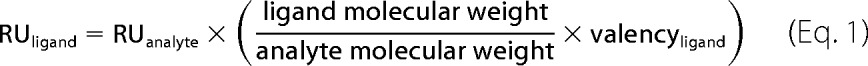 |
Note that the calculated ligand and analyte molecular weights are based on apparent molecular weight determined by SDS-PAGE.
The determination of the equilibrium dissociation binding constant (KD) was performed simultaneously on CD44 that was captured by immobilized Hermes-3 mAb (designated as “CD44·Hermes-3 mAb” complex) and PSGL-1 that was captured by immobilized KPL-1 mAb (designated as “PSGL-1·KPL-1 mAb” complex) from KG1a lysate (injected at a flow rate of 30 μl/min for 700 s). Subsequently, dE-selectin was injected at different concentrations in a sequence of low to high concentrations with no regeneration step between subsequent dE-selectin injections at a flow rate of 30 μl/min for 130 s. A 60-s buffer washing step was performed at the end of each dE-selectin injection. The maximum RU reached prior to the start of the wash with buffer (RUmax) where a steady-state condition was nearly met was blotted against the dE-selectin concentration and fitted using the steady-state model provided by the Biacore evaluation software to determine the KD value. The dissociation rate constant (koff) of the CD44 from Hermes-3 mAb or the apparent dissociation rate constant (koff-apparent) of the interaction of CD44·Hermes-3 mAb with dE-selectin (the dissociation of CD44 or CD44·dE-selectin from Hermes-3 mAb and/or the dissociation of dE-selectin from CD44·Hermes-3 mAb complex) from a single concentration type binding study was calculated by fitting the stable phase in the buffer washing step using the Biacore evaluation software. For calculating the activity of the surface-immobilized ligand to bind dE-selectin, we used Equation 1 to calculate the theoretical RUmax and compare it with that obtained experimentally from the binding isotherm.
Blot Rolling Assay
The blot rolling assay permits detection of physiological shear-dependent selectin interactions on membrane protein ligands (CD44/HCELL) resolved by SDS-PAGE; it was performed as described previously (19, 20). Briefly, Western blots of immunoprecipitated CD44 and PSGL-1 membrane proteins were stained with anti-sLex mAb (HECA-452), and the membrane was rendered translucent by immersion in HBSS containing 5 mm HEPES and 10% glycerol. CHO-E cells were resuspended (5 × 106/ml) in HBSS containing 5 mm HEPES, 2 mm CaCl2, and 10% glycerol adjusted to different NaCl concentrations (50, 150, and 200 mm). The blots were placed within a parallel plate flow chamber, and CHO-E cells were perfused at a physiologically relevant shear stress of 0.25 dyne/cm2 with an adjustment in the volumetric flow rate made to account for the increase in viscosity due to the presence of 10% glycerol in the flow medium. Molecular weight markers were used as a guide to aid placement of the flow chamber over the stained bands of interest. CHO-E cell suspensions containing 5 mm EDTA, which eliminates divalent cation-dependent E-selectin adhesion, were used as a control for defining the level of nonspecific adhesion above which CHO-E cell adhesion was considered. The results are reported from six experiments using four fields of view per experiment.
For single cell tracking, images were acquired at one frame/s in avi format and then converted to TIFF format. The background was subtracted, and the contrast was enhanced using NIH ImageJ. The processed TIFF files were imported into Imaris V7 software (Bitplane) for tracking rolling cells. The cells were treated with a “spot” function in the “Surpass” menu, and the centroid position was determined using both the minimum diameter of the cells (10 μm) and their minimum threshold intensity. The accuracy in determining the centroid position of the cells as a result of Brownian motion was determined using cells that were stuck on the surface (n = 17) and was found to be 10 ± 4 μm. The clustered cells or those that remained in the field of view for less than 4 s were eliminated, the rest of the cells were tracked using an “autoregressive motion algorithm,” and their position coordinates were extracted as an Excel file. The displacement in the X and Y direction were separated and normalized using custom MATLAB code. The X-position of each rolling cell (parallel to the flow) versus time was plotted in OriginPro. Displacements greater than four acquisition data points (4 s) were considered as rolling events, and those that were less than four acquisition data points were considered as adhesion events. Displacements that were more than 30 μm/s (3× the minimum diameter of the cell) were not considered as they were likely to arise from imperfection in the membrane. The velocity, Vx (μm/s), was calculated by fitting the slope of only the moving phase of the rolling cells (i.e. the adhesion phase where the cells did not move was not included and was considered to mark a new cell rolling event) with a linear fit in OriginPro, and the average velocity for each condition was calculated from the average of the slopes. The average distance traveled by the cells for each condition corresponds to the average of the distances traveled during the moving phase of the rolling cells.
Statistical Analysis
Data are expressed as the means ±S.E. Statistical significance of differences between means was determined by one-way analysis of variance. If means were shown to be significantly different, multiple comparisons were performed post hoc by the Tukey t test. Statistical significance was defined as p < 0.05.
Results
CD44/HCELL Supports E-selectin-dependent Interactions as Measured by Conventional Binding Assays
In this study, we used the well established E-selectin ligand CD44/HCELL (11) expressed on KG1a cells as a model ligand to develop and examine the performance of our SPR-based IP method. We first validated the recombinant-E-selectin-IgG chimeric protein (dE-selectin) interaction with CD44/HCELL by flow cytometry (Fig. 1A), confocal microscopy (Fig. 1B), and Western blot analysis (Fig. 1C). We further used the blot rolling assay (19, 20) to evaluate binding of CHO-E perfused over HECA-452-immunostained blots of immunoprecipitated CD44/HCELL from KG1a lysates. As illustrated in Fig. 1D, CHO-E cells interacted and adhered to CD44/HCELL specifically and was abrogated when either control CHO cells (CHO-Mock; no E-selectin) were used or when CHO-E cells were suspended in flow medium containing 5 mm EDTA (confirming that binding is Ca2+-dependent).
FIGURE 1.

Validation of CD44 binding to E-selectin using well established approaches. A, representative flow cytometry histograms of KG1a cells stained for the expression of CD44 (left) and the binding of dE-selectin (middle). A dot plot showing KG1a cells dually stained for CD44 and dE-selectin is shown on the right. Dotted curves show isotype (for CD44) and EDTA (20 mm; for dE-selectin) controls, and shaded curves show specific binding. B, KG1a cells were dually immunostained with CD44 mAb (red; left) and recombinant dE-selectin (green; middle), and in the merged image, colocalization of CD44 with dE-selectin is shown (orange; right). Cell surface labeling with isotype control or secondary antibody alone served as a background control (data not shown). C, CD44 was immunoprecipitated from KG1a cell lysate. The captured proteins were run on a 4–20% SDS-polyacrylamide gradient gel and transferred to a PVDF membrane for Western blot analysis. The membrane was blotted with either dE-selectin (left) or CD44 mAbs (clones 515 and 2C5; right) followed by isotype-matched HRP-conjugated antibody for visualization. D, interaction of CD44 with E-selectin by blot rolling assay was performed on immunoprecipitated CD44 from KG1a cell lysates. Immunoprecipitated protein was resolved by 4–20% SDS-PAGE and blotted for sLex binding using HECA-452 mAb. CHO-E cells were then perfused over the stained PVDF membrane at 0.25 dyne/cm2. After cell perfusion, the numbers of rolling cells/field were counted from six experiments using four distinct fields of view in each experiment. As a control, 5 mm EDTA was added to the buffer containing CHO-E cells. CHO-Mock (CHO-M) cells were used to assess the nonspecific adhesion. Results shown reflect multiple assays (n = 4) on HECA-452 blots of multiple (n = 3) membrane preparations. Data are the mean ± S.E. (error bars).
A Novel SPR-based Assay to Measure the Binding of E-selectin with Its Ligands
The scheme of the SPR-based IP assay as it relates to measuring the interaction of CD44/HCELL with E-selectin is a three-step process (Fig. 2A): 1) coupling CD44 mAb to the surface of a CM5 sensor chip, 2) injection of KG1a cell lysate under conditions where nonspecific interactions with the CM5 surface and the mAb are minimal and the ligand of interest (i.e. CD44) is specifically captured, and 3) addition of recombinant dE-selectin under flow. As illustrated in the binding sensorgrams in Fig. 2B, at the end of the lysate injection and during the wash with buffer, we observed a significant accumulation of RU in the 515 mAb channel compared with only residual RU in the control flow cells that did not contain mAb (blank) or contained an isotype control (MsIgG1) specific for the mAb of interest (isotype). We next investigated whether the captured CD44 is associated in multiligand complexes by recovering the 515 mAb-bound protein(s) as described under “Experimental Procedures” after introducing a 1- or 5-min buffer washing step at the end of the lysate injection and analyzing the recovered protein(s) by Western blotting. Immunostaining with CD44 mAb verified the presence of CD44 after both the 1- and 5-min buffer washing steps (Fig. 2B, inset). Immunostaining for the presence of PSGL-1, a highly expressed E-selectin ligand on hematopoietic cells, showed that a 5-min wash with buffer effectively removed PSGL-1 contamination (Fig. 2B, inset).
FIGURE 2.

Optimization of the SPR-based IP in studying the interaction of E-selectin with CD44/HCELL. A, an experimental schematic diagram. Step 1, mAb immobilization on CM5 chip; step 2, lysate injection to capture CD44; step 3, injection of dE-selectin. B, raw data of KG1a lysate injection on surface-immobilized mAb. Three flow cells were used in this experiment: blank (dashed line), immobilized 515 mAb (5500 RU; gray line), and immobilized isotype control (5000 RU; black line); the mAb immobilization (step 1) is not shown. Arrows mark the start and end of the lysate injection followed by washing with a running buffer (step 2). The sensorgrams are uncorrected for the bulk refractive index of the buffer and therefore display a large increase in RU during the lysate injection. Inset, Western blot analysis of 515 mAb-captured proteins from crude KG1a lysate. Captured proteins were eluted (as described under “Experimental Procedures”) following a buffer wash of either 1 or 5 min. The recovered material was analyzed by Western blotting with CD44 mAb (left panels) and/or with PSGL-1 mAb (right panels). C, flow cytometry was used to determine the ability of three different clones of CD44 mAbs (515, Hermes-3, and IM7) to stain CD44 on KG1a cells. The dotted curves represent isotype controls, and the shaded curves represent specific binding. D, Western blot analysis of the efficiency of three different clones of CD44 mAbs (515, Hermes-3, and IM7) to each immunoprecipitate CD44 from KG1a lysate; blots were stained with CD44 (515, Hermes-3, and IM-7) and HECA-452 mAbs. E, CD44 mAb clones (515 (7200 RU; gray line), Hermes-3 (5740 RU; black line), and IM7 (4100 RU; dashed line)) or isotype (4600 RU) was immobilized to capture CD44 from KG1a lysate. Subsequently, dE-selectin binding to the captured CD44 was determined. The mAb immobilization step is omitted. The sensorgrams show the lysate injection followed by dE-selectin injection (177 nm), which are normalized after subtracting the bulk refractive index and nonspecific interaction using the control isotype flow cell. koff is the dissociation rate constant for CD44 from Hermes-3 mAb or 515 mAb, and koff-apparent is for dE-selectin or CD44·dE-selectin from Hermes-3 mAb or 515 mAb and dE-selectin from CD44·Hermes-3 mAb or CD44·515 mAb. F, using Hermes-3 mAb (3670 RU) to capture CD44 and isotype control (3600 RU), the Ca2+-dependent binding of CD44 to dE-selectin was measured as in E above. dE-selectin (177 nm) was injected first in the presence of 5 mm EDTA and then in the presence of 1 mm CaCl2. This is representative of two independent experiments. G, purified CD44 from KG1a lysate (1 μm; according to “Experimental Procedures”) was injected for 200 s at 20 μl/min over either immobilized Hermes-3 mAb (left) or immobilized dE-selectin (right). 13,000 RU of dE-selectin was immobilized using an amine coupling kit on a CM5 chip. Inset, Western blot of purified CD44 from KG1a lysate stained for CD44 (using Hermes-3 mAb). All SPR binding was conducted in running buffer containing 50 mm NaCl. koff is for CD44·dE-selectin complex. This is representative of n = 2 independent experiments. For all SPR experiments, koff and koff-apparent were calculated by fitting the stable phase in the buffer washing step using the Biacore evaluation software.
The fact that a mAb works in solution-based IP does not guarantee it will work when immobilized to the CM5 surface. Flow cytometry and IP were first used to assess the ability of three different clones of CD44 mAbs (515, Hermes-3, and IM7) to interact with KG1a cells (Fig. 2, C and D). However, upon coupling to CM5, IM7 mAb completely lost its ability to capture CD44 (no accumulation of RU occurs at the end of lysate injection), whereas both 515 mAb and Hermes-3 mAb captured CD44 but with distinctive affinities (Fig. 2E); the dissociation rate constant (koff) during the washing step with buffer for CD44 binding to 515 mAb was 12-fold faster than to Hermes-3 mAb (Fig. 2E). Based on Equation 1 and the maximum RU reached at the end of the lysate injection and just prior to the start of the wash with buffer (RUmax) (1040 and 640 RU in 515 mAb and Hermes-3 mAb, respectively), we estimated that 515 mAb and Hermes-3 mAb (∼150 kDa) captured CD44 (∼80 kDa) with only ∼27 and ∼20% efficiency, respectively. In general, we observed low ligand capturing efficiency, which we attributed to the immobilization of the mAb (rendering its binding site inaccessible) and/or to the low expression level of the endogenous ligands.
We next examined the ability of the CD44·515 mAb and CD44·Hermes-3 mAb complexes to interact with dE-selectin. All SPR binding studies were conducted in running buffer containing 50 mm NaCl unless otherwise stated. A marked increase in RU was detected during dE-selectin injection (177 nm) in the case of CD44·515 mAb and CD44·Hermes-3 mAb, but in the case of IM7 mAb, which failed to capture CD44, no dE-selectin RU was detected (Fig. 2E). The high stability of the CD44·Hermes-3 mAb complex makes it ideal for quantitative analysis of dE-selectin binding to CD44. The koff-apparent of the dissociation phase, which consists of the dissociation of CD44 or CD44·dE-selectin from Hermes-3 mAb as well as the dissociation of dE-selectin from CD44·Hermes-3 mAb, was 0.9 × 10−4 s−1 (Fig. 2E). However, because the koff of CD44·Hermes-3 mAb and presumably CD44·dE-selectin from Hermes-3 mAb is only 1.5-fold slower than the koff-apparent (Fig. 2E), the estimated koff-apparent would be a close approximation of the koff of the CD44·dE-selectin complex. It should be noted that although CD44·515 mAb forms a weak complex, the significant accumulation of RU during both dE-selectin injection and buffer wash still suggest that it interacted with dE-selectin (Fig. 2E). EDTA abrogated the binding between dE-selectin (177 nm) and CD44·Hermes-3 mAb complex, and this could be reversed with the introduction of Ca2+ during the dE-selectin injection (Fig. 2F), confirming the specificity of this interaction.
We further confirmed the interaction of CD44 with dE-selectin by conducting a reciprocal binding scheme to that presented in Fig. 2A. To this end, dE-selectin was immobilized on the CM5 chip surface (instead of the mAb against the ligand), and purified CD44 was injected over the dE-selectin. We partially purified native CD44 by IP from KG1a cell lysate as described under “Experimental Procedures” and assessed binding by both SPR (Fig. 2G, left) and Western blot analysis (inset). CD44 bound dE-selectin with slow on- and off-rates as evident by the slow accumulation of RU during CD44 injection and the accumulation of RU during the buffer washing step that dissociated with slow koff-apparent (Fig. 2G, right).
Using SPR to Characterize the Glycans Required on CD44/HCELL to Bind E-selectin
CD44 possesses five conserved positions for N-linked glycosylation and multiple sites for O-linked glycosylation (21). The importance of glycosylation of E-selectin ligands is illustrated by the inability of hypoglycosylated rH-CD44 (17, 18) captured by the 2C5 mAb to either bind to dE-selectin in our SPR binding assay (Fig. 3A) or be recognized by the sLex/a mAb HECA-452, which is commonly required for E-selectin recognition, in a Western blot (Fig. 3B, top panel). It should be noted that 2C5 mAb but not 515 mAb was able to recognize rH-CD44 in SPR (Fig. 3A) and Western blot analysis (Fig. 3B, bottom panel). To characterize the contribution of glycans on CD44 to dE-selectin binding by SPR, enzymes were used to remove N-glycans (PNGase F) or O-glycans (OSGE) prior to the binding measurements. Sensorgrams of dE-selectin (354 nm) binding to CD44·Hermes-3 mAb from either PNGase F-treated lysate (treated) or control lysate that underwent the same treatment conditions (buffer and incubation times/temperatures) but in the absence of the treatment enzyme (control) are shown in Fig. 3C. To perform comparative analysis, we normalized the amount of mAb-captured ligands prior to dE-selectin injection as described under “Experimental Procedures.” As evident in Fig. 3C, removal of N-glycans resulted in an ∼9-fold reduction in RUmax. However, the residually bound dE-selectin formed a stable complex that dissociated with 16-fold slower koff-apparent than did the control lysate (Fig. 3C). Considering the higher stability of CD44·Hermes-3 mAb from treated relative to control lysates (Fig. 3C), this 16-fold difference in koff-apparent is a higher estimate. These results indicate that N-glycans enhance the association but not the stability of dE-selectin·CD44 complex. To determine whether the residual staining of CD44/HCELL by dE-selectin following PNGase F treatment reflects incomplete enzyme digestion of CD44 N-glycans (Fig. 3D), we used the anti-CD34 mAb QBend-10 to assess the extent of removal of CD34 N-glycans following PNGase F treatment. QBend-10 mAb was able to recognize control samples (not treated with PNGase F) but completely lost its epitope recognition following PNGase F treatment (Fig. 3D), which is consistent with results published previously (16). Moreover, the CD34 recognized by glycan-insensitive mAb (EP373Y; recognizes the C terminus of CD34) displayed faster mobility on SDS-PAGE, confirming the efficacy of the treatment and the presence of PNGase F-treated protein (Fig. 3D) (16).
FIGURE 3.

Characterization of glycans essential for mediating binding of CD44/HCELL to E-selectin using SPR. A, CD44 mAb clones (515 (7000 RU; black line) and 2C5 (8600 RU; gray line)) or isotype control (8000 RU) was immobilized to capture rH-CD44. The mAb immobilization step is omitted. The sensorgrams show the lysate injection followed by dE-selectin injection (177 nm), which are normalized after subtracting the bulk refractive index and nonspecific interaction using the control isotype flow cell. No binding of rH-CD44 to dE-selectin was detected. This is representative of n = 2 independent experiments. B, rH-CD44 and CD44 immunoprecipitated from KG1a lysate was run on a 4–20% SDS-polyacrylamide gradient gel and transferred to a PVDF membrane for Western blot analysis. The membrane was blotted with anti-sLex mAb (HECA-452 clone) (top panel). To confirm the presence of CD44, rH-CD44 was blotted and stained with either 2C5 or 515 anti-CD44 mAb (bottom panels). Note that rH-CD44 could be blotted with 2C5 but not with 515 (bottom panel) but lacked sLex/a, which is required for E-selectin recognition (as evident by the absence of HECA-452 mAb binding) (top panel). C, Hermes-3 mAb (3270 RU) (isotype control, 2570 RU) was immobilized to capture CD44 from KG1a lysates that had been either treated with PNGase F (Treated; black line) or not (Control; gray line). Following CD44 capture, dE-selectin was injected at 354 nm. The same surface was used for both the treated and control binding studies with a regeneration step between the two runs. The normalized (dashed line) sensorgram is the same as control but normalized to the treated sensorgram based on the ratio of accumulated CD44 RU prior to dE-selectin injection. koff and koff-apparent were calculated as described in Fig. 2E. This is representative of n = 2 independent experiments. D, for Western blot analysis, CD44 was immunoprecipitated (with 515 or Hermes-3) from equivalent amounts of KG1a lysates either treated with PNGase F (+) or not (−) and blotted with either dE-selectin (top panel) or CD44 (middle panel). Note the disappearance of the QBend-10 signal due to the N-glycan removal, whereas equal amounts of CD34 protein were recognized by the CD34 mAb EP373Y (C-terminal domain of CD34; bottom panels). E, this SPR study was performed as in C except using 515 mAb (4000 RU) and its isotype (2570 RU) in place of Hermes-3 mAb. This is representative of n = 3 independent experiments. F and H, SPR analysis of OSGE treatment was performed as in C and E, respectively, using Hermes-3 mAb (8000 RU) (isotype control, 5000 RU) (F) or 515 mAb (5900 RU) (isotype control, 5000 RU) (H). These are representative of n = 2 independent experiments for Hermes-3 mAb and n = 3 independent experiments for 515 mAb. G, CD44 was immunoprecipitated following OSGE treatment as in D and subjected to Western blotting for CD44 (top panel). Because following OSGE treatment the Hermes-3 mAb does not immunoprecipitate CD44 as efficiently as untreated CD44, CD44 immunoprecipitations were performed on adjusted amounts of OSGE-treated samples (i.e. 3 times more OSGE-treated KG1a lysate) and subjected to Western blot analysis for CD44 and dE-selectin (middle panel). Note that this is consistent with the SPR results in F and H. Qbend-10 was used as an internal control to confirm O-glycan removal as in D (bottom panel). I and J, SPR analyses of sialidase treatment were performed as in C and E using Hermes-3 mAb (5440 RU) (isotype control, 4000 RU) (I) or 515 mAb (5461 RU) (isotype control, 4036 RU) (J). These are representative of n = 3 independent experiments. K, Western blot analysis of sialidase treatment was performed as in D. All SPR binding studies were conducted in a running buffer containing 50 mm NaCl. The treated and control bindings were performed on the same flow cell with a regeneration step between the two runs. dE-selectin was injected at 354 nm unless stated otherwise.
Interestingly, removal of the N-glycan increased the antigenic activity of Hermes-3 mAb to capture CD44; 2.5-fold more CD44·Hermes-3 mAb complex was formed in the treated lysate prior to the injection of dE-selectin, and it dissociated with a 3-fold reduction in koff in comparison with the control lysate (Fig. 3C). This increase in antigenic activity was confirmed by Western blotting (Fig. 3D). Furthermore, the treatment condition itself (24-h incubation at 37 °C) affected the stability of the CD44·Hermes-3 mAb complex as evident by its 3-fold higher koff value relative to that when formed using fresh lysate (cf. Fig. 3C with Fig. 2E). Our real time SPR-IP therefore provides a simple, quick, and direct measurement of such variation in mAb antigenicity to different glycoforms that is not easily detectable using conventional Western blot analyses.
We observed similar behavior for PNGase F treatment on CD44 interactions with dE-selectin when 515 mAb was used to capture in either SPR (Fig. 3E) or Western blot analysis (Fig. 3D). The effect of treatment on the antigenic activity of 515 mAb, however, was less pronounced than on Hermes-3 mAb (Fig. 3, C–E). This further supports our conclusion that the reduced binding of dE-selectin upon PNGase F treatment is not due to reduced ability of Hermes-3 mAb or 515 mAb to capture CD44.
Treatment with OSGE to remove O-glycans reduced the RUmax of dE-selectin binding to CD44·Hermes-3 mAb by ∼50-fold (Fig. 3F). It is important to note that OSGE treatment resulted in the opposite antigenic effect (compared with PNGase F) where a 1.5-fold reduction in the amount of CD44·Hermes-3 mAb was formed prior to the injection of dE-selectin (Fig. 3F). This was further confirmed by Western blot analysis (Fig. 3G, top panel). This reduction, however, is minor relative to the significantly reduced binding of dE-selectin to CD44 observed using either Hermes-3 mAb (Fig. 3F) or 515 mAb (Fig. 3H). Finally, to even out these differences in the ability of these antibodies to capture their antigenic epitopes following treatment with OSGE, we adjusted the IP lysate amount (i.e. increased) used for the OSGE-treated condition prior to Western blot analysis (under denaturing conditions). As illustrated in Fig. 3G (middle panel), dE-selectin binding is still markedly reduced, although equivalent amounts of CD44 were represented in treated and control samples. QBend-10 mAb was used to confirm the removal of O-glycans (Fig. 3G, bottom panel) (14, 16). Finally, treatment with sialidase to remove the sialic acid (included in the sLex structure) completely abolished the interaction of dE-selectin with CD44·Hermes-3 mAb (Fig. 3I) or CD44·515 mAb (Fig. 3J) by SPR and in Western blot analysis (Fig. 3K).
Determination of Equilibrium Binding Constants of Endogenous E-selectin Ligands
We next determined the KD of dE-selectin with the stably captured CD44·Hermes-3 mAb complex in buffer containing 50 mm NaCl. dE-selectin binding displayed a dose-dependent response (Fig. 4A); using a steady-state model, the KD was calculated to be 137 ± 6 nm (n = 3; Fig. 4, A and E). The koff-apparent estimated from the dissociation rate following the last dE-selectin injection in the binding isotherm was 1.7 × 10−4 ± 0.1 × 10−4 s−1 (Fig. 4, A and E). We therefore estimated the apparent association rate constant (kon-apparent) to be 1200 ± 82 m−1 s−1 (Fig. 4E). These results demonstrated that CD44 bound dE-selectin with slow on-/slow off-rate kinetics.
FIGURE 4.

Determination of the KD for the binding of CD44/HCELL and PSGL-1 to dE-selectin at different salt concentrations. A, binding of different concentrations of dE-selectin to CD44 at 50 mm NaCl; the sensorgram shows binding of consecutive injections of dE-selectin at 30 μl/min for 130 s at concentrations of 0.78, 1.5, 3.125, 6.25, 12.5, 25, 50, 100, 200, 400, and 800 nm that are spaced by a 60-s buffer washing step to CD44 captured from a KG1a lysate injection over surface-immobilized Hermes-3 mAb (7142 RU). The lysate injection is not shown. The sensorgrams are corrected for the bulk refractive index and nonspecific interactions using isotype control (6852 RU). KD was determined by fitting the binding isotherm using a steady-state model and the RUmax values just prior to the start of the buffer injection where steady-state conditions were nearly met (inset). koff-apparent was calculated as described in Fig. 2E, and kon-apparent was calculated based on the determined KD and koff-apparent values. B, binding of different concentrations of dE-selectin to CD44 at 200 mm NaCl; the experimental conditions are similar to those in A with the following concentration range used: 25, 50, 100, 200, 400, 800, and 1600 nm dE-selectin. The RU value for Hermes-3 mAb was 4900, and that for the isotype control was 4280. C, binding of different concentrations of dE-selectin to PSGL-1 at 50 mm NaCl; the experimental conditions are similar to those in A with the same concentration range of dE-selectin used. The RU value for the KPL-1 mAb was 8200, and that for the isotype control was 6852. D, binding of different concentrations of dE-selectin to PSGL-1 at 200 mm NaCl; the experimental conditions are similar to those B with the same concentration range of dE-selectin used. The RU value for KPL-1 mAb was 5800, and that for the isotype control was 4280. E, summary of binding constants of CD44·dE-selectin and PSGL-1·dE-selectin at 50 and 200 mm NaCl concentration from three independent experiments of those shown in A–D. The binding study was performed and analyzed as described in A and B.
Western blot analysis showed that the endogenous immunoprecipitated CD44 existed in different glycoforms that interact with dE-selectin (Fig. 1C). Using Equation 1, 1:1 stoichiometry, and the binding curve in Fig. 4A, we estimated that ∼14% of the 7460 RU of the mAb (∼150 kDa) captured 550 RU of CD44 (∼80 kDa, monomer) to result in a total RUligand of 1580; 1030 RU are attributed to the 14% of the surface-immobilized mAb, and 550 RU are from the captured CD44. The expected RUmax from the binding of dE-selectin (300 kDa; dimer form of E-selectin) to 1580 RUligand (mAb in complex with CD44; ∼230 kDa) assuming 1:1 stoichiometry of CD44·dE-selectin interaction is 2060 RU. We estimated the RUmax from the steady-state fit to be 1200 RU. It is possible that not all captured CD44 is properly oriented to interact with dE-selectin because CD44 is immobilized indirectly to the CM5 surface via mAb. Therefore, we estimated that 60% of the captured monomeric CD44 was able to interact with dE-selectin. However, this percentage could be an upper estimate because we did not rule out the possibility that CD44 could bind more than one dE-selectin. The ability of SPR to quantify the percentage of active molecules provides another insight into the influence of post-translational modification on protein/protein interaction.
Previous studies showed that the selectin interaction with sLex is electrostatic in nature because it is markedly reduced at higher salt concentrations (22). We observed that the KD at 200 mm NaCl (663 ± 48 nm; n = 3) increased by 5-fold in comparison with 50 mm NaCl (137 ± 6 nm; p < 0.001) (Fig. 4, B and E). The koff-apparent estimated from the binding isotherm following the last dE-selectin injection was 1.5 × 10−4 ± 0.1 × 10−4 s−1, whereas the kon-apparent was estimated to be 226 ± 28 m−1 s−1 (Fig. 4, B and E). These results indicated that CD44 and dE-selectin binding maintained the same off-rates at 200 mm NaCl relative to 50 mm NaCl, whereas the on-rate decreased by 5-fold (p < 0.001).
Antibodies that block specific ligand binding to E-selectin have not been described, making it difficult to compare the contribution of individual ligands to the overall E-selectin binding of a cell. Furthermore, the difference in the expression levels of different ligands and in their IP efficiency, which is inherently influenced by glycosylation differences, makes comparative analysis tricky to perform. Therefore, we sought to use this SPR-based IP assay to perform a comparative analysis of E-selectin binding with another well known ligand, PSGL-1. PSGL-1 formed a highly stable complex with the surface-immobilized KPL-1 mAb that is comparable with the CD44·Hermes-3 mAb complex (Fig. 4C). At 50 mm NaCl, dE-selectin bound captured PSGL-1·KPL-1 mAb with a KD comparable to that of CD44·Hermes-3 mAb: 178 ± 22 nm (n = 3) compared with 137 ± 6 nm (Fig. 4, C and E). The estimated koff-apparent and kon-apparent from the binding isotherm were 2.0 × 10−4 ± 0.2 × 10−4 s−1 and 1100 ± 106 m−1 s−1, respectively (Fig. 4, C and E), demonstrating that PSGL-1 bound dE-selectin with slow on/slow off kinetics that are comparable with those between CD44 and dE-selectin. Using a similar calculation scheme, we estimated that the captured PSGL-1 (∼240 kDa dimer; valency = 2) was 200% active in binding to dE-selectin; i.e. each dimer of PSGL-1 bound two dE-selectin molecules (Fig. 4C). Interestingly, dE-selectin bound PSGL-1 and CD44 with comparable kon-apparent and koff-apparent despite their significant difference in stoichiometry. This indicated that different binding sites are likely to act independently.
PSGL-1 binding to dE-selectin also displayed salt dependence where the KD increased by 6-fold when the NaCl concentration was increased to 200 mm (1150 ± 118 nm; n = 3) (Fig. 4, D and E), demonstrating that PSGL-1 is slightly more sensitive to salt than CD44 (p < 0.05). The estimated koff-apparent and kon-apparent from the binding isotherm were 1.9 × 10−4 ± 0.2 × 10−4 s−1 and 165 ± 13 m−1 s−1, respectively (Fig. 4, D and E). These results indicate that increasing the salt concentration from 50 to 200 mm NaCl had no effect on the off-rate of the PSGL-1 interaction with dE-selectin but reduced its on-rate by 7-fold (p < 0.01).
The weaker interaction of dE-selectin with PSGL-1 at higher salt concentrations in our SPR experiment was corroborated by blot rolling assays comparing CD44 and PSGL-1 at varying NaCl concentrations. When the salt concentration was increased from 50 to 200 mm, a 4-fold reduction in the number CHO-E cells rolling on PSGL-1 was observed compared with only a 2-fold reduction on CD44 (Fig. 5, cf. A with B). Single cell tracking of CHO-E cells was performed as described under “Experimental Procedures” to gain more quantitative information on the CD44 and PSGL-1 interactions with dE-selectin. The trajectories presented in Fig. 5C (see supplemental V i d e o 1; at 150 mm NaCl) illustrate representative examples of the rolling behavior of cells defined as either “rolling” and/or “rolling and adhesion.” The average velocity calculated from the slope of the rolling phase of the trajectories, average rolled distance, and percentage of cells that displayed adhesion behavior during rolling were calculated under each salt concentration tested in Fig. 5, A and B. At salt concentrations between 50 and 150 mm, we observed that these parameters were similar for PSGL-1 and CD44 (Fig. 5C). As the physiological salt concentration is closer to 150 mm, CD44 supports slightly lower rolling velocity and higher numbers of adherent cells. However, when the salt concentration was increased to 200 mm, we observed that 1) the velocity of CHO-E cells rolling on PSGL-1 was significantly higher than on CD44, 2) the percentage of rolling cells that displayed adhesion behavior was more decreased on PSGL-1 compared with CD44, and 3) rolling distance maintained prior to detachment (i.e. the stability of the interaction) was longer on CD44 compared with PSGL-1 (Fig. 5C). These results are consistent with the SPR binding measurements indicating that both CD44 and PSGL-1 bound dE-selectin with comparable kinetics at lower salt concentrations and that CD44 bound more strongly at higher salt concentrations. Furthermore, the slow dissociation rates of CD44 and PSGL-1 binding to E-selectin explain the short distance traveled during tethering/rolling of CHO-E cells prior to firmly adhering.
FIGURE 5.

Comparison between the binding of CD44 and PSGL-1 to CHO-E cells by blot rolling assay at various salt concentrations. A and B, adhesion bar graph for the blot rolling assay (rolling cells/mm2) for CHO-E cells perfused over SDS-PAGE immunoblots of HECA-452-reactive membrane glycoproteins of KG1a cells at 0.25 dyne/cm2 and buffer NaCl concentrations of 50, 150, and 200 mm (black bars). Immunoprecipitates of CD44/HCELL (A) and PSGL-1 (B) from KG1a cells were resolved by SDS-PAGE and blotted for HECA-452 prior to performing the assay. To control for the specificity of CHO-E binding to membrane glycoproteins, EDTA was added to the buffer containing the CHO-E cells before use in adhesion assays (gray bars). After cell perfusion, the numbers of rolling cells/field were counted from n = 3 experiments using four distinct fields of view in each experiment. Data are reported as the mean ± S.E. (error bars). C, single cell tracking of CD44 and PSGL-1 binding to E-selectin by blot rolling assays. The experiments were performed as described in A and B. The images were acquired at one frame/s, subjected to background subtraction and contrast enhancement in NIH ImageJ, and tracked on Imaris V7.6.4 software as described under “Experimental Procedures.” The centroid position of the cells was determined with an accuracy of 10 ± 4 μm. The trajectories show the X-position of each rolling cell (parallel to the flow) versus time. Displacements that were greater than 4 μm within 4 s were considered as rolling events, and those that were less were considered as adhesion events. The slope of the rolling phase of the trajectory was fitted linearly using OriginPro (gray line), and the mean velocity, Vx, for each condition was calculated from the average of the slopes. The average velocity, percentage (%) of rolling cells that displayed adhesion behavior (as illustrated for the trajectories), and averaged traveling distance during the rolling phase of the trajectories under each condition are reported. Data are reported as the mean ± S.E.
Binding of Ligands to Monomeric E-selectin
Little is known about the actual physiological distribution of E-selectin on endothelial cells and the mechanisms by which they mediate cell adhesion under flow. Clustering and conformational changes of P-selectin resulting from force play a role in increasing the binding affinity between the endothelial cells and the flowing cells (23–25). In stark contrast to our findings, previous SPR studies of selectin binding to various ligands tended to indicate fast on/fast off kinetics (26–28). Upon closer examination of the literature (Table 1), we noticed that the form of selectin used, either dimer or monomer, plays a role. We therefore sought to characterize mE-selectin binding to CD44·Hermes-3 mAb and PSGL-1·KPL-1 mAb. Interestingly, both ligands bound mE-selectin much more weakly than dE-selectin (Fig. 6A). The binding kinetics displayed a transient nature (fast on-/fast off-rates) as evident in the curves displayed in Fig. 6A where RUmax is reached immediately after the start of the mE-selectin injection and then returns to baseline immediately after the end of the mE-selectin injection and the start of the wash with the buffer. The E-selectin monomeric and dimeric nature was confirmed by native gel analysis (Fig. 6B).
TABLE 1.
Comparison of affinities and kinetics of selectin/ligand interactions measured by SPR
Note that most measurements are reported for experiments performed at 25 °C. ND, too low to detect. Results represent the mean ± S.E.
| Interaction |
Species | [Salt] | Form of selectin | KD | kon | koff | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Selectin | Ligand | |||||||
| mm | nm | m−1 s−1 | s−1 | |||||
| E-selectin | PSGL-1 | Human | 50 | Dimer | 178 ± 22 | 1,100 ± 106 | 2.0 × 10−4 ± 0.2 × 10−4 | This study |
| 200 | Dimer | 1,150 ± 118 | 165 ± 13 | 1.9 × 10−4 ± 0.2 × 10−4 | This study | |||
| 200 | Monomer | ND | This study | |||||
| CD44 | Human | 50 | Dimer | 137 ± 6 | 1,200 ± 82 | 1.7 × 10−4 ± 0.1 × 10−4 | This study | |
| 200 | Dimer | 663 ± 48 | 226 ± 28 | 1.5 × 10−4 ± 0.1 × 10−4 | This study | |||
| 200 | Monomer | ND | This study | |||||
| ESL-1 | Mouse | 150 | Monomer | 62,000 | 48,000 | 3.0 | Wild et al. (28) | |
| P-selectin | PSGL-1 | Human | 150 | Monomer | 320 | 4,400,000 | 1.4 | Mehta et al. (26) |
| PSGL-1 | Human | 150 | Monomer | 403 | Molenaar et al. (34) | |||
| 150 | Dimer | 4.1 | Molenaar et al. (34) | |||||
| PSGL-1a | Human | 150 | Monomer | 1,500 | 930,000 | 1.4 | Klopocki et al. (35) | |
| PSGL-1 | Human | 150 | Monomerb | 826 | Somers et al. (33) | |||
| PSGL-1 | 150 | Monomer | 288 | 140,000 | 0.04 | Waldron et al. (25) | ||
| PSGL-1 | Human | 150 | Monomerb | 517 | Phan et al. (36) | |||
| PTX-3 | Human | 150 | Dimer | 123 | 23,000 | 2.9 × 10−3 | Deban et al. (37) | |
| L-selectin | GlyCAM-1 | Mouse | 150 | Monomer | 108,000 | ≥105 | ≥10 | Nicholson et al. (27) |
| L-selectin | PSGL-1a | Human | 150 | Monomer | 47,000 | 150,000 | 7.0 | Klopocki et al. (35) |
a A peptide of PSGL-1 was used (2-GSP-6).
b The selectins used for the study comprised the lectin and EGF domains only.
FIGURE 6.
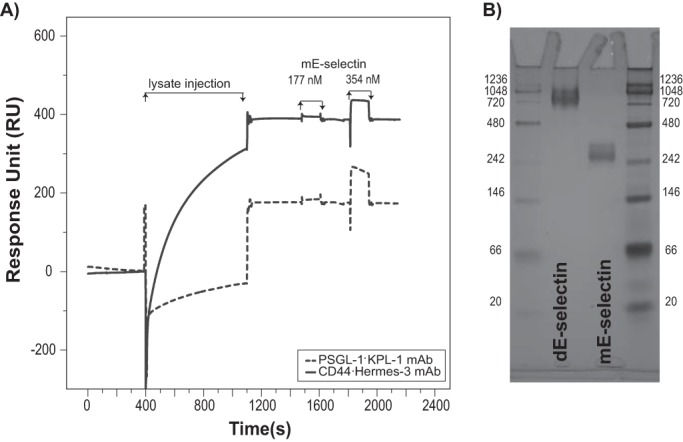
SPR binding of CD44 (black line) and PSGL-1 (dashed line) to mE-selectin. A, Hermes-3 mAb (for CD44; 3650 RU), KPL-1 mAb (for PSGL-1; 5000 RU), or isotype control (3400 RU) was used for real time immunoprecipitation from KG1a lysates. Following antigen capture, mE-selectin was injected at either 177 or 354 nm. The sensorgrams were corrected for the bulk refractive index and nonspecific interactions using the isotype control. This is a representative experiment of n = 2 independent experiments. B, native PAGE of E-selectin monomer and dimer was performed where 2 μg of mE-selectin and 4 μg of dE-selectin were diluted in 2× loading buffer (25% glycerol, 62.5 mm Tris base (pH 6.8), and 1% bromphenol blue) and then run on a 10% SDS-polyacrylamide gel without SDS in the running buffer. The gel was then visualized using SimplyBlueTM safe stain. This assay is representative of two independent experiments.
E-selectin Binding to CD44/HCELL Isolated from Hematopoietic Cells
CD44 has been shown to act as an E-selectin ligand on the surface of many hematopoietic cells and cancers (10, 15, 29–32). We therefore sought to characterize the intrinsic binding of dE-selectin to CD44 from the lysate of two additional hematopoietic cell lines, THP-1 and HL-60 cells. These cell lines express CD44 and display HECA-452-reactive sialyl Lewis structures that subsequently bound dE-selectin as assessed by flow cytometry and Western blotting (Fig. 7, A and B). Cellular extracts were prepared from THP-1, HL-60, and KG1a cells, and CD44 was captured by surface-immobilized Hermes-3 mAb. dE-selectin bound CD44 from both cellular extracts with kinetics comparable to that from KG1a (cf. Fig. 7, C and D with Fig. 4E). These results demonstrate that the intrinsic binding of CD44 from these different cells is comparable.
FIGURE 7.

Comparison of the binding affinity of CD44 captured from different cell lysates to dE-selectin. A, flow cytometric analysis of CD44 expression on KG1a, HL-60, and THP-1 cells is shown as the average geometric mean fluorescence intensity (above the isotype control) of three independent experiments. Note that KG1a expresses the highest levels of CD44 followed by THP-1 and HL-60. B, CD44 was immunoprecipitated from KG1a, HL-60, or THP-1 cell lysate normalized for total CD44 protein. The captured proteins were run on a 4–20% SDS-polyacrylamide gradient gel and transferred to a PVDF membrane for Western blot analysis. The membrane was blotted with either dE-selectin, HECA-452 mAb, or CD44 mAbs (clones 515, Hermes-3, and 2C5) followed by isotype-matched HRP-conjugated antibody for visualization. NIH ImageJ was used to quantify the intensity of Western blot bands using the gel analyzer tool; the number displayed represents the density of each band related to the KG1a band as a standard. All results are representative of three independent experiments. C, binding of different concentrations of dE-selectin to CD44 at 50 mm NaCl; the sensorgram shows binding of consecutive injections of dE-selectin at 20 μl/min for 180 s at concentrations of 0.78, 1.5, 3.125, 6.25, 12.5, 25, 50, 100, 200, 400, and 800 nm that are spaced by a 60-s buffer washing step to CD44 captured from a HL-60 (left) or THP-1 (right) lysate injection over surface-immobilized Hermes-3 mAb (7369 RU for HL-60 and 6808 RU for THP-1). The lysate injection is not shown. The sensorgrams are corrected for the bulk refractive index and nonspecific interactions using anti-MsIgG2a isotype control (7834 RU for HL-60 and 6852 RU for THP-1). KD, koff-apparent, and kon-apparent were determined as described in Fig. 4. D, summary of binding constants of CD44·dE-selectin at 50 mm NaCl captured from different cell lysates from four independent experiments. The binding study was performed and analyzed as described in Fig. 4A. E, adhesion bar graph for the blot rolling assay (rolling cells/mm2) for CHO-E cells perfused over SDS-PAGE immunoblots of HECA-452-reactive membrane glycoproteins of either KG1a, HL-60, or THP-1 cells at 0.25 dyne/cm2 and buffer NaCl concentrations of 150 mm. Immunoprecipitates of CD44/HCELL from KG1a, HL-60, or THP-1 cells were resolved by SDS-PAGE and blotted for HECA-452 prior to performing the assay (as described for Fig. 5). To control for the specificity of CHO-E binding to membrane glycoproteins, EDTA was added to the buffer containing the CHO-E cells before use in adhesion assays (gray bars). After cell perfusion, the numbers of rolling cells/field were counted in four distinct fields of view in each experiment. The adhesion bar graph is representative of three independent experiment and data reported as the mean ± S.E. (error bars).
The expression of CD44 protein differs among the three cell types as seen in the flow cytometry in Fig. 7A where the highest level of CD44 protein was found on KG1a, whereas the lowest was found on HL-60. Western blot analysis of dE-selectin binding to nearly equivalent amounts of CD44 protein isolated from each of these three hematopoietic cell lines revealed that CD44 binds dE-selectin in all cell lines albeit to varying degrees. Analysis of sLex (HECA-452) staining of CD44 by Western blotting indicated that CD44 from HL-60 and KG1a cells displays the highest signal for sLex, whereas the signal from CD44 of THP-1 cells was lower (Fig. 7B). The percentage of HECA-452 on THP-1 and HL-60 relative to the amount of bound dE-selectin after normalizing the slight difference in the amount of loaded CD44 was 50% for THP-1 and 150% for HL-60 relative to that in KG1a (Fig. 7B). In fact, the stoichiometry of binding of dE-selectin to CD44 from THP-1 and HL-60 derived from their binding isotherm (Fig. 7, C and D) as described above was 65 ± 6 and 110 ± 12%, respectively. Overall, these results demonstrate that levels of total CD44 protein are highest on KG1a cells, and the highest expression of CD44/HCELL is found on KG1a and HL-60 cells. But the intrinsic binding of CD44 is comparable in these cells.
Blot rolling studies of THP-1, HL-60, and KG1a under physiological flow conditions illustrated that CD44 isolated from KG1a and HL-60 cells supported rolling of CHO-E cells to similar levels, whereas CD44 isolated from THP-1 cells supported significantly less rolling. Interestingly, CD44 isolated from KG1a (2.1 ± 0.8 μm/min) cells led to significantly slower rolling velocities compared with that isolated from HL-60 (3.9 ± 2.3 μm/min; p < 0.001) and THP-1 (4.3 ± 1.9 μm/min; p < 0.001). These results suggest that ligand density (i.e. sLex decoration on CD44) dictates binding to E-selectin and enables slower rolling velocities.
Discussion
In this study, we demonstrated the feasibility of performing quantitative real time IP from cellular extracts on an SPR chip and applied this method to study the interaction of CD44/HCELL and PSGL-1 with E-selectin (14, 15). This novel approach allows researchers to look directly at native ligands expressed on the particular cell of interest. Purified native CD44/HCELL bound recombinant dE-selectin with a KD value of 137 ± 6 nm at 50 mm NaCl; the binding displayed remarkably slow association (1200 ± 82 m−1 s−1) and dissociation (1.7 × 10−4 ± 0.1 × 10−4 s−1) rates (Fig. 4, A and E). At 200 mm NaCl, the KD increased to 663 ± 48 nm (Fig. 4, B and E). Interestingly, kon-apparent was more sensitive to ionic strength than was koff-apparent (Fig. 4, B and E), suggesting that CD44/HCELL relies on electrostatic interactions to initiate its contact with E-selectin prior to forming more specific interactions and firm binding with E-selectin. Although the crystal structure of the lectin domain of E-selectin in complex with sLex suggests that binding is primarily electrostatically mediated (33), these interactions are extensive and able to resist the increased salt in our assay.
PSGL-1 shared many features with CD44/HCELL binding to recombinant E-selectin. The KD was similar to that of CD44 at 50 mm but was 2-fold lower at 200 mm NaCl (Fig. 4). The kon-apparent and koff-apparent were also comparable with those of CD44 and exhibited slow on- and off-rates with the on-rate more sensitive to the ionic strength than was the off-rate (Fig. 4E). These binding kinetics were manifested in the functional blot rolling assay where PSGL-1 displayed slightly more sensitivity to increasing salt than CD44 (Fig. 5, A and B). Single cell tracking of the blot rolling results revealed that the rolling parameters of PSGL-1, velocity, adherence, and rolling distance, were similar to those of CD44 at salt concentrations between 50 and 150 mm NaCl but weaker at 200 mm (Fig. 5C). Using this quantitative approach, we concluded that binding of E-selectin to CD44/HCELL and PSGL-1 is very similar, but CD44/HCELL displays slightly better binding at and above physiological salt concentrations. These results also suggest that the tendency of E-selectin to bind its ligands is more dependent on the sLex decoration.
Using our SPR approach, we further characterized glycans required to support E-selectin binding. Prior studies in which whole cell lysates were used in conjunction with functional blot rolling assays reported that N-glycans rather than O-glycans are the key contributors to CD44/HCELL binding to E-selectin (14). However, our data revealed that the removal of either N- or O-glycans resulted in a marked reduction in CD44 binding to E-selectin (Fig. 3). Nonetheless, O-glycans alone were able to form a stable complex with E-selectin albeit more weakly than when both O- and N-glycans were present (Fig. 3, C and D). Hence, these data reveal for the first time a role for O-glycans in CD44/HCELL-mediated interactions with E-selectin, indicating that O- and N-glycans are comparable scaffolds for sLex. We suggest that the reason for not detecting this previously is that the O-glycans could have been masked by other E-selectin ligands expressed on the surface (14). Indeed, a variant CD44/HCELL isoform isolated from colon carcinoma LS174T cells binds E-selectin in an O-glycan-dependent manner (9).
We also extended these studies to look at dE-selectin binding to CD44 isolated from two other leukemic cell lines, THP-1 and HL-60 cells. The current study is the first to show the expression of an E-selectin-binding form of CD44 on THP-1 cells. However, the existence of an E-selectin-binding form of CD44 on HL-60 cells is somewhat controversial. One study suggests that HL-60 cells roll on bone marrow endothelial cells likely through interactions via PSGL-1 and E-selectin because HECA-452 (anti-sLex antibody) reactivity appeared to be greatest at the molecular mass of monomeric PSGL-1 (∼120kDa) in a whole cell lysate of HL-60 cells (14). This study, however, did not look specifically at CD44 on HL-60 cells, and in fact because the expression of CD44 is indeed lower on HL-60 cells compared with KG1a cells, the sLex staining may be too faint to detect. In a later study, CD44 isolated from HL-60 cells was able to support E-selectin and some P-selectin binding but at a significantly lower level compared with KG1a cells (32). This study by Hanley et al. (32) also shows that the standard form of CD44 (CD44s; ∼80–95kDa) on HL-60 cells is recognized by the HECA-452 antibody, suggesting that it is decorated by sLex. Here we show in a direct manner that CD44 isolated from HL-60 cells is decorated with sLex as determined by HECA452 reactivity is able to bind dE-selectin using Western blotting and our SPR assay and is able to support the rolling of CHO-E cells under flow (although with a rolling velocity that is faster than that measured for CD44 isolated from KG1a cells). Hence, this is the first study to show that CD44 isolated from THP-1 and HL-60 leukemic cell lines binds E-selectin. It is important to note that on a per cell basis the expression of CD44/HCELL varies among each of these cell lines and may therefore have a more prevalent role as an E-selectin ligand on KG1a cells where higher amounts of CD44/HCELL are expressed compared with THP-1 and HL-60 cells (Fig. 7, A and B).
To our knowledge, this is the first documented binding kinetics of E-selectin to PSGL-1 or CD44 determined by SPR, although binding has been measured for another E-selectin ligand, E-selectin ligand-1 (28). E-selectin ligand-1 binds mE-selectin with a 100-fold higher KD than our reported KD for CD44 or PSGL-1 binding to the dE-selectin. We suspect this is likely the result of the use of mE-selectin.
Monomeric forms of selectins act differently than dimeric or oligomeric (i.e. clustered) forms of selectins. A review of the SPR literature with reference to whether the monomeric or dimeric selectin was used to calculate the KD values is presented in Table 1. The data presented here show that mE-selectin binds with fast on/fast off kinetics, whereas dE-selectin binds with slow on/slow off kinetics to both PSGL-1 and CD44/HCELL. Previous SPR studies using monomeric forms of selectins (P-, L-, and E-selectins) confirm that binding is transient (Table 1) (25–28, 33–37). In fact, a direct comparison of monomeric with dimeric P-selectin binding kinetics to PSGL-1 clearly indicates that by dimerizing P-selectin the KD is significantly reduced with slow on-/slow off-rates measured (34). Using a micropipette technique to analyze two-dimensional binding of mE-selectin or dE-selectin with mPSGL-1 or dPSGL-1, a recent study showed that mE-selectin binds equally to both mPSGL-1 and dPSGL-1; however, dE-selectin binds better to dPSGL-1 (38). This implies that the reason why dE-selectin binds tighter than mE-selectin is that the ligand is either a dimer (i.e. dPSGL-1) or has multiple binding sights (i.e. CD44/HCELL). Indeed, previous studies proposed that dimerization of both PSGL-1 and P-selectin stabilizes tethering and rolling likely by increasing “rebinding” within a bond cluster so if one bond dissociates there is an opportunity for it to rebind because the cell remains tethered by the second bond, and this prolongs the overall lifetime of the initial tether, allowing time for the cell to form new bonds (26, 39).
Considering their function in the adhesion cascade of mediating cell tethering and rolling, selectins are hypothesized to bind their ligands with very fast association and dissociation rates. A consequence of this significant difference in kinetics between the monomeric and dimeric forms is that the kon is substantially lower (by 100–10,000-fold), which would preclude the selectin from binding its ligand with fast association to capture cells from the flow and would bring into question its role in the first step of the adhesion cascade. A model may be thus envisioned where initially the monomeric form of the selectin mediates the fast on/fast off kinetics that helps to tether the cell, thereby leading to dimerization/oligomerization of the selectin on the endothelial cell, which subsequently mediates the slower on and off kinetics likely responsible for the slower rolling often associated with E-selectin (40, 41).
A mechanism often adopted by low affinity receptors to reinforce the avidity of their interactions with their ligands and thus to slow the koff of a biochemical interaction is dimerization/oligomerization of the receptor. Dimerization has been shown to be important in the functioning of P-selectin and L-selectin (34, 39, 42) and is suggested to create a functional advantage by influencing the rolling velocity and firm adhesion of leukocytes. The present study is the first to highlight a direct difference in the ability of the monomeric and dimeric forms of E-selectin to bind ligands and correlate with previous studies of other selectins. Indeed, early studies indicated that physiological evidence exists for both monomeric and oligomeric selectins (43, 44). Our results, in conjunction with previous work, underscore the importance of particular cell surface presentations of selectins or their ligands during leukocyte rolling on vascular surfaces under flow. Recent studies elaborated on the existence and significance of P- and E-selectin clustering and on the regulation of P-selectin clustering in leukocyte recruitment (23, 24, 45–47). These studies so eloquently show that clustering of P- and E-selectins in clathrin-coated pits and lipid rafts of endothelial cells supports slower rolling of leukocytes by forming bond clusters with PSGL-1 and that if these structures are disrupted rolling is inhibited. Furthermore, the dimerization/oligomerization of P-selectin on endothelial cells is actually mediated by a tetraspanin, CD63, which is essential for the correct organization and function of P-selectin (45). Moreover, CD63 is regulated by annexinA8, a membrane-trafficking protein required for the delivery of CD63 to Weibel-Palade bodies where P-selectin is stored (46). No publication to date has reported such mechanisms of regulation of E-selectin oligomerization.
Many studies have focused on the hypothesis that deals with the monomer form where cell adhesion is limited by its off-rate, and therefore suggestions about the mechanism to compensate this weak binding have focused on models where changes in the conformation of the selectin increase bond lifetimes or increase avidity through clustering (as discussed above). The former hypothesis refers to that of a “catch bond” where the catch bond lifetime increases under shear force. This model is largely based on the interaction of P-selectin with PSGL-1 and is supported by single molecule optical tweezer assays that show that the lifetime of the interaction between P-selectin and its ligand increases as the force increases before it starts to decrease when the force exceeds the intrinsic affinity of this interaction (48). Biochemical, structural, and SPR binding studies mapped this mechanism to an extended conformer of P-selectin that bound PSGL-1 with 5-fold lower KD (slower on- and off-rates) than the bent conformer. The catch bond model therefore proposes that P-selectin binding is limited by its low affinity and that shear force is required to promote the formation of the extended conformer. However, the extended conformer is also formed upon binding to sLex in the absence of shear force (25, 36). A recently published structure of E-selectin complexed with glycomimetic ligand (49) shows an extended structure similar to that of P-selectin with a PSGL-1 fragment, suggesting that an extended conformer may also exist for E-selectin. This could thereby operate in coordination with oligomerization of selectins to enhance their affinity to their ligands.
In addition to the oligomerization of selectins, clustering of their ligands could provide a parallel mechanism to stabilize rolling (50). This clustering is likely mediated via interactions with the cytoskeleton either directly through adaptor proteins, such as ezrin/radixin/moesin and ankyrin, or indirectly through proteins enriched within lipid rafts (51, 52). Interestingly, a recent study suggested that the clustering mediated by ankyrin binding to the cytoplasmic domain of CD44 enhances its binding to E-selectin under flow (52). Using the assay outlined here, it would be difficult, however, to address the effect of ligand oligomerization on binding to E-selectin. The oligomerization state of the captured molecules is unknown and is likely heterogeneous (52), but also the state of the ligands on the cell membrane is different from that in solution because at least in some cases this oligomerization is regulated by the cell cytoskeleton. To further understand the contribution of ligand clustering to binding kinetics using an SPR approach, future experiments may be developed to use live cells where manipulation of the ligands in the cell membrane could be tested.
The coupling of real time IP from cellular extracts with SPR enabled us to bypass the complex and challenging steps involved in expressing and purifying native glycoforms of CD44 and PSGL-1. This method is exceptionally fast and robust once optimized for a particular mAb and ligand capturing. It provides binding information that generally agrees with results derived from traditional immunoprecipitation and Western blot analysis. However, it also provides the added advantage of being quantitative, sensitive to residual and transient binding, and comparative; i.e. it factors in the differential ability of an antibody to bind its ligand in its various post-translationally modified forms.
Author Contributions
D. B. A. and A. A. performed and analyzed the experiments. S. M. H., K. S., and S. Z. G. provided key materials, knowledge, and methods. D. B. A. and J. S. M. conceived and designed the study, analyzed the results. and wrote the manuscript. All authors discussed and revised the manuscript.
Supplementary Material
Acknowledgments
We thank Luay I. Joudeh (King Abdullah University of Science and Technology) for technical support with the SPR machine, Dr. Slobodan Jergic (University of Wollongong) for discussion about and help with SPR data analysis, and Heba AbuSamra for support and discussions relating to analysis of single cell tracking experiments. We also thank Dr. Virginia A. Unkefer from Academic Writing Services at King Abdullah University of Science and Technology for editing the manuscript.
This work was supported by King Abdullah University of Science and Technology faculty baseline research funding and a university competitive research grant (to J. S. M. and S. M. H.). The authors declare no conflict of interest.

This article contains supplemental Video 1.
- sLex
- sialyl Lewis x
- SPR
- surface plasmon resonance
- IP
- immunoprecipitation
- PSGL-1
- P-selectin glycoprotein ligand-1
- mE-selectin
- monomeric E-selectin
- dE-selectin
- dimeric E-selectin
- HCELL
- hematopoietic cell E-/L-selectin ligand
- HBSS
- Hanks' balanced salt solution
- Ab
- antibody
- PNGase
- peptide:N-glycosidase
- OSGE
- O-sialoglycoprotein endopeptidase
- RU
- response units
- rH
- recombinant human
- MsIgG
- mouse IgG
- KD
- equilibrium dissociation binding constant
- koff-apparent
- apparent dissociation rate constant
- kon-apparent
- apparent association rate constant.
References
- 1. Butcher E. C. (1991) Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell 67, 1033–1036 [DOI] [PubMed] [Google Scholar]
- 2. Springer T. A. (1994) Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 76, 301–314 [DOI] [PubMed] [Google Scholar]
- 3. Frenette P. S., Mayadas T. N., Rayburn H., Hynes R. O., Wagner D. D. (1996) Susceptibility to infection and altered hematopoiesis in mice deficient in both P- and E-selectins. Cell 84, 563–574 [DOI] [PubMed] [Google Scholar]
- 4. Schweitzer K. M., Dräger A. M., van der Valk P., Thijsen S. F., Zevenbergen A., Theijsmeijer A. P., van der Schoot C. E., Langenhuijsen M. M. (1996) Constitutive expression of E-selectin and vascular cell adhesion molecule-1 on endothelial cells of hematopoietic tissues. Am. J. Pathol. 148, 165–175 [PMC free article] [PubMed] [Google Scholar]
- 5. Polley M. J., Phillips M. L., Wayner E., Nudelman E., Singhal A. K., Hakomori S., Paulson J. C. (1991) CD62 and endothelial cell-leukocyte adhesion molecule 1 (ELAM-1) recognize the same carbohydrate ligand, sialyl-Lewis x. Proc. Natl. Acad. Sci. U.S.A. 88, 6224–6228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sackstein R. (2005) The lymphocyte homing receptors: gatekeepers of the multistep paradigm. Curr. Opin. Hematol. 12, 444–450 [DOI] [PubMed] [Google Scholar]
- 7. Alon R., Feizi T., Yuen C. T., Fuhlbrigge R. C., Springer T. A. (1995) Glycolipid ligands for selectins support leukocyte tethering and rolling under physiologic flow conditions. J. Immunol. 154, 5356–5366 [PubMed] [Google Scholar]
- 8. Fuhlbrigge R. C., Kieffer J. D., Armerding D., Kupper T. S. (1997) Cutaneous lymphocyte antigen is a specialized form of PSGL-1 expressed on skin-homing T cells. Nature 389, 978–981 [DOI] [PubMed] [Google Scholar]
- 9. Hanley W. D., Burdick M. M., Konstantopoulos K., Sackstein R. (2005) CD44 on LS174T colon carcinoma cells possesses E-selectin ligand activity. Cancer Res. 65, 5812–5817 [DOI] [PubMed] [Google Scholar]
- 10. Zarbock A., Ley K., McEver R. P., Hidalgo A. (2011) Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood 118, 6743–6751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sackstein R., Dimitroff C. J. (2000) A hematopoietic cell L-selectin ligand that is distinct from PSGL-1 and displays N-glycan-dependent binding activity. Blood 96, 2765–2774 [PubMed] [Google Scholar]
- 12. Asa D., Raycroft L., Ma L., Aeed P. A., Kaytes P. S., Elhammer A. P., Geng J. G. (1995) The P-selectin glycoprotein ligand functions as a common human leukocyte ligand for P- and E-selectins. J. Biol. Chem. 270, 11662–11670 [DOI] [PubMed] [Google Scholar]
- 13. Moore K. L., Eaton S. F., Lyons D. E., Lichenstein H. S., Cummings R. D., McEver R. P. (1994) The P-selectin glycoprotein ligand from human neutrophils displays sialylated, fucosylated, O-linked poly-N-acetyllactosamine. J. Biol. Chem. 269, 23318–23327 [PubMed] [Google Scholar]
- 14. Dimitroff C. J., Lee J. Y., Rafii S., Fuhlbrigge R. C., Sackstein R. (2001) CD44 is a major E-selectin ligand on human hematopoietic progenitor cells. J. Cell Biol. 153, 1277–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Merzaban J. S., Burdick M. M., Gadhoum S. Z., Dagia N. M., Chu J. T., Fuhlbrigge R. C., Sackstein R. (2011) Analysis of glycoprotein E-selectin ligands on human and mouse marrow cells enriched for hematopoietic stem/progenitor cells. Blood 118, 1774–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hernandez Mir G., Helin J., Skarp K. P., Cummings R. D., Mäkitie A., Renkonen R., Leppänen A. (2009) Glycoforms of human endothelial CD34 that bind L-selectin carry sulfated sialyl Lewis x capped O- and N-glycans. Blood 114, 733–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baker K. N., Rendall M. H., Hills A. E., Hoare M., Freedman R. B., James D. C. (2001) Metabolic control of recombinant protein N-glycan processing in NS0 and CHO cells. Biotechnol. Bioeng. 73, 188–202 [DOI] [PubMed] [Google Scholar]
- 18. Hossler P., Khattak S. F., Li Z. J. (2009) Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology 19, 936–949 [DOI] [PubMed] [Google Scholar]
- 19. Fuhlbrigge R. C., King S. L., Dimitroff C. J., Kupper T. S., Sackstein R. (2002) Direct real-time observation of E- and P-selectin-mediated rolling on cutaneous lymphocyte-associated antigen immobilized on Western blots. J. Immunol. 168, 5645–5651 [DOI] [PubMed] [Google Scholar]
- 20. Sackstein R., Fuhlbrigge R. (2009) Western blot analysis of adhesive interactions under fluid shear conditions: the blot rolling assay. Methods Mol. Biol. 536, 343–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lesley J., Hyman R. (1998) CD44 structure and function. Front. Biosci. 3, d616–630 [DOI] [PubMed] [Google Scholar]
- 22. Torgersen D., Mullin N. P., Drickamer K. (1998) Mechanism of ligand binding to E- and P-selectin analyzed using selectin/mannose-binding protein chimeras. J. Biol. Chem. 273, 6254–6261 [DOI] [PubMed] [Google Scholar]
- 23. Kiely J. M., Hu Y., García-Cardeña G., Gimbrone M. A., Jr. (2003) Lipid raft localization of cell surface E-selectin is required for ligation-induced activation of phospholipase Cγ. J. Immunol. 171, 3216–3224 [DOI] [PubMed] [Google Scholar]
- 24. Setiadi H., McEver R. P. (2008) Clustering endothelial E-selectin in clathrin-coated pits and lipid rafts enhances leukocyte adhesion under flow. Blood 111, 1989–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Waldron T. T., Springer T. A. (2009) Transmission of allostery through the lectin domain in selectin-mediated cell adhesion. Proc. Natl. Acad. Sci. U.S.A. 106, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mehta P., Cummings R. D., McEver R. P. (1998) Affinity and kinetic analysis of P-selectin binding to P-selectin glycoprotein ligand-1. J. Biol. Chem. 273, 32506–32513 [DOI] [PubMed] [Google Scholar]
- 27. Nicholson M. W., Barclay A. N., Singer M. S., Rosen S. D., van der Merwe P. A. (1998) Affinity and kinetic analysis of L-selectin (CD62L) binding to glycosylation-dependent cell-adhesion molecule-1. J. Biol. Chem. 273, 763–770 [DOI] [PubMed] [Google Scholar]
- 28. Wild M. K., Huang M. C., Schulze-Horsel U., van der Merwe P. A., Vestweber D. (2001) Affinity, kinetics, and thermodynamics of E-selectin binding to E-selectin ligand-1. J. Biol. Chem. 276, 31602–31612 [DOI] [PubMed] [Google Scholar]
- 29. Burdick M. M., Chu J. T., Godar S., Sackstein R. (2006) HCELL is the major E- and L-selectin ligand expressed on LS174T colon carcinoma cells. J. Biol. Chem. 281, 13899–13905 [DOI] [PubMed] [Google Scholar]
- 30. Barthel S. R., Hays D. L., Yazawa E. M., Opperman M., Walley K. C., Nimrichter L., Burdick M. M., Gillard B. M., Moser M. T., Pantel K., Foster B. A., Pienta K. J., Dimitroff C. J. (2013) Definition of molecular determinants of prostate cancer cell bone extravasation. Cancer Res. 73, 942–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shirure V. S., Reynolds N. M., Burdick M. M. (2012) Mac-2 binding protein is a novel E-selectin ligand expressed by breast cancer cells. PLoS One 7, e44529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hanley W. D., Napier S. L., Burdick M. M., Schnaar R. L., Sackstein R., Konstantopoulos K. (2006) Variant isoforms of CD44 are P- and L-selectin ligands on colon carcinoma cells. FASEB J. 20, 337–339 [DOI] [PubMed] [Google Scholar]
- 33. Somers W. S., Tang J., Shaw G. D., Camphausen R. T. (2000) Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLeX and PSGL-1. Cell 103, 467–479 [DOI] [PubMed] [Google Scholar]
- 34. Molenaar T. J., Appeldoorn C. C., de Haas S. A., Michon I. N., Bonnefoy A., Hoylaerts M. F., Pannekoek H., van Berkel T. J., Kuiper J., Biessen E. A. (2002) Specific inhibition of P-selectin-mediated cell adhesion by phage display-derived peptide antagonists. Blood 100, 3570–3577 [DOI] [PubMed] [Google Scholar]
- 35. Klopocki A. G., Yago T., Mehta P., Yang J., Wu T., Leppänen A., Bovin N. V., Cummings R. D., Zhu C., McEver R. P. (2008) Replacing a lectin domain residue in L-selectin enhances binding to P-selectin glycoprotein ligand-1 but not to 6-sulfo-sialyl Lewis x. J. Biol. Chem. 283, 11493–11500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Phan U. T., Waldron T. T., Springer T. A. (2006) Remodeling of the lectin-EGF-like domain interface in P- and L-selectin increases adhesiveness and shear resistance under hydrodynamic force. Nat. Immunol. 7, 883–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deban L., Russo R. C., Sironi M., Moalli F., Scanziani M., Zambelli V., Cuccovillo I., Bastone A., Gobbi M., Valentino S., Doni A., Garlanda C., Danese S., Salvatori G., Sassano M., Evangelista V., Rossi B., Zenaro E., Constantin G., Laudanna C., Bottazzi B., Mantovani A. (2010) Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat. Immunol. 11, 328–334 [DOI] [PubMed] [Google Scholar]
- 38. Zhang Y., Jiang N., Zarnitsyna V. I., Klopocki A. G., McEver R. P., Zhu C. (2013) P-selectin glycoprotein ligand-1 forms dimeric interactions with E-selectin but monomeric interactions with L-selectin on cell surfaces. PLoS One 8, e57202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ramachandran V., Yago T., Epperson T. K., Kobzdej M. M., Nollert M. U., Cummings R. D., Zhu C., McEver R. P. (2001) Dimerization of a selectin and its ligand stabilizes cell rolling and enhances tether strength in shear flow. Proc. Natl. Acad. Sci. U.S.A. 98, 10166–10171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kunkel E. J., Ley K. (1996) Distinct phenotype of E-selectin-deficient mice—E-selectin is required for slow leukocyte rolling in vivo. Circ. Res. 79, 1196–1204 [DOI] [PubMed] [Google Scholar]
- 41. Kunkel E. J., Jung U., Ley K. (1997) TNF-alpha induces selectin-mediated leukocyte rolling in mouse cremaster muscle arterioles. Am. J Physiol. 272, H1391–H1400 [DOI] [PubMed] [Google Scholar]
- 42. Li X., Steeber D. A., Tang M. L., Farrar M. A., Perlmutter R. M., Tedder T. F. (1998) Regulation of L-selectin-mediated rolling through receptor dimerization. J. Exp. Med. 188, 1385–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barkalow F. J., Barkalow K. L., Mayadas T. N. (2000) Dimerization of P-selectin in platelets and endothelial cells. Blood 96, 3070–3077 [PubMed] [Google Scholar]
- 44. Ushiyama S., Laue T. M., Moore K. L., Erickson H. P., McEver R. P. (1993) Structural and functional characterization of monomeric soluble P-selectin and comparison with membrane P-selectin. J. Biol. Chem. 268, 15229–15237 [PubMed] [Google Scholar]
- 45. Doyle E. L., Ridger V., Ferraro F., Turmaine M., Saftig P., Cutler D. F. (2011) CD63 is an essential cofactor to leukocyte recruitment by endothelial P-selectin. Blood 118, 4265–4273 [DOI] [PubMed] [Google Scholar]
- 46. Poeter M., Brandherm I., Rossaint J., Rosso G., Shahin V., Skryabin B. V., Zarbock A., Gerke V., Rescher U. (2014) Annexin A8 controls leukocyte recruitment to activated endothelial cells via cell surface delivery of CD63. Nat. Commun. 5, 3738. [DOI] [PubMed] [Google Scholar]
- 47. Yoshida M., Westlin W. F., Wang N., Ingber D. E., Rosenzweig A., Resnick N., Gimbrone M. A. (1996) Leukocyte adhesion to vascular endothelium induces E-selectin linkage to the actin cytoskeleton. J. Cell Biol. 133, 445–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Evans E., Leung A., Heinrich V., Zhu C. (2004) Mechanical switching and coupling between two dissociation pathways in a P-selectin adhesion bond. Proc. Natl. Acad. Sci. U.S.A. 101, 11281–11286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Preston R. C., Jakob R. P., Binder F. P. C., Sager C. P., Ernst B., Maier T. (2015) E-selectin ligand complexes adopt an extended high-affinity conformation. J. Mol. Cell. Biol. 10.1093/jmcb/mjv046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McEver R. P., Zhu C. (2010) Rolling cell adhesion. Annu. Rev. Cell Dev. Biol. 26, 363–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Miner J. J., Xia L., Yago T., Kappelmayer J., Liu Z., Klopocki A. G., Shao B., McDaniel J. M., Setiadi H., Schmidtke D. W., McEver R. P. (2008) Separable requirements for cytoplasmic domain of PSGL-1 in leukocyte rolling and signaling under flow. Blood 112, 2035–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang Y., Yago T., Zhang N., Abdisalaam S., Alexandrakis G., Rodgers W., McEver R. P. (2014) Cytoskeletal regulation of CD44 membrane organization and interactions with E-selectin. J. Biol. Chem. 289, 35159–35171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Merzaban J. S., Imitola J., Starossom S. C., Zhu B., Wang Y., Lee J., Ali A. J., Olah M., Abuelela A. F., Khoury S. J., Sackstein R. (2015) Cell surface glycan engineering of neural stem cells augments neurotropism and improves recovery in a murine model of multiple sclerosis. Glycobiology 10.1093/glycob/cwv046 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.