

Background: The Burkholderia cenocepacia lipid A is hypoacylated.
Results: Aminoarabinose residues in lipid A contribute to Burkholderia lipid A binding to the TLR4·MD-2 complex.
Conclusion: A novel mode of Burkholderia lipopolysaccharide-TLR4·MD-2 interactions promotes inflammation.
Significance: Modifications of the lipid A structure enhance proinflammatory responses of hypoacylated lipopolysaccharide.
Keywords: cystic fibrosis, Gram-negative bacteria, innate immunity, lipopolysaccharide (LPS), molecular modeling, Burkholderia, TLR4/MD-2
Abstract
Lung infection by Burkholderia species, in particular Burkholderia cenocepacia, accelerates tissue damage and increases post-lung transplant mortality in cystic fibrosis patients. Host-microbe interplay largely depends on interactions between pathogen-specific molecules and innate immune receptors such as Toll-like receptor 4 (TLR4), which recognizes the lipid A moiety of the bacterial lipopolysaccharide (LPS). The human TLR4·myeloid differentiation factor 2 (MD-2) LPS receptor complex is strongly activated by hexa-acylated lipid A and poorly activated by underacylated lipid A. Here, we report that B. cenocepacia LPS strongly activates human TLR4·MD-2 despite its lipid A having only five acyl chains. Furthermore, we show that aminoarabinose residues in lipid A contribute to TLR4-lipid A interactions, and experiments in a mouse model of LPS-induced endotoxic shock confirmed the proinflammatory potential of B. cenocepacia penta-acylated lipid A. Molecular modeling combined with mutagenesis of TLR4-MD-2 interactive surfaces suggests that longer acyl chains and the aminoarabinose residues in the B. cenocepacia lipid A allow exposure of the fifth acyl chain on the surface of MD-2 enabling interactions with TLR4 and its dimerization. Our results provide a molecular model for activation of the human TLR4·MD-2 complex by penta-acylated lipid A explaining the ability of hypoacylated B. cenocepacia LPS to promote proinflammatory responses associated with the severe pathogenicity of this opportunistic bacterium.
Introduction
A central theme in innate immunity involves recognition of conserved microbial molecules (pathogen-associated molecular patterns) by surface receptors expressed on phagocytic cells (1, 2). Pathogen recognition triggers cell signaling cascades, leading to activation of transcription factors such as nuclear factor-κB (NF-κB) and interferon regulatory factors, which in turn stimulate production of inflammatory cytokines (e.g. TNF-α, IL-1β, and type I interferons) (3). Lipopolysaccharide (LPS), the major component of the Gram-negative bacterial outer membrane, elicits potent innate immune responses through interactions with a receptor complex composed of Toll-like receptor 4 (TLR4)5 and myeloid differentiation factor 2 (MD-2) (4). Depending on the amount and chemical nature of LPS released from the pathogen, TLR4·MD-2 recognition stimulates a protective immune response or leads to uncontrolled inflammation associated with high mortality (5, 6). LPS is a complex glycolipid consisting of three distinct domains (see Fig. 1) (7): lipid A, core oligosaccharide, and in many bacteria a repeating polysaccharide moiety known as the O-antigen (8–10). Lipid A, composed of an acylated glucosamine disaccharide backbone, is the LPS moiety recognized by the TLR4·MD-2 receptor complex. Lipid A bioactivity depends on its chemical structure. The number and distribution of acyl chains and the presence of the phosphate groups in the diglucosamine backbone determine the agonistic and antagonistic activities of lipid A (11–19). Most enteric bacteria such as Escherichia coli produce hexa-acylated bisphosphorylated lipid A, which has the highest cytokine-inducing capacity in mammals. In contrast, tetra-acylated lipid A (such as lipid IVA) and most penta-acylated lipid A forms lack activity on human cells (19, 20).
FIGURE 1.

Structure of B. cenocepacia LPS inner core and lipid A. B. cenocepacia lipid A is heterogeneous, being composed of a mixture of penta- and tetra-acylated species (30, 40). Lipid A fatty acids are two 3-(R)-hydroxyhexadecanoic acids, two 3-(R)-hydroxytetradecanoic acids, and one tetradecanoic acid (30, 40). The dotted lines indicate the non-stoichiometric substitution.
Lipid A interacts with a hydrophobic pocket formed by two anti-parallel β-sheets of MD-2 mediating the dimerization and activation of the TLR4·MD-2 complex (19, 21). The MD-2 hydrophobic pocket can accommodate up to five acyl chains. For agonistic hexa-acylated E. coli lipid A, five acyl chains are buried within the pocket, whereas the sixth chain lies on a channel of the MD-2 surface, building a hydrophobic region and the dimerization interface required for interaction with the TLR4 partner (referred to here as TLR4*). This arrangement enables hydrophobic interactions bridging the TLR4·MD-2·LPS heterodimer and promoting the juxtaposition of the intracellular domains, leading to activation of signal transduction (18, 22). By contrast, antagonist lipid IVA binds to human MD-2 with all four acyl chains completely buried in the hydrophobic pocket precluding dimerization and subsequent activation (23). Similarly, many penta-acylated lipid As act as an antagonist of human TLR4·MD-2 (24, 25). In contrast, lipid IVA can activate the murine TLR4·MD-2 complex (26).
Bacteria regulate the degree of lipid A acylation in response to environmental conditions. For example, Pseudomonas aeruginosa strains possess penta-acylated lipid A but can produce hexa-acylated lipid A during infection in cystic fibrosis (CF) patients (27), gaining the ability to elicit stronger inflammation. Intriguingly, the non-CF pathogen Porphyromonas gingivalis produces a high heterogeneous LPS lipid A whose penta-acylated isoform potently activates the NF-κB pathway in human gingival fibroblasts in a similar manner to the E. coli LPS (28). Therefore, the molecular mechanisms underlying TLR4 dimerization and activation cannot always be predicted based on the E. coli lipid A-TLR4·MD-2 paradigm.
Burkholderia cenocepacia is an opportunistic Gram-negative bacterium causing serious infections in CF patients (29). Understanding how B. cenocepacia infection elicits inflammation is crucial to finding new ways to improve treatment of infected CF patients. The structure of the B. cenocepacia LPS (LPSBC) was described previously (30). The lipid A moiety of LPSBC consists of a mixture of penta-acylated and tetra-acylated, bisphosphorylated diglucosamine backbone with two 4-amino-4-deoxy-l-arabinose (l-Ara4N) residues linked by phosphodiester linkages (see Fig. 1) (30). The acyl chains are made of 3-(R)-hydroxyhexadecanoic acid C16:0 (3-OH), 3-(R)-hydroxytetradecanoic acid C14:0 (3-OH), and hydroxytetradecanoic acid C14:0 (see Fig. 1) (30). The l-Ara4N decoration of lipid A is essential for B. cenocepacia viability (31) and represents the major determinant of resistance to cationic antimicrobial peptides (30, 31). The structural features of LPSBC suggest that the molecule would be a very poor TLR4·MD-2 complex agonist compared with the prototypic enterobacterial LPS. In contrast, several studies show that LPSBC as well as biological and synthetic lipid A analogues is highly proinflammatory (32–36). Therefore, the current model for binding/activation of human TLR4·MD-2 complex cannot explain why LPSBC is proinflammatory. In this work, we investigated the molecular basis of the mechanism of LPSBC recognition and report for the first time that acyl chains longer than those in enterobacterial lipid A together with the l-Ara4N residues allow B. cenocepacia lipid A (LABC) to fit into the binding pocket of MD-2 in a manner that promotes TLR4 dimerization, leading to activation of inflammatory responses in cellular and animal models.
Experimental Procedures
LPS Extraction and Purification
LPS was prepared from B. cenocepacia strains MH71 and MH75 (30). Both strains are isogenic derivatives of the K56-2 clinical isolate and contain a deletion of the wbiF gene that eliminates O-antigen production. MH75 also has a deletion removing genes involved in UDP-l-Ara4N synthesis and therefore cannot produce LPS with l-Ara4N residues (LPSBCΔAra). Both MH71 and MH75 also carry a suppressor mutation in the LPS transport gene lptG that allows for the transport of LPS devoid of l-Ara4N to the bacterial outer membrane (30). The P. aeruginosa RP73 clinical isolate was obtained from a chronically infected CF patient and kindly provided by Prof. Burkhard Tümmler (Klinische Forschergruppe, Medizinische Hochschule Hannover, Hannover, Germany) (37, 38). Strains were plated on trypticase soy agar plates and cultured in trypticase soy broth at 37 °C. For large scale LPS purification, bacteria were treated with hot phenol/water (39). After extensive dialysis against distilled water, the extracted phases were subjected to enzymatic digestions to remove nucleic acids and protein contaminants. Water and phenol fractions were analyzed by 13.5% SDS-PAGE and silver staining (40). The LPS fraction was exclusively found in the water phase.
Isolation of Lipid A
Lipid A was obtained by hydrolysis of the LPS with 100 mm sodium acetate buffer, pH 4.4 (100 °C for 3 h). The solution was extracted three times with CHCl3/MeOH/H2O (100:100:30, v/v/v) and centrifuged (4 °C at 5000 × g for 15 min). The organic phase contained lipid A, and the water phase contained the core oligosaccharide. The former was further purified through several washes with distilled water and then lyophilized. The lipid A and core oligosaccharide structures of strains K56-2, MH71, and MH75 were characterized elsewhere (Fig. 1) (30, 41).
Mouse Endotoxic Studies
Animal studies were conducted according to protocols approved by the San Raffaele Scientific Institute (Milan, Italy) Institutional Animal Care and Use Committee and adhered strictly to the Italian Ministry of Health guidelines for the use and care of experimental animals. Efforts were made to minimize the number of animals used and their suffering. C57BL/6 mice (20–22 g; male; Charles River) were challenged via intraperitoneal injection with 300 μg/mouse LPS from E. coli and P. aeruginosa RP73, LPSBC, and LPSBCΔAra. Control mice were challenged with sterile saline solution. Five hours after treatment, mice were sacrificed by CO2 administration, and blood was collected from heart puncture. The blood clot was left at room temperature for 15–30 min. The clot was removed by centrifugation at 2000 × g for 10 min at 4 °C. Sera were collected and stored at −20 °C. TNF-α concentration in sera was determined by ELISA (R&D Systems) according to the manufacturer's instructions using antibody pairs and recombinant standards from R&D Systems.
Human Embryonic Kidney (HEK) 293 Cell Activation and Luciferase Reporter Assays
Expression plasmids containing sequences of human TLR4 and MD-2 and the pELAM-1 firefly luciferase plasmid were a gift from Dr. C. Kirschning (Technical University of Munich, Munich, Germany). Expression plasmid containing sequence of mouse TLR4 was purchased from InvivoGen. Expression plasmid for mouse MD-2 was a gift from Dr. Y. Nagai (University of Tokyo, Tokyo, Japan). The Renilla luciferase phRL-TK plasmid was purchased from Promega. Recombinant MD-2 genes were cloned into pEF-BOS with FLAG and His tags on the C terminus. Recombinant TLR4 genes were cloned into pUNO with a C-terminal HA tag. Transfection reagent JetPEI was purchased from Polyplus Transfection (France) and was used according to the manufacturer's instructions. The HEK293 cells were provided by Dr. J. Chow (Eisai Research Institute, Andover, MA) and grown in DMEM supplemented with 10% FBS. The MD-2 mutants were made using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. All plasmids were sequenced to confirm the appropriate mutations. For the NF-κB-luciferase reporter assay, HEK293 cells were seeded in 96-well plates at 3 × 104 cells/well and incubated overnight in a humidified atmosphere (5% CO2) at 37 °C. The next day when cells were 60–80% confluent, they were co-transfected with the plasmids for MD-2 (10 ng), TLR4 (1 ng), NF-κB-dependent luciferase (50 ng), and constitutive Renilla luciferase (10 ng) using JetPEI transfection reagent (all amounts are in ng/well). Cells were stimulated 6 h after transfection with endotoxin preparations. Cells were lysed after 16 h of stimulation in 1× reporter assay lysis buffer (Promega) and analyzed for reporter gene activities using the Dual-Luciferase reporter assay system. Relative luciferase units were calculated by normalizing the luciferase activity of each sample with the constitutive Renilla activity measured within the same sample.
Statistical Analysis
Results were expressed as mean ± S.D. from experiments done in triplicate. Statistical calculations and tests in vitro and in vivo were performed using Student's t test considering p ≤ 0.05 as the limit of statistical significance and a p value of 0.001 as extremely significant.
Molecular Modeling: Building and Geometry Optimization of LPSBC and LPSBCΔAra
Three-dimensional coordinates were built by Maestro (42). Molecular mechanics optimization (universal force field), semiempirical calculations (AM1), and density functional theory (B3LYP/6-31G*) were subsequently applied using Gaussian 03 (43). Ammonium and carboxylic acid groups were considered as ionized. Conformational analysis on DS1, DS2, and DS3 disaccharides was performed with MacroModel (44). The parameters were as follows: MM3* force field and water solvent dielectric constant of 1.0. The charges from the force field were used with an extended cutoff. The best conformers were selected, and from them, molecular dynamics simulations (MDSs) with implicit water and MM3* as the force field were performed using Schrödinger Maestro 9.3 and Impact 5.8 (42, 45) (MM3* force field; dielectric constant, 80.0; number of MDS steps, 100; time step (ps), 0.001).
The full three-dimensional structures of LABCΔAra, LABC, and LPSBC were built with Maestro using optimized disaccharide scaffolds obtained from the conformational analysis. Fatty acid chains, DS6, and aminoarabinoses were optimized separately using Gaussian 03 (B3LYP/6-31G*). The starting conformation for lipid A optimization was obtained from E. coli lipid A from Protein Data Bank code 3fxi. The full structure of LPSBC was built by putting together core region disaccharides and lipid A with five aliphatic chains. Final MDS of the geometry was performed with implicit water by means of Impact (MM3* force field; dielectric constant, 80.0; number of MDS steps, 100; time step (ps), 0.001). Three-dimensional structures of lipid A molecules for validation were extracted from their corresponding Protein Data Bank files (E. coli lipid A was obtained from Protein Data Bank code 3fxi, and Re chemotype of E. coli lipid A was obtained from Protein Data Bank code 3vq2), refined with the help of Maestro, and finally submitted to MDS with Impact (MM3* force field and implicit water).
Molecular Modeling
For docking studies with MD-2 and TLR4 proteins, three-dimensional coordinates from MD-2 protein were obtained from Protein Data Bank codes 3fxi (human MD-2) and 3vq2 (murine MD-2) and refined and minimized with the Protein Preparation Wizard module of Maestro using the AMBER force field (46). The TLR4 structure was obtained from Protein Data Bank code 3fxi and treated following the same procedure after removing water molecules and any other non-standard residue.
Two different docking methodologies were used for docking studies of LABCΔAra and LABC on MD-2: AutoDock and AutoDock Vina. Although AutoDock is a widely used semiempirical docking method (47), the recently released AutoDock Vina combines empirical and knowledge-based scoring functions with good performance and reduced computing time (48). Each ligand was docked into human and murine MD-2 protein using AutoDock 4.2 (49) and separately using AutoDock Vina 1.1.2 (48). For human MD-2 (from Protein Data Bank code 3fxi), the Autogrid grid point spacing was set at 0.375 Å; center coordinates of the grid box were 29.00, −7.00, and 17.875 (x, y, z); and number of grid points in xyz was 41, 53, and 83. For murine MD-2 (from Protein Data Bank code 3vq2), the Autogrid grid point spacing was set at 0.375 Å and center coordinates of the grid box were −27.50, −15.50, and 22.00 (x, y, z), leading to 75 × 40 × 60 (x, y, z) grid points. The best result from each docking job with LABC was used as the starting geometry for subsequent docking calculations. Different combinations of allowed rotatable bonds were also considered for the ligands. Docking calculations with AutoDock were performed using a genetic algorithm (number of individuals in population, 150; maximum number of energy evaluations, 2,500,000–5,000,000; maximum number of generations, 27,000; number of top individuals to survive to next generation, 1; rate of gene mutation, 0.02; rate of crossover, 0.8; window size, 10; α parameter of Cauchy distribution, 0.0; β parameter of Cauchy distribution, 1.0). When docking LPSBC, flexible docking was also performed considering Asp-101, Glu-120, and Glu-122 as flexible residues. Docking calculations with AutoDock Vina were also performed. Coordinates and dimensions of grid boxes, starting geometries, and general methodology were the same as for AutoDock. When docking LPSBC, flexible docking was also performed considering Asp-101 as a flexible residue. Three-dimensional structures of the docked complexes were optimized by MDS with Impact (implicit water and AMBER* force field).
For docking experiments studies of the B. cenocepacia LPS core on human TLR4, we used AutoDock Vina 1.1.2. Torsional bonds from the ligand were allowed to rotate. Ammonium groups were considered as ionized. The grid box was built on TLR4 from Protein Data Bank code 3fxi with grid center at 26.00, −22.00, and 11.50 (x, y, z), number of grid points of 41 × 41 × 43 (x, y, z); and spacing of 0.375 Å. Final optimization of the three-dimensional structure of the docked complex was carried out by MDS with Impact (implicit water and AMBER* force field).
The full complex of human TLR4·MD-2 with B. cenocepacia LPS core was built by merging docked MD-2·LPS core complex with docked LPS inner core·TLR4 using Protein Data Bank code 3fxi as a template. The resulting structure was optimized by MDS with implicit water (AMBER* force field). Coupling of two TLR4·MD-2·LPSBC complexes was finally performed also using Protein Data Bank code 3fxi as a template. The full three-dimensional structure of the dimer complex was submitted to MDS with implicit water and AMBER* force field (charges from, force field; cutoff, extended; method, Polak-Ribière conjugate gradient; maximum iterations, 500; converge on, gradient; convergence threshold, 0.05). This final structure for the TLR4·MD-2·LPSBC complex was used to generate the mutant TLR4·MD-2·LPSBC complex (D294A, R322A, S415A*, and S416A*) by changing the corresponding side chains to Ala side chain. The resulting structure was submitted to MDS with implicit water and AMBER* force field. Energy analysis was performed by means of the MM-ISMSA method (50).
Results
LPS and Lipid A from B. cenocepacia Activate the TLR4·MD-2 Complex and Have Proinflammatory Activity in Vivo
TLR4·MD-2 activation by LPSBC and LABC was examined using the NF-κB reporter luciferase assay in transiently transfected HEK293 and HEK293T cells co-expressing murine (mMD-2) or human MD-2 (hMD-2) and murine (mTLR4) or human TLR4 (hTLR4). Initial results revealed that LPSBC and LABC activate both human and mouse TLR4·MD-2 complexes (Figs. 2 and 3, A and B). Because LABC possesses longer acyl chains than those in E. coli lipid A and constitutively carries one or two l-Ara4N residues, to further understand the structure-function relationship of LPSBC, we utilized LPS from a mutant strain lacking the ability to produce l-Ara4N (LPSBCΔAra). NF-κB reporter luciferase assays demonstrated that at 5 ng/ml LPSBC and LPSBCΔAra induced activation of TLR4·MD-2 complexes (Fig. 2). Similar results were obtained in dose-response experiments with purified LABC and LABCΔAra (at 5, 10, and 50 ng/ml) in cells co-expressing mTLR4·mMD-2 and hTLR4·hMD-2 (Fig. 3, A and B, respectively), which also demonstrated consistent agonistic activity at the lowest concentration of 5 ng/ml. However, NF-κB activation by mouse (Fig. 3A) and human (Fig. 3B) complexes was significantly lower with LABCΔAra, suggesting that the l-Ara4N modification of the lipid A plays a role in bioactivity of the LPS molecule.
FIGURE 2.

LPSBC activates the murine and human TLR4·MD-2 complexes. NF-κB activation upon stimulation of HEK293 mTLR4·mMD-2 and hTLR4·hMD-2 after 6 h with 5 ng/ml LPSBC and LPSBCΔAra. Stimulation for 6 h with E. coli LPS was used as the control. The data are pooled from three independent experiments done in triplicate. Bars indicate S.D.; significance was calculated in comparison with stimulation with E. coli LPS (*, p < 0.05; **, p < 0.01). Curly brackets indicate significance calculated comparing LPSBC and LPSBCΔAra (*, p < 0.05; **, p < 0.01). NS, no stimulation.
FIGURE 3.

Lipid A activation of murine and human TLR4·MD-2 complexes. A, NF-κB activation upon stimulation of HEK293 mTLR4·mMD-2 after 6 h with LABC and LABCΔAra. Stimulations for 6 h with E. coli LPS, hexa-acylated lipid A, and lipid IVA were used as controls. The data are pooled from three independent experiments done in triplicate. Bars indicate S.D.; significance was calculated in comparison with stimulation with hexa-acylated E. coli lipid A (*, p < 0.05; **, p < 0.01; ***, p < 0.001); curly brackets indicate significance calculated comparing LPSBC and LPSBCΔAra (**p < 0.01). B, NF-κB activation upon stimulation of HEK293 hTLR4·hMD-2 after 6 h with LABC and LABCΔAra. Stimulations for 6 h with E. coli LPS, hexa-acylated lipid A, and lipid IVA were used as controls. The data are pooled from three independent experiments done in triplicate. Bars indicate S.D.; significance was calculated in comparison with stimulation with hexa-acylated E. coli lipid A (*, p < 0.05; ***, p < 0.001); curly brackets indicate significance calculated comparing LPSBC and LPSBCΔAra (*, p < 0.05). NS, no stimulation.
Comparisons with NF-κB activation elicited by synthetic E. coli-type hexa-acylated lipid A revealed significantly less activation by LPSBC and LABCΔAra (all at 50 ng/ml) (Fig. 3, A and B). Lower activation by LPSBC and LPSBCΔAra could depend on weaker binding to MD-2 or less efficient activation of TLR4. Therefore, we examined whether LPSBC and LPSBCΔAra could interfere with TLR4 signaling elicited by the E. coli LPS by a competition assay in which HEK293 cells transfected with hTLR4·hMD-2 were preincubated with LPSBC or LPSBCΔAra for 1 h and then restimulated with E. coli LPS for 4 h (Fig. 4). Preincubation of cells with 10 ng/ml LPSBC followed by stimulation by 1 ng/ml E. coli LPS revealed weaker NF-κB activation than that with E. coli LPS alone (p < 0.05; Fig. 4). Interestingly, a 2-fold reduction in NF-κB activation was evident in the case of preincubation with 10 ng/ml LPSBCΔAra (p < 0.01; Fig. 4). Furthermore, a significant difference was found in activation by 10 ng/ml E. coli LPS alone compared with that obtained after preincubation of cells with 100 ng/ml LPSBC and LPSBCΔAra (p < 0.01; Fig. 4). In contrast, this inhibitory effect was reduced by increasing the concentration of the preincubated LPSBC and LPSBCΔAra (100 ng/ml) added to the 1 ng/ml E. coli LPS; such effect is probably due to the contribution of a weak activation of MD-2·TLR4 by LPSBC and LPSBCΔAra. Collectively, these results suggest that LPSBCΔAra efficiently binds to TLR4·MD-2 but is a weaker agonist compared with the E. coli LPS and LPSBC.
FIGURE 4.

B. cenocepacia LPSBCΔAra effects on the E. coli LPS agonist activity. The potential antagonist activity of LPSBC and LPSBCΔAra on hexa-acylated E. coli LPS was assayed. NF-κB activation upon stimulation of HEK293 hTLR4 after 1 h with LPSBC (1, 10, and 100 ng/ml) and LPSBCΔAra (1, 10, and 100 ng/ml) followed by exposure to E. coli LPS (1 and 10 ng/ml) for 4 h is shown. The data are pooled from three independent experiments done in triplicate. Bars indicate S.D.; significance was calculated in comparison with stimulation with E. coli LPS (1 and 10 ng/ml) (*, p < 0.05; **, p < 0.01). NS, no stimulation.
The proinflammatory activity of LPS from B. cenocepacia was assessed using an established in vivo model of endotoxic shock. C57Bl/6 mice were challenged by intraperitoneal injection of LPS from E. coli and P. aeruginosa RP73 (a CF strain expressing penta- and tetra-acylated lipid A (37)), LPSBC, and LPSBCΔAra. After 5 h of treatment, TNF-α levels in sera were measured to evaluate proinflammatory and endotoxic potential. Compared with the saline solution control, penta-acylated LPSBC induced robust TNF-α production (LPSBC versus control, p < 0.01; Fig. 5), whereas LPSBCΔAra induced 3-fold less systemic release of TNF-α in sera (LPSBCΔAra versus LPSBC, p < 0.01; Fig. 5), suggesting a role for l-Ara4N residues in the endotoxic LPS response in agreement with the in vitro results (Fig. 2).
FIGURE 5.

Proinflammatory and endotoxic potential of LPSBC in C57Bl/6 mice. From three to five mice per group were challenged via intraperitoneal injection with 300 μg/mouse LPS from E. coli and P. aeruginosa RP73, LPSBC, and LPSBCΔAra. TNF-α levels in sera were quantified after 5 h of treatment. Treatment with sterile saline solution was used as control (Ctrl). The data are pooled from two independent experiments. Bars indicate mean ± S.D. Statistical analysis was made for pair wise comparisons (*, p < 0.05; **, p < 0.01).
Identification of MD-2 Amino Acid Residues Involved in the Interaction with LPSBC
To further investigate the molecular recognition of LPSBC and LPSBCΔAra by TLR4·MD-2, we constructed targeted hMD-2 and mMD-2 mutants that were co-expressed with hTLR4 or mTLR4 in transfected HEK293 cells. We focused on residues at the dimerization interface of the MD-2 protein (22, 51). In particular, we examined Val-82 located in proximity to the pocket entrance at a loop between β-strands 5 and 6. This region contains several conserved, solvent-exposed hydrophobic residues that contribute to crucial hydrophobic interactions in the activated receptor complex (22). Val-82 was replaced by phenylalanine (V82F) to augment the hydrophobic interactions of the MD-2·lipid A complex with TLR4. In comparison with HEK293 cells transfected with parental MD-2, hTLR4·hMD-2 V82F led to increased activation upon stimulation with LPSBC and LPSBCΔAra, whereas E. coli LPS activation was not significantly modified (Fig. 6). This agrees with the notion that hexa-acylated lipid A provides stronger hydrophobic interactions with TLR4, and therefore increased hydrophobicity at position 82 augments the activation by penta-acylated LPS. No significant differences were detected between stimulation with LPSBC and LABCΔAra on hTLR4·hMD-2 V82F. Hydrophobic interactions between mTLR4·mMD-2 and LA are expected to be similar to those involving the hTLR4·hMD-2 complex because nearly all the hydrophobic residues are conserved (51). However, the electrostatic interactions that contribute to receptor selectivity differ between mMD-2 and hMD-2 proteins. Particularly, hMD-2 has a positively charged lysine at position 122 near the pocket entrance, whereas mMD-2 has a negatively charged glutamic acid (51). Moreover, hMD-2 has a positively charged lysine at position 125, whereas mMD-2 has a hydrophobic leucine (51). In the complex, lysine residues 122 and 125 are close to the disaccharide backbone of LA (52) and are important for the species-specific differences in the recognition of lipid IVA between hMD-2 and mMD-2. We therefore introduced amino acid replacements at positions 122 and 125 in both MD-2 homologues to test whether the substitutions could affect LA binding and influence the selectivity of TLR4 activation by the LABC variants. The E122K replacement in mMD-2 introducing the equivalent lysine of hMD-2 had little or no effect on the activation induced by LABC, LABCΔAra, or the hexa-acylated lipid A. In contrast, this replacement significantly decreased activation by lipid IVA (p < 0.001; Fig. 7) likely due to removal of the repulsive forces toward its bisphosphorylated disaccharide backbone. These repulsive interactions have been proposed to be important for positioning of lipid IVA in an appropriate orientation for TLR4 dimerization (52). Thus, the unchanged LABC/LABCΔAra responsiveness suggests that the single residue at position 122 of the murine MD-2 protein is not critical for LABC/LABCΔAra signaling.
FIGURE 6.
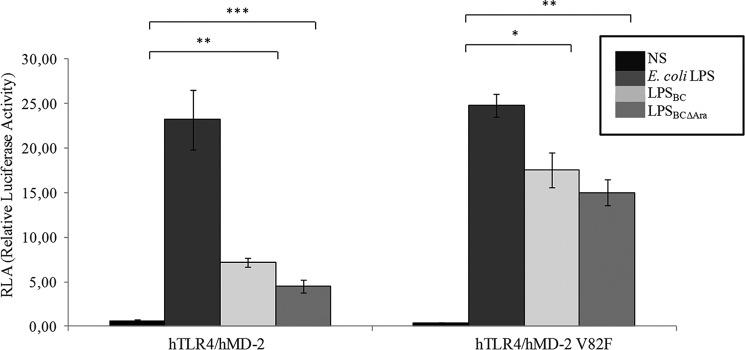
B. cenocepacia LPS acylation pattern is responsible for the NF-κB activation. NF-κB activation upon stimulation of HEK293 hTLR4·hMD-2 after 6 h with LPSBC and LPSBCΔAra is shown. Stimulation for 6 h with E. coli LPS was used as the control. The same protocol was used to stimulate HEK293 hTLR4·hMD-2 V82F cells. The data are pooled from three independent experiments done in triplicate. Bars indicate S.D.; significance was calculated in comparison with E. coli LPS (*, p < 0.05; **, p < 0.01; ***, p < 0.001). NS, no stimulation.
FIGURE 7.

Lys-122 and Lys-125 in the B. cenocepacia LPS signaling on murine and human TLR4·MD-2 complexes. An NF-κB-luciferase reporter assay stimulating HEK293 hTLR4·hMD-2, HEK293 mTLR4·mMD-2, and mTLR4·hMD-2 after 6 h with LPSBC and LPSBCΔAra was carried out. Stimulations for 6 h with E. coli LPS and with lipid IVA were used as controls. The same protocol was used to stimulate HEK293 mTLR4·mMD-2 E1222K and HEK293 mTLR4·mMD-2 E122K/L125K cells. The data are pooled from two independent experiments done in triplicate. Bars indicate S.D.; statistical analysis was calculated for pairwise comparisons (*, p < 0.05; ***, p < 0.001). NS, no stimulation.
Stimulation of cells expressing the double E122K/L125K mMD-2 replacement (Fig. 7) showed substantially increased activity of the mutant MD-2 regardless of the LPS variant (hexa-acylated lipid A versus hexa-acylated lipid A, p < 0.05; LABC versus LABC, p < 0.001; LABCΔAra versus LABCΔAra, p < 0.001, respectively, on mTLR4·mMD-2 and mTLR4·mMD-2 E122K L125K; Fig. 7) except with lipid IVA whose activity was strongly impaired (p < 0.001; Fig. 7). Conversely, hMD-2 in combination with mTLR4 exhibited significantly stronger activation by LPSBCΔAra (p < 0.05; Fig. 7), demonstrating that the l-Ara4N affects interaction with the TLR4 ectodomain. Moreover, the hMD-2 K122E mutant had an additive effect in combination with mTLR4 on the activation induced by LABCΔAra, showing an increment of the NF-κB activity (LABCΔAra mTLR4·hMD-2 K122E versus LABCΔAra hTLR4·hMD-2, p < 0.001; LABCΔAra mTLR4·hMD-2 K122E versus LABCΔAra mTLR4·hMD-2, p < 0.05; Fig. 7). This probably reflects the presence of repulsive forces toward the phosphate groups of the lipid A that might facilitate the orientation toward the Cys-95/Cys-105 loop (52, 53).
Together, these experiments using hMD-2 and mMD-2 mutants support the notion that the peculiar structure of the LABC plays a key role in its interaction with MD-2 as well as in the dimerization process. Our data suggest that the l-Ara4N molecules on the diglucosamine backbone influence the binding of lipid A to the TLR4·MD-2 complex, although their absence is not sufficient to impair signaling.
Molecular Modeling of LPSBC Binding to TLR4·MD-2
Our previous results suggested that different MD-2 binding affinities drive the agonist activity of LPS. To provide a model for the interactions of LPSBC and LPSBCΔAra, docking calculations were undertaken, and the two ligands were docked into hMD-2 and mMD-2 proteins. Validation of the docking protocols was performed with AutoDock and AutoDock Vina by docking E. coli lipid A into the MD-2 protein from their corresponding crystallographic structures (Protein Data Bank codes 3fxi (human) and 3vq2 (murine)). Values of root mean square deviation and predicted free energy of binding values indicated an excellent performance of both programs in predicting the crystallographic binding pose for both hMD-2 and mMD-2 proteins (Table 1).
TABLE 1.
Theoretical free energy of binding for the docking calculations with AutoDock 4.2 and AutoDock Vina (in bold)
Energy values are in kcal mol−1. Root mean square deviation (r.m.s.d.) values are in Å.
| Human MD-2 | Murine MD-2 | |
|---|---|---|
| LABC | −20.02/−25.30 | −17.73/−23.60 |
| LABCΔAra | −18.22/−18.60 | −13.09/−17.20 |
| E. coli lipid A | −24.41/−28.3 (r.m.s.d. = 0.282/0.599) | −24.73/−27.9 (r.m.s.d. = 0.647/0.394) |
LABC and LABCΔAra were predicted to bind both MD-2 homologues with binding poses that agreed with the crystallographic binding poses for E. coli lipid A (superimposition with E. coli lipid A in Figs. 8 and 9). Docking poses consisted of four fatty acid chains deeply immersed into the hydrophobic pocket of MD-2, establishing van der Waals and CH-π interactions with the side chains of most of the lipophilic residues of the MD-2 pocket, mainly consisting of aliphatic Leu, Ile, and Val residues; aromatic Phe (numbering 76, 104, 119, 121, 126, 147, and 151); and Tyr (numbering 102 and 131) residues. All docked poses were found to participate in hydrophobic interactions with most of these residues. Higher efficiency in establishing lipophilic interactions can be deduced given the length of these four fatty acid chains. The fifth chain is placed into the groove or channel defined by Phe-126, Leu-87, Val-82, and Arg-90. This channel has been identified in the x-ray structures of the complex of TLR4·MD-2 with E. coli lipid A (18) as the allocation site for one lipid chain, allowing completion of the hydrophobic interface required for dimerization with the second TLR4·MD-2·ligand partner (referred to here as TLR4*·MD-2*). This precludes formation of the activated TLR4·MD-2·ligand multimer. Also Phe-126 has been proposed as a switch controlling the agonist/antagonist conformation of MD-2 because Phe-126 mutation prevents dimerization and abolishes downstream signaling (18, 53).
FIGURE 8.

Predicted binding mode of the LPSBC core to hTLR4·MD-2. The computational model from docking followed by MDS is shown. LPSBC docked to MD-2 is shown in Corey-Pauling-Koltun colors. Superimposed fatty acids chains from E. coli lipid A are shown in different colors. Some representative residues from the MD-2 binding site are represented with carbon atoms in green.
FIGURE 9.

Predicted binding mode of the LPSBC core to hTLR4·MD-2. Details of LPSBC (Corey-Pauling-Koltun colors) docked to MD-2 are shown. Superimposed fatty acids chains from E. coli lipid A are shown with carbon atoms in blue.
Therefore, the model predicts that the greater length of the five fatty acid chains compared with E. coli lipid A places the fifth chain outside the MD-2 pocket, thus building the dimerization interface in the Phe-126, Leu-87, Val-82, and Arg-90 groove, analogously to the sixth fatty acid of E. coli lipid A, whereas the other four chains remain inside the MD-2 pocket, providing stabilizing interactions.
Furthermore, the disaccharide scaffolds of LABC and LABCΔAra together with the phosphate groups are predicted to be in the outer region and to establish electrostatic interactions with the polar residues that define the rim of the MD-2 pocket (Fig. 10). The higher predicted affinity for LPSBC could be explained in terms of additional anchorage points arising from the l-Ara4N residues. In particular, these l-Ara4N molecules can establish additional interactions with MD-2. The hydroxyl (OH) group from Tyr-102 establishes bridged H-bonding with the OH at position 2 of l-Ara4N and the OH from the acyl chain (Fig. 10), whereas OH at position 3 from the same l-Ara4N establishes a hydrogen bond with the Ser-118 OH group. In the case of l-Ara4N-2, the OH group from one of the acyl chains establishes a hydrogen bond with the ys-122 CO group, whereas OH-3 and ammonium groups, although of the H-bond distance range (4.5 Å), are well oriented toward carbonyl groups from Lys-122 and Gly-123 and may enable a putative H-bond networking at this site. These interactions could also be identified in the complex with mMD-2 (data not shown). This predicted binding mode of LABC could thus explain the unexpected significant level of activation in the biological assays. The lack of some lipophilic interactions arising from the absent sixth fatty acid chain in comparison with E. coli lipid A could also be counterbalanced by these extra polar interactions from the l-Ara4N residues.
FIGURE 10.

Predicted binding mode of the LPSBC core to hTLR4·MD-2. Detail of some H-bond interactions between the LPSBC inner core and TLR4·MD-2 involving Tyr-102 is shown. LPSBC is represented in Corey-Pauling-Koltun colors, and MD-2 protein is represented in green.
Molecular Model of the hTLR4·MD-2 dimer in Complex with LPSBC
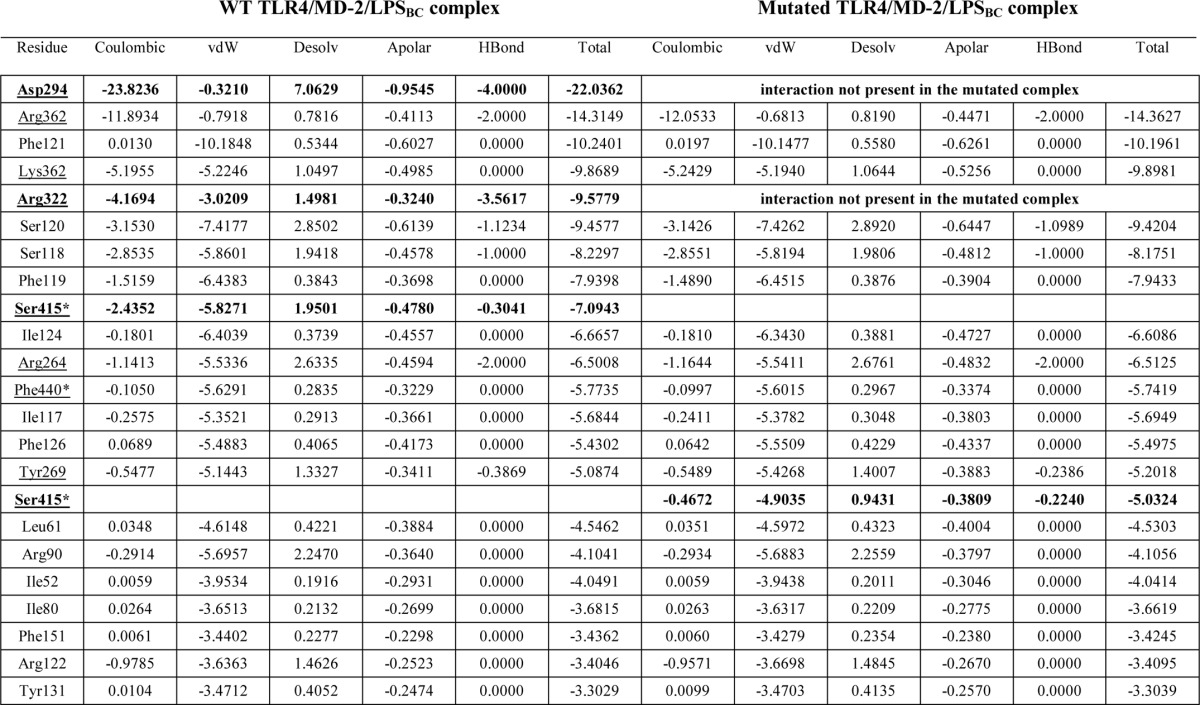
To develop a structural model for the TLR4·MD-2 dimer in complex with LPSBC, we first docked the inner core oligosaccharide moiety of LPSBC (Fig. 1) on hTLR4 using as a guide the region where E. coli LPS inner core binds. The best binding pose was selected (theoretical binding free energy of −2.8 kcal mol−1 by AutoDock Vina) taking also into account a proper orientation for the building of the full complex. The docked pose superimposed with E. coli LPS core from Protein Data Bank code 3fxi is shown in Fig. 11. The hTLR4·core complex was then assembled to the MD-2·LABC complex (best result from AutoDock Vina calculations), leading to a full hTLR4·MD-2·LPSBC complex. The receptor dimer was built using Protein Data Bank code 3fxi as a template. The full three-dimensional structure of the dimer complex (Fig. 12) was subjected to MDS, which did not show meaningful differences with the docked complexes (data not shown). The three-dimensional model of the full complex supports the key interactions for the molecular recognition of LPSBC that were described above and also confirms that the l-Ara4N residues (including the one present in the LPS core region; Fig. 1) play a fundamental role in the complex formation as they participate in H-bonding at the dimerization interfaces (Fig. 13). First, both ammonium groups from l-Ara4N-2 and -3 (Fig. 13) (this latter is attached to a d-glycero-d-talo-2-octulosonic acid in the core moiety; Fig. 1) establish hydrogen bonds with the TLR4 Arg-264 and Asp-294 carboxylate groups. Second, the ammonium group of l-Ara4N-1 is close to the Asp-395 carboxylate and the Ser-416 OH group, which belong to the opposite TLR4*. Third, the OH-3 from l-Ara4N-1 establishes an H-bond with the Ser-415 CO group of TLR4* (Fig. 13). The three-dimensional models from docking could suggest higher activation for LPSBC versus LPSBCΔAra arising from the extra anchorage points from arabinoses. This agrees with the higher endotoxic potential of LPSBC in the biological assays. Based on this TLR4·MD-2·LPSBC model, we built a new model with the following four mutations: D294A, R322A, S415A*, and S416A*. These mutations involve the main residues establishing interactions with the ammonium groups from the three Ara4N residues and the OH group from the terminal glucose (Fig. 13). (i) Asp-294 side chain establishes polar interactions reinforced by hydrogen bonds with the ammonium group from Ara4N-3, and is in the proximity of the ammonium group from Ara4N-2, so this residue can be considered as a main anchorage point for TLR4·MD-2·LPSBC; Asp-294 was mutated to Ala to abolish this interaction. (ii) Ser-415* and Ser-416* are placed in the partner TLR4* and provide an anchorage point for the ammonium group from Ara4N-1 through polar interactions with the side chain of Ser-415* and the backbone CO groups; both Ser residues were mutated to Ala. (iii) Although not directly in contact with any of the Ara4N moieties, the Arg-322 side chain establishes a hydrogen bond with the OH group at position 3 from the terminal glucose, and we mutated it to compare its contribution to the global energy of the system; this interaction is absent in the complex of TLR4·MD-2 with E. coli LPS (Protein Data Bank code 3fxi) and could be considered as a distinctive interaction for LPSBC core. MDS and energy calculations of both systems (TLR4·MD-2·LPSBC and mutant TLR4·MD-2·LPSBC) have shown an important difference in the global energy: the wild type complex (TLR4·MD-2·LPSBC model) is around 32 kcal mol−1 more stable than the mutant counterpart mainly due to the coulombic term, pointing to the presence of important polar interactions (Table 2). Analysis of the contributions of each residue to the ligand binding energy reveals that this energy difference is mainly due to the lack of the interactions involving the mutated residues. (i) Interaction with Asp-294 is the strongest interaction in the TLR4·MD-2·LPSBC system (around 22 kcal mol−1) and is absent in the mutant. (ii) Also the interaction with Arg-322 is missing; this is one of the main interactions in the TLR4·MD-2·LPSBC system (around 9.6 kcal mol−1). (iii) The lower coulombic contribution to the interaction energy in the mutant TLR4·MD-2·LPSBC complex is due to the absence of the interaction between the Ser-415* side chain and the ammonium group. These results give reasonable evidence that the Ara4N ammonium groups provide additional anchorage interactions accounting for the final stability of the TLR4·MD-2·LPSBC complex. Abolishing these interactions leads to a less stable complex, suggesting that they are crucial for the binding.
FIGURE 11.
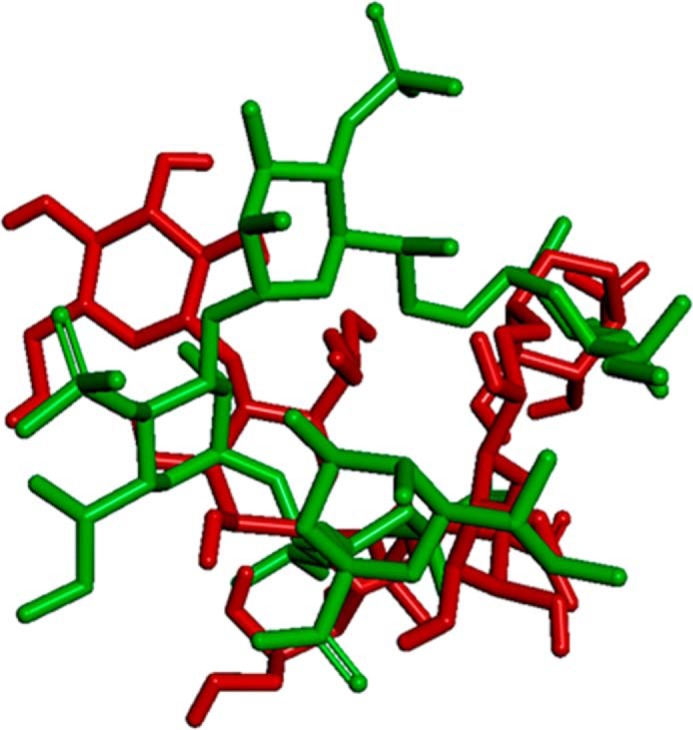
Superimposition of best dock results of LPSBC core. The docking was obtained using AutoDock Vina (depicted in red) and E. coli LPS core from Protein Data Bank code 3fxi (depicted in green). TLR4 is not shown for the sake of clarity.
FIGURE 12.

Predicted binding mode of the LPSBC core to hTLR4·MD-2. The computational model from docking followed by MDS is shown. Left panel, three-dimensional model of the dimer of LPSBC core in complex with TLR4·MD-2. Right panel, detail of the dimer of LPSBC inner core in complex with TLR4·MD-2.
FIGURE 13.
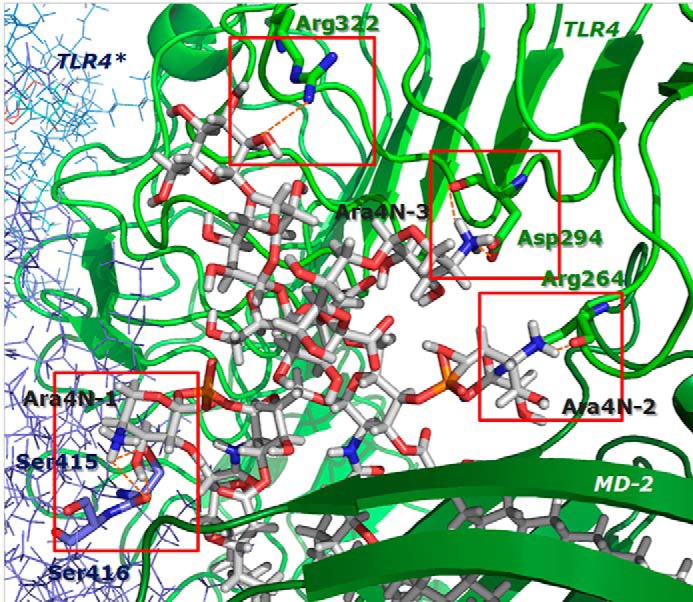
Predicted binding mode of the LPSBC core to hTLR4·MD-2. Details of some interactions between the LPSBC inner core and TLR4·MD-2 from the computational model are shown.
TABLE 2.
Analysis of the per residue contributions to the total ligand binding energy for both complexes TLR4·MD-2·LPSBC and the mutated TLR4·MD-2·LPSBC (D294A, R322A, S415A*, and S416A*)
Energy calculations come from the MDS. Only the top 22 contributions are shown (top 20 in the case of the mutant). Residues from TLR4 are underlined. Residues from the partner TLR4 are also marked with *. vdW, van der Waals; Desolv, desolvated.
Discussion
Lipid A is a major determinant of cytokine induction in host immune cells (11–19, 54). Generally, the highest immunostimulatory activity of lipid A correlates with its hexa-acylation pattern (11–19, 54) because the sixth acyl chain protrudes from the MD-2 binding pocket, bridging TLR4·MD-2 complex dimerization. On the contrary, underacylated lipid A molecules are not (or are poorly) sensed by human TLR4·MD-2 as they are potentially accommodated within the MD-2 pocket. Here, we demonstrate a role for l-Ara4N residues in lipid A that explains why the B. cenocepacia LPS (naturally consisting of a mixture of penta- and tetra-acylated forms) acts as a strong TLR4·MD-2 agonist. Our results agree with a recent report by Hollaus et al. (36) demonstrating that a synthetic Burkholderia lipid A substituted with l-Ara4N exclusively at the anomeric phosphate acts as a potent human TLR4·MD-2 agonist.
Our in vitro and in vivo results explain and expand the above conclusions and show that B. cenocepacia penta-acylated lipid A (both LABC and LABCΔAra) elicits an inflammatory response activating the TLR4·MD-2 complex, suggesting that the model of the receptor complex activation should also include other structural components. Furthermore, molecular modeling experiments indicated that the increased length of the two amide-linked acyl chains of the B. cenocepacia lipid A moiety (3-(R)-hydroxyhexadecanoic acid C16:0 (3-OH) versus 3-(R)-hydroxytetradecanoic acid C14:0 (3-OH) from the E. coli lipid A) compensates for the lack of one fatty acid chain filling the hydrophobic binding pocket of MD-2 protein, allowing the placement into the channel of the fifth acyl chain responsible for TLR4 dimerization. The replacement in E. coli lipid A of the secondary dodecanoic acid C12:0 with a hexadecanoic acid C16:0 results in a weaker LPS agonist (55). However, our data indicate that not only the increased length of the acyl chain but also its position is important for agonist activity.
Furthermore, our mutagenesis studies using two B. cenocepacia lipid A forms, LABC and LABCΔAra, uncovered differential responsiveness on transfected HEK293 cells, indicating an important role also for the l-Ara4N residues in the TLR4·LABC binding process. Particularly, the significantly different immunostimulatory activity of LABC compared with LABCΔAra (Fig. 7) observed in the presence of the human MD-2 K122E mutation might be related to the occurrence on LABC of the l-Ara4N residues, which could mask the negatively charged phosphate groups and then reduce the repulsive forces introduced with the K122E mutation. In contrast, the absence of l-Ara4N in LABCΔAra might explain its increased agonistic activity when tested on HEK293 cells transfected with mTLR4·hMD-2 K122E because the repulsive forces introduced might facilitate the orientation toward the Cys-95/Cys-105 loop (52, 53). Therefore, it is reasonable to assume that the l-Ara4N modification with its positively charged ammonium group could favor the electrostatic interactions allowing receptor/lipid A binding. Intriguingly, recent work by Maeshima et al. (56) revealed several key charged amino acid residues in TLR4 and MD-2 mediating host-specific responses to the glucosamine-modified penta-acylated lipid A from Bordetella pertussis and its unmodified counterpart by human and mouse TLR4·MD-2 complexes. This further underscores the importance of positively charged residues decorating penta-acylated lipid A and interacting with human TLR4·MD-2 charged amino acid residues to promote complex activation (56).
Our molecular modeling experiments further supported the notion that l-Ara4N residues provide additional polar interactions affecting the LABC binding to the TLR4·MD-2 and contribute to anchoring the lipid A into the receptor complex. The network of hydrogen bonds and polar interactions contributing to anchoring the lipid A into the TLR4·MD-2 involves not only the sugar residues from the core oligosaccharide (for example, hydrogen bonds involving Arg-322; Fig. 13) but also the three l-Ara4N residues and the partner TLR4*. As stated above, mutagenesis studies demonstrate that several Lys residues involved in LPS binding play indispensable roles through the polar interactions with the phosphate groups (56–59). In our dimer model, Lys-122 is involved in binding the lipid A moiety of the LPSBC through polar interactions with the core; as for V82F, its role in building the groove that accommodates the acyl chain from lipid A, completing the interaction surface required for dimerization, has already been mentioned. The replacement of Val-82 by phenylalanine involves changes of van der Waals interactions into CH-π interactions, which may favor the binding of this chain, which is longer than the corresponding chain on E. coli LPS. Thus, the structural peculiarities of B. cenocepacia, i.e. its acylation pattern and the presence of the l-Ara4N residues in the lipid A region, exert a synergistic effect in the activation and dimerization of the LPS receptor, opening new insights into the comprehension of the molecular recognition of LPS by TLR4·MD-2.
Author Contributions
A. M. designed the research. F. D. L. executed the chemistry and immunochemistry experiments. Ł. K. and S. M.-S. performed the molecular dynamics experiments. F. D. L., A. M., Ł. K., S. M.-S., M. A. V., A. O., N. I. L., C. G., R. L., M. P., M. A. H., A. D. S., A. S., R. J., and A. B. contributed with analytical tools, reagents, or particular experiments. F. D. L., M. A. V., S. M.-S., and A. M. analyzed data and wrote the paper. F. D. L., A. M., Ł. K., S. M.-S., M. A. V., A. O., N. I. L., C. G., R. L., M. P., M. A. H., A. D. S., A. S., R. J., and A. B. analyzed the results and approved the final version of the manuscript.
This work was supported in part by Spanish Ministry of Economy and Competitiveness Grant CTQ2011-22724, Universidad CEU San Pablo Grants PC14/2011 and PC13/2012, the Slovenian Research Agency, Centre of Excellence NMR - Future Innovation for Sustainable Technologies, European structural funds, Cystic Fibrosis Canada, European Cooperation in Science and Technology Actions BM1003 and CM1102, and Marie Curie Initial Training Networks (ITN) GLYCOPHARM PITN-GA-2012-317297 and H2020-MSCA-ITN-2014-ETN TOLLerant. The authors declare that they have no conflict of interest with the content of this article.
- TLR4
- Toll-Like receptor 4
- CF
- cystic fibrosis
- LA
- lipid A
- l-Ara4N
- 4-amino-4-deoxy-l-arabinose
- LABC
- B. cenocepacia lipid A
- LABCΔAra
- B. cenocepacia lipid A lacking l-Ara4N
- LPSBC
- B. cenocepacia LPS
- LPSBCΔAra
- B. cenocepacia LPS lacking l-Ara4N
- MD-2
- myeloid differentiation factor 2
- MDS
- molecular dynamics simulation
- DS
- disaccharide
- mMD-2
- murine MD-2
- hMD-2
- human MD-2
- hTLR4
- human TLR4
- mTLR4
- murine TLR4.
References
- 1. Janeway C. A., Jr., Medzhitov R. (2002) Innate immune recognition. Annu. Rev. Immunol. 20, 197–216 [DOI] [PubMed] [Google Scholar]
- 2. Gay N. J., Gangloff M. (2007) Structure and function of Toll receptors and their ligands. Annu. Rev. Biochem. 76, 141–165 [DOI] [PubMed] [Google Scholar]
- 3. Broz P., Monack D. M. (2013) Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 13, 551–565 [DOI] [PubMed] [Google Scholar]
- 4. Nagai Y., Akashi S., Nagafuku M., Ogata M., Iwakura Y., Akira S., Kitamura T., Kosugi A., Kimoto M., Miyake K. (2002) Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat. Immunol. 3, 667–672 [DOI] [PubMed] [Google Scholar]
- 5. Angus D. C., Linde-Zwirble W. T., Lidicker J., Clermont G., Carcillo J., Pinsky M. R. (2001) Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 29, 1303–1310 [DOI] [PubMed] [Google Scholar]
- 6. Rangel-Frausto M. S. (2005) Sepsis: still going strong. Arch. Med. Res. 36, 672–681 [DOI] [PubMed] [Google Scholar]
- 7. Raetz C. R., Whitfield C., (2002) Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holst O., Molinaro A. (2009) in Microbial Glycobiology: Structures Relevance and Applications (Moran A. P., Holst O., Brennan P. J., von Itzstein M., eds) pp. 29–56, Elsevier, San Diego, CA [Google Scholar]
- 9. Knirel Y. (2009) in Microbial Glycobiology: Structures Relevance and Applications (Moran A. P., Holst O., Brennan P. J., von Itzstein M., eds) pp. 57–74, Elsevier, San Diego, CA [Google Scholar]
- 10. Silipo A., Erbs G., Shinya T., Dow J. M., Parrilli M., Lanzetta R., Shibuya N., Newman M. A., Molinaro A. (2010) Glyco-conjugates as elicitors or suppressors of plant innate immunity. Glycobiology 20, 406–419 [DOI] [PubMed] [Google Scholar]
- 11. Brandenburg K., Mayer H., Koch M. H., Weckesser J., Rietschel E. T., Seydel U. (1993) Influence of the supramolecular structure of free lipid A on its biological activity. Eur. J. Biochem. 218, 555–563 [DOI] [PubMed] [Google Scholar]
- 12. Rietschel E. T., Kirikae T., Schade F. U., Mamat U., Schmidt G., Schmidt G., Loppnow H., Ulmer A. J., Zähringer U., Seydel U., Di Padova F., Schreier M., Brade H. (1994) Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 8, 217–225 [DOI] [PubMed] [Google Scholar]
- 13. Brandeburg K., Seydel U., Schromm A. B., Loppnow H., Koch M. H., Rietschel E. T. (1996) Conformation of lipid A, the endotoxic centre of the bacterial lipopolysaccharide. J. Endotoxin Res. 3, 173–178 [Google Scholar]
- 14. Seydel U., Oikawa M., Fukase K., Kusumoto S., Brandenburg K. (2000) Intrinsic conformation of lipid A is responsible for agonistic and antagonistic activity. Eur. J. Biochem. 267, 3032–3039 [DOI] [PubMed] [Google Scholar]
- 15. Schromm A. B., Brandenburg K., Loppnow H., Moran A. P., Koch M. H., Rietschel E. T., Seydel U. (2000) Biological activities of lipopolysaccharides are determined by the shape of their lipid A portion. Eur. J. Biochem. 267, 2008–2013 [DOI] [PubMed] [Google Scholar]
- 16. Fukuoka S., Brandenburg K., Müller M., Lindner B., Koch M. H., Seydel U. (2001) Physico-chemical analysis of lipid A fractions of lipopolysaccharide from Erwinia carotovora in relation to bioactivity. Biochim. Biophys. Acta 1510, 185–197 [DOI] [PubMed] [Google Scholar]
- 17. Oikawa M., Shintaku T., Fukuda N., Sekljic H., Fukase Y., Yoshizaki H., Fukase K., Kusumoto S. (2004) NMR conformational analysis of biosynthetic precursor-type lipid A: monomolecular state and supramolecular assembly. Org. Biomol. Chem. 2, 3557–3565 [DOI] [PubMed] [Google Scholar]
- 18. Park B. S., Song D. H., Kim H. M., Choi B. S., Lee H., Lee J. O. (2009) The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195 [DOI] [PubMed] [Google Scholar]
- 19. Maeshima N., Fernandez R.C. (2013) Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front. Cell. Infect. Microbiol. 3, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Golenbock D. T., Hampton R. Y., Qureshi N., Takayama K., Raetz C. R. (1991) Lipid A-like molecules that antagonize the effects of endotoxins on human monocytes. J. Biol. Chem. 266, 19490–19498 [PubMed] [Google Scholar]
- 21. Klett J., Reeves J., Oberhauser N., Pérez-Regidor L., Martín-Santamaria S. (2014) Modulation of toll-like receptor 4. Insights from x-ray crystallography and molecular modeling. Curr. Top. Med. Chem. 14, 2672–2683 [DOI] [PubMed] [Google Scholar]
- 22. Resman N., Vasl J., Oblak A., Pristovsek P., Gioannini T. L., Weiss J. P., Jerala R. (2009) Essential roles of hydrophobic residues in both MD-2 and toll-like receptor 4 in activation by endotoxin. J. Biol. Chem. 284, 15052–15060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ohto U., Fukase K., Miyake K., Satow Y. (2007) Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVA. Science 316, 1632–1634 [DOI] [PubMed] [Google Scholar]
- 24. Ernst R. K., Yi E. C., Guo L., Lim K. B., Burns J. L., Hackett M., Miller S. I. (1999) Specific lipopolysaccharide found in cystic fibrosis airway Pseudomonas aeruginosa. Science 286, 1561–1565 [DOI] [PubMed] [Google Scholar]
- 25. Lohmann K. L., Vandenplas M. L., Barton M. H., Bryant C. E., Moore J. N. (2007) The equine TLR4/MD-2 complex mediates recognition of lipopolysaccharide from Rhodobacter sphaeroides as an agonist. J. Endotoxin Res. 13, 235–242 [DOI] [PubMed] [Google Scholar]
- 26. Saitoh S., Akashi S., Yamada T., Tanimura N., Kobayashi M., Konno K., Matsumoto F., Fukase K., Kusumoto S., Nagai Y., Kusumoto Y., Kosugi A., Miyake K. (2004) Lipid A antagonist, lipid IVA, is distinct from lipid A in interaction with Toll-like receptor 4 (TLR4)-MD-2 and ligand-induced TLR4 oligomerization. Int. Immunol. 16, 961–969 [DOI] [PubMed] [Google Scholar]
- 27. Hajjar A. M., Ernst R. K., Tsai J. H., Wilson C. B., Miller S. I. (2002) Human Toll-like receptor 4 recognizes host-specific LPS modifications. Nat. Immunol. 3, 354–359 [DOI] [PubMed] [Google Scholar]
- 28. Herath T. D., Darveau R. P., Seneviratne C. J., Wang C.Y., Wang Y., Jin L. (2013) Tetra- and Penta-acylated lipid A structures of Porphyromonas gingivalis LPS differentially activate TLR4-mediated NF-κB signal transduction cascade and immuno-inflammatory response in human gingival fibroblasts. PLoS One 8, e58496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Speert D. P. (2002) Advances in Burkholderia cepacia complex. Paediatr. Respir. Rev. 3, 230–235 [DOI] [PubMed] [Google Scholar]
- 30. Hamad M. A., Di Lorenzo F., Molinaro A., Valvano M. A. (2012) Aminoarabinose is essential for lipopolysaccharide export and intrinsic antimicrobial peptide resistance in Burkholderia cenocepacia. Mol. Microbiol. 85, 962–974 [DOI] [PubMed] [Google Scholar]
- 31. Ortega X. P., Cardona S. T., Brown A. R., Loutet S. A., Flannagan R. S., Campopiano D. J., Govan J. R., Valvano M. A. (2007) A putative gene cluster for aminoarabinose biosynthesis is essential for Burkholderia cenocepacia viability. J. Bacteriol. 189, 3639–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zughaier S. M., Ryley H. C., Jackson S. K. (1999) Lipopolysaccharide (LPS) from Burkholderia cepacia is more active than LPS from Pseudomonas aeruginosa and Stenotrophomonas maltophilia in stimulating tumor necrosis factor alpha from human monocytes. Infect. Immun. 67, 1505–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. De Soyza A., Ellis C. D., Khan C. M., Corris P. A., Demarco de Hormaeche R. (2004) Burkholderia cenocepacia lipopolysaccharide, lipid A, and proinflammatory activity. Am. J. Respir. Crit. Care Med. 170, 70–77 [DOI] [PubMed] [Google Scholar]
- 34. Bamford S., Ryley H., Jackson S. K. (2007) Highly purified lipopolysaccharides from Burkholderia cepacia complex clinical isolates induce inflammatory cytokine responses via TLR4-mediated MAPK signalling pathways and activation of NFκB. Cell. Microbiol. 9, 532–543 [DOI] [PubMed] [Google Scholar]
- 35. Silipo A., Molinaro A., Ieranò T., De Soyza A., Sturiale L., Garozzo D., Aldridge C., Corris P. A., Khan C. M., Lanzetta R., Parrilli M. (2007) The complete structure and pro-inflammatory activity of the lipooligosaccharide of the highly epidemic and virulent gram-negative bacterium Burkholderia cenocepacia ET-12 (strain J2315). Chem. Eur. J. 13, 3501–3511 [DOI] [PubMed] [Google Scholar]
- 36. Hollaus R., Ittig S., Hofinger A., Haegman M., Beyaert R., Kosma P., Zamyatina A. (2015) Chemical synthesis of Burkholderia lipid A modified with glycosyl phosphodiester-linked 4-amino-4-deoxy-β-L-arabinose and its immunomodulatory potential. Chem. Eur. J. 21, 4102–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Di Lorenzo F., Silipo A., Bianconi I., Lore' N. I., Scamporrino A., Sturiale L., Garozzo D., Lanzetta R., Parrilli M., Bragonzi A., Molinaro A. (2015) Persistent cystic fibrosis isolate Pseudomonas aeruginosa strain RP73 exhibits an under-acylated LPS structure responsible of its low inflammatory activity. Mol. Immunol. 63, 166–175 [DOI] [PubMed] [Google Scholar]
- 38. Jeukens J., Boyle B., Bianconi I., Kukavica-Ibrulj I., Tümmler B., Bragonzi A., Levesque R. C. (2013) Complete genome sequence of persistent cystic fibrosis isolate Pseudomonas aeruginosa strain RP73. Genome Announc. 1, e00568–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Westphal O. J., Jann K. (1965) Bacterial lipopolysaccharides: extraction with phenol-water and further applications of the procedure. Methods Carbohydr. Chem. 5, 83–91 [Google Scholar]
- 40. Kittelberger R., Hilbink F. (1993) Sensitive silver-staining detection of bacterial lipopolysaccharides in polyacrylamide gels. J. Biochem. Biophys. Methods 26, 81–86 [DOI] [PubMed] [Google Scholar]
- 41. Ortega X., Silipo A., Saldías M. S., Bates C. C., Molinaro A., Valvano M. A. (2009) Biosynthesis and structure of the Burkholderia cenocepacia K56-2 lipopolysaccharide core oligosaccharide: truncation of the core oligosaccharide leads to increased binding and sensitivity to polymyxin B. J. Biol. Chem. 284, 21738–21751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schrödinger (2012) Suite 2012: Maestro, version 9.3, Schrödinger, LLC, New York [Google Scholar]
- 43. Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Montgomery J. A., Jr., Vreven T., Kudin K. N., Burant J. C., Millam J. M., Iyengar S. S., Tomasi J., Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G. A., Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J. E., Hratchian H. P., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Ayala P. Y., Morokuma K., Voth G. A., Salvador P., Dannenberg J. J., Zakrzewski V. G., Dapprich S., Daniels A. D., Strain M. C., Farkas O., Malick D. K., Rabuck A. D., Raghavachari K., Foresman J. B., Ortiz J. V., Cui Q., Baboul A. G., Clifford S., Cioslowski J., Stefanov B. B., Liu G., Liashenko A., Piskorz P., Komaromi I., Martin R. L., Fox D. J., Keith T., Al-Laham M. A., Peng C. Y., Nanayakkara A., Challacombe M., Gill P. M. W., Johnson B., Chen W., Wong M. W., Gonzalez C., Pople J. A. (2004) Gaussian 03, Revision E.01, Gaussian, Inc., Wallingford, CT [Google Scholar]
- 44. Schrödinger (2012) Suite 2012: MacroModel, version 9.9, Schrödinger, LLC, New York [Google Scholar]
- 45. Schrödinger (2012) Suite 2012: Impact, version 5.8, Schrödinger, LLC, New York, NY [Google Scholar]
- 46. Schrödinger (2012) Suite 2012: Protein Preparation Wizard: Epik, version 2.3; Impact, version 5.8; Prime, version 3.1, Schrödinger, LLC, New York [Google Scholar]
- 47. Goodsell D. S., Morris G. M., Olson A. J. (1996) Automated docking of flexible ligands: applications of AutoDock. J. Mol. Recognit. 9, 1–5 [DOI] [PubMed] [Google Scholar]
- 48. Trott O., Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Klett J., Núñez-Salgado A., Dos Santos H. G., Corte′s-Cabrera A., Perona A., Gil-Redondo R., Abia D., Gago F., Morreale A. (2012) MM-ISMSA: an ultrafast and accurate scoring function for protein-protein docking. J. Chem. Theory Comput. 9, 3395–3408 [DOI] [PubMed] [Google Scholar]
- 51. Meng J., Drolet J. R., Monks B. G., Golenbock D. T. (2010) MD-2 residues tyrosine 42, arginine 69, aspartic acid 122, and leucine 125 provide species specificity for lipid IVA. J. Biol. Chem. 285, 27935–27943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ohto U., Fukase K., Miyake K., Shimizu T., (2012) Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc. Natl. Acad. Sci. U.S.A. 109, 7421–7426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kobayashi M., Saitoh S., Tanimura N., Takahashi K., Kawasaki K., Nishijima M., Fujimoto Y., Fukase K., Akashi-Takamura S., Miyake K. (2006) Regulatory roles for MD-2 and TLR4 in ligand-induced receptor clustering. J. Immunol. 176, 6211–6518 [DOI] [PubMed] [Google Scholar]
- 54. Molinaro A., Holst O., Di Lorenzo F., Callaghan M., Nurisso A., D'Errico G., Zamyatina A., Peri F., Berisio R., Jerala R., Jiménez-Barbero J., Silipo A., Martín-Santamaría S. (2015) Chemistry of lipid A: at the heart of innate immunity. Chem. Eur. J. 21, 500–519 [DOI] [PubMed] [Google Scholar]
- 55. Bainbridge B. W., Coats S. R., Pham T. T., Reife R. A., Darveau R. P. (2006) Expression of a Porphyromonas gingivalis lipid A palmitylacyltransferase in Escherichia coli yields a chimeric lipid A with altered ability to stimulate interleukin-8 secretion. Cell. Microbiol. 8, 120–129 [DOI] [PubMed] [Google Scholar]
- 56. Maeshima N., Evans-Atkinson T., Hajjar A. M., Fernandez R. C. (2015) Bordetella pertussis lipid A recognition by Toll-like receptor 4 and MD-2 is dependent on distinct charged and uncharged interfaces. J. Biol. Chem. 290, 13440–13453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gruber A., Mancek M., Wagner H., Kirschning C. J., Jerala R. (2004) Structural model of MD-2 and functional role of its basic amino acid clusters involved in cellular lipopolysaccharide recognition. J. Biol. Chem. 279, 28475–28482 [DOI] [PubMed] [Google Scholar]
- 58. Re F., Strominger J. L. (2003) Separate functional domains of human MD-2 mediate Toll-like receptor 4-binding and lipopolysaccharide responsiveness. J. Immunol. 171, 5272–5276 [DOI] [PubMed] [Google Scholar]
- 59. Visintin A., Latz E., Monks B. G., Espevik T., Golenbock D. T. (2003) Lysines 128 and 132 enable lipopolysaccharide binding to MD-2, leading to Toll-like receptor-4 aggregation and signal transduction. J. Biol. Chem. 278, 48313–48320 [DOI] [PubMed] [Google Scholar]