

Background: TPCs are regulated by NAADP and other factors.
Results: NAADP-induced Ca2+ release from acidic stores evokes depolarizing currents in pancreatic β cells. Inhibition of NAADP signaling or TPC knock out attenuates Ca2+ signaling and insulin secretion.
Conclusion: NAADP-evoked Ca2+ release enhances β cell excitability and insulin secretion in response to glucose or sulfonylureas.
Significance: NAADP signaling pathways offer novel therapeutic targets for diabetes treatment.
Keywords: diabetes, endosome, insulin, lysosome, nicotinic acid adenine dinucleotide phosphate (NAADP), TPC1, TPC2
Abstract
Pancreatic β cells are electrically excitable and respond to elevated glucose concentrations with bursts of Ca2+ action potentials due to the activation of voltage-dependent Ca2+ channels (VDCCs), which leads to the exocytosis of insulin granules. We have examined the possible role of nicotinic acid adenine dinucleotide phosphate (NAADP)-mediated Ca2+ release from intracellular stores during stimulus-secretion coupling in primary mouse pancreatic β cells. NAADP-regulated Ca2+ release channels, likely two-pore channels (TPCs), have recently been shown to be a major mechanism for mobilizing Ca2+ from the endolysosomal system, resulting in localized Ca2+ signals. We show here that NAADP-mediated Ca2+ release from endolysosomal Ca2+ stores activates inward membrane currents and depolarizes the β cell to the threshold for VDCC activation and thereby contributes to glucose-evoked depolarization of the membrane potential during stimulus-response coupling. Selective pharmacological inhibition of NAADP-evoked Ca2+ release or genetic ablation of endolysosomal TPC1 or TPC2 channels attenuates glucose- and sulfonylurea-induced membrane currents, depolarization, cytoplasmic Ca2+ signals, and insulin secretion. Our findings implicate NAADP-evoked Ca2+ release from acidic Ca2+ storage organelles in stimulus-secretion coupling in β cells.
Introduction
Pancreatic β cells are electrically excitable, and in response to elevated blood glucose concentrations, oscillatory bursts of Ca2+ action potentials mediated by VDCCs5 are elicited. These drive cytosolic Ca2+ ([Ca2+]i) oscillations that, in turn, induce pulsatile insulin release (1), and defects in their generation may be associated with the loss of glucose homeostasis in type-2 diabetes (2). Glucose-evoked membrane depolarization results from the closure of ATP-dependent potassium (KATP) channels, octameric complexes of sulfonylurea receptor 1 (SUR1) and inwardly rectifying Kir6.2 potassium channel subunits (3), and inactivating or activating mutations in the KATP channel (where the KATP channel is the ATP-dependent potassium channel) subunits lead to congenital hyperinsulinemia (4) or neonatal diabetes (5), respectively. However, KATP channel closure alone is not sufficient to depolarize the membrane to threshold, and activation of an additional depolarizing current has also been postulated (6, 7). The existence of an additional glucose-regulated membrane current in β cells is suggested by the finding that mice lacking functional KATP channels (8), like Sur1 or Kir6.2 knock-out mice), are not hypoglycemic, and islets from adult knock-out mice are still capable of responding to glucose with electrical activity, [Ca2+]i oscillations, and insulin secretion (9–11). The identity and regulation of this membrane conductance remain an enigma.
In contrast to the Ca2+ influx across the plasma membrane that plays a critical role in effecting insulin granule exocytosis, Ca2+ release from intracellular stores has been thought to play a modulatory rather than a triggering role in stimulus-secretion coupling in the pancreatic β cell. [Ca2+]i oscillations in response to glucose are modulated by the uptake and release of Ca2+ from endoplasmic reticulum (ER) Ca2+ stores (12) and also from acidic Ca2+ storage organelles (13). In addition, several incretins, such as glucagon-like peptide 1 and acetylcholine, are thought to enhance insulin secretion by mechanisms that are, in part, dependent on Ca2+ release from intracellular stores via intracellular messengers such as cAMP and inositol trisphosphate (IP3) (14, 15). However, recent studies have suggested that the newly discovered Ca2+-mobilizing messenger NAADP might play an important role in β cell Ca2+ signaling (16–24).
NAADP, the most potent of the Ca2+-mobilizing messengers described, has been shown to mediate local Ca2+-signaling events by releasing Ca2+ from acidic, endolysosomal Ca2+ stores in several vertebrate and invertebrate cells (25–27), and appears to be a critical trigger for many Ca2+-signaling events (26–28). The most prominent target Ca2+ release channels for NAADP have recently been identified as the two members of the endolysosomal two-pore channel family, TPC1 and TPC2 (29–37). Some studies report a lack of NAADP sensitivity in isolated lysosomes (23, 38), which may reflect technical issues, but also may be due in part to loss of NAADP binding to an accessory protein (39–42) forming part of a multiprotein signaling complex in endolysosomal membranes (27, 43–45). NAADP-induced Ca2+ release in MIN6 cells can be disrupted by the lysomotropic agent glycyl-l-phenylalanine-β-naphthylamide (GPN) or bafilomycin, which disrupts acidic store Ca2+ storage implicating lysosomally related organelles as the principal target for NAADP in these cells (19, 20, 23). In the pancreatic β cell line MIN6, and primary mouse β cells, glucose increases NAADP synthesis and hence intracellular levels (18, 20, 22), consistent with its role as an intracellular messenger. NAADP introduced into mouse pancreatic β cells via a patch pipette was found to evoke a series of oscillatory plasma membrane currents, which were blocked by the NAADP antagonist Ned-19 (21) and were abolished in pancreatic β cells prepared from Tpcn2−/− mice (29). Furthermore, increasing concentrations of Ned-19 abolished glucose-evoked Ca2+ spiking in mouse pancreatic β cells, suggesting an important role for NAADP in stimulus-response coupling in these cells (21). This finding is consistent with our earlier study showing that prior desensitization of NAADP-sensitive Ca2+ release mechanisms block subsequent glucose-evoked Ca2+ signals in MIN6 cells (18).
Glucose (18) and glucagon like-peptide 1 (18, 20) have both been reported to increase β cell NAADP levels, effects that may be partially dependent on the ADP-ribosyl cyclase, CD38 (20, 22). At present, ADP-ribosyl cyclases, including CD38, are the only characterized enzymes that have been demonstrated to catalyze the synthesis of NAADP, using NADP and nicotinic acid as substrates by a base-exchange mechanism (46, 47). It has been suggested that glucose stimulation increases the internalization of CD38 involving cytoskeletal changes (22) with NAADP synthetic sites associated with acidic organelles (20). Furthermore, glucose-evoked Ca2+ signals and insulin secretion are impaired in mouse Cd38−/− pancreatic β cells, and Cd38−/− mice show glucose intolerance (48), and human CD38 autoantibodies and CD38 mutations have been shown to be associated with type-2 diabetes (49, 50). Recently, extracellular NAADP was found to be transported into mouse pancreatic β cells where it evoked Ca2+ release from acidic stores (24). Remarkably, intraperitoneal injections of NAADP were found to restore glucose-evoked insulin secretion in the db/db mouse model of type-2 diabetes and to ameliorate blood glucose regulation (24).
Here, we have used the cell-permeant analogue of NAADP, NAADP-AM (51), the selective cell-permeant NAADP antagonist Ned-19 (21), Tpcn1−/− and Tpcn2−/− mice (29), to explore a possible role for TPC-dependent NAADP-induced Ca2+ release from acidic stores in glucose-induced [Ca2+]i increases and insulin secretion in primary mouse β cells.
Experimental Procedures
Preparation of Islets of Langerhans and Islet β Cell Clusters
Islets of Langerhans were aseptically isolated by collagenase digestion of the pancreases of 8–10-week-old male mice of the following strains: CD1, Tpcn2+/+ and Tpcn2−/− (29), Tpcn1+/+ and Tpcn1−/− (52), with Tpcn mice in a B6;129 background. All mice were killed by cervical dislocation and age- and sex-matched (and for the latter two, background strain-matched). Except for the hormone release measurements (for which intact islets were used), clusters of islet β cells and single β cells were prepared by dispersing islets in a Ca2+-free medium and cultured on circular coverslips for 1–4 days in RPMI 1640 culture medium (GIBCO, Paisley, UK) containing 10% heat-inactivated fetal calf serum, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 10 mm glucose.
[Ca2+]i Measurements
Cultured clusters of islet cells were loaded with 1 μm Fura PE3-AM or Fura 2-AM (Teflabs, Austin, TX) for 60 min at 37 °C in a bicarbonate-buffered solution containing 10 mm glucose. The coverslip was then used as the bottom of a temperature-controlled perifusion chamber (Bioscience Tools, San Diego) mounted on the stage of an inverted microscope. The flow rate was 1.5 ml/min, and the temperature within the chamber was 37 °C. [Ca2+]i was measured at dual-wavelength (340 and 380 nm) excitation spectrofluorimetry, using a CCD camera (Photon Technologies International, Princeton, NJ) to capture the emitted fluorescence at 510 nm. When [Ca2+]i was simultaneously measured in a voltage-clamped single cell, the patch pipette contained 100 μm Fura 2 pentapotassium salt, and the emitted fluorescence was captured at 510 nm using a photomultiplier (Photon Technologies International, Princeton, NJ).
Measurement of Flavine Adenine Dinucleotide (FAD) Fluorescence
Cultured clusters of β cells were preincubated for 60 min at 37 °C in a control medium containing 3 mm glucose and then transferred to the stage of an LSM 510 confocal microscope. After a further 10 min of perfusion by 3 mm glucose, the recording was started. The oxidized form of the FAD was excited at 488 nm. Emitted fluorescence was collected with a 505-nm long-pass filter.
Electrophysiology
All patch clamp measurements were carried out using a multiclamp 700B patch clamp amplifier and the software pClamp 9 (Axon Instruments, Foster City, CA). When using the perforated whole-cell mode of the patch clamp technique, the electrical contact was established by adding the pore-forming antibiotic, amphotericin B, to the pipette solution. Amphotericin (stock solution of 60 mg/ml in DMSO) was used at a final concentration of 300 μg/ml. The tip of the pipette was filled with antibiotic-free solution, and the pipette was then back-filled with amphotericin-containing solution. The voltage clamp was considered satisfactory when the access resistance was <30 megohms and stable. In the standard whole-cell configuration, the access resistance was <15 megohms. Patch pipettes were pulled from borosilicate glass capillaries (World Precision Instruments, Hertfordshire, UK); they had resistances of 3–5 megohms when filled with intracellular solution.
All experiments were carried out on single β cells. Two criteria were used to identify β cells. The capacitance of mouse α-, δ-, and β cells has been reported to be 4.4, 5, and 7.4 picofarads, respectively. Therefore, only large cells with a capacitance of >5 picofarads were chosen for this study. The average capacitance was 7.6 ± 0.2 picofarads. After verification of the capacitance, a depolarizing protocol was applied to identify the properties of the voltage-dependent Na+ current, which is known to be largely inactivated at resting potential in β cells but not in α and δ cells. Thus, cells in which a large Na+ current could be activated by a small depolarizing pulse from a holding potential of −70 mV were discarded. By contrast, cells that displayed a Na+ current only after a hyperpolarizing pulse to −140 mV were considered to be β cells and were used for the experiments.
The whole-cell KATP channel current (IK(ATP)) was monitored by 100-ms duration pulses of ±20 mV from a holding potential of −70 mV. Whole-cell Ca2+ currents were recorded by depolarizing the plasma membrane with a 100-ms pulse from −80 to 10 mV.
Solutions
The medium used for the isolation of islets and for all experiments was a bicarbonate-buffered solution containing (in mm) the following: 120 NaCl, 4.8 KCl, 2.5 CaCl2, 1.2 MgCl2, and 24 NaHCO3. It was gassed with O2/CO2 (94:6) to maintain pH 7.4 at 37 °C. Except for the electrophysiological experiments, it was supplemented with 1 mg/ml BSA (fraction V, Roche Applied Science, Mannheim, Germany). When the concentration of KCl was increased, the concentration of NaCl was correspondingly decreased to keep the osmolarity of the medium unchanged.
For electrophysiological measurements of IK (ATP), the standard extracellular solution contained (in mm) the following: 140 NaCl, 4.8 KCl, 2.5 CaCl2, 1.2 MgCl2, 5 HEPES (pH adjusted to 7.40 with NaOH), and 10 mm glucose. These solutions were gassed with O2/CO2 (94:6%). For the perforated patch measurements of membrane currents and potential, the pipette solution contained (in mm) the following: 70 K2SO4, 10 NaCl, 10 KCl, 3.7 MgCl2, and 5 HEPES (pH adjusted to 7.1 with KOH). For whole Ca2+ current, the pipette solution consisted of (in mm) the following: Cs2SO4 substituted for K2SO4. For NAADP infusion experiments, the pipette solution contained (in mm) the following: 125 K+ gluconate, 10 KCl, 10 NaCl, 10 KCl, 1 MgCl2, 3 Mg-ATP, 0.1 Na-GTP, and 5 HEPES (pH adjusted to 7.1 with KOH). In Fig. 3A, 100 μm Fura 2 pentapotassium was added.
FIGURE 3.
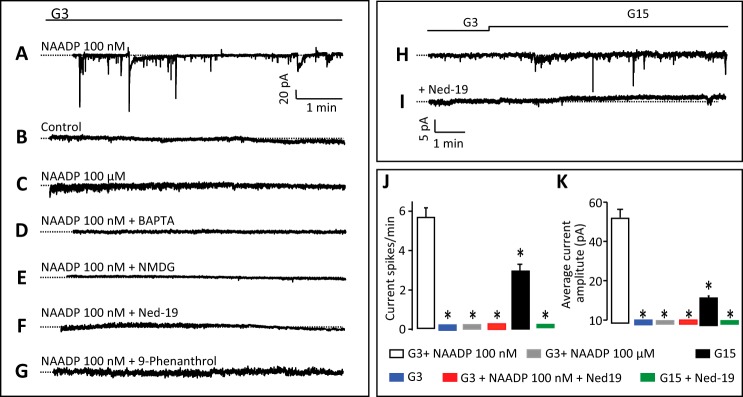
NAADP evokes Ca2+-dependent inward currents in pancreatic β cells. A, inward currents at −70 mV evoked by the infusion of 100 nm NAADP through a patch pipette in the standard whole-cell configuration. B–G, currents are absent under control conditions (i.e. in the absence of NAADP in the pipette solution (B); in the presence of 100 μm NAADP in the pipette solution (C); in the presence of 10 mm Ca2+ chelator BAPTA alongside 100 nm NAADP in the patch pipette (D); when the positive ions in the extracellular solution were replaced with N-methyl-d-glucamine (E); in the presence of extracellular Ned-19 (100 μm) (F); and the presence of the TRPM4 channel inhibitor 9-phenanthrol (10 μm) (G)). The dotted lines represent the zero current level. H and I, glucose-induced inward currents in single cells clamped at −70 mV in the absence (H) or presence (I) of Ned-19 (100 μm). The dotted lines represent the zero current level. J and K, quantification of data from A to I and showing frequencies (J) and amplitudes (K) of currents. Traces/histograms are representative of or were obtained from 10 (A), 18 (B), 11 (C), 8 (D), 9 (E), 11 (F), 5 (G), 4 (H), 4 (I). *, p < 0.05.
Gene Expression Analysis
Total RNA was extracted from mice pancreas and liver following the RNeasy QiaRNA extraction procedure, including a DNase treatment (Qiagen). RT-PCR was performed in a reaction containing extracted RNA, the SuperScriptII One-Step RT-PCR system with Platinum Taq (Invitrogen), and the following gene-specific primers: Tpcn2 exons 4–8 amplicon (forward, 5′-gggcttcatcattttcctga-3′; reverse, 5′-ttgttggaagtcgtcagcag-3′). The following parameters were used for RT: 50 °C (30 min), 94 °C (2 min); PCR, 30 cycles of 94 °C (15 s), 57 °C (30 s), and 68 °C (1 min), followed by a final extension at 68 °C (10 min). For gene expression analysis in β cells, cDNA was produced from total RNA using the High Capacity cDNA reverse transcription kit (Applied Biosystems), and PCR was performed with the following gene-specific primers for Tpcn2 exons 22–25 amplicon (forward, 5′-aacgtgatggtggtgaacaat-3′; reverse, 5′-gtctgccaaagctacaccttg-3). The following parameters were used for PCR: 30 cycles of 95 °C (30 s), 53 °C (30 s), and 72 °C (1 min), followed by a final extension at 72 °C (10 min). Tpcn1 mRNA expression was analyzed as described previously (52).
Insulin Secretion
Islets were isolated from mice and cultured in RPMI 1640 medium overnight before insulin secretion was assessed. Insulin secretion was measured during 1-h static incubations in Krebs-Ringer Buffer (KRB) containing (in mm) the following: 18.5 NaCl, 2.54 CaCl2, 1.19 KH2PO4, 4.74 KCl, 25 NaHCO3, 1.19 MgSO4, and 10 HEPES (pH 7.4). Samples of the supernatant were assayed for insulin using a mouse insulin ELISA kit (Mercodia, Sweden). Where Ned-19 was used, islets were preincubated for 5 min with the drug prior to the addition of secretagogues.
Glucose Tolerance Tests
Male Tpcn1−/− (Tpcn1tm1Dgen) (52) and Tpcn2−/− (Tpcn2Gt(YHD437)Byg) (29) mice and strain-matched wild types aged 66–76 days were fasted overnight and then given 2 g/kg intraperitoneal glucose (in the form of an autoclaved 20% glucose solution). Blood samples taken from the tail vein at 0, 15, 30, 60, and 120 min were analyzed with an Accu-Chek Compact Plus glucose monitor. Mean values for each time were compared using the Student's t test, with p < 0.05 taken as significant.
Insulin Secretion from Whole Pancreata
Pancreatic perfusions were performed within 15 min of cervical dislocation in 80–100-day-old mice essentially as described elsewhere (53). At the end of the perfusion, the pancreas was dissected and transferred in acid/ethanol (ethanol/H2O/HCl, 52:17:1). All samples were then stored at −20 °C. Only experiments with an output rate greater than 200 ml/min were assayed for insulin. The hormone assay was done using a commercially available RIA kit (Millipore, Watford, UK).
TPC2 Localization Studies
Human islets were prepared from beating heart donors with appropriate ethical permission and consents as described previously (54). Islets were dissociated into single cells and, after fixation in 4% (w/v) paraformaldehyde, were treated with antibodies as below (55). Rabbit anti-TPC2 antibody (1:150) was revealed with Alexa 568-conjugated secondary antibody (1:1500, Invitrogen, Paisley, UK). Guinea pig anti-insulin (1:300, DAKO, Ely, UK), goat anti-EEA1 (1:150, Santa Cruz Biotechnology, Santa Cruz, CA), and rat anti-LAMP-1 (1:150, Santa Cruz Biotechnology) were revealed with Alexa 488 secondary antibodies (1:1500, Invitrogen). Murine MIN6 clonal β cells (56) were transfected with plasmid encoding TPC2-mCherry using Lipofectamine 2000, and 48 h later were fixed, stained, and imaged as above. Images were captured using a Zeiss Axiovert 200 M spinning disc confocal imaging system (×40 oil immersion objective corrected for chromatic aberration; Hamamatsu ImageEM 9100-13 back-illuminated EM-CCD camera) with illumination (491 and 568 nm) provided by solid state lasers (Crystal Laser, NV) using a laser merge module (Spectral Applied Physics, Ontario, Canada) (57).
Electron Microscopy
The islet preparations were fixed in 2% paraformaldehyde and 2% glutaraldehyde in cacodylate buffer in the presence of calcium chloride, washed, post-fixed in 1% osmium tetroxide, and contrast-enhanced by staining en bloc with uranyl acetate. Sections were further contrasted with Reynold's lead.
Chemicals
Ned-19, Ned-20 (21), and NAADP-AM (51) were synthesized in-house as described previously. Bafilomycin was from LC Laboratories, and other chemicals were from Sigma.
Results
Characterization of NAADP-AM-evoked Ca2+ Release
We first investigated the effects of NAADP on intracellular Ca2+ concentrations ([Ca2+]i) in primary mouse pancreatic β cells using the membrane-permeant NAADP analogue NAADP-AM (51). Given that the concentration-response curve is bell-shaped in mammalian cells (58), different NAADP-AM concentrations were tested to optimize the response (Fig. 1A) (58). 10 nm NAADP-AM gave only a small response, whereas 10 μm NAADP-AM gave no response at all. An intermediate concentration of 60 nm was found to give the most consistent and largest Ca2+ response and was used for subsequent studies. In 8/10 clusters of β cells, extracellular application of NAADP-AM (60 nm), in the presence of low glucose (3 mm), evoked delayed [Ca2+]i increases (Fig. 1A). The peak was reached >15 min after application of NAADP-AM (Fig. 1F). To assess the role of the ER in the action of NAADP-AM (59), we treated the cells with the SERCA pump inhibitor thapsigargin in the absence of extracellular Ca2+ to remove functional ER Ca2+ stores. In keeping with earlier observations (13), the NAADP-AM-evoked [Ca2+]i transients were larger and occurred more rapidly after thapsigargin treatment (Fig. 1, B, E, and F) (13). Previous studies have implicated acidic Ca2+ stores as the principal target organelles for NAADP in pancreatic β cells and other cells (19, 60). Accordingly, bafilomycin (which inhibits Ca2+ uptake into acidic stores dependent on V-H+-ATPase activity (60)) blocked Ca2+ release in response to NAADP-AM treatment (Fig. 1, C and E). In addition, the membrane-permeant NAADP antagonist Ned-19 (100 μm) (Fig. 2F) (21) also completely abolished the Ca2+ transient evoked by NAADP-AM (Fig. 1, D and E). Collectively, these data suggest that NAADP targets acidic Ca2+ stores rather than the ER in mouse pancreatic β cells.
FIGURE 1.

NAADP mobilizes Ca2+ from acidic Ca2+ stores in mouse primary β cells. A, clusters of β cells were superfused with 3 mm glucose and stimulated by NAADP-AM, 10 nm (red line), 60 nm (black line), and 10 μm (blue line), and glucose (10 mm) as indicated. B, clusters of β cells were pre-treated with 1 μm thapsigargin (TG) for 1 h. They were then stimulated by NAADP-AM (60 nm) in the absence of extracellular Ca2+ and in the presence of a nonstimulatory glucose concentration (3 mm). C, NAADP-AM-induced [Ca2+]i response observed in B is prevented by bafilomycin (Baf, 3 μm) treatment. D, pretreatment of β cells with Ned-19 (100 μm) blocks the NAADP-AM-induced [Ca2+]i rise observed in B. E and F, quantification of the results from A to D. Traces are representative of results obtained in 8 (A), 7 (B), 6 (C), and 6 (D) clusters of islet β cells.
FIGURE 2.

Glucose-induced [Ca2+]i response in pancreatic β cells are dependent on NAADP-evoked Ca2+ release from acidic stores. A, typical [Ca2+]i oscillations induced by the stimulation of clusters of islet β cells by an increase of glucose from 3 to 10 mm as indicated. B, clusters of β cells were challenged with 10 mm glucose, and NAADP-AM (60 nm) was added acutely as indicated. C, clusters of islet β cells were challenged with 10 mm glucose in the continuous presence of Ned-19. High K+ was applied as indicated. D, clusters of β cells were stimulated by an increase of glucose from 3 to 10 mm glucose, and Ned-19 was applied acutely as indicated. E, clusters of β cells were stimulated by an increase of glucose from 3 to 10 mm, and Ned-20 was added as indicated. Representative trace was obtained in five separate clusters of islet β cells. F and G, structures of Ned-19 and Ned-20, a close structural analogue in which the fluorine atom is para on the benzene ring. H, clusters of islet β cells were pretreated with 3 μm bafilomycin and challenged with glucose or K+ (45 mm) as indicated by horizontal bars (by opening the KATP channels, diazoxide (Dz) (100 μm) prevents the direct effect of glucose on the membrane potential). I, glucose-induced [Ca2+]i oscillations are reversibly abolished by acute addition of 3 μm bafilomycin (Baf). J, quantification of the frequency of [Ca2+]i oscillations in I. K, clusters of pancreatic β cells were challenged with 10 mm glucose, and concanamycin (6 μm) was added acutely as indicated by the horizontal bar. The trace is representative of results obtained from eight clusters of islet β cells. Other traces are representative of results obtained in 11 (A), 5 (B), 8 (C), 9 (D), 8 (H), and 10 (I) clusters of islet β cells (*, p < 0.05, Student's t test).
Modulation of Glucose-evoked Ca2+ Spiking by Ned-19 and Vacuolar Proton Pump Inhibitors
We next examined whether NAADP signaling plays a role in glucose-mediated responses in primary β cells as suggested previously in MIN6 cells (18). Stimulation of mouse pancreatic β cells by 10 mm glucose resulted in [Ca2+]i oscillations that were superimposed upon a sustained plateau (Fig. 2A). Acute application of 60 nm NAADP-AM first enhanced Ca2+ spiking from 5.8 ± 0.7 to 11.6 ± 1.3 spikes/min (n = 5; p < 0.01) and then abolished glucose-evoked [Ca2+]i oscillations after about 20–25 min (Fig. 2B). This finding is consistent with the bell-shaped concentration-response curve to NAADP in mammalian cells (18, 51, 61); the initial stimulatory effect was mediated by low concentrations, and the subsequent inhibition reflected the build-up of higher self-desensitizing concentrations of NAADP (16, 18). The antagonist (18, 51, 61) Ned-19 was also found to inhibit the glucose-induced [Ca2+]i rise, and it abolished the Ca2+ oscillations, without affecting the initial [Ca2+]i decrease due to ATP-enhanced Ca2+ uptake by the ER (Fig. 2C) (62). When Ned-19 was applied after commencement of glucose-evoked Ca2+ responses, the glucose-induced [Ca2+]i plateau was abolished (Fig. 2D). The structurally related analogue Ned-20, which is not an NAADP antagonist (Fig. 2G) (21), was without effect (Fig. 2E).
Because NAADP mobilized Ca2+ from acidic organelles (Fig. 1C) (60), we examined whether selective pharmacological interference of Ca2+ storage by acidic organelles with agents that affect Ca2+ uptake into these stores (60) modulates glucose-evoked Ca2+ signaling. Preincubation of β cells with bafilomycin prevented the glucose-induced [Ca2+]i rise (Fig. 2H), as observed previously in the MIN6 β cell line (19). When bafilomycin was applied acutely, it reduced glucose-induced [Ca2+]i oscillations (Fig. 2I). Similar results were obtained with concanamycin, another V-type-H+-ATPase blocker (Fig. 2K). The effect of bafilomycin on the frequency of glucose-evoked [Ca2+]i transients is summarized in Fig. 2J. These observations are consistent with the hypothesis that NAADP-sensitive acidic Ca2+ stores play a key role in sustaining glucose-induced [Ca2+]i oscillations in mouse pancreatic β cells (63).
NAADP Modulation of Plasma Membrane Currents and Membrane Potential
We have previously shown that intracellular application of NAADP in β cells evokes oscillatory currents (21, 29). To examine the impact of NAADP-evoked Ca2+ release on the excitability of pancreatic β cells in more detail, we infused NAADP (10–100 nm) through a patch pipette in the standard whole-cell configuration while simultaneously measuring the membrane currents. In β cells clamped at −70 mV, intracellular application of 100 nm NAADP evoked intermittent inward currents of varying amplitudes (Fig. 3A). These currents contrast with the periodic outward hyperpolarizing K+ currents evoked by IP3-evoked Ca2+ release in β cells (64), and they underscore the differential actions of these two Ca2+-mobilizing messengers. The inward currents were absent in control cells where no NAADP was added to the intracellular solution (Fig. 3B), as well as when highly desensitizing concentrations (>1 μm) of NAADP were used (Fig. 3C), consistent with the bell-shaped concentration-response curve due to self-desensitization of the NAADP receptor (18, 61). Moreover, these currents were also prevented by addition of the Ca2+ chelator BAPTA to the pipette solution, demonstrating that they are likely dependent on Ca2+ release (Fig. 3D and see Fig. 7A). When N-methyl-d-glucamine replaced extracellular K+ and Na+, no inward currents could be seen in response to 100 nm NAADP (Fig. 3E). Taken together with the BAPTA studies, these data are consistent with the inward currents being due to opening of Ca2+-activated cation channels. The NAADP antagonist Ned-19 (100 μm) also abolished the NAADP-evoked currents, as shown previously (Fig. 3F) (21). NAADP-evoked Ca2+ release has recently been reported to activate TRPM4 channels in HeLa cells (65), and because this Ca2+-activated nonselective cation channel has been proposed to control insulin secretion (66) along with TRPM5 (67–69), we tested the effect of 9-phenanthrol, a TRPM4 blocker (70), on the NAADP-evoked currents in β cells. We found that the currents were inhibited by this drug (Fig. 3G), suggesting a possible involvement of TRPM4 channels.
FIGURE 7.

NAADP evokes membrane currents and Ca2+ signals in pancreatic β cells from wild-type but not Tpc2−/− mice. A and B, simultaneous [Ca2+]i (black trace, upper) and whole-cell current recording (gray trace, lower) in response to the infusion of 100 nm NAADP through a patch pipette from a wild type (A) or Tpcn2−/− (B) single β cells voltage clamped at −70 mV. Traces are representative of results obtained from four (A) and four (B) single β cells. C, electron micrographs of mouse pancreatic β cells from wild-type and Tpcn2−/− mice. EM sections are shown of β cells from pancreatic islet preparations from Tpcn2−/− knock-out mice as indicated. Scale bar, 1 μm.
Importantly, like NAADP, high glucose (15 mm) concentrations also evoked spontaneous inward currents in cells voltage-clamped at −70 mV (Fig. 3H). These glucose-induced inward currents were also blocked by Ned-19 (100 μm) (Fig. 3I). The frequencies and amplitudes of the above NAADP- and glucose-evoked currents from Fig. 3, A–I, are summarized in Fig. 3, J and K, respectively. Taken together, these data raise the exciting possibility that NAADP-mediated Ca2+ release from acidic stores may modulate the glucose-mediated membrane currents that in turn initiate β cell electrical activity and insulin secretion.
We next examined the effect of intracellular NAADP on the pancreatic β cell plasma membrane potential. A nonstimulatory level of glucose (3 mm) alone (Fig. 4A) had no effect on resting membrane potential (around −70 mV). In the presence of 3 mm glucose, NAADP-AM (60 nm) evoked low amplitude voltage oscillations that did reach the threshold for action potential firing (Fig. 4B). At 10 mm glucose, glucose-induced electrical activity consisting of 30–40-mV action potentials was observed. Under these conditions, the blockade of NAADP action with Ned-19 (100 μm) resulted in membrane hyperpolarization and suppression of electrical activity (Fig. 4C).
FIGURE 4.
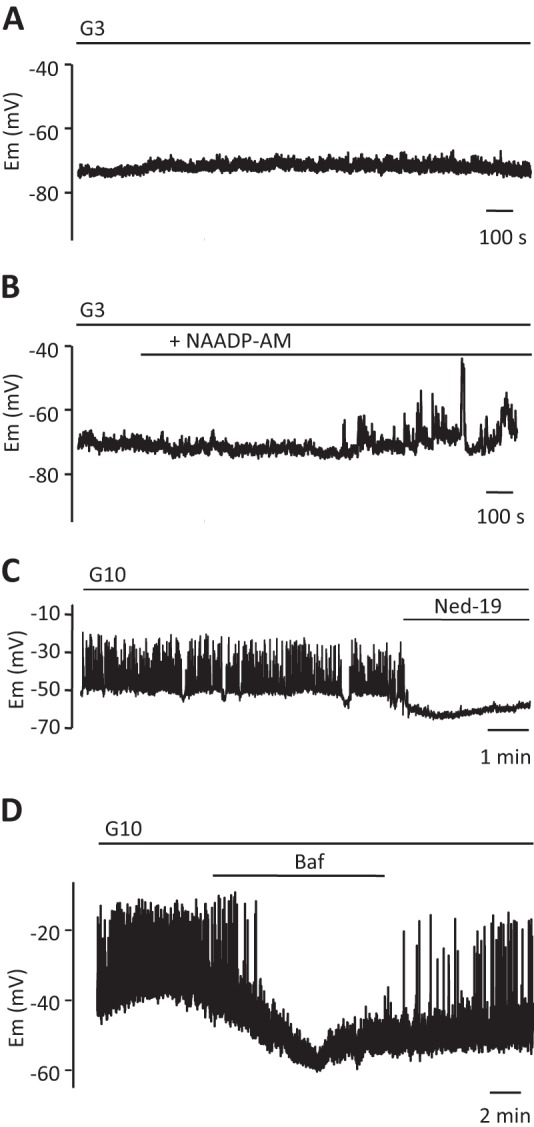
NAADP-evoked Ca2+ release from acidic stores modulates membrane potential. A and B, membrane potential (Em) recordings from β cells in small clusters exposed to 3 mm glucose. NAADP-AM (60 nm) was applied as indicated in B. C, Ned-19 (100 μm) abolishes the typical electrical activity evoked by 10 mm glucose in a single pancreatic β cell. D, typical electrical activity evoked by 10 mm glucose in a single β cell is reversibly abolished by bafilomycin (3 μm). Traces are representative of results obtained from three (A), six (B), and seven (C and D) single β cells. All traces represent different cells. *, p < 0.05, Student's t test.
If NAADP-evoked Ca2+ release is involved in the regulation of the membrane potential by glucose, pharmacological manipulation of ion fluxes across the endomembranes involved would be predicted to impact on glucose-mediated changes in membrane excitability. It is therefore of interest that bafilomycin reversibly suppressed glucose-evoked action potentials (Fig. 4D).
Lack of Effects of Ned-19 and Vacuolar Proton Pump Inhibitors on Plasma Membrane Currents and Cell Metabolism and Validation of Compound Used
Although the profound effects of Ned-19 and bafilomycin on glucose-mediated Ca2+ signaling and electrical changes above were ascribed to antagonism of NAADP and abrogation of acidic organelle Ca2+ storage, it was important to rule out other targets that could potentially account for the effects of these two agents on glucose action.
Ned-19 exerted no significant effects upon glucose metabolism, as indicated by the persistence of glucose-induced decrease in mitochondrial FAD fluorescence (Fig. 5A). The inhibitor was also without effect on KATP channel activity; in the presence of 10 mm glucose, the whole-cell conductance averaged 0.21 ± 0.04 and 0.20 ± 0.06 nanosiemens in the absence or presence of Ned-19 (Fig. 5B). By contrast, the combination of the KATP channel activator diazoxide and the mitochondrial inhibitor sodium azide (1 mm) resulted in a large increase in KATP channel activity. Similarly, there was no inhibitory effect of Ned-19 on the voltage-gated Ca2+ currents; the peak current during depolarization from −70 to 0 mV averaged 110 ± 1 pA (n = 10) and 114 ± 1 pA (n = 10) in the absence and presence of Ned-19, respectively (Fig. 5C). Thus, the suppression of electrical activity cannot simply be attributed to activation of KATP channels or inhibition of VDCCs. In addition, Ned-19 was without effect on Ca2+ release induced by the stimulation of muscarinic receptors with acetylcholine (100 μm) (Fig. 5D), which leads to the opening of IP3 receptors and discharge of ER stores (71). These data are therefore consistent with a high degree of selectivity of Ned-19 as an antagonist of NAADP and demonstrate that its effects are consistent with a major role for NAADP-induced Ca2+ release in glucose-induced Ca2+ signaling.
FIGURE 5.

Selectivity of pharmacological compounds employed. Ned-19 does not affect mitochondrial metabolism, plasma membrane currents, or IP3-induced Ca2+ release. A–C, changes in flavin adenine dinucleotide (FAD) fluorescence in response to glucose, which reflect glucose metabolism (A), the whole-cell KATP current (B), and the whole-cell voltage-dependent Ca2+ current (C) produced by Ned-19 (100 μm). D, no effect of Ned-19 on [Ca2+]i increases evoked by the muscarinic agonist acetylcholine (ACh). Traces are representative of results obtained in seven (B) and six (C) single β cells or six (A) and six (D) clusters of islet β cells. Bafilomycin (Baf) does not impair either glucose metabolism or whole-cell Ca2+ or K+-ATP currents. E, top, clusters of islets β cells were loaded for 10 min with 10 nm of the potentially sensitive probe for measuring membrane potential changes in mitochondria, tetramethylrhodamine ethyl ester. An image was taken 5 min after washout of the dye. Middle, an image was taken after 15 min of incubation with 3 μm bafilomycin. Bottom, an image was taken after application of 2 μm of the protonophore, carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP), which depolarizes the mitochondrial membrane. Either pretreatment (F) or acute addition (G) of bafilomycin does not alter glucose metabolism reflected by changes in flavin adenine dinucleotide (FAD) fluorescence in response to glucose. Neither whole-cell Ca2+ current (H) nor whole-cell K+-ATP current (I) was affected by bafilomycin (in H the Ca2+ current was recorded from the same cell before (black trace) and after bafilomycin application (red trace)). Images in E are representative of results obtained in six separate experiments. Traces are representative of results obtained in four (F and G) clusters of β cells and seven (H) and five (I) single β cells. Bafilomycin-sensitive Ca2+ stores are separate from the ER. J and K, clusters of islet β cells were transferred to a Ca2+-free solution before 1 μm thapsigargin (Tg), and 3 μm bafilomycin was added as indicated by horizontal bars. Representative traces of results obtained in seven (J) and six (K) clusters of islet β cells. L, lysosomes are essential for glucose-induced [Ca2+]i oscillations. Clusters of islet β cells were stimulated by 10 mm glucose, and 50 μm glycyl-l-phenylalanine 2-naphthylamide (GPN) was applied as indicated. The GPN is a cathepsin C substrate, which permeabilizes the lysosomes by osmotic swelling. Trace is representative of results obtained in seven clusters of islet β cells. Dz, diazoxide; ACh, acetylcholine.
We next analyzed the effects of bafilomycin on the same functional parameters. The effects of the latter inhibitor were independent of alterations in glucose-evoked changes in mitochondrial membrane potential or cell metabolism (Fig. 5, E–G), VDCCs (Fig. 5H), or modulation of KATP channels (Fig. 5I). Moreover, in the absence of extracellular Ca2+, bafilomycin increased [Ca2+]i after thapsigargin treatment (Fig. 5J), and vice versa (Fig. 5K), confirming that bafilomycin-sensitive Ca2+ stores are distinct from the ER. The application of the lysomotropic agent GPN, a lysosomotropic agent that abrogates Ca2+ storage by lysosomes (19, 25), exerted effects that resembled those of bafilomycin (Fig. 5L), again indicating that a lysosome-related organelle is the likely source of Ca2+ release.
TPC2 Expression and Subcellular Localization in Endocrine Pancreas
Turning to the molecular targets for NAADP, we have recently identified TPC2, encoded by the Tpcn2 gene, as a critical mediator of the NAADP response (29), and we have shown that it is required to couple stimuli to Ca2+ release from acidic stores (72). This channel is localized on acidic stores, but not at the ER or plasma membrane, and in particular it co-localizes with organelles of the endolysosomal system, most prominently the lysosomes (29). RT-PCR analysis of mouse islets indicates that Tpcn2 is expressed in mouse islets (Fig. 6A). Affirming the localization of TPC2 to acidic stores in β cells, immunolocalization of the endogenous TPC2 in primary human β cells revealed substantial overlap with immunoreactivity of LAMP1, a major lysosomal marker (Fig. 6B). There was substantially less overlap with immunoreactivity for insulin or EEA1, used as markers for insulin granules (73) and endosomes (29), respectively. In a complementary approach, TPC2-mCherry was expressed in MIN6 cells (Fig. 6C). A similar lysosomal localization of TPC2-mCherry transfected into this murine β cell line was seen, but some co-localization with insulin granules could also be seen.
FIGURE 6.
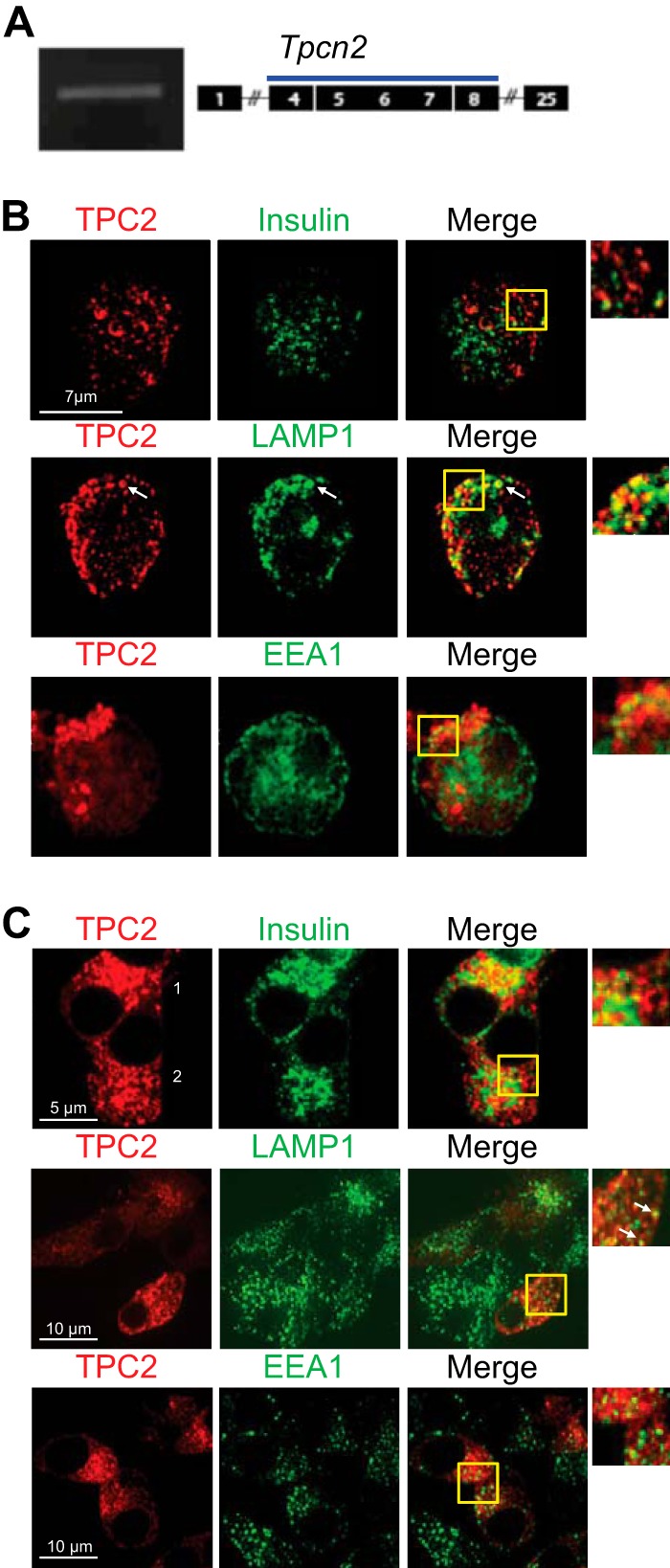
Subcellular localization of TPC2 in human β cells and mouse MIN6 β cells. A, RT-PCR product corresponding to TPC2 mRNAs in the pancreas from WT mice with expected product size of 501 bp. B, co-labeling of endogenous TPC2 and organelle markers in single human β cells. Single human β cells were fixed and imaged as described under “Experimental Procedures.” Zoomed images show areas within the yellow boxes. Although little if any co-localization was observed between TPC2 and insulin or EEA1 staining, clear overlap was apparent between TPC2 and LAMP1 in well defined subcellular structures (arrows). C, colabeling of overexpressed TPC2-mCherry and organelle markers in clonal mouse β cells. MIN6 cells were transfected with a TPC2-mCherry construct (see under “Experimental Procedures”) and subsequently fixed and permeabilized. Guinea pig anti-insulin, goat anti-EEA1, and rat anti-LAMP-1 were revealed as in A. Overlap between TPC2 and LAMP1-labeled structures is clearly apparent (inset, arrows). Limited overlap between TPC2 and insulin was observed only in cells expressing high levels of TPC2-mcherry (cell 1), but little if any co-labeling of insulin and TPC2 was observed in the majority of cells (cell 2) where TPC2-mCherry levels were lower.
Glucose and Tolbutamide-evoked Ca2+ Responses Are Reduced in Isolated Pancreatic β Cells from Tpcn2−/− Mice
We next examined the effects of infusing NAADP (100 nm) into mouse pancreatic β cells from age-, sex-, and background-matched wild-type and Tpcn2−/− mice via the patch pipette during standard whole-cell recordings while simultaneously measuring intracellular Ca2+ concentrations (by Fura 2) and inward membrane currents. In wild-type β cells, NAADP (100 nm) evoked [Ca2+]i transients, some of which associated with transient inward currents (Fig. 7A). However, both NAADP-evoked Ca2+ transients and currents were absent in cells prepared from Tpcn2−/− mice (Fig. 7B), mirroring the effect of Ned-19 on the wild-type cells (Fig. 3F). Detailed morphological comparison of β cells from Tpcn2−/− and wild-type mice by electron microscopy indicated no substantial differences in morphology, organelle number, or distribution, including those of insulin granules (Fig. 7C), making it unlikely that changes in these parameters were the underlying cause for alterations in NAADP responses observed in Tpcn2−/− null β cells.
Next, we examined the Ca2+ responses to glucose of β cells prepared from wild-type and Tpcn2−/− mice (Fig. 8, A and B). In Tpcn2−/− β cells, glucose-evoked Ca2+ transients were either abolished, reduced in amplitude, or delayed (Fig. 8, B and C) compared with the robust responses observed in wild-type cells (Fig. 8, A and C). The average [Ca2+]i rises evoked by high glucose were substantially reduced (but not abolished) in all Tpcn2−/− β cells studied (Fig. 8C), although activation of VDCCs by membrane depolarization by K+ (45 mm) in the presence of the KATP channel opener, diazoxide (100 μm), still evoked a large [Ca2+]i response (Fig. 8B). Similar results were obtained from Tpcn1−/− β cells (Fig. 8, D–F). These studies indicate that the lysosomal TPC2 and endosomal TPC1 channels play a significant role in the generation of glucose-induced Ca2+ signals in pancreatic β cells, likely through their modulation of β cell electrical activity.
FIGURE 8.

Glucose-evoked Ca2+ signals are impaired in pancreatic β cells from Tpcn2−/− and Tpcn1−/− mice. A and B, [Ca2+]i oscillations in response elevating glucose from a basal 3 mm to 10 mm glucose in wild-type (A) and Tpcn2−/− β cells (B). B, experiments were concluded by addition of high extracellular K+ (45 mm) in combination with diazoxide (Dz) (100 μm). C, averaged R/R0 changes in WT and Tpcn2−/− clusters of β cells (four different patterns were observed in Tpcn2−/− cells) over a period of 20 min after the rise of glucose concentration from 3 to 10 mm. The changes in [Ca2+]i are displayed as the normalized ratio R/R0 (R0 is the basal level before the stimulation with high glucose). Traces are representative of results obtained from four (A) and four (B) single β cells and 23 (A) and 29 (B) clusters of β cells. (*, p < 0.05, Student's t test.) D–F, averaged R/R0 changes in clusters of β cells from WT and Tpcn1−/− mice (two different patterns were observed in Tpcn1−/− cells) over a period of 20 min after the rise of glucose concentration from 3 to 10 mm. Traces are representative of results obtained from 11 (D) and 13 (E) clusters of β cells. (*, p < 0.05, Student's t test.) G, clusters of β cells were bathed in different conditions as indicated by different colors, and tolbutamide (25 μm) was applied as indicated. The sulfonylurea was unable to evoke a [Ca2+]i rise in the absence of glucose (black trace, n = 5), although a significant [Ca2+]i rise is observed in the presence of 3 mm glucose (green trace, n = 7). 30 min pretreatment with low concentrations of NAADP-AM (60 nm) (blue line, n = 6) permitted a [Ca2+]i response to tolbutamide in the absence of glucose. H, NAADP-AM permits 25 μm tolbutamide to rise [Ca2+]i in the absence of glucose. The change in ratio is expressed in %, the baseline before addition of tolbutamide being 100%. **, p < 0.01. I, clusters of pancreatic β cells were isolated from wild-type (black trace, representative of n = 5) and Tpcn2−/− mice (red trace, representative of n = 7) and bathed in media containing 3 mm glucose. Tolbutamide (25 μm) was added as indicated by the horizontal bar. (*, p < 0.05, Student's t test.)
It has been noted that a minimum concentration of glucose “fuel” is required for threshold concentrations of the oral hypoglycemic agent and KATP inhibitor tolbutamide to mimic the electrical effects of raised glucose levels in mouse β cells (74). In agreement, we also found that in the absence of glucose (0 mm), tolbutamide (25 μm) treatment failed to increase [Ca2+]i, in contrast to the effect in the presence of 3 mm glucose. However, pretreatment with NAADP-AM (60 nm) partially reconstitutes the Ca2+ signal with tolbutamide in the absence of glucose (Fig. 8, G and H). Furthermore, we found that in wild-type mouse β cells exposed to 3 mm glucose, tolbutamide (25 μm) evoked a rise in [Ca2+]i (Fig. 8I), whereas cells from Tpcn2−/− mice failed to respond to tolbutamide.
Role of NAADP and TPC2 in Glucose-induced Insulin Secretion
Having implicated a key role for NAADP, TPC2, and lysosomal Ca2+ stores in Ca2+ signaling and electrical activity, we finally examined the effect of disrupting NAADP signaling upon insulin secretion itself. Insulin secretion was measured from isolated whole islets in response to glucose or 45 mm K+. Prior treatment of islets with Ned-19 (100 μm) for 5 min substantially inhibited insulin secretion induced by 15 mm glucose but not by 45 mm K+ (Fig. 9A). The absence of an effect of Ned-19 on insulin secretion induced by high K+ makes it unlikely that there is a relevant off-target effect of the inhibitor upon the exocytotic machinery.
FIGURE 9.

Role of the NAADP-sensitive Ca2+ stores and the two-pore channel 2 (TPC2) in stimulus-secretion coupling in pancreatic β cells. A, Ned-19 blocks glucose-evoked insulin secretion. Insulin secretion from control intact islets of Langerhans was triggered by glucose (15 mm) or K+ (45 mm). When Ned-19 was used, the islets were pretreated for 5 min prior to stimulation with the secretagogues. Data are means ± S.E. obtained from three different islet preparations with batches of 10 islets. * indicates p < 0.05 preparations with batches of 10 islets. B, insulin secretion from perfused pancreata from WT and Tpcn1−/− mice in response to a rise of glucose from 3 to 20 mm at 8 min. The secretion is expressed as percentage of pancreatic content/min. Wild-type trace (WT) is shown in black and knock-out (KO) animals in red. C, insulin secretion from perfused pancreata from WT and Tpcn2−/− mice in response to a rise of glucose from 3 to 20 mm at 8 min. The secretion is expressed as percentage of pancreatic content/min. Wild-type trace (WT) is shown in black and knock-out (KO) animals in red. D, area under the curve (AUC) for 1st and 2nd phase insulin secretion. The traces show the average ± S.E. of n = 6 WT and n = 6 for Tpcn1−/− and Tpcn2−/−. * indicates a p < 0.07 significance level between WT and KO using a one-sided t test. E and F, glucose tolerance tests. Glucose tolerance test was performed on WT (n = 5) and Tpcn1−/− and Tpcn2−/− (n = 6) animals by intraperitoneal injection of 2 g/kg glucose solution after an overnight fasting. Results are expressed as concentrations of blood glucose at time 0 for each animal. Values are means ± S.E. of results obtained with mice for each group.
We then studied insulin secretion evoked by 20 mm glucose using the perfused pancreas preparation in wild-type, Tpcn1−/−, and Tpcn2−/− mice. In the Tpcn1−/− and Tpcn2−/− pancreata, both the 1st (the response during the initial 7 min) and 2nd phase release (insulin secretion once the 1st phase release had ended) were reduced by ∼50% (Fig. 9, B–D).
Finally, we performed intraperitoneal blood glucose tests in fasted TPC knock-out mice in comparison with the corresponding strain-matched wild-type animals to examine the effect of perturbing TPC expression upon glucose homeostasis in the whole animal. This revealed that in Tpcn1−/− mice (Fig. 9E) the blood glucose levels peaked at significantly higher levels than WT, and the time course revealed an impaired glucose tolerance capacity. In contrast, Tpcn2−/− mice (Fig. 9F) were less affected.
Discussion
This study highlights the importance of NAADP-sensitive acidic stores and the newly identified endolysosomal channels TPC1 and TPC2 in Ca2+ signaling during stimulus-secretion coupling in mouse pancreatic β cells. Since its discovery as a potent Ca2+-mobilizing agent in sea urchin egg homogenates (75), NAADP has been widely demonstrated to evoke Ca2+ signals in an extensive range of mammalian cells, including those of both the endocrine and exocrine pancreas (76). NAADP is an alternative product of multifunctional ADP-ribosyl cyclase enzymes, which is also responsible for the synthesis of the ryanodine receptor-regulating messenger cADPR (46). Building on early studies suggesting that cADPR is an important regulator of Ca2+ signaling during secretion-coupling in pancreatic β cells (77), we now reported that NAADP also mobilizes Ca2+ in pancreatic β cells (16–18).
In contrast to the other two principal mobilizing messengers IP3 and cADPR, the major target organelles for NAADP in sea urchin eggs are acidic stores rather than the ER. Pharmacological approaches and cell fractionation studies revealed that NAADP releases Ca2+ from a separate organelle to the ER (78, 79), identified as acidic lysosomally related organelles (25). This principle was later extended to mammalian cells and NAADP release from acidic stores and has now been established in a large number cell types (60, 80). In many cells, signaling domains at lysosome-ER junctions have been observed (81). Thus, NAADP-evoked Ca2+ release from acidic stores may trigger further Ca2+ release from larger ER Ca2+ stores through the mediation of IP3 receptors and ryanodine receptors (59). Here, we have found that Ca2+ signals evoked by the membrane-permeant NAADP analogue NAADP-AM are from intracellular stores because they persist in the absence of extracellular Ca2+ and that the NAADP antagonist Ned-19 blocks this effect (Fig. 1).
Comparison of the effects of drugs that effect Ca2+ uptake and storage in different organelles supports a role for acidic stores rather than the ER as the target of NAADP. Bafilomycin selectively inhibits vacuolar H+ pumps that acidify acidic stores, and it has been shown that Ca2+ uptake into acidic organelles is pH-dependent and probably mediated by Ca2+/H+ exchange (60). Bafilomycin treatment was thus found to abolish NAADP-AM-evoked Ca2+ release (Fig. 1C). In contrast, thapsigargin (a SERCA pump inhibitor that blocks Ca2+ uptake into the ER) was found to enhance NAADP-AM-induced Ca2+ release. This suggests that NAADP-evoked Ca2+ release in the β cell does not trigger further Ca2+ release through ER mechanisms. Rather the predominant role of the ER here is to act to buffer Ca2+ rather than as a source for release, and the functional removal of the ER decreases Ca2+ buffering, allowing Ca2+ release from acidic stores to increase further in the cytoplasm. The role of the ER to buffer Ca2+ during signaling has also been noted for glucose-evoked Ca2+ signals where glucose first decreases cytoplasmic Ca2+ due to increased ATP generation and stimulation of SERCA pumps (12, 82, 83). Previous studies also support acidic stores as targets for NAADP. Ca2+ indicators targeted to acidic granules or ER in MIN6 cells showed that NAADP releases Ca2+ from acidic organelles but not the ER (17). Bafilomycin and the lysosomotropic agent GPN abolishes Ca2+ release by photolysis of caged NAADP in MIN6 cells, but it does not affect IP3-evoked Ca2+ release (19). In primary mouse β cells, NAADP-evoked Ca2+ release was inhibited by GPN, which was shown to lyse acidic stores selectively (13, 23). The delay in Ca2+ responses seen with NAADP-AM (Figs. 1 and 4) may also be determined partly by the time for hydrolysis of ester groups by intracellular endogenous esterases, which varies between cells (51), but the delay is probably largely due to initial buffering of Ca2+ by the ER because it was decreased by thapsigargin treatment. In addition, by analogy with the situation in the ER (83), uptake of Ca2+ into acidic stores might be enhanced by glucose-stimulated ATP production, and because luminal Ca2+ sensitizes TPCs to low NAADP concentrations, this could promote Ca2+ release from these stores (84, 85), an effect that would be enhanced by removing competing ER stores.
Since the initial reports linking NAADP-evoked Ca2+ release to two-pore channels (29–31), there have been numerous reports of TPCs playing an essential role in mediating NAADP-evoked Ca2+ release from acidic stores (58, 86). However, recent evidence points to a separate NAADP-binding protein that interacts with TPCs to confer NAADP sensitivity (39). Indeed, the requirement of NAADP-binding proteins may suggest that under certain circumstances these proteins may interact with multiple channel types (87). This may explain a recent report of the loss of sensitivity of isolated lysosomes to NAADP (23, 38), and the finding that glucose may apparently still evoke Ca2+ signals in β cells from Tpcn1−/−/Tpcn2−/− mice (23), although it should be noted that the concentrations of NAADP-AM used in the report by Wang et al. (23) are more than 3 orders of magnitude greater than those found to be effective here. However, consistent with the data presented here, these authors (23) also reported that NAADP mobilizes Ca2+ from acidic stores in the INS-1 β cell line, an effect blocked by Ned-19, and evoke membrane depolarization and spike generation in this cell line. We show here that NAADP-evoked Ca2+ transients in β cells are abolished in cells prepared from Tpcn1−/− and Tpcn2−/− mice (Fig. 7, A and B). We found that endogenous TPC2 proteins in human β cells co-localized with lysosomal markers (Fig. 6B). Interestingly, TPC2 did not appear to colocalize with insulin granules, which have also been proposed to function as NAADP-sensitive Ca2+ stores in β cells (13, 17).
In addition to mobilizing Ca2+ from acidic stores, NAADP was also found here to evoke plasma membrane cation currents and to depolarize the plasma membrane. NAADP applied through the patch pipette at low concentrations evoked a series of inward current transients (Fig. 3, A–G). Application of higher NAADP (100 μm) gave no response, consistent with the bell-shaped concentration-response curve for NAADP in mammalian systems and paralleling the concentration dependence of NAADP for Ca2+ release in β cells (Fig. 1A) (16, 18, 20) and other mammalian cells (58). These currents were blocked by Ned-19 and by BAPTA, suggesting that they are Ca2+-activated. Their abolition by replacing Na+ with N-methyl-d-glucamine suggests that the currents are cation currents largely carried by Na+ ions. Interestingly, a nonselective cation current has also been reported to be activated by GLP-1 (where GLP-1 is glucagon-like peptide 1), an agonist that has also been reported to elevate NAADP levels (20), in the HIT-T15 β cell line (88). The identity of the channels responsible has not been established, but our results with 9-phenanthrol may tentatively point to some involvement of the TRPM4 channels. NAADP-evoked Ca2+ release has recently been shown to activate TRPM4 channels in HeLa cells (65). Moreover, TRPM4 and TRPM5 channels have been proposed to mediate in part a Ca2+-dependent depolarization of the plasma membrane in the INS-1 β cell line (66–68, 89) and may play a general role as key components of membrane-based Ca2+ oscillators providing initial cell membrane depolarization for cell activation (90). The NAADP-evoked currents were found to be coincident with small NAADP-evoked Ca2+ transients (Fig. 7A), and neither NAADP-evoked Ca2+ transients nor currents were observed in cells from Tpcn2−/− mice (Fig. 7B). We propose that these currents are due to NAADP-evoked Ca2+ release from endolysosomal stores via TPC2 channels and that this, in turn, via elevation of [Ca2+]i, leads to Ca2+-dependent activation of plasma membrane cation channels, possibly TRPM4 or TRPM5. Activation of these channels would then result in membrane depolarization. The finding that application of NAADP-AM elicited a series of membrane potential spikes (Fig. 4B) is consistent with this scenario and in agreement with a report that NAADP causes membrane depolarization in INS1 cells (23).
To investigate the role of NAADP-mediated Ca2+ signaling in glucose-induced electrical activity and [Ca2+]i, four different approaches were used to block NAADP signaling. These were as follows: (i) abrogation of Ca2+ storage by acidic stores with vacuolar proton pump inhibitors and GPN; (ii) inhibition of the NAADP receptor by Ned-19; (iii) self-desensitization of the NAADP receptor by NAADP; and (iv) knock-out of Tpcn2 and Tpcn1, genes encoding proposed NAADP target channels. Intriguingly, high glucose was also found to evoke small Ned-19-sensitive currents similar to those evoked by pipette application of NAADP (Fig. 3H). Thus, NAADP signaling may contribute, at least partly, to bringing the membrane potential from rest to the threshold for activation of VDCCs (Fig. 10). As has been recognized for a long time, closure of KATP channels is not sufficient to explain how glucose depolarizes the pancreatic β cell; a depolarizing membrane current is also required (7). We propose that NAADP/TPC1/2-dependent mobilization of Ca2+ from an acidic intracellular store results in activation of depolarizing cation-conducting plasmalemmal ion channels and that this brings the membrane potential to the threshold for action potential firing. This is consistent with our finding that in the absence of NAADP-evoked Ca2+ signals in cells from Tpcn2−/− mice, the KATP channel blocker, tolbutamide, at threshold concentrations fails to evoke Ca2+ signals as seen in wild-type cells (Fig. 8I). Indeed, it is remarkable that tolbutamide cannot by itself mimic glucose-induced Ca2+ signals but requires NAADP/TPCs. Indeed, tolbutamide will only evoke Ca2+ signals when the acidic vesicle pathway is co-stimulated either with subthreshold concentrations of NAADP/AM or with a permissive subthreshold glucose (3 mm) concentration (Fig. 8, G and H).
FIGURE 10.
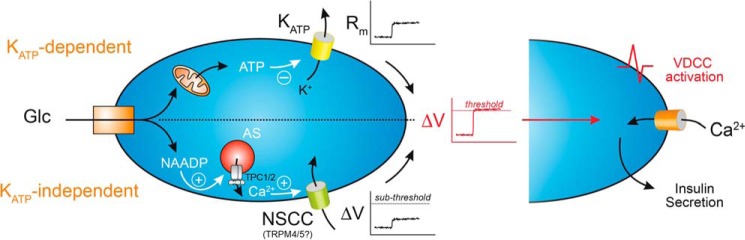
Proposed model for synergistic effects of NAADP-regulated TPC2 and KATP channels synergizes in glucose-evoked insulin secretion. NAADP-induced Ca2+ release synergizes with the KATP-dependent pathway to depolarize the plasma membrane and activate VDCCs. The ATP-mediated closure of KATP channels increases membrane resistance, which together with Ca2+-dependent depolarizing currents (possibly TRPM4/5 channels), activated by NAADP-induced Ca2+-release via TPC2 expressed on acidic stores, may depolarize the plasma membrane to threshold for VDCC activation (AS, acidic stores; NSCC, nonselective cation channel; Rm, membrane resistance; VDCC, voltage-dependent Ca2+ channels; Glc, glucose). In addition, NAADP-induced Ca2+-release together with VDCC-mediated Ca2+ influx are both required for exocytosis of insulin granules.
The final step in the stimulus-secretion coupling is the exocytosis of insulin-containing granules. In isolated islets, Ned-19 completely blocked glucose-evoked insulin secretion. Ned-19, however, had no effect on secretion evoked by depolarizing islet cells with high extracellular K+, which bypasses electrical activity and depolarizes the membrane potential to ∼−10 mV and opens the VDCCs. This finding makes it possible to discard the explanation that Ned-19 inhibits insulin secretion by an off-target effect on the exocytotic machinery. In a more in vivo setting, glucose-evoked insulin secretion from perfused whole pancreata from Tpcn2−/− and Tpcn1−/− mice was investigated. Insulin secretion stimulated by glucose (20 mm) was substantially reduced compared with that from wild-type animals (Fig. 9B). Thus, we provide evidence that NAADP signaling is an important regulator of stimulus-secretion coupling in pancreatic β cells (Fig. 10).
Surprisingly, Tpcn−/− mice are only mildly diabetic as assessed by glucose tolerance tests (Fig. 9), with a significant impairment in Tpcn1−/− mice. However, a recent study has implicated TPC2 as a novel gene for diabetic traits in mice, rats, and humans (91), with a decrease in fasting glucose and insulin levels reported in Tpcn2−/− mice. The effects of knocking out Tpcn genes in mice may result in complex phenotypes, including compensatory mechanisms, with regard to blood glucose and insulin levels because NAADP-mediated Ca2+ release has been implicated in GLUT4 translocation in murine skeletal muscle (92). Furthermore, NAADP signaling has been implicated in the action of peroxisome proliferator-activated receptor γ agonists in their insulin-sensitizing actions to ameliorate insulin resistance (93). Future studies with tissue-specific inactivation of these genes will be required to address these questions.
Although NAADP levels in β cells have been reported to be increased in response to elevated glucose, and also in response to the incretin hormone GLP-1 (18, 20), the mechanisms are not well understood. However, ADP-ribosyl cyclases have been implicated in NAADP synthesis in β cells (20). Although CD38 is a membrane-bound ecto-enzyme, glucose treatment of β cells induces endocytosis of CD38 that requires cytoskeletal changes (22). Inhibition of CD38 internalization with jasplakinolide, which promotes actin polymerization, blocks glucose-stimulated NAADP levels and impairs glucose-evoked Ca2+ signaling (22). NAADP synthesis is found to be associated with lysosomal membrane fractions (20). We have previously argued that cADPR (and NAADP) synthesis may occur within the acidic organelles (94). The luminal acidic pH of acidic organelles would provide an optimal environment for NAADP synthesis by ADP-ribosyl cyclases (46). We showed that pyridine nucleotides are transported into organelles, and second messenger products are transported into the cytoplasm to their site of action (94).
Antibodies to CD38 (95) and a missense mutation in the CD38 gene (50) have been linked to type 2 diabetes, and this could potentially be accounted for by reduced NAADP synthesis. Remarkably, in a recent study, it was found that intraperitoneal injections of NAADP could restore defective insulin secretion and blood glucose regulation in db/db mice, an animal model of type 2 diabetes (24), presumably via the NAADP transport mechanisms described above.
Conclusions
We propose that NAADP-evoked Ca2+ release from acidic stores via TPC2 or TPC1 channels evokes a small local Ca2+ signal that activates Ca2+-dependent cation currents in the plasma membrane. The finding that the membrane currents evoked by intracellular Ca2+ mobilization are blocked by 9-phenanthrol implicates TRPM4 channels in this process. Additional direct effects of NAADP-evoked Ca2+ release on exocytosis itself cannot be excluded at this stage, with a possible contribution from exocytotic granules themselves (17, 96). The NAADP/TPC/acidic organelle pathway represents a new component of the glucose-evoked trigger to depolarize the plasma membrane upon KATP channel closure resulting in VDCC-mediated Ca2+ influx and insulin secretion (Fig. 10). This new pathway may offer new targets for novel diabetic therapies.
Author Contributions
A. A. and A. G. designed the experiments, and A. A. conducted the project. A. A., J. P., G. A. R., and A. G. wrote the manuscript. M. R., L. T., K. R., and J. P. produced and characterized the Tpcn2−/− mice. F. C., T. P., and G. S. C. performed some of the [Ca2+]i measurement experiments. K. C. performed the gene expression experiments. R. P., A. M. L., and G. C. C. synthesized and characterized NAADP-AM, Ned-19, and Ned-20. A. J. M. designed experiments and produced Fig. 7. G. A. R. and E. A. B. performed immunocytochemical studies. K. S. performed insulin secretion experiments. P. J. supplied and prepared human islets. P. R., S. C. C., M. B., W. S., and Q. Z. performed secretion and cell physiological measurements. P. M. H. and P. W. T. performed the electron microscopy. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We gratefully acknowledge the help from the staff at the University of Oxford Biomedical Science Facility for their help in breeding and maintaining the Tpcn2−/− mice. We also thank Professor Frances Ashcroft for comments on the manuscript.
This work was supported by grants from The Wellcome Trust, the Medical Research Council, and The Royal Society. The authors declare that they have no conflicts of interest with the contents of this article.
We dedicate this paper to the memory of Dr. Matthias Braun who died during the preparation of the manuscript.
- VDCC
- voltage-dependent Ca2+ channel
- cADPR
- cyclic adenosine diphosphate ribose
- ER
- endoplasmic reticulum
- GPN
- glycyl-l-phenylalanine-β-naphthylamide
- IP3
- inositol 1,4,5-trisphosphate
- NAADP-AM
- nicotinic acid adenine dinucleotide phosphate acetoxymethyl ester
- TPC
- two-pore channel
- SERCA
- sarco/endoplasmic reticulum Ca2+-ATPase; cytosolic Ca2+
- NAADP
- nicotinic acid adenine dinucleotide phosphate
- BAPTA
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid.
References
- 1. Henquin J. C., Ravier M. A., Nenquin M., Jonas J. C., Gilon P. (2003) Hierarchy of the beta-cell signals controlling insulin secretion. Eur. J. Clin. Invest. 33, 742–750 [DOI] [PubMed] [Google Scholar]
- 2. Ashcroft F. M., Rorsman P. (2012) Diabetes mellitus and the beta cell: the last 10 years. Cell 148, 1160–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ashcroft S. J. (2000) The beta-cell KATP channel. J. Membr. Biol. 176, 187–206 [DOI] [PubMed] [Google Scholar]
- 4. Dunne M. J., Cosgrove K. E., Shepherd R. M., Aynsley-Green A., Lindley K. J. (2004) Hyperinsulinism in infancy: from basic science to clinical disease. Physiol. Rev. 84, 239–275 [DOI] [PubMed] [Google Scholar]
- 5. Girard C. A., Wunderlich F. T., Shimomura K., Collins S., Kaizik S., Proks P., Abdulkader F., Clark A., Ball V., Zubcevic L., Bentley L., Clark R., Church C., Hugill A., Galvanovskis J., et al. (2009) Expression of an activating mutation in the gene encoding the KATP channel subunit Kir6.2 in mouse pancreatic beta cells recapitulates neonatal diabetes. J. Clin. Invest. 119, 80–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Best L. (2005) Glucose-induced electrical activity in rat pancreatic beta cells: dependence on intracellular chloride concentration. J. Physiol. 568, 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Henquin J. C., Nenquin M., Ravier M. A., Szollosi A. (2009) Shortcomings of current models of glucose-induced insulin secretion. Diabetes Obes. Metab. 11, 168–179 [DOI] [PubMed] [Google Scholar]
- 8. Islam M. S. (2010) Calcium signaling in the islets. Adv. Exp. Med. Biol. 654, 235–259 [DOI] [PubMed] [Google Scholar]
- 9. Miki T., Nagashima K., Tashiro F., Kotake K., Yoshitomi H., Tamamoto A., Gonoi T., Iwanaga T., Miyazaki J., Seino S. (1998) Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 95, 10402–10406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Düfer M., Haspel D., Krippeit-Drews P., Aguilar-Bryan L., Bryan J., Drews G. (2004) Oscillations of membrane potential and cytosolic Ca2+ concentration in SUR1−/− beta cells. Diabetologia 47, 488–498 [DOI] [PubMed] [Google Scholar]
- 11. Szollosi A., Nenquin M., Henquin J. C. (2007) Overnight culture unmasks glucose-induced insulin secretion in mouse islets lacking ATP-sensitive K+ channels by improving the triggering Ca2+ signal. J. Biol. Chem. 282, 14768–14776 [DOI] [PubMed] [Google Scholar]
- 12. Arredouani A., Henquin J. C., Gilon P. (2002) Contribution of the endoplasmic reticulum to the glucose-induced [Ca2+]c response in mouse pancreatic islets. Am. J. Physiol. Endocrinol. Metab. 282, E982–E991 [DOI] [PubMed] [Google Scholar]
- 13. Duman J. G., Chen L., Palmer A. E., Hille B. (2006) Contributions of intracellular compartments to calcium dynamics: implicating an acidic store. Traffic 7, 859–872 [DOI] [PubMed] [Google Scholar]
- 14. Holz G. G., Holz G. (2004) New insights concerning the glucose-dependent insulin secretagogue action of glucagon-like peptide-1 in pancreatic beta cells. Horm. Metab. Res. 36, 787–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Drucker D. J. (2006) The biology of incretin hormones. Cell Metab. 3, 153–165 [DOI] [PubMed] [Google Scholar]
- 16. Johnson J. D., Misler S. (2002) Nicotinic acid-adenine dinucleotide phosphate-sensitive calcium stores initiate insulin signaling in human beta cells. Proc. Natl. Acad. Sci. U.S.A. 99, 14566–14571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mitchell K. J., Lai F. A., Rutter G. A. (2003) Ryanodine receptor type I and nicotinic acid adenine dinucleotide phosphate receptors mediate Ca2+ release from insulin-containing vesicles in living pancreatic beta cells (MIN6). J. Biol. Chem. 278, 11057–11064 [DOI] [PubMed] [Google Scholar]
- 18. Masgrau R., Churchill G. C., Morgan A. J., Ashcroft S. J., Galione A. (2003) NAADP: a new second messenger for glucose-induced Ca2+ responses in clonal pancreatic beta cells. Curr. Biol. 13, 247–251 [DOI] [PubMed] [Google Scholar]
- 19. Yamasaki M., Masgrau R., Morgan A. J., Churchill G. C., Patel S., Ashcroft S. J., Galione A. (2004) Organelle selection determines agonist-specific Ca2+ signals in pancreatic acinar and beta cells. J. Biol. Chem. 279, 7234–7240 [DOI] [PubMed] [Google Scholar]
- 20. Kim B. J., Park K. H., Yim C. Y., Takasawa S., Okamoto H., Im M. J., Kim U. H. (2008) Generation of NAADP and cADPR by glucagon-like peptide-1 evokes Ca2+ signal that is essential for insulin secretion in mouse pancreatic islets. Diabetes 57, 868–878 [DOI] [PubMed] [Google Scholar]
- 21. Naylor E., Arredouani A., Vasudevan S. R., Lewis A. M., Parkesh R., Mizote A., Rosen D., Thomas J. M., Izumi M., Ganesan A., Galione A., Churchill G. C. (2009) Discovery of a drug-like, small-molecule antagonist of the second messenger NAADP enabled by accessible virtual screening. Nat. Chem. Biol. 5, 220–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shawl A. I., Park K. H., Kim B. J., Higashida C., Higashida H., Kim U. H. (2012) Involvement of actin filament in the generation of Ca2+ mobilizing messengers in glucose-induced Ca2+ signaling in pancreatic beta cells. Islets 4, 145–151 [DOI] [PubMed] [Google Scholar]
- 23. Wang X., Zhang X., Dong X. P., Samie M., Li X., Cheng X., Goschka A., Shen D., Zhou Y., Harlow J., Zhu M. X., Clapham D. E., Ren D., Xu H. (2012) TPC proteins are phosphoinositide-activated sodium-selective ion channels in endosomes and lysosomes. Cell 151, 372–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Park K. H., Kim B. J., Shawl A. I., Han M. K., Lee H. C., Kim U. H. (2013) Autocrine/paracrine function of nicotinic acid adenine dinucleotide phosphate (NAADP) for glucose homeostasis in pancreatic beta cells and adipocytes. J. Biol. Chem. 288, 35548–35558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Churchill G. C., Okada Y., Thomas J. M., Genazzani A. A., Patel S., Galione A. (2002) NAADP mobilizes Ca2+ from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell 111, 703–708 [DOI] [PubMed] [Google Scholar]
- 26. Galione A. (2006) NAADP, a new intracellular messenger that mobilizes Ca2+ from acidic stores. Biochem. Soc. Trans. 34, 922–926 [DOI] [PubMed] [Google Scholar]
- 27. Galione A. (2015) A primer of NAADP-mediated Ca signalling: from sea urchin eggs to mammalian cells. Cell Calcium 58, 27–47 [DOI] [PubMed] [Google Scholar]
- 28. Guse A. H., Lee H. C. (2008) NAADP: a universal Ca2+ trigger. Sci. Signal. 1, re10. [DOI] [PubMed] [Google Scholar]
- 29. Calcraft P. J., Ruas M., Pan Z., Cheng X., Arredouani A., Hao X., Tang J., Rietdorf K., Teboul L., Chuang K. T., Lin P., Xiao R., Wang C., Zhu Y., Lin Y., et al. (2009) NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459, 596–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zong X., Schieder M., Cuny H., Fenske S., Gruner C., Rotzer K., Griesbeck O., Harz H., Biel M., Wahl-Schott C. (2009) The two-pore channel TPCN2 mediates NAADP-dependent Ca2+-release from lysosomal stores. Pflugers Arch. 458, 891–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brailoiu E., Churamani D., Cai X., Schrlau M. G., Brailoiu G. C., Gao X., Hooper R., Boulware M. J., Dun N. J., Marchant J. S., Patel S. (2009) Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J. Cell Biol. 186, 201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ruas M., Rietdorf K., Arredouani A., Davis L. C., Lloyd-Evans E., Koegel H., Funnell T. M., Morgan A. J., Ward J. A., Watanabe K., Cheng X., Churchill G. C., Zhu M. X., Platt F. M., Wessel G. M., et al. (2010) Purified TPC isoforms form NAADP receptors with distinct roles for Ca2+ signalling and endolysosomal trafficking. Curr. Biol. 20, 703–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jha A., Ahuja M., Patel S., Brailoiu E., Muallem S. (2014) Convergent regulation of the lysosomal two-pore channel-2 by Mg2+, NAADP, PI(3,5)P2 and multiple protein kinases. EMBO J. 33, 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grimm C., Holdt L. M., Chen C. C., Hassan S., Müller C., Jörs S., Cuny H., Kissing S., Schröder B., Butz E., Northoff B., Castonguay J., Luber C. A., Moser M., Spahn S., et al. (2014) High susceptibility to fatty liver disease in two-pore channel 2-deficient mice. Nat. Commun. 5, 4699. [DOI] [PubMed] [Google Scholar]
- 35. Ogunbayo O. A., Zhu Y., Shen B., Agbani E., Li J., Ma J., Zhu M. X., Evans A. M. (2015) Organelle-specific subunit interactions of the vertebrate two-pore channel family. J. Biol. Chem. 290, 1086–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sakurai Y., Kolokoltsov A. A., Chen C. C., Tidwell M. W., Bauta W. E., Klugbauer N., Grimm C., Wahl-Schott C., Biel M., Davey R. A. (2015) Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 347, 995–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ruas M., Davis L. C., Chen C. C., Morgan A. J., Chuang K. T., Walseth T. F., Grimm C., Garnham C., Powell T., Platt N., Platt F. M., Biel M., Wahl-Schott C., Parrington J., Galione A. (2015) Expression of Ca2+-permeable two-pore channels (TPC) rescues NAADP-signalling in TPC-deficient cells. EMBO J. 34, 1743–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cang C., Zhou Y., Navarro B., Seo Y. J., Aranda K., Shi L., Battaglia-Hsu S., Nissim I., Clapham D. E., Ren D. (2013) mTOR regulates lysosomal ATP-sensitive two-pore Na+ channels to adapt to metabolic state. Cell 152, 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Walseth T. F., Lin-Moshier Y., Jain P., Ruas M., Parrington J., Galione A., Marchant J. S., Slama J. T. (2012) Photoaffinity labeling of high affinity nicotinic acid adenine dinucleotide phosphate (NAADP)-binding proteins in sea urchin egg. J. Biol. Chem. 287, 2308–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lin-Moshier Y., Walseth T. F., Churamani D., Davidson S. M., Slama J. T., Hooper R., Brailoiu E., Patel S., Marchant J. S. (2012) Photoaffinity labeling of nicotinic acid adenine dinucleotide phosphate (NAADP) targets in mammalian cells. J. Biol. Chem. 287, 2296–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morgan A. J., Galione A. (2014) Two-pore channels (TPCs): current controversies. BioEssays 36, 173–183 [DOI] [PubMed] [Google Scholar]
- 42. Marchant J. S., Patel S. (2013) Questioning regulation of two-pore channels by NAADP. Messenger 2, 113–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marchant J. S., Lin-Moshier Y., Walseth T. F., Patel S. (2012) The molecular basis for Ca2+ signalling by NAADP: two-pore channels in a complex? Messenger 1, 63–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin-Moshier Y., Keebler M. V., Hooper R., Boulware M. J., Liu X., Churamani D., Abood M. E., Walseth T. F., Brailoiu E., Patel S., Marchant J. S. (2014) The two-pore channel (TPC) interactome unmasks isoform-specific roles for TPCs in endolysosomal morphology and cell pigmentation. Proc. Natl. Acad. Sci. U.S.A. 111, 13087–13092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hockey L. N., Kilpatrick B. S., Eden E. R., Lin-Moshier Y., Brailoiu G. C., Brailoiu E., Futter C. E., Schapira A. H., Marchant J. S., Patel S. (2015) Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J. Cell Sci. 128, 232–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aarhus R., Graeff R. M., Dickey D. M., Walseth T. F., Lee H. C. (1995) ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J. Biol. Chem. 270, 30327–30333 [DOI] [PubMed] [Google Scholar]
- 47. Palade P. (2007) The hunt for an alternate way to generate NAADP. Am. J. Physiol. Cell Physiol. 292, C4–C7 [DOI] [PubMed] [Google Scholar]
- 48. Kato I., Yamamoto Y., Fujimura M., Noguchi N., Takasawa S., Okamoto H. (1999) CD38 disruption impairs glucose-induced increases in cyclic ADP-ribose, [Ca2+]i, and insulin secretion. J. Biol. Chem. 274, 1869–1872 [DOI] [PubMed] [Google Scholar]
- 49. Ikehata F., Satoh J., Nata K., Tohgo A., Nakazawa T., Kato I., Kobayashi S., Akiyama T., Takasawa S., Toyota T., Okamoto H. (1998) Autoantibodies against CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) that impair glucose-induced insulin secretion in noninsulin-dependent diabetes patients. J. Clin. Invest. 102, 395–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yagui K., Shimada F., Mimura M., Hashimoto N., Suzuki Y., Tokuyama Y., Nata K., Tohgo A., Ikehata F., Takasawa S., Okamoto H., Makino H., Saito Y., Kanatsuka A. (1998) A missense mutation in the CD38 gene, a novel factor for insulin secretion: association with type II diabetes mellitus in Japanese subjects and evidence of abnormal function when expressed in vitro. Diabetologia 41, 1024–1028 [DOI] [PubMed] [Google Scholar]
- 51. Parkesh R., Lewis A. M., Aley P. K., Arredouani A., Rossi S., Tavares R., Vasudevan S. R., Rosen D., Galione A., Dowden J., Churchill G. C. (2008) Cell-permeant NAADP: a novel chemical tool enabling the study of Ca2+ signaling in intact cells. Cell Calcium 43, 531–538 [DOI] [PubMed] [Google Scholar]
- 52. Ruas M., Chunag K-T., Davis L. C., Al-Douri A., Tynan P. W., Tunn R., Teboul L., Galione A., Parrington J. (2014) TPC1 has two variant isoforms and their removal has different effects on endolysosomal functions compared to loss of TPC2. Mol. Cell. Biol. 34, 3981–3992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Q., Ramracheya R., Lahmann C., Tarasov A., Bengtsson M., Braha O., Braun M., Brereton M., Collins S., Galvanovskis J., Gonzalez A., Groschner L. N., Rorsman N. J., Salehi A., Travers M. E., et al. (2013) Role of KATP channels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab. 18, 871–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cross S. E., Hughes S. J., Partridge C. J., Clark A., Gray D. W., Johnson P. R. (2008) Collagenase penetrates human pancreatic islets following standard intraductal administration. Transplantation 86, 907–911 [DOI] [PubMed] [Google Scholar]
- 55. Meur G., Simon A., Harun N., Virally M., Dechaume A., Bonnefond A., Fetita S., Tarasov A. I., Guillausseau P. J., Boesgaard T. W., Pedersen O., Hansen T., Polak M., Gautier J. F., Froguel P., et al. (2010) Insulin gene mutations resulting in early-onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes 59, 653–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Miyazaki J., Araki K., Yamato E., Ikegami H., Asano T., Shibasaki Y., Oka Y., Yamamura K. (1990) Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 127, 126–132 [DOI] [PubMed] [Google Scholar]
- 57. Rutter G. A., Loder M. K., Ravier M. A. (2006) Rapid three-dimensional imaging of individual insulin release events by Nipkow disc confocal microscopy. Biochem. Soc. Trans. 34, 675–678 [DOI] [PubMed] [Google Scholar]
- 58. Galione A. (2011) NAADP Receptors. Cold Spring Harb. Perspect. Biol. 3, a004036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Patel S., Churchill G. C., Galione A. (2001) Coordination of Ca2+ signalling by NAADP. Trends Biochem. Sci. 26, 482–489 [DOI] [PubMed] [Google Scholar]
- 60. Morgan A. J., Platt F. M., Lloyd-Evans E., Galione A. (2011) Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 439, 349–374 [DOI] [PubMed] [Google Scholar]
- 61. Cancela J. M., Churchill G. C., Galione A. (1999) Coordination of agonist-induced Ca2+-signalling patterns by NAADP in pancreatic acinar cells. Nature 398, 74–76 [DOI] [PubMed] [Google Scholar]
- 62. Worley J. F., 3rd, McIntyre M. S., Spencer B., Mertz R. J., Roe M. W., Dukes I. D. (1994) Endoplasmic reticulum calcium store regulates membrane potential in mouse islet beta cells. J. Biol. Chem. 269, 14359–14362 [PubMed] [Google Scholar]
- 63. Arredouani A., Evans A. M., Ma J., Parrington J., Zhu M. X., Galione A. (2010) An emerging role for NAADP-mediated Ca2+ signaling in the pancreatic beta-cell. Islets 2, 323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ammälä C., Larsson O., Berggren P. O., Bokvist K., Juntti-Berggren L., Kindmark H., Rorsman P. (1991) Inositol trisphosphate-dependent periodic activation of a Ca2+-activated K+ conductance in glucose-stimulated pancreatic beta cells. Nature 353, 849–852 [DOI] [PubMed] [Google Scholar]
- 65. Ronco V., Potenza D. M., Denti F., Vullo S., Gagliano G., Tognolina M., Guerra G., Pinton P., Genazzani A. A., Mapelli L., Lim D., Moccia F. (2015) A novel Ca2+-mediated cross-talk between endoplasmic reticulum and acidic organelles: implications for NAADP-dependent Ca2+ signalling. Cell Calcium 57, 89–100 [DOI] [PubMed] [Google Scholar]
- 66. Cheng H., Beck A., Launay P., Gross S. A., Stokes A. J., Kinet J. P., Fleig A., Penner R. (2007) TRPM4 controls insulin secretion in pancreatic beta cells. Cell Calcium 41, 51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Colsoul B., Schraenen A., Lemaire K., Quintens R., Van Lommel L., Segal A., Owsianik G., Talavera K., Voets T., Margolskee R. F., Kokrashvili Z., Gilon P., Nilius B., Schuit F. C., Vennekens R. (2010) Loss of high-frequency glucose-induced Ca2+ oscillations in pancreatic islets correlates with impaired glucose tolerance in Trpm5−/− mice. Proc. Natl. Acad. Sci. U.S.A. 107, 5208–5213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Brixel L. R., Monteilh-Zoller M. K., Ingenbrandt C. S., Fleig A., Penner R., Enklaar T., Zabel B. U., Prawitt D. (2010) TRPM5 regulates glucose-stimulated insulin secretion. Pflügers Arch. 460, 69–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Krishnan K., Ma Z., Björklund A., Islam M. S. (2014) Role of transient receptor potential melastatin-like subtype 5 channel in insulin secretion from rat beta cells. Pancreas 43, 597–604 [DOI] [PubMed] [Google Scholar]
- 70. Grand T., Demion M., Norez C., Mettey Y., Launay P., Becq F., Bois P., Guinamard R. (2008) 9-Phenanthrol inhibits human TRPM4 but not TRPM5 cationic channels. Br. J. Pharmacol. 153, 1697–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gilon P., Henquin J. C. (2001) Mechanisms and physiological significance of the cholinergic control of pancreatic beta-cell function. Endocr. Rev. 22, 565–604 [DOI] [PubMed] [Google Scholar]
- 72. Tugba Durlu-Kandilci N., Ruas M., Chuang K. T., Brading A., Parrington J., Galione A. (2010) TPC2 proteins mediate NAADP and agonist-evoked contractions of smooth muscle. J. Biol. Chem. 285, 24925–24932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mitchell K. J., Pinton P., Varadi A., Tacchetti C., Ainscow E. K., Pozzan T., Rizzuto R., Rutter G. A. (2001) Dense core secretory vesicles revealed as a dynamic Ca2+ store in neuroendocrine cells with a vesicle-associated membrane protein aequorin chimaera. J. Cell Biol. 155, 41–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Henquin J. C. (1998) A minimum of fuel is necessary for tolbutamide to mimic the effects of glucose on electrical activity in pancreatic beta cells. Endocrinology 139, 993–998 [DOI] [PubMed] [Google Scholar]
- 75. Lee H. C., Aarhus R. (1995) A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J. Biol. Chem. 270, 2152–2157 [DOI] [PubMed] [Google Scholar]
- 76. Zhao Y., Graeff R., Lee H. C. (2012) Roles of cADPR and NAADP in pancreatic cells. Acta Biochim. Biophys. Sin. 44, 719–729 [DOI] [PubMed] [Google Scholar]
- 77. Takasawa S., Nata K., Yonekura H., Okamoto H. (1993) Cyclic ADP-ribose in insulin secretion from pancreatic beta cells. Science 259, 370–373 [DOI] [PubMed] [Google Scholar]
- 78. Genazzani A. A., Galione A. (1996) Nicotinic acid-adenine dinucleotide phosphate mobilizes Ca2+ from a thapsigargin-insensitive pool. Biochem. J. 315, 721–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lee H. C., Aarhus R. (2000) Functional visualization of the separate but interacting calcium stores sensitive to NAADP and cyclic ADP-ribose. J. Cell Sci. 113, 4413–4420 [DOI] [PubMed] [Google Scholar]
- 80. Galione A., Morgan A. J., Arredouani A., Davis L. C., Rietdorf K., Ruas M., Parrington J. (2010) NAADP as an intracellular messenger regulating lysosomal calcium-release channels. Biochem. Soc. Trans. 38, 1424–1431 [DOI] [PubMed] [Google Scholar]
- 81. Morgan A. J., Davis L. C., Wagner S. K., Lewis A. M., Parrington J., Churchill G. C., Galione A. (2013) Bidirectional Ca2+ signaling occurs between the endoplasmic reticulum and acidic organelles. J. Cell Biol. 200, 789–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gilon P., Arredouani A., Gailly P., Gromada J., Henquin J. C. (1999) Uptake and release of Ca2+ by the endoplasmic reticulum contribute to the oscillations of the cytosolic Ca2+ concentration triggered by Ca2+ influx in the electrically excitable pancreatic B-cell. J. Biol. Chem. 274, 20197–20205 [DOI] [PubMed] [Google Scholar]
- 83. Arredouani A., Guiot Y., Jonas J. C., Liu L. H., Nenquin M., Pertusa J. A., Rahier J., Rolland J. F., Shull G. E., Stevens M., Wuytack F., Henquin J. C., Gilon P. (2002) SERCA3 ablation does not impair insulin secretion but suggests distinct roles of different sarcoendoplasmic reticulum Ca2+ pumps for Ca2+ homeostasis in pancreatic beta cells. Diabetes 51, 3245–3253 [DOI] [PubMed] [Google Scholar]
- 84. Pitt S. J., Funnell T. M., Sitsapesan M., Venturi E., Rietdorf K., Ruas M., Ganesan A., Gosain R., Churchill G. C., Zhu M. X., Parrington J., Galione A., Sitsapesan R. (2010) TPC2 is a novel NAADP-sensitive Ca2+ release channel, operating as a dual sensor of luminal pH and Ca2+. J. Biol. Chem. 285, 35039–35046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rybalchenko V., Ahuja M., Coblentz J., Churamani D., Patel S., Kiselyov K., Muallem S. (2012) Membrane potential regulates nicotinic acid adenine dinucleotide phosphate (NAADP) dependence of the pH- and Ca2+-sensitive organellar two-pore channel TPC1. J. Biol. Chem. 287, 20407–20416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Patel S., Ramakrishnan L., Rahman T., Hamdoun A., Marchant J. S., Taylor C. W., Brailoiu E. (2011) The endolysosomal system as an NAADP-sensitive acidic Ca2+ store: role for the two-pore channels. Cell Calcium 50, 157–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Guse A. H. (2012) Linking NAADP to ion channel activity: a unifying hypothesis. Sci. Signal. 5, pe18. [DOI] [PubMed] [Google Scholar]
- 88. Leech C. A., Habener J. F. (1997) Insulinotropic glucagon-like peptide-1-mediated activation of nonselective cation currents in insulinoma cells is mimicked by maitotoxin. J. Biol. Chem. 272, 17987–17993 [DOI] [PubMed] [Google Scholar]
- 89. Marigo V., Courville K., Hsu W. H., Feng J. M., Cheng H. (2009) TRPM4 impacts on Ca2+ signals during agonist-induced insulin secretion in pancreatic beta cells. Mol. Cell. Endocrinol. 299, 194–203 [DOI] [PubMed] [Google Scholar]
- 90. Guinamard R., Demion M., Launay P. (2010) Physiological roles of the TRPM4 channel extracted from background currents. Physiology 25, 155–164 [DOI] [PubMed] [Google Scholar]
- 91. Tsaih S. W., Holl K., Jia S., Kaldunski M., Tschannen M., He H., Andrae J. W., Li S. H., Stoddard A., Wiederhold A., Parrington J., Ruas da Silva M., Galione A., Meigs J., Meta-analyses of Glucose and Insulin-related Traits Consortium (MAGIC) Investigators, et al. (2014) Identification of a novel gene for diabetic traits in rats, mice, and humans. Genetics 198, 17–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Park D. R., Park K. H., Kim B. J., Yoon C. S., Kim U. H. (2015) Exercise ameliorates insulin resistance via Ca2+ signals distinct from those of insulin for GLUT4 translocation in skeletal muscles. Diabetes 64, 1224–1234 [DOI] [PubMed] [Google Scholar]
- 93. Song E. K., Lee Y. R., Kim Y. R., Yeom J. H., Yoo C. H., Kim H. K., Park H. M., Kang H. S., Kim J. S., Kim U. H., Han M. K. (2012) NAADP mediates insulin-stimulated glucose uptake and insulin sensitization by PPARγ in adipocytes. Cell Rep. 2, 1607–1619 [DOI] [PubMed] [Google Scholar]
- 94. Davis L. C., Morgan A. J., Ruas M., Wong J. L., Graeff R. M., Poustka A. J., Lee H. C., Wessel G. M., Parrington J., Galione A. (2008) Ca2+ signaling occurs via second messenger release from intraorganelle synthesis sites. Curr. Biol. 18, 1612–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Pupilli C., Giannini S., Marchetti P., Lupi R., Antonelli A., Malavasi F., Takasawa S., Okamoto H., Ferrannini E. (1999) Autoantibodies to CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) in Caucasian patients with diabetes: effects on insulin release from human islets. Diabetes 48, 2309–2315 [DOI] [PubMed] [Google Scholar]
- 96. Davis L. C., Morgan A. J., Chen J. L., Snead C. M., Bloor-Young D., Shenderov E., Stanton-Humphreys M. N., Conway S. J., Churchill G. C., Parrington J., Cerundolo V., Galione A. (2012) NAADP activates two-pore channels on T cell cytolytic granules to stimulate exocytosis and killing. Curr. Biol. 22, 2331–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]