

Background: Amylomaltase MalQ catalyzes the transglycosylation of maltose and maltodextrins in E. coli.
Results: Three different x-ray structures and the product equilibrium concentrations for different substrates were determined.
Conclusion: MalQ undergoes major conformational changes during catalysis, and its product spectrum depends on substrate chain length.
Significance: Novel insights into the MalQ mechanism and its key role for maltose metabolism in E. coli were obtained.
Keywords: carbohydrate metabolism, enzyme mechanism, Escherichia coli (E. coli), protein crystallization, x-ray crystallography, acarbose, enzymatically derived inhibitor, maltose/maltodextrin metabolism
Abstract
Amylomaltase MalQ is essential for the metabolism of maltose and maltodextrins in Escherichia coli. It catalyzes transglycosylation/disproportionation reactions in which glycosyl or dextrinyl units are transferred among linear maltodextrins of various lengths. To elucidate the molecular basis of transglycosylation by MalQ, we have determined three crystal structures of this enzyme, i.e. the apo-form, its complex with maltose, and an inhibitor complex with the transition state analog acarviosine-glucose-acarbose, at resolutions down to 2.1 Å. MalQ represents the first example of a mesophilic bacterial amylomaltase with known structure and exhibits an N-terminal extension of about 140 residues, in contrast with previously described thermophilic enzymes. This moiety seems unique to amylomaltases from Enterobacteriaceae and folds into two distinct subdomains that associate with different parts of the catalytic core. Intriguingly, the three MalQ crystal structures appear to correspond to distinct states of this enzyme, revealing considerable conformational changes during the catalytic cycle. In particular, the inhibitor complex highlights the requirement of both a 3-OH group and a 4-OH group (or α1–4-glycosidic bond) at the acceptor subsite +1 for the catalytically competent orientation of the acid/base catalyst Glu-496. Using an HPLC-based MalQ enzyme assay, we could demonstrate that the equilibrium concentration of maltodextrin products depends on the length of the initial substrate; with increasing numbers of glycosidic bonds, less glucose is formed. Thus, both structural and enzymatic data are consistent with the extremely low hydrolysis rates observed for amylomaltases and underline the importance of MalQ for the metabolism of maltodextrins in E. coli.
Introduction
Uptake and metabolism of α1–4-linked glucose polymers in E. coli are accomplished by the maltose system (1). Within the cell, maltose/maltodextrins are mutually converted by the three enzymes MalP, MalQ, and MalZ, whereas MalP and MalQ are the major players for the catabolic degradation of maltodextrins. MalP cleaves glucose moieties by phosphorolysis from the nonreducing end to yield glucose-1-P, with maltopentaose constituting the shortest substrate (4, 5). Cells lacking MalP accumulate large amounts of linear maltodextrins, due to the action of MalQ (6).
MalQ converts short maltodextrins, including maltose and maltotriose, to longer maltodextrins and glucose. Bacterial strains lacking MalQ cannot grow on maltose and maltotriose (7). Concerted action of MalP and MalQ via maltodextrins as common intermediates leads to the formation of glucose and glucose-1-P. Both are subsequently converted to glucose-6-P by glucokinase and phosphoglucomutase, respectively, to enter glycolysis. Previous investigations indicated that increased internal glucose levels reduce the activity of MalQ (8). Furthermore, maltodextrin metabolism is linked to that of glycogen via the shared intermediate maltotriose, which is not only a substrate of MalQ but also constitutes the inducer of the maltose system (9, 10). Thus, degradation of glycogen leads to a basal induction of mal genes even in the absence of external maltodextrins. In contrast, in the presence of maltose and maltotriose, MalQ can compensate for a lack of the glycogen synthase GlgA (11).
MalQ cleaves any linear maltodextrins, releases their reducing end, and transfers the resulting nonreducing dextrinyl moiety (donor) onto another maltodextrin or glucose (acceptor) (12, 13). This results in a series of maltodextrins that differ in length by one monosaccharide unit as well as glucose (Fig. 1). MalQ from Escherichia coli was the first amylomaltase to be isolated and biochemically characterized (12–15). Much later, the malQ gene was cloned, and its encoded amino acid sequence of 694 residues (78.5 kDa) was elucidated (16). MalQ is a member of the amylomaltases (EC 2.4.1.25) or 4-α-glucanotransferases, which are structurally and mechanistically related to α-amylases. Both belong to the glycoside hydrolase clan H. However, based on sequence characteristics, amylomaltases have been classified as a separate GH family (GH77) (17).
FIGURE 1.
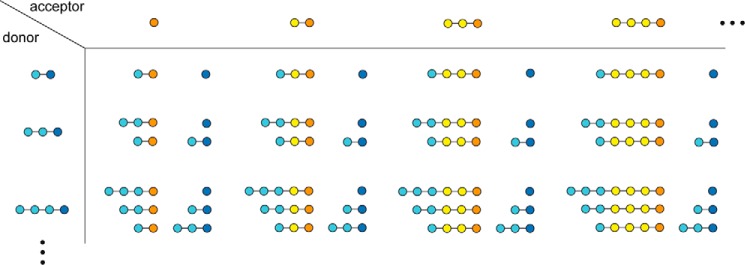
Schematic representation of possible MalQ reactions, starting with maltose as the first possible donor for a sugar unit. For clarity, only glucose and the shorter saccharides maltose, maltotriose, and maltotetraose are considered here. Donor and acceptor molecules are depicted in light blue and yellow, respectively, with their reducing ends highlighted in blue and orange. Notably, the primary reaction products are substrates for subsequent reaction cycles (cf. supplemental material).
In contrast to α-amylases, which mostly catalyze hydrolysis of glycosidic bonds, amylomaltases strongly favor the transglycosylation or disproportionation reaction with retention of the α-anomeric configuration (18). Structures of amylomaltases from three Thermus species were previously described (19–21). Notably, with ∼500 residues the thermophilic homologs are considerably shorter than MalQ from E. coli. The larger part of the additional ∼190 residues forms an ∼140-residue N-terminal extension of previously unknown structure and function.
Here, we present three distinct crystal structures of MalQ from E. coli: the apo-form, the complex with maltose, and the complex with the heptasaccharide transition state analog acarviosine-glucose-acarbose (AGA),2 which represent different states of the catalytic cycle. Furthermore, we have employed an HPLC-based assay to monitor the MalQ product spectrum at equilibrium, revealing the dependence on the number of glycosidic bonds in the substrate(s).
Experimental Procedures
Enzyme Cloning, Expression, and Purification
The C-terminal 10 residues of MalQ (UniProt ID P15977) represent an unusual repeat of amino acids, i.e. Arg4-Ala3-Lys3, which were most likely structurally disordered. To decrease the flexibility and positive charge at the C terminus of MalQ, the six C-terminal residues were replaced by Ser-Ala-His6, thus appending a His6 tag for affinity purification. To this end, the malQ gene was amplified from the genomic DNA of the E. coli strain XL1-blue by PCR with Pfu Ultra II DNA polymerase (Agilent) using the primer pair 5′-GA GAT ATA CAT ATG GAA AGC AAA CGT CTG GA-3′/5′-GCT TCT GCG CCG TCT GTC CAA ATC CTT CAG CAA-3′ and cloned on pASK75-his (22) via NdeI and AfeI (Fermentas). The resulting expression vector encodes the 78.9-kDa protein MalQ(1–688)-SAHHHHHH (MalQ-His) under the control of a tetracycline-dependent promoter. The correct sequence of the vector was confirmed by DNA sequencing.
MalQ-His was produced in the E. coli strain BL21 in LB medium. To incorporate l-selenomethionine (SeMet) into the recombinant protein for experimental phasing of the diffraction data, bacteria were cultivated in M63 medium (22 mm KH2PO4, 40 mm K2HPO4, 15 mm (NH4)2SO4, 0.2% (w/v) glucose, 1 mm MgSO4, 4.5 μm FeSO4) supplemented with 50 mg/liter l-SeMet (Acros Organics), 0.02 mg/liter tryptophan, 0.4 mg/liter of the other proteinogenic amino acids, as well as 0.04 mg/liter 4-aminobenzoic acid, 0.04 mg/liter 4-hydroxybenzoic acid, 0.4 mg/liter xanthine, 0.4 mg/liter uracil, 0.01 mg/liter biotin, 0.01 mg/liter nicotinamide, 1 μg/liter riboflavin, and 0.01 mg/liter thiamine. Bacteria were cultivated at the 2-liter scale using shake flasks at 22 °C. Protein expression was induced by adding 0.2 mg/liter anhydrotetracycline at OD550 = 0.6. After 6 h (16 h for SeMet incorporation), the cells were harvested by centrifugation, and the pellets were stored at −20 °C.
After thawing on ice, the bacteria were resuspended in 0.5 m NaCl, 50 mm HEPES/Na, pH 8.0, 10 mm imidazole, and 2 mm β-mercaptoethanol (BME) and disrupted in a French pressure cell (Aminco). The cell suspension was cleared by centrifugation at 35,000 × g, and the supernatant was applied onto an iminodiacetic acid-Sepharose column charged with Zn2+ ions. Unbound protein was rinsed with 0.5 m NaCl, 50 mm HEPES/Na, pH 8.0, 2 mm BME, and then MalQ-His was eluted with a linear gradient of 0–0.3 m imidazole/HCl in the same buffer. Appropriate fractions were pooled, dialyzed against 0.5 m NaCl, 20 mm Tris/HCl, pH 8.0, 2 mm BME and further purified by size exclusion chromatography (SEC) using a preparative Superdex200 26/60 column (GE Healthcare) equilibrated with the same buffer. SEC fractions corresponding to monomeric MalQ were pooled, supplemented with 2 mm tris(2-carboxethyl)phosphine, and concentrated to 13 mg/ml for crystallization. The final yield per liter culture was 12 mg of MalQ.
Protein Crystallization, Data Collection, and Refinement
Crystals of MalQ-His were obtained by sitting drop vapor diffusion at 20 °C using an Evo robotic system (Tecan). Drops containing 200 nl of protein and 200 nl of reservoir solution were equilibrated against 100 μl of reservoir. Crystals of apo-MalQ formed in the presence of 1.2 m Na3citrate. Complexes of MalQ with maltose (MalQ·maltose) and acarviosine-glucose-acarbose (MalQ·AGA) were obtained by incubation of MalQ-His with 2 mm maltotriose (Applichem) and 2 mm acarbose (LKT Laboratories), respectively, prior to crystallization. Diffraction quality crystals of MalQ·maltose grew in the presence of 20% (w/v) PEG-3350, 0.2 m KI, whereas MalQ·AGA and SeMet-derivatized MalQ·AGA crystals were obtained in 20% (w/v) PEG-3350, 0.2 m NaH2PO4, and 20% (w/v) PEG-3350, 0.2 m NaCl, respectively. Cryoprotection of the crystals for flash freezing in liquid nitrogen was achieved by adding 3 μl of the corresponding reservoir solution supplemented with 25% (v/v) ethylene glycol, 0.25 m NaCl, and 10 mm Tris/HCl, pH 8.0.
X-ray diffraction data were collected at beamline 14.2 of the BESSY synchrotron (Berlin-Adlershof, Germany) operated by the Helmholtz-Zentrum Berlin (23). Data sets were processed with the XDS package (Table 1) (24). Crystallographic phases were determined by multiple wavelength anomalous dispersion with a SeMet-derivatized MalQ·AGA crystal. To this end, datasets at four different wavelengths, e.g. high energy remote, peak, inflection point, and low energy remote, were collected (Table 1). Phases were determined with HKL2MAP (25). All 21 expected selenium sites could be identified, resulting in a well interpretable electron density map. The initial structure was automatically built with BUCCANEER (26) and completed in repeating cycles of manual model building with COOT (27) and refinement with REFMAC5 (Table 1) (28). Translation, libration, and screw (TLS) groups were determined with TLSMD (29) and used during refinement with REFMAC5. Subsequently, the structures of apo-MalQ and MalQ·maltose were solved by molecular replacement with the refined MalQ·AGA structure using PHASER (30). Again, model building and refinement were accomplished with COOT and REFMAC5. Structures were validated with COOT and MolProbity (31). Molecular graphics were prepared with PyMOL (Schrödinger). Atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 4S3P, 4S3Q, and 4S3R.
TABLE 1.
Data collection, phasing, and refinement statistics
Values in parentheses correspond to the highest resolution shell. Values of the Ramachandran plot were calculated with MolProbity (31). Other corresponds to ions and small organic molecules that should not be confused with biologically relevant ligands of MalQ. rh, high energy remote; pk, peak; ip, inflection point; rl, low energy remote.
| Apo-MalQ | MalQ·maltose | MalQ(SeMet)·AGA | ||||
|---|---|---|---|---|---|---|
| Data collection | ||||||
| Space group | P2221 | P1 | P212121 | |||
| Unit cell (Å); (°) | 85.3, 106.1, 217.7; 90, 90, 90 | 75.1, 77.0, 128.4; 76.1, 75.5, 66.3 | 65.0, 75.8, 163.7; 90, 90, 90 | |||
| Wavelength (Å) | 0.9184 | 0.9184 | 0.9786 (rh) | 0.9798 (pk) | 0.9800 (ip) | 0.9821 (rl) |
| Resolution (Å) | 34.0–2.80 (2.90–2.80) | 34.2–2.10 (2.20–2.10) | 50.0–2.30 (2.44–3.30) | 50.0–2.30 (2.44–2.30) | 50.0–2.30 (2.44–2.30) | 50.0–2.05 (2.17–2.05) |
| Completeness (%) | 99.5 (99.9) | 94.3 (96.4) | 99.4 (98.5) | 99.4 (98.3) | 99.4 (98.2) | 99.6 (98.7) |
| Unique reflections | 49,608 (4885) | 138,781 (18,524) | 69,132 (11,085) | 69,158 (11,064) | 69,155 (11,067) | 97,865 (15,684) |
| Multiplicity | 5.2 (5.3) | 2.1 (2.1) | 3.2 (3.2) | 3.2 (3.2) | 3.2 (3.2) | 3.9 (3.8) |
| Mean I/σI | 13.5 (2.8) | 12.0 (3.0) | 9.7 (1.8) | 9.5 (1.7) | 10.2 (1.9) | 12.0 (2.1) |
| Rmeas (%) | 14.6 (74.5) | 7.9 (53.5) | 12.0 (76.0) | 12.1 (78.1) | 11.2 (71.2) | 8.7 (76.1) |
| Wilson B-factor (Å2) | 38.2 | 36.5 | 40.2 | 40.1 | 40.2 | 38.9 |
| Experimental phasing | ||||||
| No. of selenium sites | 21/21 | |||||
| Map correlation (%) | 66.7 | |||||
| Figure of merit (%) | 63.5 | |||||
| Refinement | ||||||
| Resolution (Å) | 34.35–2.80 (2.87–2.80) | 34.23–2.10 (2.16–2.10) | 47.25–2.10 (2.15–2.10) | |||
| No. of reflections working | 47,198 (3412) | 131,810 (9981) | 45,668 (3322) | |||
| No. of reflections test | 2410 (194) | 6970 (516) | 2433 (168) | |||
| Rcryst (%) | 18.0 (29.4) | 22.1 (29.4) | 16.9 (24.3) | |||
| Rfree (%) | 22.4 (34.0) | 25.8 (33.1) | 20.5 (24.4) | |||
| Molecules/ligands | 2/− | 3/1 | 1/1 | |||
| No. of atoms, protein/ligand/waters/other | 10,994/−/187/− | 16,350/23/467/28 | 5421/76/226/4 | |||
| B values (Å2) protein/ligand/waters/other | 42.3/−/25.6/− | 39.5/55.3/33.9/43.7 | 39.4/39.6/38.5/40.8 | |||
| Ramachandran plot, favored/outlier | 1342/0 | 1988/0 | 663/0 | |||
| r.m.s.d. bonds (Å)/angles (°) | 0.01/1.09 | 0.01/1.27 | 0.01/1.40 | |||
Enzyme Assay
A MalQ enzymatic assay was performed with the substrates maltose (Applichem), maltotriose, as well as the inhibitor acarbose. Also, different ratios of maltose/glucose and maltotriose/glucose were investigated. Each sugar was dissolved at 0.1 m in 100 μl of 0.5 m NaCl, 20 mm Tris/HCl, pH 8.0, 2 mm BME. The reaction was started at 30 °C by adding 1.5 μl of a 1.3 mg/ml solution of MalQ in the same buffer. All setups were performed in triplicate. Reactions were stopped at different time points between 3 min and 16 h by inactivation of MalQ at 95 °C for 3 min. Denatured enzyme was removed by centrifugation, and the supernatant was mixed with 100 μl of trichloroacetic acid and 200 μl of 1% (w/v) dansylhydrazine in ethanol and incubated at 80 °C for 10 min (32). 5 μl of the resulting sugar derivatives were injected into a 1200 HPLC system (Agilent) equipped with a VP 260/10 NUCLEODUR C18 column (Machery-Nagel). Elution of the sugar hydrazones was performed with a gradient of 10–30% (v/v) acetonitrile in 10 mm NH4HCO3, pH 8.5, and monitored at 340 nm. The C18 column was calibrated with dansylhydrazones of glucose, maltose, maltotriose, and acarbose. Obviously, dansylhydrazones of maltotetraose and maltopentaose could also be resolved on this column. The retention times of the sugar hydrazones were 23.6, 22.4, 20.7, 20.6, 18.6, and 17.2 min for glucose, maltose, maltotriose, acarbose, maltohexaose, and maltopentaose, respectively. The corresponding peak areas were integrated, and the relative amount of each sugar was plotted against time with KaleidaGraph software (Synergy).
Statistical Simulation of the Maltodextrin Product Profile from the MalQ Reaction
Maltodextrin equilibrium concentrations were calculated by means of a contingency table comprising all possible MalQ reactions up to maltodecaose using Microsoft Office Excel 2007 (see supplemental Table 1). For practical reasons, the set of donor and acceptor molecules was limited to a maximal length of 10 glucose units. Attack of each glycosidic bond within a donor oligosaccharide was considered equally likely, except for glucose, which cannot act as a donor. In each cell of the table, the maltodextrin product concentration was calculated as the mathematical product of donor and acceptor concentrations divided by the number of glycosidic bonds of the donor (to account for the equal distribution of possible reaction products). The resulting maltodextrin product concentrations were summed up for each chain length. These sums were then used as starting concentrations for the next round of calculation. Final equilibrium concentrations were approximated after 10 iterations (cf. supplemental Table 1).
Results
Purification and Structural Determination of MalQ
After overexpression in E. coli, MalQ was purified to homogeneity by immobilized metal affinity chromatography and SEC. Attempts to transfer MalQ into a buffer of moderate ionic strength, i.e. 150 mm NaCl or less, resulted in soluble oligomers of the enzyme as judged by SEC. Therefore, MalQ was always kept in a buffer containing 500 mm NaCl during purification, crystallization, and enzymatic assays (cf. “Experimental Procedures”). Crystals of apo-MalQ, MalQ·maltose, and MalQ·AGA belonged to space groups P2221, P1, and P212121, respectively, containing 2, 3, and 1 polypeptide chains per asymmetric unit (Table 1).
Both chains of apo-MalQ showed a continuously interpretable electron density for residues 2–690, including residues Ser and Ala of the linker between the protein and C-terminal His6 tag. In addition, molecule 1 showed density for the N-terminal Met residue as well as the first His of the tag. The MalQ·maltose complex was obtained by co-crystallization of MalQ with maltotriose. The asymmetric unit contained three polypeptide chains, which showed electron density for residues 2–43 and 52–690, whereas residues 44–51 were disordered in all three chains. One and five C-terminal His residues of the affinity tag were observed in molecules 1 and 2, respectively. In the following, we will refer to protein chain 1 of each asymmetric unit for the apo-MalQ and the MalQ·maltose complex, respectively. MalQ crystallized in the presence of acarbose showed interpretable electron density for residues 2–42 and 52–690. Again, the connecting residues 43–51 were not resolved, although unambiguous density was visible for a ligand (see below).
Overall Structure of MalQ
The tertiary structure of MalQ can be subdivided into three major domains as follows: A, B, and N (Fig. 2). Domain A represents the catalytic core and adopts a TIM barrel fold (33), comprising a central barrel of eight parallel β-strands, each flanked by an α-helix, in a counter-clockwise manner. Noteworthy, in MalQ the sixth α-helix is missing and replaced by a 310-helix with a single turn. The (β/α)8 core harbors several insertions at the carboxyl-end of the β-strands. Together, these insertions are referred to as domain B (21) and include the three distinct subdomains B1 to B3 (Fig. 2). Contrasting with previous reports, these subdomains are here sequentially numbered.
FIGURE 2.

Structural overview of MalQ. A, schematic representation of apo-MalQ. The TIM barrel domain A, subdomains N1, N2, and B1 to B3 are colored gray, blue, light blue, and green, yellow/orange, pink/salmon, respectively. B, corresponding topology diagram colored as above. Residue numbers indicate the domain boundaries. Red dots mark catalytically important residues.
B1 represents the largest of the three subdomains, with 166 residues. It shows a purely α-helical architecture and is a characteristic feature of the amylomaltase family GH77 (17, 21). B1 includes residues 186–351, which are inserted after strand β2 of domain A (Aβ2). Basically, this subdomain can be viewed as a pair of 4-helical bundles that wrap around the N-terminal part of helix Aα2. The functionally most important subdomain is B2, which forms a lid for the active site during catalysis and harbors the 400-s loop (250-s loop in the thermophilic homologs; see below). In MalQ, this subdomain consists of two insertions after strands Aβ3 and Aβ4 and includes residues 377–427 (B2a) and 449–473 (B2b). B2 shows a mixed α/β structure with a central four-stranded, antiparallel twisted β-sheet flanked by short α-helices. B2 interacts with B1 via the two helical segments of B2a, which precede the first β-strand.
Finally, two shorter helical insertions, forming subdomain B3 and comprising residues 545–613 (B3a) and 637–672 (B3b) are found downstream of strands Aβ7 and Aβ8, respectively. B3a carries the transition state-stabilizing side chain of Asp-548. Subdomain B3 interacts with B1 via B3b but does not form any contacts with B2 in the absence of a substrate. In contrast, substrate binding induces conformational changes that lead to extensive contacts between B3a and B2a, as seen in the MalQ·AGA structure. A short insertion occurs after strand Aβ1, where it binds at the interface of B1 and B3. The longer loop that follows Aβ6, which interacts with B3a, is a result of the missing helix Aα6 mentioned above.
In addition to the catalytic domains A and B, MalQ exhibits a peculiar N-terminal extension of 128 residues, referred to as domain N. These residues show no significant sequence homology to other known protein structures. This extension is present in most Enterobacteraceae but is missing in other γ-Proteobacteria. Domain N folds into the subdomains N1 (1–42) and N2 (52–128), both connected by a flexible nine-residue linker that is only resolved in the apo-MalQ structure (Fig. 2).
Subdomain N1 includes two helices, connected by a short two-stranded antiparallel β-sheet, that pack against each other at an angle of 52°. This subdomain tightly associates with the two insertions of subdomain B2 and thus undergoes the same domain movements as B2 in the course of the catalytic cycle (see below). Notably, the αββα topology of N1 is similar to that of B2a (residues 390–425) (cf. Fig. 2). Nevertheless, because of the structural flexibility and the low sequence identity of this motif (12% over 40 residues), it is difficult to infer an evolutionary relationship.
Subdomain N2 resembles a β-sandwich consisting of a five-stranded mixed β-sheet and a short two-stranded antiparallel β-sheet. N2 is rather loosely attached to domain A via its C-terminal portion (cf. Fig. 2). At first glance, the N2 structure is reminiscent of carbohydrate-binding modules (CBMs). However, a comparison with structurally known CBMs did not reveal significant similarities. In fact, CBMs that bind α1–4-linked sugars (tribes CBM-A to C) (34) show different topologies.
N-terminal extensions similar to N2 of MalQ were also observed in other sugar-metabolizing enzymes, such as cellulase CelA from Alicyclobacillus acidocaldarius (PDB code 3H2W) (35) or the structurally and mechanistically related maltosyltransferase GlgE from Streptomyces coelicolor (PDB code 4C4N) (36), but could not be attributed to a discrete function. Basically, subdomain N2 adopts a fibronectin type III (FnIII)-like fold (37). A distinct feature of subdomain N2 is a variation of the characteristic FnIII β-sandwich of two antiparallel β-sheets abe and c′cfg, in which β-strand a associates with β-strand g of the latter sheet in a parallel manner.
Comparison of the Crystal Structures of MalQ in Three Different Ligand States
In the crystal structure of apo-MalQ the active site did not show any electron density for a ligand molecule. Residues of the catalytic triad comprising the nucleophile Asp-448, the acid/base catalyst Glu-496, and the putative transition state stabilizer Asp-548 interact with other residues of the active site pocket as follows: Asp-448 forms a salt bridge with Arg-446, Glu-496 is hydrogen-bonded to the main chain nitrogens of His-449 and Leu-498, and Asp-548 is hydrogen-bonded to Asn-648.
In contrast, interpretable ligand density was observed in the substrate pocket of molecule 1 of the MalQ·maltose complex, which was in accordance with a maltose molecule that occupies subsites −2 and −1 (Fig. 3) (38). Apparently, the observed maltose resulted from the MalQ-catalyzed conversion of maltotriose, the compound that was actually added to the crystallization solution (see “Experimental Procedures” and further below). Crystallographic B values and occupancy refinement of the maltose suggest a ligand occupancy of about 70%. Overall, the substrate pocket is slightly narrower in this case than in the apo structure (Fig. 4). Binding of the first glucose moiety to subsite −2 involves two hydrogen bonds with Asn-648 and a water molecule, as well as van der Waals contacts with Pro-650 and Gly-651. The second glucose in subsite −1 is hydrogen-bonded to Asp-548 and His-547 and also undergoes hydrophobic interactions with Trp-413 and Tyr-201 (Fig. 3A). Notably, with a distance of 4.2 Å the catalytic nucleophile Asp-448 is too far away for a potential attack of C1 in subsite −1. Also, the acid/base catalyst Glu-496 and the putative transition state stabilizer Asp-548 are with 6.0 and 4.8 Å too distant from the O1 atom in subsite −1. Thus, the MalQ·maltose complex represents a catalytically incompetent conformation.
FIGURE 3.

Ligand interactions in the MalQ·maltose (A) and MalQ·AGA complexes (B). Schematic representations of the ligands (black) and all interacting residues (colored as in Fig. 2). MalQ residues that form hydrogen bonds or van der Waals contacts with the respective ligand are indicated as sticks and ellipses, respectively. Water molecules that interact with both the ligand and the protein are depicted as small blue spheres. Numbers inside the sugar rings label the corresponding subsites. Contacts between the sugar substrate and the nucleophilic side chain of Asp-448 (2.6 Å), the acid/base catalyst Glu-496 (2.9 Å), and the putative transition state stabilizer Asp-548 (3.0 Å) are highlighted as red dashed lines. In the MalQ·maltose structure these residues are too far apart (Asp-448, 4.2 Å; Glu-496, 6.0 Å) for contact formation with the substrate. Illustration of the omit FO − FC electron density map (green mesh; contoured at 2.0 and 3.0 σ for maltose and AGA, respectively) with superimposed ligand (stick model) is depicted underneath.
FIGURE 4.
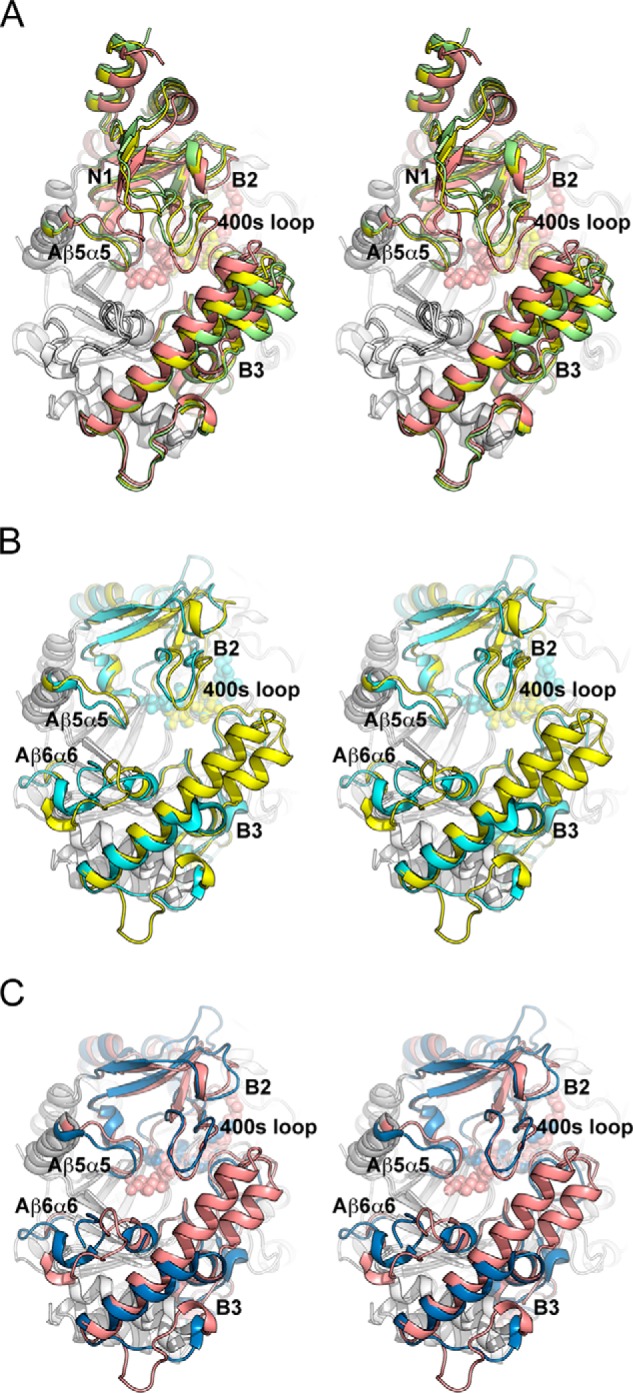
Structural comparison (stereo view) of MalQ with amylomaltase from Thermus sp. A, superposition of the MalQ structures in schematic representation as follows: apo-MalQ (green), MalQ·maltose (yellow), and MalQ·AGA (salmon). Subdomain N2 was omitted for clarity. B, superposition of MalQ·maltose (yellow) and amylomaltase from T. aquaticus (PDB code 1ESW, cyan) (40). Subdomains N1 and N2 of MalQ are not depicted, due to their absence in the Thermus structures. C, similar superposition of MalQ·AGA (salmon) and the amylomaltase of T. thermophilus (PDB code 2OWW, blue) (19). For superposition, the 36 Cα positions of the eight β-strands within the central TIM barrel were used, corresponding to residues 143–148, 181–184, 370–375, 444–447, 492–495, 516–519, 540–543, and 632–635 of MalQ. Ligands are shown as spheres in the same color as the schematics.
Surprisingly, in the crystal structure of MalQ obtained with the tetrasaccharide ligand acarbose electron density revealed seven connected carbohydrate moieties in the active site. This observation can be explained by a MalQ-catalyzed conversion of two acarbose molecules to the elongated (and even more potent) inhibitor AGA upon release of one glucose molecule (see below). The excellent density and the B values of AGA after refinement indicate 100% occupancy of this heptasaccharide analog. However, the B values of the individual sugar units are not uniform. At subsites −2 to +2, the sugar moieties show the lowest B values, with an average of ∼33 Å2, followed by subsites −3 and +3 with ∼41 Å2. The highest B value with 65 Å2 was observed for the cyclohexene moiety at subsite −4, i.e. at the periphery of the substrate pocket.
The MalQ·AGA complex offers the unique opportunity to identify seven sugar-binding subsites of MalQ, four at the nonreducing and three at the reducing end of the substrate pocket (Fig. 3). A large part of subsite −4 is solvent-accessible. Interaction with the first cyclohexene moiety of AGA involves only residues of subdomain B2, a hydrogen bond with Glu-381 and van der Waals (vdW) contacts with Gly-414. Another hydrogen bond is formed with a protein-coordinated water molecule. In subsite −3, the first 6-deoxyglucose moiety of AGA interacts with residues Asn-412 and Thr-652 of subdomains B2 and B3, which form a hydrogen bond and vdW contacts, respectively. Again, a protein-coordinated water molecule forms a hydrogen bond. An OH group at C6 of a natural maltodextrin substrate would probably also permit hydrogen bond formation with Ser-202 of subdomain B1. In subsite −2, the glucose moiety is engaged in five hydrogen bonds, i.e. with Ser-199 and Ser-202 of subdomain B1, Asn-648 and Gly-651 of subdomain B3, as well as a coordinated water molecule. In addition, vdW contacts are formed with Trp-413 and Pro-650 in subdomains B2 and B3, respectively.
The second cyclohexene moiety of AGA is bound to subsite −1. It is sandwiched by Tyr-201 and Trp-413 of subdomains B1 and B2, respectively. Three hydrogen bonds are formed with side chains of His-547 and Asp-548 from subdomain B3. The nucleophile Asp-448 in domain A is with 2.6 Å distance in close contact to the C1 atom of this cyclohexene ring, thus mimicking the transition state of the transglycosylation reaction. Furthermore, Asp-548 is with 3 Å in close contact to the N′ atom of the nonhydrolyzable bond (Fig. 3). Subsite +1 binds the second 6-deoxyglucose moiety of AGA. Four hydrogen bonds are formed with residues Gln-411 and His-449 of subdomain B2 as well as Glu-496 and Leu-498 of domain A. Another hydrogen bond is contributed by a water molecule. An OH group at C6 would most likely also lead to hydrogen bond formation with Gln-411.
Strikingly, in this structure the side chain of Glu-496 adopts a catalytically competent conformation (see further below) accepting two hydrogen bonds, one from the HN′ group of the nonhydrolyzable bond and one from 3-OH of the second 6-deoxyglucose moiety. Asp-404 and Leu-406 of subdomain B2 form subsite +2, where they bind the glucose moiety via hydrogen bonds and vdW contacts, respectively. In addition, two water molecules form hydrogen bonds. The last glucose moiety is bound at subsite +3, which is lined by residues of domain A and subdomain B2. The side chain of Phe-522 nicely stacks the glucose ring, whereas Gly-499 forms a hydrogen bond. Additional vdW contacts are contributed by Ile-405 and Leu-406.
The three crystal structures solved for apo-MalQ, MalQ·maltose, and MalQ·AGA provide a detailed view of the conformational changes that occur during a catalytic cycle (Fig. 4A). To allow qualitative assessment of these changes, the A domains of the three structures were superimposed using the eight β-strands of the TIM barrel. In the second step, the relative positions of the subdomains, treated as rigid bodies, were calculated with respect to the apo structure. In the MalQ·maltose complex subdomains B2+N1 and B3 are rotated by 3° and 2°, respectively, toward the ligand-binding site, thus narrowing the pocket. Nevertheless, the bound maltose in subsites −2 and −1 is still exposed to the solvent. As this conformation of MalQ is neither fully open nor fully closed, we refer to it as “intermediate.”
In contrast, the MalQ·AGA complex shows more pronounced structural differences. Here, subdomains B2+N1 and B3 move by 9° and 7° (relative to domain A in the apo-form) toward the bound transition state analog, which leads to the “closed” conformation (Fig. 4). The central part of the bound inhibitor (subsites −2 to +2) is enclosed by domain A and subdomains B2+B3. This can be described as a V-shaped tunnel, with the catalytic residues at its bottom, where sugar moieties are tightly bound. Although subdomain N1 undergoes the same conformational changes as subdomain B2, it does not form any contacts to the ligand in the MalQ·AGA complex. Minor differences that seem not relevant for the catalytic cycle are also observed for domain N2, which is only loosely attached to the catalytic core.
Comparison of MalQ with Homologous Amylomaltase Structures
A DALI search (39) for similar tertiary structures revealed four amylomaltases from thermophilic Gram-negative bacteria as the closest matches, i.e. those from Thermus aquaticus (PDB codes 1ESW and 1CWY) (21), Thermus brockianus (2X1I) (20), Thermus thermophilus (2OWC) (19), and Aquifex aeolicus (1TZ7). All of these enzymes include ∼500 residues, with ∼24% sequence identity, and show r.m.s.d. values upon superposition with MalQ of 2.2–2.5 Å. The next similar structure is that of the disproportionating enzyme (D-enzyme) from potato (PDB code 1X1N), with 21% identical residues. Thereafter, the sequence identity indicator sharply drops to below 15%, corresponding to members of the GH13 family of glycosyl hydrolases.
The mesophilic MalQ is considerably larger than its thermophilic homologs. Most obvious is its N-terminal extension, i.e. residues 1–138, which do not occur in the Thermus enzymes. The first 128 amino acids belong to domain N, followed by 10 residues of domain A that pack against the N-terminal side of the TIM barrel. The first structurally equivalent residues in domain A occupy positions 142 and 5 in MalQ and the Thermus enzymes, respectively. Furthermore, domain A of MalQ carries a couple of insertions. Like in MalQ, helix Aα6 of the TIM barrel is missing in the Thermus enzymes, whereas the loop that connects strands Aβ6 and Aβ7 is three residues shorter in MalQ and adopts a significantly different conformation.
Nonetheless, the largest structural differences are found in domain B. Considerable insertions are located in the MalQ subdomains B1 and B3, which are extended by 20 and 37 residues, respectively, compared with the Thermus amylomaltases. In contrast, because of the two shorter loops, the catalytically most important subdomain B2 is shorter by seven residues in MalQ, i.e. six residues in B2a and one residue in B2b. These loops interact with each other as well as with the N-terminal subdomain N1. However, both loops are located at the surface of MalQ and do not contribute to the active site (Fig. 2A).
The amylomaltase from T. aquaticus (TaAM) in complex with bound acarbose (PDB code 1ESW) (40) is most similar to the MalQ·maltose structure with an r.m.s.d. of 2.2 Å over 475 Cα positions. Larger differences are only observed in loops preceding strands Aβ5 and Aβ6 as well as in the 400-s (250-s) loop (Fig. 4B). This is in agreement with the previous notion that the crystallized TaAM was trapped in a conformation that does not permit catalysis (19). As in the MalQ·maltose complex, the nucleophile Asp-448 (Asp-293) and the acid/base catalyst Glu-496 (Glu-340) are too far away from the bound substrate/inhibitor for interaction (Fig. 5).
FIGURE 5.
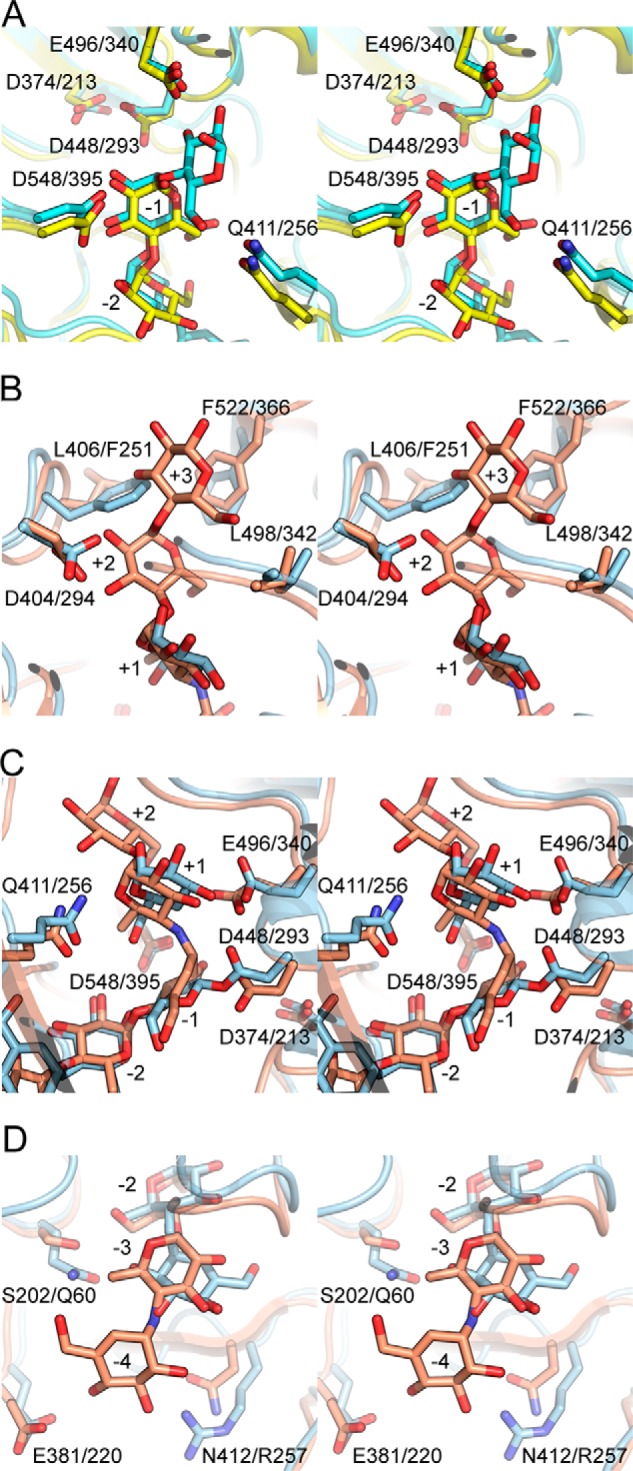
Comparison of MalQ·ligand complexes (stereo view) with those of amylomaltases from Thermus sp. A, MalQ·maltose (yellow) versus TaAM·acarbose (cyan, 1ESW), subsites −2 to −1. B–D, comparison of the subsites of MalQ·AGA (salmon) versus TtAM·AG·4G (blue, 2OWW), subsites +3 to +1 (B), subsites +2 to −2 (C), and subsites −2 to −4 (D). Ligands and relevant residues are shown as sticks in the same color.
In contrast, the two complexes of T. thermophilus with acarviosine-glucose (TtAM·AG; PDB entry 2OWC) and with AG plus 4-deoxyglucose (TtAM·AG/4G; PDB entry 2OWW) (19) are most similar to the MalQ·AGA complex, with an r.m.s.d. of 2.2 Å over 474 Cα positions in each case (Fig. 4C). Differences around subsite −1 between MalQ·AGA and TtAM·AG/4G can be attributed to the fact that these structures represent different states of the catalytic cycle, i.e. with bound transition state analog or covalent intermediate. However, differences around subsite +1 are likely caused by the missing OH group of 4-deoxyglucose, which prevents proper interaction with the acid/base catalyst Glu-496 (Glu-340), resulting in its catalytically incompetent orientation (Fig. 5C).
Even though the backbone of the 400-s (250-s) loop adopts a conformation in MalQ·AGA that seems to narrow subsite +3 compared with TtAM, subsite +3 of MalQ·AGA is even larger and binds a glucose moiety owing to the following effects: (i) an alternative conformation of the Leu-406 (Phe-251) side chain (Fig. 5B) and (ii) a different conformation of loop Aβ5α5 (Fig. 4C). At the opposite end of the substrate pocket, subsite −3 is occupied in a different way in the TtAM·AG structure than in the MalQ·AGA complex; the cyclohexene ring in subsite −3 of TtAM·AG is rotated by 180° with respect to the 6-deoxyglucose of the MalQ·AGA complex. Therefore, the conformation of AG in TtAM would not allow occupation of subsite −4 as observed in MalQ·AGA (Fig. 5D).
Substrate Dependence of Maltodextrin Equilibrium Concentrations
MalQ catalyzes a peculiar transglycosylation reaction among maltodextrins in which, given that hydrolytic side reactions are negligible, the concentration of (i) monosaccharide units, (ii) glycosidic bonds, and (iii) total number of molecules each stays constant over time. Thus, the concentration distribution of all maltodextrin products at chemical equilibrium should only depend on the length of the initial substrate. Examples of MalQ reactions are schematically illustrated in Fig. 1, whereas a larger set of possible MalQ reactions is provided in the supplemental material. To investigate this notion, we performed enzymatic assays with the two substrates maltose and maltotriose as well as with the inhibitor acarbose.
To this end the resulting sugar mixtures were derivatized with dansylhydrazine and analyzed by HPLC at different time points. This method allowed the individual quantification of maltodextrins up to maltopentaose, whereas longer maltodextrins could not be separated from the dansylhydrazine peak. Thus, the relative amount of each oligosaccharide was determined as a fraction of all detectable sugars (Fig. 6). Notably, the measured maltodextrin equilibrium concentrations for maltose as a substrate were found comparable with those obtained previously for radiolabeled maltose (6).
FIGURE 6.

MalQ enzymatic assay. Evolution of maltodextrin product concentrations including glucose, maltose, maltotriose, maltotetraose, and maltopentaose starting from the substrates maltose (A) and maltotriose (B) are plotted over time. C, illustrates the very slow conversion of acarbose by MalQ. The product mixtures were derivatized with dansylhydrazine, separated by HPLC and their relative peak areas were quantified.
Early work on the MalQ reaction using purified maltose as substrate demonstrated a considerable lag phase that led to the conclusion that maltotriose is the shortest possible donor (12), an observation that was not confirmed by others (6, 8). Likewise, such a lag phase was not observed in our enzyme assay. Although this might be explained by the presence of trace amounts of maltotriose or longer maltodextrins in the maltose substrate, addition of 1 μm maltotriose did not speed up the enzymatic reaction.
As could be expected, after the start of the reaction the concentration of the substrate steadily decreased, whereas both glucose and maltodextrin product concentrations increased until the equilibrium was reached (Fig. 6). Comparing the results with the different substrates, this enzyme assay indeed demonstrated that the equilibrium concentrations of glucose and maltodextrins are dependent on the initial average number of glycosidic bonds per substrate molecule (b). It appeared that the amount of glucose produced under these conditions is related to the substrate concentration by the ratio 1 − (b/(b + 1)) (equaling ½ or 50% for maltose and ⅓ or 33% for maltotriose). Intriguingly, in this series the concentration of each maltodextrin product declines by the same factor for each additional glucose unit (Fig. 7). An empirical description of this behavior is given by Equation 1,
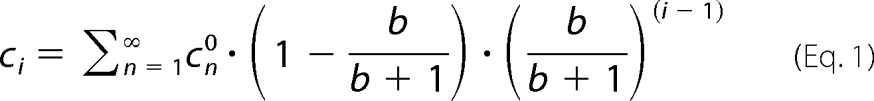 |
where n is the number of glucose units of the substrate, and i is the number of glucose units in each product molecule. Of note, this equation also predicts maltodextrin equilibrium concentrations resulting from substrate mixtures, for example maltose + glucose, maltotriose + glucose, maltose + maltotriose, etc. (data not shown). The same relationship has been described for the disproportionating enzyme 1 (DPE1) from Arabidopsis thaliana (41).
FIGURE 7.

Equilibrium concentrations for different maltodextrin substrates. Experimentally determined maltodextrin equilibrium concentrations comprising G1 (glucose) to G8 (maltooctaose) for the substrates 100 mm maltose, 100 mm maltotriose, and a mixture of 50 mm maltose + 50 mm glucose are shown as gray bars. A, comparison of experimental values with values according to the empirical formula (cf. “Results”; white bars). B, predicted statistical product distribution (cf. “Experimental Procedures”; white striped bars) in comparison with the experimental values (same as in A).
Use of the MalQ inhibitor acarbose as a putative substrate did not yield any detectable reaction product after 4 h in this enzyme assay. However, small amounts of glucose were measured after overnight incubation, although no maltodextrins were detected (Fig. 6). This indicates that AGA of the MalQ·AGA complex can be replaced by acarbose, which is present at high concentrations, and thus new AGA and glucose are synthesized from two acarbose molecules, eventually resulting in slow turnover.
Discussion
The three-dimensional structure of MalQ represents the first example of a mesophilic bacterial amylomaltase. It reveals a very similar catalytic core as the previously solved amylomaltase structures from Thermus species (19–21), with a catalytic triad comprising the nucleophile Asp-448, the acid/base catalyst Glu-496, and the transition state stabilizer Asp-548. In addition, MalQ exhibits an N-terminal extension, consisting of subdomains N1 and N2, which seems unique to amylomaltases from Enterobacteriaceae. However, their function remains elusive.
Interestingly, the three crystal structures of MalQ in different ligand states solved here represent distinct conformations assumed during the catalytic cycle, from empty, over substrate binding/release to the transition state of the reaction. In particular, the MalQ·AGA complex, the first structure of an amylomaltase with a bound transition state analog, has provided crucial insights into the reaction mechanism. First of all, it was possible to elucidate seven subsites for carbohydrate binding that play a role in the recognition of maltodextrins. Notably, binding of glucose units to subsites −3 to −1 involves residues from the three subdomains B1 to B3 (assuming a 6-OH group for the 6-deoxyglucose moiety observed at subsite −3), whereas the remaining subsites are dominated by subdomain B2.
As expected, the number of ligand contacts with subsites −2 to +1, which surround the reactive glycosidic bond, is considerably higher than for the remaining subsites, whose affinities appear to decrease toward the periphery (Fig. 3). The distribution of contacts suggests that longer maltodextrins should have higher affinities for the active site than shorter ones. Moreover, because of the larger number of interactions, the donor subsites seem to be preferentially occupied, which indicates that glycosidic bonds at the center and toward the reducing end of the donor substrate are more likely to be positioned at the active site for transglycosylation than those at the nonreducing end. This notion is also supported bythe structure of the MalQ·maltose complex, in which maltose occupies subsites −2 to −1.
In the apo structure and one of the MalQ·maltose complexes (like in all previously published thermophilic amylomaltase structures), the acid/base catalyst Glu-496 (Glu-340) is hydrogen-bonded to the main chain amides of Leu-498 (Leu-342) and His-449 (His-294), adopting a catalytically incompetent orientation (Fig. 5A). Upon binding of another substrate to acceptor site +1, the side chain carboxylate rotates into its proper position for catalysis. Therefore, the hydrogen bond with Leu-498 is released, and two new ones are formed with the bridging oxygen of the glycosidic bond (equivalent to the N′ nitrogen in AGA) and the 3-OH of the glucose moiety bound to subsite +1, while the hydrogen bond to His-449 is maintained. In contrast, a 4-deoxyglucose as seen bound in the T. thermophilus structure (PDB code 2OWW) seems not sufficient to trigger reorientation of the acid/base catalyst (Fig. 5C). The requirement of a 4-OH group (or glycosidic bond) for activation of the catalytic center explains the extremely low hydrolysis rate that has been measured for MalQ (42), like for the maltodextrin glycosyltransferase from Thermotoga maritima (43), and is also consistent with the results of our enzymatic assay.
In addition, a significant conformational difference of the loop connecting Aβ5 and Aα5 was observed in the MalQ·AGA structure (cf. Fig. 4). The reorientation of Glu-496 (Glu-340) and therefore the breakage of its hydrogen bond with the carbonyl oxygen of Leu-498 (Leu-342) seems to cause this conformational change of the Aβ5-Aα5 loop by up to 3 Å toward subdomain B2. Alternatively, it is possible that binding of a glucose unit to subsite +2 induces the loop movement, which may favor reorientation of Glu-496 (Glu-340) even further than the mere presence of a glucose in subsite +1; this effect could enhance preference for maltose and longer maltodextrins over glucose as the acceptor molecule.
Unexpectedly, functional inhibition of MalQ by acarbose is achieved by the chemical formation of AGA, which can be explained by a double displacement mechanism (Fig. 8). First, an acarbose molecule binds to subsites −3 to +1, such that Glu-496 can protonate the glycosidic bond between the two glucose units (subsites −1 and +1). Then, Asp-448 performs a nucleophilic attack of glucose C1 in subsite −1. This results in a covalent ester intermediate between the acarviosine-glucose in subsites −3 to −1 and Asp-448. Subsequently, the liberated glucose molecule at the reducing end exchanges with a second acarbose molecule, which initially is present at much higher concentrations and binds to subsites +1 to +4. The free 4′-OH group of its cyclohexene moiety at subsite +1 becomes deprotonated by Glu-496, allowing attack of the covalent intermediate at C1; in this way, the second acarbose molecule displaces Asp-448 to finally form acarviosine-glucose-acarbose via a glycosidic bond. This modified heptasaccharide product, which initially occupies subsites −3 to +4, finally seems to reposition within the MalQ active site to occupy subsites −4 to +3. In this new position the acarviosine moiety that stems from the second acarbose molecule is tightly bound to subsites −1 to +1 where it mimics the transition state with its cyclohexene moiety and the nonhydrolyzable secondary amino group (cf. Fig. 8).
FIGURE 8.
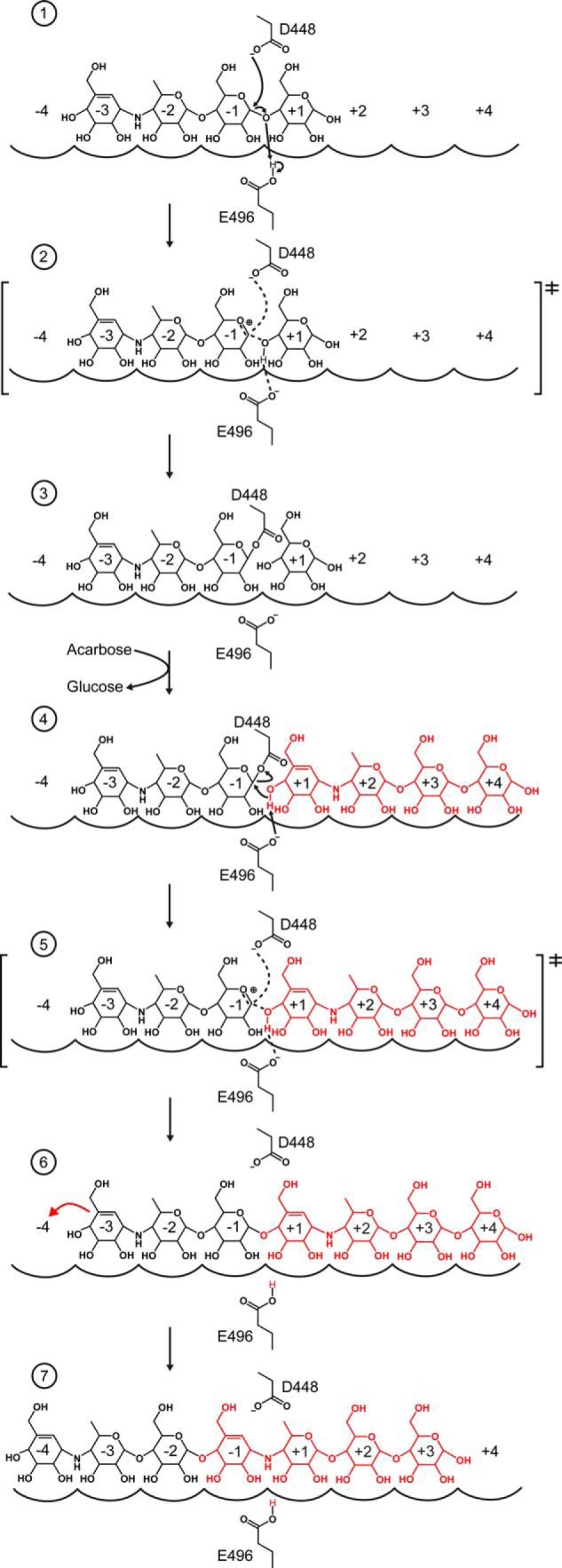
Mechanism of MalQ inhibition by acarbose. The two acarbose molecules that are converted to AGA by MalQ are colored black and red. MalQ subsites are indicated by arches and numbered −4 to +4. The catalytic residues Asp-448 and Glu-496 are shown as sticks. The reaction proceeds in seven stages (see text), each shown in one panel, which can be deduced from distinct crystal structures. Panel 1, MalQ·maltose and TaAM·acarbose (PDB code 1ESW); panel 2, MalQ·AGA; panels 3 and 4, TtAM·AG and TtAM·AG·4G (PDB codes 2OWC and 2OWW); panel 5, MalQ·AGA; panel 6, MalQ·maltose and TaAM·acarbose (PDB code 1ESW); and panel 7, MalQ·AGA.
Our enzyme assay demonstrates that the maltodextrin product spectrum of MalQ critically depends on the initial total number of glycosidic bonds in the substrate molecules. The more glycosidic bonds a substrate contains the further is the equilibrium of the reaction shifted toward longer maltodextrins and lower amounts of the glucose monosaccharide. Notably, a simple statistical simulation of the MalQ catalyzed reaction revealed significantly different equilibrium concentrations for glucose and maltodextrins (Fig. 7B and supplemental Table 1). In all experiments, the measured amount of glucose was lower, whereas the amounts of maltodextrins were higher than expected from a purely statistical distribution. First, this is consistent with an extremely low hydrolysis rate of the covalent intermediate. Second, increased concentrations of longer maltodextrins such as maltotetraose and maltopentaose suggest differences in the Gibbs free energies of maltodextrin glycosidic bonds. However, it has been demonstrated before that the enthalpy of all glycosidic bonds should be comparable (44). Thus, according to the second law of thermodynamics, the observed equilibria must be driven by maximization of entropy, which is in line with previous investigations of 4-α-glucanotransferase from potato and the DPE1 from A. thaliana (41, 45).
According to Equation 1, the longer a maltodextrin substrate, the less glucose is formed at equilibrium, which also makes sense from a physiological perspective. To quickly build up longer maltodextrins inside the cell, substrates of MalQ should already contain as many glycosidic bonds as possible, which, assuming subsequent degradation via phosphorolysis through MalP, eventually leads to a higher yield of ATP. The periplasmic α-amylase MalS preferentially cleaves maltohexaose units from longer maltodextrins to allow efficient cytoplasmic import by the maltose/maltodextrin ATP-binding cassette transporter MalFGK2 (2). Therefore, after intracellular MalQ action, the glucose concentration at equilibrium should be only 1/6 of the original maltohexaose concentration, in contrast to an expected ratio of ½ for maltose as a substrate. Apart from that, if glucose is withdrawn via phosphorylation by glucokinase, the equilibrium shifts toward even longer maltodextrins (higher degree of polymerization), which further favors phosphorolysis by MalP.
E. coli strains with a deletion of the phosphotransferase system for glucose (−ptsG) and glucokinase (−glk) are unable to phosphorylate glucose (8). Under these circumstances, phosphoglucose can only be formed by MalP-catalyzed phosphorolysis of maltodextrins. Interestingly, such cells are unable to grow on maltose, which was previously interpreted as an inhibition of MalQ by high internal glucose levels and, consequently, lack of substrate for MalP. In our MalQ enzyme assay, the final concentration of glucose raises up to ∼130 mm at equilibrium (65% if a mixture of 100 mm glucose and 100 mm maltose is used as substrate), without detectable enzyme inhibition. This glucose concentration is even higher than physiological intracellular glucose concentrations (56 mm) measured in a comparable genetic background (3). Hence, our interpretation for the inability of these mutant strains to grow on maltose as the sole carbon source is the unfavorable equilibrium concentration of maltopentaose, the shortest substrate of MalP (4), as well as longer maltodextrins, eventually leading to high intracellular glucose concentrations.
Author Contributions
A. Sc. conceived and coordinated the research. S. C. W. performed the experiments. S. C. W., A. Sc., and A. Sk. analyzed the data, wrote the paper, and approved the final version of the manuscript.
Supplementary Material
Acknowledgment
We thank the Helmholtz-Zentrum Berlin for allocation of synchrotron radiation beamtime.
This work was supported by Deutsche Forschungsgemeinschaft Grant SCHI1105/2-1 and Helmholtz-Zentrum Berlin. The authors declare that they have no conflict of interest with the contents of this article.

This article contains supplemental material.
The atomic coordinates and structure factors (codes 4S3P, 4S3Q, and 4S3R) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- AGA
- acarviosine-glucose-acarbose
- AG
- acarviosine-glucose
- BME
- β-mercaptoethanol
- r.m.s.d.
- root mean square deviation
- SEC
- size exclusion chromatography
- SeMet
- l-selenomethionine
- TaAM
- amylomaltase from Thermus aquaticus
- TtAM
- amylomaltase from Thermus thermophilus
- 4G
- 4-deoxyglucose
- PDB
- Protein Data Bank
- vdW
- van der Waals.
References
- 1. Boos W., Shuman H. (1998) Maltose/maltodextrin system of Escherichia coli: transport, metabolism, and regulation. Microbiol. Mol. Biol. Rev. 62, 204–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Freundlieb S., Ehmann U., Boos W. (1988) Facilitated diffusion of p-nitrophenyl-α-d-maltohexaoside through the outer membrane of Escherichia coli. Characterization of LamB as a specific and saturable channel for maltooligosaccharides. J. Biol. Chem. 263, 314–320 [PubMed] [Google Scholar]
- 3. Xia T., Han Q., Costanzo W. V., Zhu Y., Urbauer J. L., Eiteman M. A. (2015) Accumulation of d-glucose from pentoses by Escherichia coli. Appl. Environ. Microbiol. 81, 3387–3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Becker S., Palm D., Schinzel R. (1994) Dissecting differential binding in the forward and reverse reaction of Escherichia coli maltodextrin phosphorylase using 2-deoxyglucosyl substrates. J. Biol. Chem. 269, 2485–2490 [PubMed] [Google Scholar]
- 5. O'Reilly M., Watson K. A., Schinzel R., Palm D., Johnson L. N. (1997) Oligosaccharide substrate binding in Escherichia coli maltodextrin phosphorylase. Nat. Struct. Biol. 4, 405–412 [DOI] [PubMed] [Google Scholar]
- 6. Dippel R., Boos W. (2005) The maltodextrin system of Escherichia coli: metabolism and transport. J. Bacteriol. 187, 8322–8331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schwartz M. (1987) in Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology (Neidhardt F. C., ed) pp. 1482–1502, American Society for Microbiology, Washington, D. C. [Google Scholar]
- 8. Lengsfeld C., Schönert S., Dippel R., Boos W. (2009) Glucose- and glucokinase-controlled mal gene expression in Escherichia coli. J. Bacteriol. 191, 701–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dippel R., Bergmiller T., Böhm A., Boos W. (2005) The maltodextrin system of Escherichia coli: glycogen-derived endogenous induction and osmoregulation. J. Bacteriol. 187, 8332–8339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ehrmann M., Boos W. (1987) Identification of endogenous inducers of the mal regulon in Escherichia coli. J. Bacteriol. 169, 3539–3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Park J. T., Shim J. H., Tran P. L., Hong I. H., Yong H. U., Oktavina E. F., Nguyen H. D., Kim J. W., Lee T. S., Park S. H., Boos W., Park K. H. (2011) Role of maltose enzymes in glycogen synthesis by Escherichia coli. J. Bacteriol. 193, 2517–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Palmer T. N., Ryman B. E., Whelan W. J. (1976) The action pattern of amylomaltase from Escherichia coli. Eur. J. Biochem. 69, 105–115 [DOI] [PubMed] [Google Scholar]
- 13. Wiesmeyer H., Cohn M. (1960) The characterization of the pathway of maltose utilization by Escherichia coli. II. General properties and mechanism of action of amylomaltase. Biochim. Biophys. Acta 39, 427–439 [DOI] [PubMed] [Google Scholar]
- 14. Monod J., Torriani A. M. (1950) De l'amylomaltase d'Escherichia coli. Ann. Inst. Pasteur. 78, 65–77 [PubMed] [Google Scholar]
- 15. Wiesmeyer H., Cohn M. (1960) The characterization of the pathway of maltose utilization by Escherichia coli. I. Purification and physical chemical properties of the enzyme amylomaltase. Biochim. Biophys. Acta 39, 417–426 [DOI] [PubMed] [Google Scholar]
- 16. Pugsley A. P., Dubreuil C. (1988) Molecular characterization of malQ, the structural gene for the Escherichia coli enzyme amylomaltase. Mol. Microbiol. 2, 473–479 [DOI] [PubMed] [Google Scholar]
- 17. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. MacGregor E. A., Janecek S., Svensson B. (2001) Relationship of sequence and structure to specificity in the α-amylase family of enzymes. Biochim. Biophys. Acta 1546, 1–20 [DOI] [PubMed] [Google Scholar]
- 19. Barends T. R., Bultema J. B., Kaper T., van der Maarel M. J., Dijkhuizen L., Dijkstra B. W. (2007) Three-way stabilization of the covalent intermediate in amylomaltase, an α-amylase-like transglycosylase. J. Biol. Chem. 282, 17242–17249 [DOI] [PubMed] [Google Scholar]
- 20. Jung J. H., Jung T. Y., Seo D. H., Yoon S. M., Choi H. C., Park B. C., Park C. S., Woo E. J. (2011) Structural and functional analysis of substrate recognition by the 250s loop in amylomaltase from Thermus brockianus. Proteins 79, 633–644 [DOI] [PubMed] [Google Scholar]
- 21. Przylas I., Tomoo K., Terada Y., Takaha T., Fujii K., Saenger W., Sträter N. (2000) Crystal structure of amylomaltase from Thermus aquaticus, a glycosyltransferase catalysing the production of large cyclic glucans. J. Mol. Biol. 296, 873–886 [DOI] [PubMed] [Google Scholar]
- 22. Skerra A. (1994) Use of the tetracycline promoter for the tightly regulated production of a murine antibody fragment in Escherichia coli. Gene 151, 131–135 [DOI] [PubMed] [Google Scholar]
- 23. Mueller U., Darowski N., Fuchs M. R., Förster R., Hellmig M., Paithankar K. S., Pühringer S., Steffien M., Zocher G., Weiss M. S. (2012) Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron. Radiat. 19, 442–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pape T., Schneider T. R. (2004) HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J. Appl. Crystallogr. 37, 843–844 [Google Scholar]
- 26. Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 27. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Painter J., Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 30. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Avigad G. (1977) Dansylhydrazine as a fluorimetric reagent for thin-layer chromatographic analysis of reducing sugars. J. Chromatogr. 139, 343–347 [DOI] [PubMed] [Google Scholar]
- 33. Alber T., Banner D. W., Bloomer A. C., Petsko G. A., Phillips D., Rivers P. S., Wilson I. A. (1981) On the three-dimensional structure and catalytic mechanism of triose-phosphate isomerase. Philos. Trans. R. Soc. Lond. B Biol. Sci. 293, 159–171 [DOI] [PubMed] [Google Scholar]
- 34. Carvalho C. C., Phan N. N., Chen Y., Reilly P. J. (2015) Carbohydrate-binding module tribes. Biopolymers 103, 203–214 [DOI] [PubMed] [Google Scholar]
- 35. Eckert K., Vigouroux A., Lo Leggio L., Moréra S. (2009) Crystal structures of A. acidocaldarius endoglucanase Cel9A in complex with cello-oligosaccharides: strong −1 and −2 subsites mimic cellobiohydrolase activity. J. Mol. Biol. 394, 61–70 [DOI] [PubMed] [Google Scholar]
- 36. Syson K., Stevenson C. E., Rashid A. M., Saalbach G., Tang M., Tuukkanen A., Svergun D. I., Withers S. G., Lawson D. M., Bornemann S. (2014) Structural insight into how Streptomyces coelicolor maltosyl transferase GlgE binds α-maltose 1-phosphate and forms a maltosyl-enzyme intermediate. Biochemistry 53, 2494–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bork P., Holm L., Sander C. (1994) The immunoglobulin fold. Structural classification, sequence patterns and common core. J. Mol. Biol. 242, 309–320 [DOI] [PubMed] [Google Scholar]
- 38. Davies G. J., Wilson K. S., Henrissat B. (1997) Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 321, 557–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Przylas I., Terada Y., Fujii K., Takaha T., Saenger W., Sträter N. (2000) X-ray structure of acarbose bound to amylomaltase from Thermus aquaticus. Implications for the synthesis of large cyclic glucans. Eur. J. Biochem. 267, 6903–6913 [DOI] [PubMed] [Google Scholar]
- 41. Kartal O., Mahlow S., Skupin A., Ebenhöh O. (2011) Carbohydrate-active enzymes exemplify entropic principles in metabolism. Mol. Syst. Biol. 7, 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ruzanski C., Smirnova J., Rejzek M., Cockburn D., Pedersen H. L., Pike M., Willats W. G., Svensson B., Steup M., Ebenhöh O., Smith A. M., Field R. A. (2013) A bacterial glucanotransferase can replace the complex maltose metabolism required for starch to sucrose conversion in leaves at night. J. Biol. Chem. 288, 28581–28598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liebl W., Feil R., Gabelsberger J., Kellermann J., Schleifer K. H. (1992) Purification and characterization of a novel thermostable 4-α-glucanotransferase of Thermotoga maritima cloned in Escherichia coli. Eur. J. Biochem. 207, 81–88 [DOI] [PubMed] [Google Scholar]
- 44. Goldberg R. N., Bell D., Tewari Y. B., McLaughlin M. A. (1991) Thermodynamics of hydrolysis of oligosaccharides. Biophys. Chem. 40, 69–76 [DOI] [PubMed] [Google Scholar]
- 45. Nakatani H. (1999) Monte Carlo simulation of 4-α-glucanotransferase reaction. Biopolymers 50, 145–151 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.