

Background: The evolutionary history of family 2 polysaccharide lyases is unknown.
Results: Functional analysis highlights a key lysine-tryptophan transition involved in exolysis.
Conclusion: Subtle changes in amino acid structure can transform enzyme activity.
Significance: Combinatorial use of ancestral sequence reconstruction, gene resurrection, and structure-function analysis is valuable for elucidating the function and evolutionary history of polysaccharide lyases.
Keywords: enzyme kinetics, enzyme structure, magnesium, manganese, protein evolution, exolysis, polysaccharide lyase
Abstract
Family 2 polysaccharide lyases (PL2s) preferentially catalyze the β-elimination of homogalacturonan using transition metals as catalytic cofactors. PL2 is divided into two subfamilies that have been generally associated with secretion, Mg2+ dependence, and endolysis (subfamily 1) and with intracellular localization, Mn2+ dependence, and exolysis (subfamily 2). When present within a genome, PL2 genes are typically found as tandem copies, which suggests that they provide complementary activities at different stages along a catabolic cascade. This relationship most likely evolved by gene duplication and functional divergence (i.e. neofunctionalization). Although the molecular basis of subfamily 1 endolytic activity is understood, the adaptations within the active site of subfamily 2 enzymes that contribute to exolysis have not been determined. In order to investigate this relationship, we have conducted a comparative enzymatic analysis of enzymes dispersed within the PL2 phylogenetic tree and elucidated the structure of VvPL2 from Vibrio vulnificus YJ016, which represents a transitional member between subfamiles 1 and 2. In addition, we have used ancestral sequence reconstruction to functionally investigate the segregated evolutionary history of PL2 progenitor enzymes and illuminate the molecular evolution of exolysis. This study highlights that ancestral sequence reconstruction in combination with the comparative analysis of contemporary and resurrected enzymes holds promise for elucidating the origins and activities of other carbohydrate active enzyme families and the biological significance of cryptic metabolic pathways, such as pectinolysis within the zoonotic marine pathogen V. vulnificus.
Introduction
Polysaccharide lyases (PLs)4 are a class of carbohydrate active enzymes (i.e. “CAZymes”) (1, 2) that have proven useful for investigating convergent enzyme evolution (3–5). PLs deploy a β-elimination mechanism to cleave glycosidic linkages within uronic acids, such as homogalacturonan (HG), a homopolymer of galacturonic acid and a primary component of pectin within the cell wall of plants (6). This reaction generates products with a 4,5-unsaturation at the non-reducing end (Fig. 1A). To perform β-elimination, unrelated PL families are dependent on three convergent structural features: a Brønstead base (most commonly an arginine) to deprotonate the C5 carbon, a catalytic metal cofactor (most often Ca2+) to acidify the departing C5 proton and stabilize the oxyanion intermediate, and a stabilizing arginine residue to interact with O2 and O3 of the modified GalA residue (3–5). Cleavage can occur indiscriminately at internal linkages throughout the polysaccharide (i.e. endolysis) or exclusively at the terminus of the substrate (i.e. exolysis; Fig. 1B).
FIGURE 1.
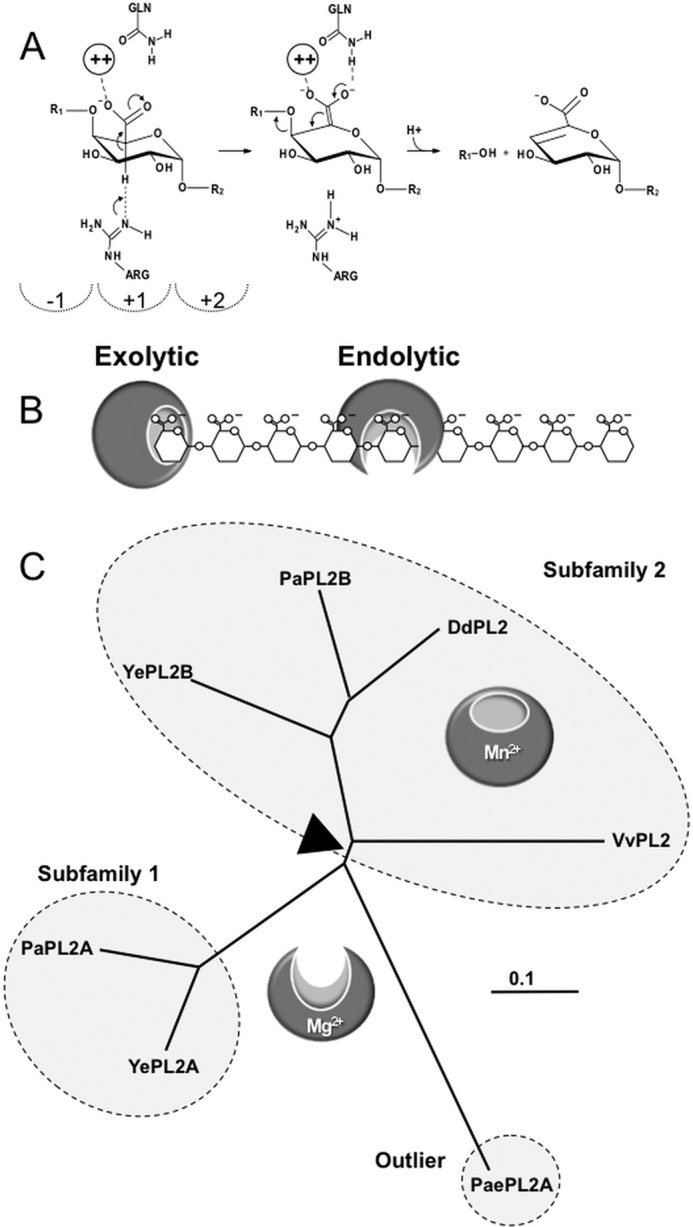
Function and phylogeny of PL2s. A, β-elimination reaction coordinate resulting in a 4,5-unsaturated product. The dashed semicircles indicate subsites in the positive (toward the reducing end) and negative (toward the non-reducing end) of the scissile bond. B, schematic of exolytic and endolytic modes of activity on HG. Exolytic enzymes digest HG strictly from the terminus of the polysaccharide and release a single defined product. Endolytic enzymes cleave internal glycosidic linkages to generate a mixed product profile. C, representative unrooted tree highlighting the phylogeny of subfamily 1 and 2 members discussed in this study. Boundaries identified by CAZy are indicated with dashed circles. The associated modes of activity are represented with endolytic and exolytic models. The presumed transitional sequence space between these activities is shown with a black triangle. Ye, Y. enterocoliticus; Dd, D. dadantii; Pa, P. atrosepticum; Pae, Paenibacillus sp.
The majority of PL family 2 members (PL2s) partition into one of two functionally distinct subfamilies. Intriguingly, many species contain two paralogous PL2 copies that appear to have arisen by gene duplication and functional divergence (i.e. neofunctionalization). Insights into the functional landscape of these two subfamilies of PL2 have identified a correlation between cellular localization, mode of activity, and metal selectivity (3, 7). Subfamily 1 (e.g. YePL2A) contains secreted endolytic members, whereas subfamily 2 members (e.g. YePL2B) are intracellular and exolytic and preferentially harness Mn2+ during catalysis (3, 8). Interestingly, PaePL2 from Paenibacillus sp. Y412MC10, an outlier that is endolytic and preferentially utilizes Mg2+ (Fig. 1C) (7), has provided a snapshot into the evolution of PL2s and the activity of the progenitor enzyme (7) (Table 1). A similar relationship has been described for the structurally unrelated PL22 cytoplasmic lyase family (Table 1) (4). Preferential use of transition metals in PL2s and PL22s is mediated by histidines (PL2 coordination pockets display two histidines; PL22 coordination pockets display three histidines), which displace acidic residues found within Ca2+-selective PLs (3, 4). Nitrogen ligands provide more favorable coordination chemistries for transition metals (9).
TABLE 1.
Catalytic properties of characterized PL2s and PL22s
SF, subfamily; pHΔ, experimentally determined pH optimum; ND, not determined; NA, not applicable.
| Enzyme | SF | pHΔ | Activity | Location | Metala | Reference |
|---|---|---|---|---|---|---|
| DdPL2B (PelW) | 2 | ND | Exo | Cytoplasm | Co2+, Mn2+, Ni2+ | Ref. 8, this studyb |
| PaePL2 | NA | 7.4 | Endo | Cytoplasm | Mg2+ | Ref. 7, this studyb |
| PaPL2B | 2 | ND | Exo | Cytoplasm | ND | This studyb |
| VvPL2 | 2 | 9.3 | Endo | Secreted | Mg2+ | This studyb |
| YePL2A | 1 | 9.6 | Endo | Secreted | Mg2+ | Ref. 3, this studyb |
| YePL2B | 2 | 8.6 | Exo | Cytoplasm | Mn2+ | Ref. 3, this studyb |
| DdPL22 (Ogl) | 1 | ND | Exo | Cytoplasm | Mn2+ | Ref. 8 |
| YePL22 | 1 | 7.6 | Exo | Cytoplasm | Mn2+ | Ref. 4 |
a The preferential metal that provides maximal activity in recovery assays.
b These assays were done with the exhaustive dialysis technique as opposed to the depletion-supplementation approach.
The earliest diverging outgroup of PL2s is strictly cytoplasmic (4, 7), which suggests that transition metals are a prerequisite for intracellular β-elimination. Ca2+ is an intracellular signaling molecule, and it is present at limiting levels in the cytoplasm of bacteria (0.1–2 μm) to prevent signaling interference and modification of subcellular structures (10, 11). In contrast, the periplasm is believed to be a more heterogeneous metallo-environment because extracellular ions are free to passively diffuse across the outer membrane (12). The β-helix PLs (PL1, PL3, and PL9), the largest group of PLs most commonly associated with phytopathogens and saprophytes, are secreted into the periplasm or extracellular environment. β-Helix PL families active on HG preferentially coordinate Ca2+ (13) and appear to have evolved for colonization and modification of the plant cell wall. Ca2+ plays a crucial role in the maintenance of plant cell wall integrity, and its levels are high in this environment (10 μm to 10 mm) (14).
Pectins are ubiquitous nutrients for environmental saprophytes, target substrates for macerating phytopathogens (e.g. soft rot), and components of dietary fibers that are digested by symbiotic microbes within the intestines of animals. Perhaps surprisingly, HG utilization and functional pectinases have also been reported for several human enteric pathogens, including Yersinia spp. (4, 15, 16) (Fig. 1C). Although the biological significance of pectinolysis within human pathogens is not clearly understood, several possible roles have been hypothesized, including environmental persistence, colonization of agricultural crops as vectors for transmission, and utilization of pectic nutrients within the intestine of an infected animal host (17). In this light, the presence of a pectinolytic pathway, complete with transport machinery (KdgM-like porin and solute binding protein), polysaccharide lyases (PL2, PL9, and PL22), and a homologue of a unique HG-binding protein (CBM32) (18), has been identified within the genome of Vibrio vulnificus (Fig. 2A). V. vulnificus is a marine-borne bacterium most commonly associated with gastroenteritis caused by the consumption of contaminated seafood or septicemia resulting from wading in contaminated water with open wounds (19). Correspondingly, pectin represents a nutrient niche that is not consistent with its lifestyle (20). This pathway is not strictly conserved within Vibrionaceae, and whether it represents a historical remnant of a pectinolytic ancestor of V. vulnificus or evolved by horizontal gene transfer in response to its coastal water-zoonotic infectious life cycle remains to be determined.
FIGURE 2.

Characterization of HG modification by VvPL2. A, the V. vulnificus HG utilization locus is displayed as a schematic with representative gene sizes shown to scale. Genes that have been classified to assigned CAZy families (CBM, CBM32) and genes predicted in be involved in transport (KdgM and SBP) are labeled. Gene IDs are displayed below. B, product profile of HG digestion by VvPL2 over time. A saturated marker of GalA-GalA3 is shown on the left, and unsaturated products (degree of polymerization 2–5) are displayed on the right. C, pH profile of VvPL2. The temperature optima of VvPL2 (D), YePL2A (E), and YePL2B (F) are shown. Shown are representative kinetic plots for YePL2A (G), YePL2B (H), and VvPL2 (I) on GalA3. Product formation represents the detection of ΔGalA by UV absorbance at 232 nm. J, metal exchange assays for YePL2A, YePL2B, and VvPL2 using the dialysis method and quantified using initial velocities with 1 mg ml−1 HG and equivalent enzyme concentrations. The two y axis scales represent product values for YePL2B and VvPL2 (left) and YePL2A (right). Shown are Michaelis-Menten plots for YePL2A (K) and YePL2B (L) in the presence of various catalytic metals using the dialysis method. Error bars, S.D.
Further insights into the evolutionary history of PL2s after the gene duplication event will help illuminate the adaptations required for metal-dependent activity and cellular specialization of pectin utilization, in addition to the biological significance of pectinolysis for various enteric pathogens. This study describes the structure and function of VvPL2, which is the first reported pectinase from V. vulnificus. Based upon its phylogenetic position within subfamily 2, VvPL2 represents a potential endolytic-exolytic transitional remnant. Additionally, we perform ancestral sequence reconstruction (ASR) of the PL2 family and resurrect progenitor PL2s to compare their activities with contemporary enzymes from subfamilies 1 and 2. We propose that ASR is an underexploited approach within the CAZyme field that will assist in streamlining the characterization of unknown enzyme activities and illuminating the evolutionary basis of substrate recognition and modification in other PL and CAZyme families.
Experimental Procedures
Biochemical Characterization of VvPL2
Purification of Enzymes
Synthesized codon-optimized VvPL2, DdPL2, and PaPL2B genes were subcloned in pET28 (BioBasic Inc., Mississauga, Canada), and YePL2A and YePL2B plasmids (3) were transformed into Escherichia coli BL21 Star (DE3) cells and grown in LB broth containing 50 μg ml−1 kanamycin sulfate. Cells were grown at 37 °C with agitation at 180 rpm until cell density reached an A600 of ∼0.8. Cultures were cooled to 16 °C, agitation was reduced to 120 rpm, and genes were induced with a final concentration of 200 μm isopropyl 1-thio-β-d-galactopyranoside. Overnight cultures were centrifuged at 7,000 × g for 10 min. Cells were chemically lysed by resuspension in a solution of 8% (w/v) sucrose, 0.65% (v/v) deoxycholate, 0.65% (v/v) Triton X-100, 30 mm NaCl, 350 μg ml−1 lysozyme, 6 μg ml−1 DNase, 30 mm Tris, pH 8.0. After lysis, lysate was centrifuged at 13,000 × g for 45 min. The clarified supernatant was passed through a 0.45-μm filter and applied to a gravity flow nickel affinity chromatography column and eluted with 0.5 m NaCl, 20 mm Tris, pH 8.0, with a stepwise increase in imidazole concentration of 5, 10, 100, and 500 mm. Samples containing the protein of interest were concentrated with an Amicon ultrafiltration cell (EMD Millipore) and passed through a HiPrep 16/60 Sephacryl S-200 HR size exclusion chromatography column (GE Healthcare) in 20 mm Tris-HCl, pH 8.0. Pure samples were pooled and concentrated.
Generation of Loop Swap Mutants
Two mutants were created by replacing residues 601–654 of YePL2A with 562–632 of YePL2B as described previously (48). Native YePL2A and YePL2B in pET28a were used as templates. 3′-Regions were grafted to 5′-regions in a secondary PCR, and full gene sequences were ligated into NheI and XhoI restriction enzyme cut sites in pET28a. Ligated transformants were sequenced. Enzymes were produced and purified as above.
Enzyme Assays
Optimal pH was determined by dialyzing samples of enzyme overnight into buffers: BisTris, pH 6.6–7.2; Tris, pH 7.1–9.0; CAPSO, pH 8.9–10.3; CAPS, pH 9.7–11.1; and CABS, pH 10.0–11.0. After equilibration, enzyme was incubated at 37 °C in 1 mg ml−1 HG, 50 mm buffer, and the reaction was monitored at 232 nm. Optimal temperature was determined by incubating samples of enzyme in water baths at temperatures ranging from 5 to 60 °C for 15 min. Enzyme was then added to 1 mg ml−1 HG, 20 mm CAPSO, pH 9.0, pre-equilibrated to temperature. Reactions were run for 3 min and monitored at 232 nm. Divalent metal cation preference was determined by dialyzing samples of enzyme into 2 mm EDTA in 20 mm CAPSO, pH 9.0, to remove divalent metal cations from solution. Fractions were then dialyzed into deionized water and incubated with 1 mg ml−1 HG, 50 mm CAPSO, pH 9.0, at 37 °C to ensure that activity had been ablated. Fractions were further dialyzed into solutions containing 1 mm CaCl2, MgCl2, MnCl2, CoCl2, NiCl2, or CuCl2, CAPSO, pH 9.0. After equilibration, samples were incubated in 1 mg ml−1 HG, 50 mm CAPSO, pH 9.0, and monitored at 232 nm.
Time course experiments were performed to determine product profiles. Enzymes were incubated in 1 mg ml−1 HG, 50 mm CAPSO, pH 9.0, 1 mm MnCl2 at 37 °C. Reactions were stopped by heating the samples to 95 °C for 10 min followed by flash freezing in liquid nitrogen. Samples were then resolved by thin layer chromatography with 1-butanol/distilled water/acetic acid (5:3:2, v/v/v) running buffer and visualized with 1% orcinol in a solution of ethanol sulfuric acid (70:3, v/v), followed by heating at 110 °C for 10 min. Samples were compared with samples of GalA (Sigma, catalog no. 48280), GalA2 (Sigma, catalog no. D4288), and GalA3 (Sigma, catalog no. T7407).
Enzyme Kinetics
PLs (100 nm to 1 μm) were incubated in increasing concentrations of HG and GalA3 with 50 mm CAPSO, pH 9.0, 1 mm MnCl2. Samples were monitored in real time at 232 nm, and product formation was determined using the extinction coefficient 5,200 m−1 cm−1. Data were analyzed, and kinetic values were determined using GraphPad Prism version 6.
Crystallization and Structure Solution of VvPL2
Crystals of VvPL2 were developed via the hanging drop vapor diffusion method at a protein concentration of 15 mg ml−1 by mixing 1.0 μl of the protein solution with an equal volume of mother liquor consisting of 16% (w/v) polyethylene glycol 3350, 0.14 m Na/K tartrate, and 0.1 m HEPES (pH 7.0) at 19 °C. Crystals were cryoprotected by brief crystal immersion into reservoir solution supplemented with 25% ethylene glycol and subsequently frozen in a liquid nitrogen stream prior to diffraction experiments.
VvPL2 crystallized in space group P65 with one protein molecule in the asymmetric unit. Diffraction data for VvPL2 in complex with two molecules of tartrate were collected at the beamline SSRL 12-2 of the Stanford Synchrotron Radiation Lightsource. The data set was processed with XDS and scaled with Scala (21). The correct phases were derived via molecular replacement with the program Phaser (22) using the Yersinia enterocolitica PL2 structure (YePL2A, Protein Data Bank code 2V8J) as a search model (3). The structure of VvPL2 was rebuilt with the program Buccanneer and iteratively improved with cycles of manual building with Coot and positional refinement with Refmac (23–25). Data collection, processing, and refinement statistics were generated by Molprobity (26) and are presented in Table 2. Ramachandran statistics were generated using Rampage (27). Mapping of the degree of residue conservation was performed with the program Consurf (28), and figures were produced using PyMOL (Schrödinger, LLC, New York).
TABLE 2.
X-ray data collection, processing, and VvPL2 model refinement statistics
| Tartrate-bound | |
|---|---|
| Data collection statistics | |
| Wavelength | 0.97949 |
| Beamline | SSRL 12-2 |
| Space group | P65 |
| Resolution | 46–1.90 (1.95–1.90)a |
| Cell dimension | 141.1, 141.1, 72.4 |
| α, β, γ (Å) | 90.0, 90.0, 120.0 |
| Rmerge | 0.096 (0.419) |
| Completeness (%) | 99.8 (100) |
| 〈I/σI〉 | 29.1 (4.6) |
| Redundancy | 4.8 (5.0) |
| Total reflections | 311,612 |
| Unique reflections | 64,421 |
| Refinement statistics | |
| Rwork (%), Rfree (%) | 15.6, 19.5 |
| Root mean square deviation | |
| Bond lengths (Å) | 0.010 |
| Bond angles (degrees) | 1.909 |
| Average B-factors (Å2) | |
| Protein molecule | 26.4 |
| Transition metal | 21.0 |
| Solvent atoms | 40.3 |
| No. of atoms | |
| Protein atoms | 4,538 |
| Transition metal | 1 |
| Solvent atoms | 596 |
| Ramachandran statisticsb | |
| Most favored (%) | 97.9 (550) |
| Additional allowed (%) | 2.1 (12) |
| Disallowed (%) | 0.0 (0) |
a Values in parentheses are for the highest resolution shell.
b Ramachandran statistics were calculated by Rampage (27).
Phylogenetic Analysis of the PL2 Family
PL2 sequences were retrieved from the CAZy database (2) and curated to remove truncated or duplicated sequences. An initial amino acid sequence alignment was built using Gblocks (29), and a guide tree was subsequently generated using PhyML (30). This guide tree was then utilized as described (31) to align the full-length sequences. A maximum likelihood (ML) phylogeny was constructed using GARLI version 2.0 (32) and the appropriate model of evolution (LG + I + G), as determined by ProtTest version 3.4 (33). A Bayesian phylogeny was also generated using MrBayes version 3.2.4 (34) and a mixed amino acid model with two parallel runs in order to ensure convergence. Both phylogenies were rooted on the branch between the γ-proteobacteria and outgroup sequences. Bootstrapping was performed in GARLI 2.0 using 1,024 replicates and a 10% burn-in. All trees were visualized using Geneious version 6.1.8 (35).
Ancestral Inference
Maximum likelihood ancestral inference was performed in PAML 4.3 (Ziheng Yang) on the basis of nucleotide, codon, and amino acid sequences using the ML phylogeny constructed for PL2. For nucleotide inference in BASEML, a nucleotide alignment exactly matching the PL2 amino acid alignment generated by PRANK was constructed using Geneious version 6.1.8, and the appropriate model of nucleotide substitution was implemented as determined by jModelTest version 2 (36). For codon and amino acid inference in CODEML, the WAG amino acid rate file was employed. The sequences inferred by the three methods were compiled, and a majority-rules approach was taken, with any remaining ambiguous sites resolved after consideration of the physicochemical properties of the inferred amino acids, their frequency among the contemporary sequences, and the inference made by the codon method (which is considered to be the most accurate). Bayesian ancestral inference was performed in MrBayes version 3.2.4 using a mixed amino acid model. Ancestral gaps were inferred using PRANK and incorporated into the ancestral sequences inferred by PAML and MrBayes.
Biochemistry of Ancestral PL2s
Node 49, 52, 54, and 74 sequences were codon-optimized, synthesized, and subcloned into a pET28 (Novagen, catalog no. 69864) expression vector using NheI and XhoI directional restriction sites (Biobasic). The Node 52 gene was subsequently subcloned into pET32 (Novagen, catalog no. 69015) to increase soluble yields. Gene products were purified by immobilized metal affinity chromatography, eluted with a 0–500 mm imidazole gradient, and dialyzed into 20 mm Tris-HCl, pH 8.0. Digests were performed using 0.1 μm enzyme and 1 mg ml−1 HG at 37 °C. Nodes 52 and 54 were performed at pH 8.0 (4 mm Tris-HCl), and Node 74 was performed at pH 9.4 (4 mm CAPS). Direct method metal recovery assays were performed by adding EDTA to a final concentration of 1 mm and supplementing with 10 mm CaCl2, MgCl2, or MnCl2. Reactions were heat-killed by boiling for 5 min and clarified by centrifugation at 13,000 × g. Products were analyzed directly or following a 10-fold concentration by TLC (as above) or high performance anion exchange with pulsed amperometric detection. This analysis was performed with a Dionex ICS-3000 chromatography system (Thermo Scientific) equipped with an autosampler as well as a pulsed amperometric detector for total carbohydrates and a UV-visible detector for unsaturated galacturonides. Aqueous sample (typically 10 μl) was injected into an analytical (4 × 250-mm) CarboPac PA1 column (Thermo Scientific) and eluted at a 0.4-ml min−1 flow rate with a sodium acetate gradient (0–1 min, 250 mm; 1–17.5 min, 250–1,000 mm; 17.5–20 min, 1,000 mm; 20–21 min, 1,000 to 250 mm; 21–35 min, 250 mm) in a constant background of 100 mm NaOH.
Site-directed Mutagenesis of YePL2B
Nucleotide substitutions were generated via PCR-mediated site-directed mutagenesis. Mutagenic primer sets were extended with KOD polymerase (Novagen, catalog no. 71086) using pETPL2B (3), encoding YePL2B as template. The entire reaction mixture was then digested with DpnI (New England BioLabs, catalog no. R0176), and one-tenth of the reaction mixture was transformed into DH5α-competent cells. Plasmid was extracted (Omega, catalog no. D6945), and constructs were verified by Sanger sequencing (McGill University and Génome Québec Innovation Centre).
Results and Discussion
Biochemical Characterization of PL2 Subfamily 2 Enzyme
To explore the full profile of PL2 subfamily 2 activities, sequence entries from its four major clades were analyzed. These representative enzymes include Y. enterocolitica subsp. enterocolitica 8081 (YePL2B; gene ID YE1886), Pectobacterium atrosepticum SCRI1043 (PaPL2B; gene ID ECA2402), Dickeya dadantii 3937 (DdPL2; gene ID Dda3937_03361), and V. vulnificus YJ016 (VvPL2; gene ID VVA1383; residues 18–556). In addition, the endolytic YePL2A (gene ID YE4069) was purified to enable comparisons with a previously characterized subfamily 1 member (3) (Fig. 1C). As anticipated, YePL2B, PaPL2B, and DdPL2 exclusively released unsaturated digalacturonate (ΔGalA2, where 2 represents the degree of polymerization), which is consistent with an exolytic mode of activity (not shown). Unexpectedly, VvPL2 displayed an endolytic product profile (Fig. 2B) and pH optimum reminiscent of subfamily 1 enzymes (Fig. 2C); however, its temperature optimum reflected a similar distribution to YePL2B (Fig. 2, D–F).
In order to compare specificities and activities, we have performed a comparative kinetic analysis between VvPL2, YePL2A, and YePL2B on both HG and the pectic fragment GalA3 (Fig. 2 (G–I) and Table 3). YePL2A is preferentially active on HG over GalA3 (∼11-fold), whereas its paralog YePL2B is preferentially active on GalA3 over HG (∼13-fold). This inverse relationship is consistent with their assigned roles in a degradative pathway. YePL2A is secreted and endolytic, which is tailored for upstream activity on polymeric HG, and YePL2B is intracellular and exolytic and performs a downstream role in oligogalacturonide depolymerization (17). In comparison, VvPL2 displays a relatively high catalytic rate on both GalA3 and HG, with preferential activity on GalA3 (∼5-fold). This plasticity may be explained by V. vulfinicus containing only one PL2 copy. In this context, VvPL2 appears to take on the roles of both YePL2A and YePL2B, and its position within the PL2 family tree suggests that it represents a transitional member based upon both sequence relatedness and function (Fig. 1C).
TABLE 3.
Kinetics of VvPL2, YePL2A, YePL2B, and YePL2A-B on GalA3 and HG
Values are shown (±) S.D. from three independent reactions.
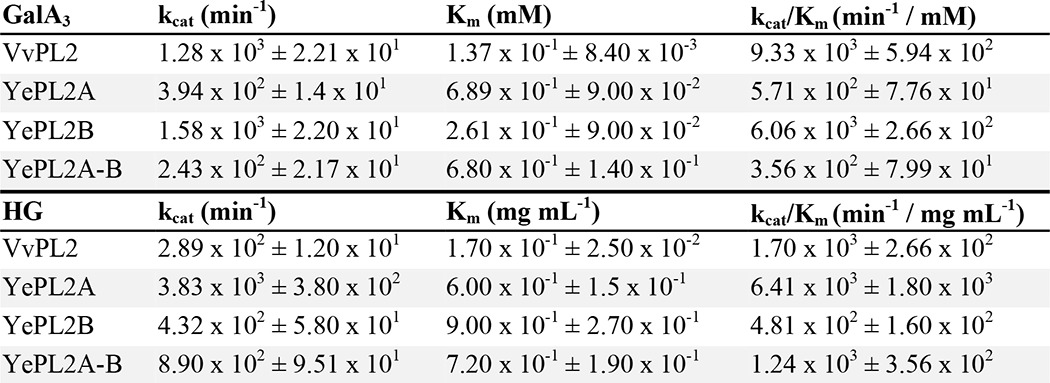
Previously, PL2s have been reported to preferentially utilize transition metals over Ca2+ during catalysis (3, 7, 8). To further explore a differential relationship between PL2 subfamilies and metal specificity, YePL2A, YePL2B, and VvPL2 were subjected here to a new preparative treatment that exchanges metal cofactors by performing an exhaustive dialysis against EDTA-buffered solutions, followed by exhaustive dialysis in divalent metal-buffered solutions. This alternative method was developed to promote gradual exchange of cofactors and supplant the direct depletion-supplementation method that has been routinely used previously (3, 8). The direct approach introduces highly concentrated metallo-microenvironments and can be deleterious to protein stability. For example, the characterization of YePL2A metal dependence was not previously possible using the direct method (3). With the dialysis substitution method implemented here, YePL2A displayed very little precipitation during its preparation with all metals tested. Initial velocities for YePL2A, YePL2B, and VvPL2 in the presence of Co2+, Mn2+, Ni2+, Mg2+, Ca2+, and Cu2+ were determined to compare related metal dependence of HG modification (Fig. 2J). For YePL2A and VvPL2, the highest catalytic rates were observed in the presence of Mg2+ (at 1 mg ml−1 HG), which agrees with what was reported for PaePL2 and supports a prominent role for Mg2+ as a metal cofactor in endolytic PL2s (3, 8). In contrast, YePL2B displayed the highest activity when supplemented with Mn2+.
To investigate the mechanistic contributions of various metals and potentially the biological significance of metal selectivity, full Michaelis-Menten kinetics were determined for YePL2A and YePL2B using the same spectrum of cofactors (Table 4 and Fig. 2 (K and L)). Mg2+ and Co2+ were confirmed to promote the highest turnover rate for YePL2A. Mn2+, however, was associated with the lowest Km, which translated into a 4-fold higher catalytic specificity constant for Mn2+ than for Mg2+ (Table 4). This suggests that Mn2+ is optimal under limiting concentrations of substrate; regardless, YePL2A demonstrates remarkable plasticity in cofactor selection. This property reflects the adaptation of secreted PL2s to the heterogeneous ionicity of the periplasm (12). The results for YePL2B indicate that the cytoplasmic enzyme has more selectivity for Mn2+ in both the rate of substrate turnover (kcat) and catalytic efficiency (kcat/Km) (Table 4). Higher specificity (Km) for and catalytic turnover (kcat) with Mn2+ translates into a 8-fold increase over Mg2+ and a ∼3-order of magnitude increase over Ca2+ in catalytic rate. This finding highlights that within the confines of the cell, cofactor selection by PL2s for Mn2+ is more stringent. In the presence of Mn2+, YePL2B appears to adopt a substrate inhibition profile when active on HG (Mn2+*; Ki = 2.25 ± 1.07 mg ml−1; Fig. 2L and Table 3). When fit to this model, the catalytic efficiency is lowered into the range of Mg2+; however, this result should be interpreted with caution because the error values increase, which may compensate for this effect. Although this observation underpins the complexity of the metal-protein-HG interaction for YePL2B, such inhibitory effects are probably negligible in nature because HG concentrations would be limited inside the cell.
TABLE 4.
Kinetics of YePL2A and YePL2B on HG in the presence of varying metal cofactors
∗ Values were calculated to account for substrate inhibition effects. Mn2+∗= substrate inhibition kinetics; Ki = 2.25 ± 1.07 mg mL-1. Values are shown (±) S.D. from three independent reactions.
Structural Analysis of VvPL2, an Endolytic Member of Subfamily 2
In order to provide insight into the molecular determinants of subfamily 2 PL2 activities, we attempted to crystalize DdPL2, PaPL2B, YePL2B, and VvPL2. Although we were able to produce high levels of each recombinant protein, they varied in solubility, stability, and crystallizability. Of the four proteins, only VvPL2 produced diffraction quality crystals, which were used to solve the protein structure by molecular replacement to 1.90 Å using YePL2A as a homology model (Protein Data Bank code 2V8K) (3). VvPL2 adopts an (α/α)7 barrel fold with an extensive active site cleft that is characteristic of endolytic enzymes (Fig. 3A).
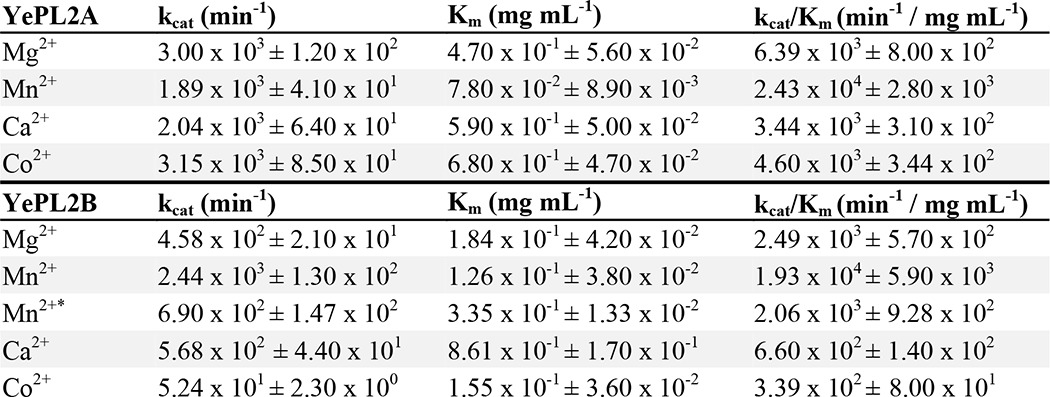
FIGURE 3.

Three-dimensional structure of VvPL2. A, schematic model of VvPL2 color-ramped blue (N terminus) to red (C terminus) and with its catalytic metal modeled as a Mn2+ shown as a purple sphere. B, superimposition of GalA from the +1 site of the YePL2A complex (Protein Data Bank entry 2V8K) within the active center of VvPL2. The backbone of VvPL2 is shown as a gray schematic with the metal-binding residues displayed as gray sticks, ordered waters as red spheres, Mn2+ as a purple sphere, and the stabilizing residue (Arg-304) and Brønstead base (Arg-191) as cyan sticks. The distances between the 2-OH and 3-OH of GalA and Arg-304, C5 and Arg-191, and the Mn2+ ion and the uronate group oxygens are labeled and shown as red dashes. C, the metal binding pocket of VvPL2 with N-terminal His tag. The map of the active center residues coordinating the transition metal is presented as maximum likelihood/σA weighted 2Fo −Fc densities, contoured at 1.0 σ and carved at 1.5 Å. The coordinated Mn2+ and ordered water are displayed as silver and red spheres, respectively. The presence of two tartrate molecules within the N-terminal His tag complex are rendered as yellow ball-and-stick models. D, alignment of the YePL2A (Protein Data Bank entry 2V8J; gray) and VvPL2 (green) metal coordination pocket. Residues are modeled as sticks, Mn2+ as purple spheres, and waters as red spheres. Residues are labeled using VvPL2/YePL2A numbering. Bond distances are indicated with yellow dashed lines. Consurf mapping displaying the conserved (magenta) and divergent (cyan) surface features of VvPL2 with subfamily 1 (E) and subfamily 2 (F) members. The highly conserved residues (Lys-152, Arg-191, Arg-304, and metal pocket) and location of the lysine-tryptophan site (K300W) are labeled.
Superimposition of VvPL2 (residues 26–566) onto YePL2A using PDBeFold highlighted the close structural similarity between the two lyases (root mean square deviation = 1.39 Å over 509 residues) (37). Significantly, the Brønstead base (Arg-191), catalytic pocket, and stabilizing residue (Arg-304) are structurally conserved, with Arg-304 ∼13.8 Å from the metal center with reasonable geometry for interacting with 2-OH and 3-OH of the GalA in the +1 subsite (Fig. 3B). These three convergent features have been suggested to be critical for β-elimination in unrelated PL folds (3–5). Based on an inspection of B-factors and residual electron density, the metal coordinated in the VvPL2 structure appears to be Ni2+ or Mn2+ (Fig. 3C). Although this may be influenced by the purification conditions, VvPL2 function was modest in the presence of Ni2+ (Fig. 2J). Therefore, whereas the natural metalloenzyme complex is assumed to contain Mn2+, we cannot rule out the possibility with the available data that the metal cofactor presented in the crystal structure has not been substituted with Ni2+. Intriguingly, the uncleaved N-terminal histidine tag from VvPL2 introduced two artifactual interactions between the Nϵ2 of VvPL2 residues His-4 and His-6, and the bound metal assisted in stabilizing the coordination sphere with a perfect octahedral symmetry (Fig. 3C). Other interactions involve His-129 (Nϵ2), Glu-150 (Oϵ2), His-192 (Nϵ2), and an ordered water molecule (HOH 383) that is activated by a 2.8-Å hydrogen bond with the Nδ1 imidazole nitrogen of His-546.
A sequence comparison between the metal binding pockets of YePL2A and VvPL2 reveals the presence of a histidine residue in VvPL2 (His-546) that replaces a glutamate in YePL2A (Glu-515), which was presumed to contribute to Mn2+-selective chemistries for subfamily 2. Structural superimpositions of the metal binding pockets, however, reveal that His-546 and Glu-515 are spatially and functionally conserved (Fig. 3D). Both residues interact with an ordered water molecule, charging it for coordination of the metal cofactor. To investigate whether the Nϵ of the imidazole group provided any definitive function, we performed both a single substitution (H530E) and insertion of the tripeptide sequence from YePL2A (Phe-512/Thr-513/Glu-514) into the equivalent sequence space on YePL2B (Tyr-528/Ile-529/His-530). These mutations did not reverse the metal selectivity in YePL2B because optimal digestion was still observed in the presence of Mn2+ after EDTA treatment; however, it did appear to be deleterious for the utilization of Ca2+ (not shown). This observation does not rule out a differential role for His-546 in exolytic enzymes but does suggest that the more stringent metal selectivity observed in the catalytic activity of YePL2B probably depends on other structural features within the metal binding site, such as residue geometry and distance, which would be supported by a greater network of interactions within the enzyme scaffold.
Based upon its activity (Table 3) and position within subfamily 2 (Fig. 1C), VvPL2 appears to represent a transitional sequence within the phylogeny of PL2. Therefore, we examined the surface of VvPL2 to identify any structural features near the active site cleft that might help illuminate the structural transition between the endolytic and exolytic subfamilies. Subfamily 1 and subfamily 2 sequences were independently mapped onto the structure of VvPL2 using Consurf (28) (Fig. 3, E and F). This program scales the conservation (magenta) and divergence (cyan) of residues to identify similar and distinct structural elements. Near its catalytic center, VvPL2 displays a high level of structural similarity with subfamily 1 sequences, which is consistent with its observed activity (Fig. 3E). Apart from the core catalytic residues, there is notably less conservation with PL2 subfamily 2 sequences (Fig. 3F). One such region includes a hallmark lysine residue (Lys-300) that is poised near the exit of the active site cleft. This lysine is invariant in subfamily 1 and is replaced with a tryptophan through the majority of subfamily 2 sequences (Fig. 4). Intriguingly, the small outgroup of early diverging sequences in subfamily 2 that includes two Marinomonas spp. and Acholeplasma brassicae display a phenylalanine and glycine, respectively, at this position (not shown).
FIGURE 4.

Ancestral sequence reconstruction of the PL2 family. A, phylogeny of the PL2 family with calculated ancestor sequence nodes highlighted (●) and labeled. The node sequences (49, 52, 54, and 74) that were targeted for gene synthesis and biochemical characterization are indicated with a black triangle. Previously characterized enzyme activities are boxed, activities reported in this study are noted with a single asterisk (see Table 1 for references), and family members with solved three-dimensional structures are designated with a double asterisk (3). B, primary structure alignment of YePL2A, YePL2B, VvPL2, Node 52, Node 54, and Node 74. Residues involved in catalysis and metal coordination are indicated with black and white triangles, respectively. The putative lysine to tryptophan switch is highlighted with a black circle.
Ancestral Sequence Reconstruction of Family 2 PLs
The biochemical properties of VvPL2 have revealed that subfamily boundaries assigned within the CAZy database do not provide enough sequence resolution to elucidate the evolutionary history of endolytic to exolytic transition in the PL2 family. Therefore, to trace lineage at the sequence level, we constructed a robust ML phylogeny of all available PL2 sequences and used this analysis to infer the sequences of ancestral PL2s positioned at a range of branch points (Fig. 4A). Almost all of the contemporary PL2 sequences available are from members of the γ-proteobacteria, with the exception of two sequences from Paenibacillus sp., a member of the Firmicutes, and Haloterrigena turkmenica, an archaeon, which were used as an outgroup and to root the tree. The topology of the ML phylogeny shown in Fig. 4A is supported by high bootstrap percentages; furthermore, a Bayesian phylogeny was also constructed for comparison and found to display identical topology (not shown). The contemporary PL2 sequences form two major clades, subfamily 1 and subfamily 2, with subfamily 1 positioned closest to the root. The ancestral nodes 49, 52, 54, and 74 were selected for ancestral inference and reconstruction, given their positions at major branch points within the phylogeny (Fig. 4A). Node 49 represents the last common ancestor of all PL2 sequences, including the outgroup sequences, whereas Node 74 represents the last common ancestor of the subfamily 1 PL2s alone. Nodes 52 and 54 both represent ancestors of subfamily 2 after divergence of VvPL2, with Node 54 being the ancestor of all subfamily 2 PL2s from plant pathogens and Node 52 being the ancestor of these same enzymes plus the endolytic VvPL2 and PL2s from Vibrio furnissi and Acholeplasma brassicae. The positions of all four of these PL2 ancestors are supported by bootstrap percentages ≥98%. Ancestral inference was performed under the ML criterion, and the average posterior probability for each of the four ancestors was >0.7 (this increases to >0.8 for Nodes 52, 54, and 74 when inference at ancestral gaps is not considered).
The four ancestral PL2s vary from their closest contemporary descendant by at least 15% (∼83 amino acids). As expected from its phylogenetic position, the closest contemporary descendant of Node 74 is a subfamily 1 PL2 from Pectobacterium wasabiae (84% sequence identity), and it possesses a lysine residue (Lys-286) conserved within the endolytic subfamily 1. In contrast, Node 54 shares the greatest sequence identity with a subfamily 2 enzyme from Yersinia pseudotuberculosis (82%) and contains the conserved tryptophan residue (Trp-286) at this same position in the active site cleft. Node 52 shares only 60% sequence identity with its closest contemporary descendant (VvPL2). Despite Node 54 sharing only 45% sequence identity with VvPL2, Nodes 52 and 54 share 65% sequence identity. Interestingly, Node 52 does not contain either the conserved lysine found in subfamily 1 or the conserved tryptophan found in subfamily 2; rather, it contains an arginine residue (Arg-283) (Fig. 4B). The most divergent of the inferred ancestral PL2s is Node 49, sharing just 56% sequence identity with its closest contemporary descendant (a subfamily 1 PL2 from P. wasabiae). Based upon sequence alignments, Node 49 does not appear to contain a lysine or tryptophan residue at this position; however, structural modeling determined that this ancestor has a truncated loop, and a lysine (Lys-268) is spatially conserved (not shown). This observation suggests that a lysine at this position is correlated with endolytic activity and that the last common ancestor of PL2s was endolytic, which would be consistent with what was previously proposed for the early diverging PaePL2 (7).
Characterization of Resurrected Ancestral PL2s
From their inferred sequences, it appears that Node 74 is endolytic and Node 54 is exolytic, but, as we have observed with VvPL2, sequence information alone cannot fully predict function. Furthermore, Node 52 contains a divergent amino acid in place of the highly conserved lysine or tryptophan residue associated with endolytic and exolytic activity, respectively. Therefore, we resurrected and characterized the enzymes from Nodes 49, 52, 54, and 74 in vitro using gene synthesis and enzyme product profiling. Nodes 52, 54, and 74 were produced as soluble protein in E. coli and found to be active on HG (Fig. 5A); however, Node 49 was not produced and could not be studied further. In agreement with its phylogenetic position as the last common ancestor of subfamily 1 and the presence of Lys-286, Node 74 displayed characteristic endolytic activity on HG, with detected products ranging in size from ΔGal2 to ΔGal4. Similarly, as an ancestor of subfamily 2, Node 54 displayed an exolytic profile and almost exclusively generated ΔGal2. Node 52 also generated an exolytic-like digestion profile of HG despite possessing an arginine at the Lys-268 position. This residue may represent a key transition in the evolution of subfamily 2 sequences because, despite having related charge potentials, arginine has more steric bulk than lysine. Intriguingly, both residues can display identical adenine bases in their first and third codon positions (lysine, AAA/AAG; arginine, AGA/AGG), which suggests that substitutions can arise by in-frame substitutions. Analysis of the nucleotide sequence of YePL2A reveals that Lys-291 is encoded by a triadenine codon, and the second position of Trp-286 in YePL2B contains a guanine. Therefore, it seems plausible that the AAA-lysine-encoding position may have evolved first to an AGA-arginine and subsequently to a TGG-tryptophan (Fig. 5B). Alternatively, it could have proceeded through an AAA → AAG silent mutation and then an AGG-arginine intermediate. In either case, this pathway would suggest that exolysis arose in part through increases in the steric bulk of this positional residue (lysine → arginine → tryptophan) and neutralization of its charge (positive → neutral) (Fig. 5B).
FIGURE 5.

Product profiling and biochemical characterization of ancestral PL2 sequences. A, high performance anion exchange with pulsed amperometric detection analysis of HG digestion profiles generated by Node 74, Node 52, and Node 54 ancestral enzymes. Pulsed amperometric detection (left axis) of oligogalacturonide elution is displayed as a black trace. The presence of ΔGalA oligosaccharides with a degree of polymerization >2 are indicated with black triangles. B, model for nucleotide progression of lysine-arginine-tryptophan stepwise mutation. C, metal cofactor analysis of Node 74 relative activity with the inset showing the product generation in the presence of Mn2+ and Mg2+. D, putative subsite structure of PL2 endolytic (top) and exolytic (bottom) active sites displaying the molecular basis of ΔGalA2 generation by introduction of a tryptophan (black bar). R, reducing end; N, non-reducing end. Brackets indicate terminal residues within the active site that can be extended. Shown are HPLC-pulsed amperometric detection analysis of YePL2B digestions of HG with Trp-286 substitution with lysine and alanine in the absence (E) and presence (F) of EDTA. The large peak after 14 min represents the primary ΔGalA2 products, and the black arrows indicate the appearance of ΔGalA3 and ΔGalA4 products.
Node 74 purified in high yields and was therefore used to further probe ancestral functions and relationships between PL2 subfamilies. This enzyme displays a pH profile (not shown) similar to that of YePL2A and maximal enzyme recovery with Mg2+ (Fig. 5C). These data indicate that ASR can accurately determine the functional relatedness of PL2s back to the subfamily divergence in their lineage and suggest that ASR will have utility for helping to define the phylogenetic relationships in other CAZyme families.
Evolution of Exolytic and Endolytic Activities within the PL2 Family
Despite numerous attempts (e.g. YePL2B, DdPL2, and PaPL2B) we have been unable to solve the structure of an exolytic PL2, and currently the molecular basis of exolytic activity in this family remains to be determined. Previously, an endolytic-exolytic switch was proposed to be the result of a loop insertion near the catalytic center (residues 200–218 of YePL2A and 188–212 of YePL2B) (3). Loop insertions have commonly been observed to be responsible for exolytic-endolytic transformations within CAZymes, including polygalacturonases (3) and family 11 PLs (38). In YePL2B and, by extension, other exolytic PL2s, the catalytic cleft would need to be remodeled to accommodate the reducing end of HG with subsites +1 and +2 for the exclusive release of ΔGalA2 (39) (Fig. 5D). Therefore, we attempted to define the structural role of the loop in YePL2B by performing loop-swapping experiments between YePL2A and YePL2B to generate the hybrid enzymes YePL2A-B (containing the B-loop) and YePL2B-A (containing the A-loop). Swapping the loops between the two enzymes lowered the rate of HG digestion but did not alter their respective product profiles (Table 3) (data not shown), which suggests that the predicted YePL2B loop is not the molecular determinant of exolysis. Intriguingly, the YePL2B loop shifted the pH optimum of the YePL2A toward YePL2B (not shown) and reduced the affinity for HG but not GalA3 (Table 3), which indicates that the loop may contain residues that contribute to the formation of distal subsites for accommodating polymerized HG.
In order to identify more subtle features that contribute to the structural basis of exolytic activity, we next performed a thorough examination of the primary structure alignments of the node enzymes and a homology model of YePL2B (not shown). Consistent with what was revealed through the ASR analysis, there is a surface-exposed tryptophan conserved within all contemporary exolytic enzymes (YePL2B, Trp-286) and Node 54 (Trp-286), which underpins that it may have a functional role. To test this possibility, we performed substitutive mutagenesis on this tryptophan (W286K and W286A). The product profile of YePL2B-W286K and YePL2B-W286A contained several populations, which suggests that the mutants had become endolytic (Fig. 5E). This effect was enhanced in the presence of EDTA (Fig. 5F). It appears that Trp-286 functions to stabilize the exolytic cleft, perhaps by occluding access to the active site and restricting interactions with polymerized HG to the reducing end (Fig. 5D). Additionally, the role of EDTA in generating this phenotype suggests that the modified cleft structure of YePL2B may be fortified by interactions with the catalytic metal. In the absence of a structure from an exolytic PL2, these results shed new light on how subtle transitions in primary structure can transform enzyme activity within closely related enzyme families.
Biological Significance and Evolution of HG Utilization Pathways within Human Enteric Pathogens
PL2s are disproportionately found in human enteric pathogens, and there are often paralogous copies within a genome that partition into subfamilies 1 and 2 (Figs. 1C and 4A) (7). The presence of two distinct PL2 activities that display differential cellular localization highlights that they are contributing to upstream endolytic (secreted) and downstream exolytic (cytoplasmic) stages of HG depolymerization (Fig. 6) (20). Several examples of species with a single copy of either an exolytic or endolytic PL2 entry do exist (Fig. 4A) (7); however, in these cases, alternative pathways for HG saccharification have evolved (17, 20). The pectinolytic pathway from V. vulnificus is one such example (Fig. 6). V. vulnificus is predicted to deploy an extracellular PL9, a periplasmic HG binding protein (endoVvCBM32) and endoVvPL2, and an intracellular oligogalacturonte lyase (exoVvPL22). HG transport is facilitated through a KdgM-like anionic porin (40, 41), and intracellular transport is predicted to be facilitated by a solute-binding protein and an ABC transporter that is distally located in the genome but under similar regulation (20). This pathway differs from what has been biochemically defined for Y. enterocolitica (Fig. 6) (17). Y. enterocolitica deploys an extracellular pectin methylesterase (YeCE8) (15); periplasmic HG-binding protein (endoYeCBM32) (18), endolytic PL2 (endoYePL2A) (3), and exolytic polygalacturonase (exoYeGH28) (16) and two intracellular depolymerases, which cleave ΔGalA2 (exoYePL2B) (3) and ΔGalA (exoYePL22) (4), respectively, from oligogalacturonide substrates. The signature architectures of these pathways may reveal subtle variances in the structure of pectic nutrients and symbioses (e.g. marine versus terrestrial) or differential roles in environmental persistence and colonization of competitive ecosystems, such as the gastrointestinal tract of animals. Further investigation into biochemical function and evolution of pectin utilization pathways containing PL2s will be foundational for understanding the roles of these enzymes in the life cycle, and potentially in the pathogenesis, of human enteric pathogens.
FIGURE 6.
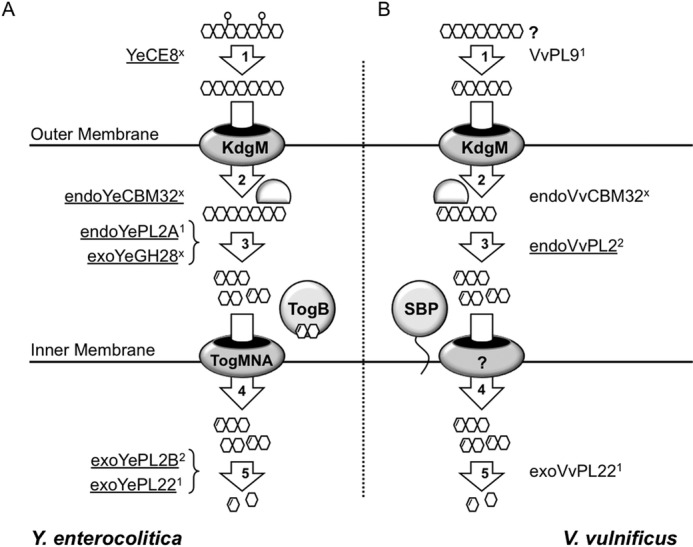
Alternative pathways (A and B) for HG utilization by human enteric pathogens that colonize distinct ecosystems. Key differences between the pathways from Y. enterocolitica and V. vulnificus include the following: extracellular, secreted pectin methylesterase (YeCE8) in Y. enterocolitica and pectate lyase (VvPL9) in V. vulnificus; periplasmic, Y. enterocolitica possesses two depolymerases (endoYePL2A and exoYeGH28), and V. vulnificus has only a single enzyme (endoVvPL2); intracellular, intracellular transport appears to be mediated through unrelated systems; cytoplasmic, Y. enterocolitica possesses two exolytic enzymes (exoYePL2B and exoYePL22), whereas V. vulnificus has only one (exoVvPL22). The key conserved features include the KdgM-anionic porin and the HG-binding protein endoCBM32 that is proposed to retain polymerized substrates within the periplasm (3). In Enterobacteriaceae, this cluster exists as a KdgM-endoPL2-CBM32 operon, which is not architecturally conserved in V. vulnificus (Fig. 2A) (18). Enzyme activities that have been biochemically characterized are underlined. #, subfamily for proteins that belong to annotated subfamilies; ×, those families that have not been assigned.
Conclusion
Assigning ancestry and biological function to sequence-based CAZyme families has been complicated by the realization that many families display great diversity in substrate specificity or mode of activity. In this light, recent efforts to partition CAZyme families into subfamilies (2, 42–44) and develop predictive tools to inform function based upon structural signatures (45–47) have helped to facilitate the sequence-to-function-based characterization of protein-carbohydrate interactions and carbohydrate-modifying enzymes. We have demonstrated here, however, that a higher level of resolution may be required to define the functional boundaries and evolution of activities within some CAZyme subfamilies. Within PL2s, the progenitor enzyme appears to have been endolytic and to preferentially harness Mg2+ for β-elimination (PaePL2 and Node 74); however, it is clear that contemporary endolytic PL2s (subfamily 1) display plasticity in metal selectivity, which can be explained by the heterogeneous metallo-environment of the periplasm and extracellular environment of the niches that these bacteria colonize. In contrast, intracellular PL2s (subfamily 2) display the highest rate of substrate turnover in the presence of Mn2+ and are exolytic.
Insights into the structure of VvPL2, which represents a “transitional” enzyme that exhibits some properties of both subfamilies, and the biochemical characterization of resurrected enzymes from the lineages of both subfamilies have revealed that the molecular basis of an endolysis to exolysis transition is not loop-dependent but rather relies on subtle changes to functional groups at the opening to the active site cleft. For example, it appears that a lysine to tryptophan transition is in part responsible for the emergence of exolysis, and this mutation may have evolved through a lysine (AAA) → arginine (AGA) → tryptophan (TGG) transition. Future research aimed at illuminating the evolution of CAZyme subfamily substrate specificity and mode of activity will be central to defining general properties in the evolution of pectin recognition and modification and the colonization of intriguing nutrient niches by microbes, such as pectinolysis by food-borne pathogens.
Author Contributions
R. M. performed and processed enzyme kinetics and metal supplementation assays, performed YePL2A/B swapping mutagenesis, crystallized VvPL2, and assisted in figure preparation; J. K. H. performed ancestral sequence reconstruction, provided comparative analysis of contemporary and resurrected enzyme sequences, and assisted in manuscript writing and figure preparation; M. D. S. solved the structure of VvPL2, refined and analyzed the model, and assisted in figure preparation; S. T. T. performed the product profiling of PLs and assisted in figure preparation; D. R. J. performed site-directed mutagenesis and enzyme digestions; A. B. B. assisted in structural analysis and project conceptualization; D. W. A. conceived and coordinated the study, wrote the paper, and prepared figures. All authors analyzed results and approved of the final version of the manuscript.
Acknowledgments
We are grateful to the beamline staff at Stanford Synchrotron Lightsource beamline 12-2 for support and technical assistance.
This work was supported by Agriculture and Agri-Food Canada (Agri-Flex #2668) and the Beef and Cattle Research Council (FDE.15.13) (to D. W. A.) and a Natural Sciences and Engineering Research Council of Canada Discovery Grant (FRN 04355) (to A. B. B.).
The atomic coordinates and structure factors (code 5A29) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PL
- polysaccharide lyase
- HG
- homogalacturonan
- ASR
- ancestral sequence reconstruction
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ML
- maximum likelihood
- CAPSO
- 3-(cyclohexylamino)-2-hydroxy-1-propanesulfonic acid
- CAPS
- N-cyclohexyl-3-aminopropanesulfonic acid.
References
- 1. Lombard V., Bernard T., Rancurel C., Brumer H., Coutinho P. M., Henrissat B. (2010) A hierarchical classification of polysaccharide lyases for glycogenomics. Biochem. J. 432, 437–444 [DOI] [PubMed] [Google Scholar]
- 2. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Abbott D. W., Boraston A. B. (2007) A family 2 pectate lyase displays a rare fold and transition metal-assisted β-elimination. J. Biol. Chem. 282, 35328–35336 [DOI] [PubMed] [Google Scholar]
- 4. Abbott D. W., Gilbert H. J., Boraston A. B. (2010) The active site of oligogalacturonate lyase provides unique insights into cytoplasmic oligogalacturonate β-elimination. J. Biol. Chem. 285, 39029–39038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Charnock S. J., Brown I. E., Turkenburg J. P., Black G. W., Davies G. J. (2002) Convergent evolution sheds light on the anti-β-elimination mechanism common to family 1 and 10 polysaccharide lyases. Proc. Natl. Acad. Sci. U.S.A. 99, 12067–12072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Caffall K. H., Mohnen D. (2009) The structure, function, and biosynthesis of plant cell wall pectic polysaccharides. Carbohydr. Res. 344, 1879–1900 [DOI] [PubMed] [Google Scholar]
- 7. Abbott D. W., Thomas D., Pluvinage B., Boraston A. B. (2013) An ancestral member of the polysaccharide lyase family 2 displays endolytic activity and magnesium dependence. Appl. Biochem. Biotechnol. 171, 1911–1923 [DOI] [PubMed] [Google Scholar]
- 8. Shevchik V. E., Condemine G., Robert-Baudouy J., Hugouvieux-Cotte-Pattat N. (1999) The exopolygalacturonate lyase PelW and the oligogalacturonate lyase Ogl, two cytoplasmic enzymes of pectin catabolism in Erwinia chrysanthemi 3937. J. Bacteriol. 181, 3912–3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harding M. M. (2001) Geometry of metal-ligand interactions in proteins. Acta Crystallogr. D Biol. Crystallogr. 57, 401–411 [DOI] [PubMed] [Google Scholar]
- 10. Gangola P., Rosen B. P. (1987) Maintenance of intracellular calcium in Escherichia coli. J. Biol. Chem. 262, 12570–12574 [PubMed] [Google Scholar]
- 11. Yanyi C., Shenghui X., Yubin Z., Jie Y. J. (2010) Calciomics: prediction and analysis of EF-hand calcium binding proteins by protein engineering. Sci. China Chem. 53, 52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ferguson A. D., Deisenhofer J. (2004) Metal import through microbial membranes. Cell 116, 15–24 [DOI] [PubMed] [Google Scholar]
- 13. Garron M. L., Cygler M. (2010) Structural and mechanistic classification of uronic acid-containing polysaccharide lyases. Glycobiology 20, 1547–1573 [DOI] [PubMed] [Google Scholar]
- 14. Hepler P. K., Winship L. J. (2010) Calcium at the cell wall-cytoplast interface. J. Integr. Plant Biol. 52, 147–160 [DOI] [PubMed] [Google Scholar]
- 15. Boraston A. B., Abbott D. W. (2012) Structure of a pectin methylesterase from Yersinia enterocolitica. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 129–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abbott D. W., Boraston A. B. (2007) The structural basis for exopolygalacturonase activity in a family 28 glycoside hydrolase. J. Mol. Biol. 368, 1215–1222 [DOI] [PubMed] [Google Scholar]
- 17. Abbott D. W., Boraston A. B. (2008) Structural biology of pectin degradation by Enterobacteriaceae. Microbiol. Mol. Biol. Rev. 72, 301–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Abbott D. W., Hrynuik S., Boraston A. B. (2007) Identification and characterization of a novel periplasmic polygalacturonic acid binding protein from Yersinia enterolitica. J. Mol. Biol. 367, 1023–1033 [DOI] [PubMed] [Google Scholar]
- 19. Oliver J. D. (2013) Vibrio vulnificus: death on the half shell: a personal journey with the pathogen and its ecology. Microb. Ecol. 65, 793–799 [DOI] [PubMed] [Google Scholar]
- 20. Rodionov D. A., Gelfand M. S., Hugouvieux-Cotte-Pattat N. (2004) Comparative genomics of the KdgR regulon in Erwinia chrysanthemi 3937 and other γ-proteobacteria. Microbiology 150, 3571–3590 [DOI] [PubMed] [Google Scholar]
- 21. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cowtan K. (2012) Completion of autobuilt protein models using a database of protein fragments. Acta Crystallogr. D Biol. Crystallogr. 68, 328–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., Murshudov G. N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 60, 2184–2195 [DOI] [PubMed] [Google Scholar]
- 26. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lovell S. C., Davis I. W., Arendall W. B., 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., Richardson D. C. (2003) Structure validation by Cα geometry: φ,ψ and Cβ deviation. Proteins 50, 437–450 [DOI] [PubMed] [Google Scholar]
- 28. Ashkenazy H., Erez E., Martz E., Pupko T., Ben-Tal N. (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Talavera G., Castresana J. (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577 [DOI] [PubMed] [Google Scholar]
- 30. Guindon S., Dufayard J. F., Lefort V., Anisimova M., Hordijk W., Gascuel O. (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 [DOI] [PubMed] [Google Scholar]
- 31. Löytynoja A. (2014) Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 1079, 155–170 [DOI] [PubMed] [Google Scholar]
- 32. Zwickl D. J. (2006) Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. Ph.D. thesis, University of Texas, Austin, TX [Google Scholar]
- 33. Darriba D., Taboada G. L., Doallo R., Posada D. (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ronquist F., Huelsenbeck J. P. (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 [DOI] [PubMed] [Google Scholar]
- 35. Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C., Thierer T., Ashton B., Meintjes P., Drummond A. (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Darriba D., Taboada G. L., Doallo R., Posada D. (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krissinel E., Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 38. Ochiai A., Itoh T., Mikami B., Hashimoto W., Murata K. (2009) Structural determinants responsible for substrate recognition and mode of action in family 11 polysaccharide lyases. J. Biol. Chem. 284, 10181–10189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Davies G. J., Wilson K. S., Henrissat B. (1997) Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 321, 557–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hutter C. A., Lehner R., Wirth C., Condemine G., Peneff C., Schirmer T. (2014) Structure of the oligogalacturonate-specific KdgM porin. Acta Crystallogr. D Biol. Crystallogr. 70, 1770–1778 [DOI] [PubMed] [Google Scholar]
- 41. Blot N., Berrier C., Hugouvieux-Cotte-Pattat N., Ghazi A., Condemine G. (2002) The oligogalacturonate-specific porin KdgM of Erwinia chrysanthemi belongs to a new porin family. J. Biol. Chem. 277, 7936–7944 [DOI] [PubMed] [Google Scholar]
- 42. Aspeborg H., Coutinho P. M., Wang Y., Brumer H., 3rd, Henrissat B. (2012) Evolution, substrate specificity and subfamily classification of glycoside hydrolase family 5 (GH5). BMC Evol. Biol. 12, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. St John F. J., González J. M., Pozharski E. (2010) Consolidation of glycosyl hydrolase family 30: a dual domain 4/7 hydrolase family consisting of two structurally distinct groups. FEBS Lett. 584, 4435–4441 [DOI] [PubMed] [Google Scholar]
- 44. Stam M. R., Danchin E. G., Rancurel C., Coutinho P. M., Henrissat B. (2006) Dividing the large glycoside hydrolase family 13 into subfamilies: towards improved functional annotations of α-amylase-related proteins. Protein Eng. Des. Sel. 19, 555–562 [DOI] [PubMed] [Google Scholar]
- 45. Abbott D. W., van Bueren A. L. (2014) Using structure to inform carbohydrate binding module function. Curr. Opin. Struct. Biol. 28, 32–40 [DOI] [PubMed] [Google Scholar]
- 46. Terrapon N., Lombard V., Gilbert H. J., Henrissat B. (2015) Automatic prediction of polysaccharide utilization loci in Bacteroidetes species. Bioinformatics 31, 647–655 [DOI] [PubMed] [Google Scholar]
- 47. Yin Y., Mao X., Yang J., Chen X., Mao F., Xu Y. (2012) dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 40, W445–W451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Horton R. M., Hunt H. D., Ho S. N., Pullen J. K., Pease L. R. (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77, 61–68 [DOI] [PubMed] [Google Scholar]