

Background: Gram-positive bacteria secrete pilins through the Sec translocon in unfolded states.
Results: Disruption of pilus disulfide bonds or genetic disruption of oxidoreductase-encoding genes mdbA and vkor abrogates pilus assembly in Actinomyces oris.
Conclusion: MdbA and VKOR constitute a disulfide bond-forming machine in A. oris.
Significance: Oxidative protein folding may be common in Actinobacteria and an attractive target for antimicrobials.
Keywords: actinobacteria, crystal structure, crystallography, disulfide, secretion, Actinomyces, coaggregation, oxidative protein folding, pili, sortase
Abstract
Export of cell surface pilins in Gram-positive bacteria likely occurs by the translocation of unfolded precursor polypeptides; however, how the unfolded pilins gain their native conformation is presently unknown. Here, we present physiological studies to demonstrate that the FimA pilin of Actinomyces oris contains two disulfide bonds. Alanine substitution of cysteine residues forming the C-terminal disulfide bridge abrogates pilus assembly, in turn eliminating biofilm formation and polymicrobial interaction. Transposon mutagenesis of A. oris yielded a mutant defective in adherence to Streptococcus oralis, and revealed the essential role of a vitamin K epoxide reductase (VKOR) gene in pilus assembly. Targeted deletion of vkor results in the same defects, which are rescued by ectopic expression of VKOR, but not a mutant containing an alanine substitution in its conserved CXXC motif. Depletion of mdbA, which encodes a membrane-bound thiol-disulfide oxidoreductase, abrogates pilus assembly and alters cell morphology. Remarkably, overexpression of MdbA or a counterpart from Corynebacterium diphtheriae, rescues the Δvkor mutant. By alkylation assays, we demonstrate that VKOR is required for MdbA reoxidation. Furthermore, crystallographic studies reveal that A. oris MdbA harbors a thioredoxin-like fold with the conserved CXXC active site. Consistently, each MdbA enzyme catalyzes proper disulfide bond formation within FimA in vitro that requires the catalytic CXXC motif. Because the majority of signal peptide-containing proteins encoded by A. oris possess multiple Cys residues, we propose that MdbA and VKOR constitute a major folding machine for the secretome of this organism. This oxidative protein folding pathway may be a common feature in Actinobacteria.
Introduction
Actinomyces spp. were an early model to study fimbriae (or pili) in Gram-positive bacteria (1). Actinomyces oris produce two types of fimbriae covalently linked to the cell wall (2). Type 1 fimbriae, comprised of the fimbrial shaft FimP and tip fimbrillin FimQ, mediate bacterial adherence to the tooth surface via FimQ interactions with salivary proline-rich protein deposits (3). Type 2 fimbriae, made of the fimbrial shaft FimA and tip fimbrillin FimB or CafA, are required for bacterial adherence to host cells, biofilm formation, and bacterial coaggregation (4–6). Like many other Gram-positive bacteria, the A. oris fimbriae are assembled by membrane-bound transpeptidase enzymes known as pilus-specific sortases, which were first discovered in the SpaA pili of Corynebacterium diphtheriae (7). Following their synthesis in the cytoplasm, it is proposed that the pilus precursors are transported across the membrane likely in unfolded states and inserted into the membrane via a cell wall sorting signal (CWSS) (8). Here, pilus-specific sortase enzymes join pilin subunits into pilus structures by cross-linking the pilin motif and the cell wall sorting signal (2, 9). A second sortase enzyme called the housekeeping sortase ultimately anchors the pilus polymers to the cell wall (10).
Critically, how pilin precursors are folded during pilus assembly is not known. One clue was revealed from structural studies of A. oris and C. diphtheriae pilins, which predicted that FimA, FimP, and SpaA contain disulfide bonds within their IgG-like domains (4, 11, 12). The tip pilin CafA contains 12 cysteine residues throughout the protein. Significantly, alanine substitution of two cysteine residues within the Cna 1 domain of CafA leads to CafA degradation and eliminates CafA pilus polymerization and cell-to-cell adherence (6). These observations suggest that disulfide bond formation is important for pilin folding.
Disulfide bond formation is catalyzed by thioredoxin-like proteins in non-reducing compartments like the eukaryotic endoplasmic reticulum, the inner membrane space of mitochondria, and the periplasm of Gram-negative bacteria (13). The protein-disulfide isomerase was the first disulfide bond-forming enzyme discovered by Anfinsen and colleagues (14, 15) that contains two redox-active CXXC motifs. A thiol-disulfide oxidoreductase DsbA in Escherichia coli also harbors a reactive disulfide bond within a CXXC motif and catalyzes disulfide bond formation by donating this linkage to reduced Cys residues in nascent polypeptides delivered to the periplasm by the Sec translocon (16, 17). Following catalysis, the CXXC catalytic site of DsbA is reduced and requires re-oxidation by the membrane-bound DsbB (18, 19). DsbB recycles DsbA activity by shuttling electrons to quinone, a component of the electron transport chain (20). Interestingly, a protein in the actinobacterium Mycobacterium tuberculosis annotated as vitamin K epoxide reductase (VKOR)3 was shown to be functionally similar to E. coli DsbB (21). Although DsbA-like proteins have been found in M. tuberculosis (22–24), to date their role as thiol-disulfide oxidoreductase enzymes has not been demonstrated in vivo. Nonetheless, oxidoreductase-encoding genes have been identified in the genomes of other actinobacteria including Corynebacterium glutamicum (21, 25), supporting the existence of an oxidative folding pathway in these organisms, which lack periplasmic spaces.
Here, we describe an oxidative folding pathway for pilus proteins in the actinobacterium A. oris. We show that A. oris pilins contain disulfide bonds that are required for pilus assembly, cell-to-cell adhesion, and biofilm formation. The oxidative folding of A. oris pilus proteins is catalyzed by a membrane-bound thiol-disulfide oxidoreductase, which we named MdbA (mdb for monoderm disulfide bond-forming). Re-oxidation of MdbA requires a second membrane-spanning oxidoreductase called VKOR. This disulfide bond-forming machine could be reconstituted in vitro using a native substrate. Importantly, a vkor mutant fails to assemble pili and is unable to form biofilms or interact with other bacteria. These defects are rescued by expression of A. oris MdbA or a comparable enzyme from C. diphtheriae. Consistent with a broad role of MdbA in protein folding, deletion of mdbA is lethal. Since over 60% of signal peptide-containing proteins encoded by these bacteria harbor two or more Cys residues, we posit that intramolecular disulfide bond formation constitutes a major folding pathway for Actinobacteria, and as such, is an important target for therapeutic development.
Experimental Procedures
Recombinant Plasmids pVKOR
To construct pVKOR, primers VKOR_F_Ndel and VKOR_R_KpnI (supplemental Table S1) were used to PCR amplify the promoter and coding regions of A. oris vkor along with flanking Ndel and KpnI sites. The resulting PCR product was digested with these enzymes, cloned into pJRD215 precut with NdeI and KpnI, and then used to transform E. coli DH5α. The resulting plasmid was electroporated into MR108.
pMdbAAo
The promoter and open reading frame (ORF) of A. oris mdbA were PCR amplified using primers (MdbAAO_F_Xbal and MdbAAO_R_Ecorl; supplemental Table S1) designed to append XbaI and EcoRI sites. The PCR product was digested with XbaI and EcoRI, and cloned into pJRD215 precut with similar enzymes. The resulting plasmid was electroporated into MR108.
pAraC-MdbAAo
Using primers mdbAAo_F_ATG and mdbAAo_R_Ecorl (supplemental Table S1), the ribosome binding site and ORF of A. oris mdbA was PCR-amplified using Phusion Polymerase® (New England Biolabs) to generate blunt ends. The resulting product was 5′ phosphorylated with polynucleotide kinase (New England Biolabs) and then digested with EcoRI. Using pBad33 as a template, primers araC_F_KpnI and araC_R amplified araC and the corresponding arabinose-inducible promoter, which was then digested with KpnI. Finally, the PCR fragments were cloned into pJRD215 precut with EcoRI and KpnI, and the resulting plasmid was electroporated into MR111.
pJRD-MdbACd
Using primers mdbACd_F and mdbACd_R_HindIII, the ORF for C. diphtheriae mdbA was PCR amplified using Phusion DNA Polymerase® (New England Biolabs) to generate blunt ends. The resulting product was 5′ phosphorylated and cut with HindIII. The promoter and ribosome binding site of A. oris mdbA were amplified with primers PmdbAAO_F_KpnI and PmdbAAO_R, and then digested with KpnI. Both DNA fragments then were ligated with pJRD215 precut with KpnI and HindIII to construct the recombinant plasmid for electroporation into MR108.
Recombinant Vectors Using pMCSG7
To generate recombinant, His-tagged MdbA proteins, primers (see supplemental Table S1) were designed to amplify the extracellular-coding regions of A. oris and C. diphtheriae mdbA. The resulting PCR products were cloned into pMCSG7 using ligation-independent cloning (26). Purified DNA fragments were treated with LIC-component T4 DNA polymerase (Novagen) and 2.5 μm dCTP. Meanwhile, pMCSG7, precut with SspI, was treated with LIC-competent T4 polymerase and dGTP. These reactions resulted in the formation of complementary overhangs between the mdbA genes and linearized vector. The products were incubated over a gradient of temperatures (3 min at 70°C, 2 min at 65°C, 2 min at 60°C, 2 min at 55°C, 1 min at 50°C, 1 min at 45°C, 1 min at 40°C, 1 min at 35°C, 1 min at 30°C, and 5 min at 25°C) for annealing. The resulting plasmids were used to transform E. coli DH5α and the insert was confirmed by DNA sequencing. The plasmids were then introduced into E. coli BL21 (DE3) for protein expression.
Site-directed Mutagenesis of Recombinant Plasmids
To construct Cys-to-Ala mutations within FimA, overlapping primers (supplemental Table S1) carrying the target mutations were used in PCR amplification using pCR2.1-FimA (4) as a template. The PCR products were digested overnight at 37 °C with DpnI to remove the parental template, and the resulting DNA samples were used to transform DH5α. The generated mutations were confirmed by sequencing, and fimA was removed from pCR2.1 by digestion with XbaI and EcoRI. The DNA fragment was cloned into pJRD508FimB precut with similar restriction enzymes. The resulting plasmids were electroporated into the A. oris ΔfimA mutant (AR4) (4).
To generate cysteine to alanine mutations within MdbACd and MdbAAo, inverse PCR was utilized using recombinant plasmids as templates (supplemental Table S2). Appropriate primers (supplemental Table S1) carrying the desired mutations were 5′ phosphorylated and used to PCR the plasmid templates with Phusion HF DNA polymerase. Purified products were treated with ligase to reform the circular plasmids, which were introduced into E. coli DH5α. The mutations were confirmed by DNA sequencing, and the plasmids were introduced to appropriate strains.
Generation of Deletion Mutants in A. oris
Nonpolar, in-frame deletion mutants in A. oris were generated using the GalK counterselection method established previously (5). Briefly, 1-kb fragments up- and downstream of a targeted gene were cloned pCWU2, an integrative plasmid expressing Kan resistance and galK genes (5). The resulting plasmid was electroporated into A. oris CW1, which lacks a functional galK. Co-integrants resulting from a single crossover event were selected for growth on Kan. To promote a recombination event, cells were grown in HI broth in the absence of Kan. Loss of the integrative plasmid was selected for growth on HI agar plates containing 0.2% 2-deoxygalactose. A conditional mdbA deletion mutant was made with the inducible plasmid pAraC-MdbAAo using a previously published protocol (27). Generated mutants were further characterized by PCR and/or Western blot analysis.
Tn5 Transposon Mutagenesis of A. oris
A library of roughly 3,000 Tn5 mutants was created using the Tn5 transposon system recently developed for A. oris (27). To identify factors required for fimbrial assembly and bacterial coaggregation, we set up a cell-based screen that is dependent on type 2 fimbriae for interaction with Streptococcus oralis. In this screen, Actinomyces Tn5 mutants grown in 96-well plates were mixed equally with S. oralis 34 in coaggregation buffer (5). Coaggregation was visually scored using an inverted microscope by comparing with both positive (A. oris MG-1 and S. oralis 34) and negative controls (S. oralis OC1 lacking RPS receptors or A. oris ΔfimA). Four coaggregation-deficient mutants obtained from this screen were further confirmed by the coaggregation and fimbrial assembly assays. Chromosomal DNA of the mutants was then isolated, and genes disrupted by Tn5 were identified by TAIL PCR and DNA sequencing (28).
Protein Purification
Cultures of E. coli BL21 (DE3) harboring individual recombinant plasmids (Table S2; pMCSG7s) were grown at 37°C in LB until A600 of ∼0.7. Protein expression was induced by addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside at 30 °C for 3 h. Cell pellets were harvested by centrifugation and resuspended in EQ buffer (50 mm Tris-HCl, pH 7.5, 100 mm NaCl). Cell lysis was achieved by using a French Press cell. Clear lysates obtained by centrifugation were subject to affinity chromatography, and purified His-tagged proteins were dialyzed in dialysis buffer (50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 10% glycerol) at 4 °C and stored at −20 °C.
For crystallization studies, E. coli cells harboring pMCSG7-MdbAAo were cultured in M9 medium supplemented with ampicillin (100 μg/ml). A selenomethionine derivative of the expressed MdbA protein was prepared as previously described (29). Cells were harvested by centrifugation, disrupted by sonication, and the insoluble cellular material was removed by centrifugation. The protein was purified by affinity chromatography using nickel-nitrilotriacetic acid (Qiagen) with the addition of 5 mm β-mercaptoethanol in all buffers. The protein was digested with 0.15 mg of tobacco etch virus protease per 20 mg of purified protein for 16 h at 4 °C, and then passed through a nickel-nitrilotriacetic acid column to remove both the tobacco etch virus protease and cleaved N-terminal tags. The final step of purification was gel-filtration on a HiLoad 16/60 Superdex 200pg column (GE Healthcare) in 10 mm HEPES buffer, pH 7.5, 200 mm NaCl and 1 mm DTT. The protein was concentrated on Amicon Ultracel 10K centrifugal filters (Millipore) approximately to 45 mg/ml.
Protein Crystallization, Data Collection, Structure Determination and Refinement
The initial crystallization condition was determined with a sparse crystallization matrix at 4 and 16 °C temperatures using the sitting-drop vapor-diffusion technique in 96-well plates with MCSG crystallization suite (Microlytic), Pi-minimal and Pi-PEG screens (Jena Bioscience) (30). Several conditions yielded diffraction quality crystals. The best crystals (rhomboidal shape, 0.2 × 0.2 × 0.15 μm) were directly obtained from G12 conditions of Pi-minimal screen (37.1% PEG 8000, 40 mm sodium iodide, 150 mm CAPSO buffer, pH 9.5) using the protein concentration of 45 mg/ml at 4 °C after 1 week. Crystals selected for data collection were flash-cooled in liquid nitrogen without addition of any cryo-protectant.
Single-wavelength x-ray diffraction data were collected at 100 K at the 19-ID beamline of the Structural Biology Center (31) at the Advanced Photon Source at Argonne National Laboratory using the program SBCcollect. The intensities were integrated and scaled with the HKL3000 suite (32).
The structure was determined by single-wavelength anomalous dispersion phasing using the HKL3000 suite (33) incorporating SHELXC, SHELXD, SHELXE (34), MLPHARE, and SOLVE/RESOLVE (35) programs. Several rounds of manual adjustments of structure models using COOT (36) and refinements with the Refmac program (37) from the CCP4 suite (38) were performed. The stereochemistry of the structure was validated with the PHENIX suite (33) incorporating MOLPROBITY (39) tools. A summary of data collection and refinement statistics is presented in supplemental Tables S1 and S2.
Cell Fractionation and Western Blotting
Equivalent overnight cultures of A. oris strains were used to inoculate fresh cultures (1:50 dilution). Cells grown to early- or mid-log phase at 37°C were normalized to an A600 of 1.0, and subject to cell fractionation as previously described (27). Proteins in culture medium (S), cell wall (W), membrane (M), and cytoplasm (C) fractions were TCA precipitated and acetone washed. Protein samples were heated in sample buffer containing SDS at 60 °C for 10 min prior to SDS-PAGE analysis using 3–12 or 3–20% Tris glycine gradient gels. Proteins were detected by immunoblotting with specific antibodies (1:5,000 for α-FimA; 1:4,000 for α-MdbAAo; and 1:10,000 for α-FimP).
Determination of the Redox Status of Pilus Proteins and MdbA by Alkylation
For A. oris, FimA monomers were isolated from A. oris AR4 pFimA-K198A, a mutant strain that expresses cell wall-anchored monomeric FimA (4). Bacteria grown overnight on HI agar plates, washed, and re-suspended in SMM buffer (500 mm sucrose, 10 mm MgCl2, 10 mm maleate, pH 6.8) were treated with 300 units ml−1 of mutanolysin for 3 h at 37 °C. The soluble cell wall fractions were isolated by centrifugation, TCA precipitated, and acetone washed.
Obtained FimA Proteins Were Alkylated by Similar Methods
Proteins were reduced in DTT-containing buffer (100 mm Tris-HCl, 1% SDS, 100 mm DTT, pH 8) at room temperature for 1 h, followed by TCA precipitation and acetone wash to remove DTT. The resulting pellets were treated with Mal-PEG in alkylation buffer (100 mm Tris-HCl, pH 6.8, 1% SDS, 20 mm Mal-PEG 2 kDa) at room temperature for 1 h, followed by TCA precipitation and acetone wash. Protein samples were then dissolved in SDS-loading buffer and separated by 3–20% Tris glycine gels for immunoblotting with α-FimA.
To investigate the redox status of MdbA, equal cell numbers of wild-type A. oris and Δvkor grown to mid-log phase were collected, washed, and re-suspended in PBS, and then lysed by mechanical disruption with glass beads (5 cycles of shaking for 30 s, followed by incubation on ice for 10 min). The protein samples were acid-trapped and precipitated with TCA, washed with acetone, and then re-suspended in alkylation buffer with Mal-PEG. Following a 1-h incubation at room temperature, the samples were TCA precipitated, re-suspended in SDS-loading buffer, and separated by 3–20% Tris glycine gels for immunoblotting with antibodies against A. oris MdbA (α-MdbAAo).
Coaggregation and Biofilm Assays
Coaggregation and biofilm assays were performed according to a previous procedure with minor modifications (5). Briefly, stationary cultures of A. oris and S. oralis were normalized to an A600 of 1.5. Bacterial cells were harvested by centrifugation, washed 3 times in TBS buffer (200 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.1 mm CaCl2), and then re-suspended in 500 μl of TBS. A. oris and S. oralis suspensions were then mixed in 12-well plates, and coaggregation was imaged with bacterial aggregates sedimented at the bottom of the wells.
For biofilm growth, equivalent overnight cultures of A. oris strains were used to inoculate fresh cultures (1:100 dilution) in 24-well plates containing 1% sucrose at 37 °C with 5% CO2 for 48 h. Biofilms were washed gently 3 times with phosphate-buffered saline (PBS), air-dried, stained with 0.5% crystal violet for 30 min, and quantified by optical density (A580) using crystal violet extracts in 80% ethanol. The results were presented as an average of three independent experiments performed in triplicate.
Reconstitution of Disulfide Bond Formation in Vitro
Recombinant FimA isolated from E. coli was reduced overnight at room temperature with 100 mm DTT in 50 mm Tris-HCl, pH 8.0. After reduction and acid-trapping of free thiols by HCl, DTT was removed by centrifugation using 3-kDa Amicon centrifugal filters and exchanged with 50 mm Tris-HCl, pH 3.5. 3 μm reduced FimA was incubated in redox buffer (100 mm Tris-HCl, pH 8.0, 2 mm EDTA, 0.2 mm GSSG, 1 mm GSH) in the presence of 1.8 μm recombinant wild-type or MdbA mutant enzymes at 37 °C. Similar reactions without enzymes were used as controls. At time intervals of 0, 5, 10, 15, and 30 min, reactions were stopped with the addition of Mal-PEG buffer (20 mm Mal-PEG, 1% SDS, 100 mm Tris-HCl, pH 6.8). After incubation at room temperature for 1 h, glycerol was added to each reaction to a final concentration of 20% before SDS-PAGE using 3–20% Tris glycine gels and detection by Coomassie stain.
Electron Microscopy
Immunogold labeling of bacterial pili was followed accordingly (40). In brief, ∼7 μl of bacterial suspension in PBS was placed onto carbon-coated nickel grids and washed with PBS containing 1% BSA, followed by incubation with 0.1% gelatin in PBS plus 1% BSA. Samples were stained with primary antibodies diluted in PBS, 1% BSA (1:100 for α-FimA and α-FimP; 1:25 for α-FimB and α-FimQ), followed by staining with secondary antibody conjugated to 12- or 18-nm gold particles diluted 1:20 in PBS, 1% BSA. Finally, samples were then washed 5 times with sterile water and stained with 1% uranyl acetate. The samples were viewed by a JEOL JEM-1400 electron microscope.
Results
Essentiality of Disulfide Bonds in Pilus Assembly, Biofilm Formation, and Interspecies Interactions
FimP and FimA, the pilus shaft proteins of types 1 and 2 fimbriae of A. oris, respectively, contain pairs of cysteine (Cys) residues within the N and C termini (see Fig. 1A for FimA). Genetics and structural studies have predicted that these residues are oxidized to form disulfide bonds (4, 12). Within FimA, thiol linkages are predicted to form between Cys116-Cys157 and Cys394-Cys445 (Fig. 1A). To test if these bonds form in vivo, we employed alkylation using methoxypolyethylene glycol-maleimide (Mal-PEG), a 2-kDa agent that reacts with free sulfhydryls to form stable thioether bonds (41). FimA monomers were collected from A. oris upon muramidase treatment (see “Experimental Procedures”), re-suspended in buffer with or without DTT, and incubated with Mal-PEG. The treated FimA was detected by Western blotting. As shown, Mal-PEG treatment did not change migration of FimA monomers in SDS-PAGE compared with the untreated protein (Fig. 1B, compare lanes 1 and 3). However, pretreatment of FimA with DTT produced a visible up-shift of the FimA band after Mal-PEG alkylation (Fig. 1B, lane 4), signifying the attachment of Mal-PEG to the protein via the formation of thioether bonds. Because pretreatment with DTT was required for protein alkylation, we conclude that the Cys residues in FimA are linked by disulfide bonds in vivo.
FIGURE 1.

Requirement of disulfide bonds in pilus assembly, biofilm formation, and coaggregation. A, the C-terminal disulfide bond of FimA is predicted to form between Cys394 and Cys445. The modeled IgG-fold domain (yellow) at the N terminus, absent from the original structure, was proposed to contain a disulfide bond between Cys116 and Cys157. B, FimA monomers isolated from the cell wall were treated or mock treated with DTT, followed by Mal-PEG. The protein samples were analyzed by immunoblotting with α-FimA. Reduced and oxidized forms of FimA are indicated. C, culture medium (M) and cell wall (W) fractions were collected from the A. oris MG1 strain (WT) and its isogenic derivatives. Equivalent protein samples harvested by TCA precipitation were analyzed by immunoblotting with α-FimA. Monomeric and polymeric forms of FimA, as well as molecular mass markers (kDa) are indicated. D, A. oris biofilms were cultivated in 12-well plates at 37 °C with 5% CO2 for 48 h. Biofilms were stained with crystal violet, and biofilm production was quantified by measuring absorbance at 580 nm. E, for coaggregation, A. oris and RPS-positive S. oralis (So34) cells were mixed in equal numbers for coaggregation were imaged by an AlphaImager. S. oralis OC1 strain lacking RPS was used as a negative control.
We next addressed whether disulfide bonds play a role in pilus assembly by substituting Cys residues in FimA individually to Ala by site-directed mutagenesis using pFimA and testing if the constructs could rescue a fimA deletion mutant (ΔfimA). To examine pilus polymerization, subcellular fractions of A. oris grown to mid-log phase were immunoblotted with α-FimA. As expected, polymerized FimA (marked P) was detected in both medium (M) and cell wall (W) fractions collected from the parental (MG-1) strain, but no FimA was detected in the ΔfimA mutant (Fig. 1C, lanes 1–4). Pilus assembly was apparent in cells complemented with pFimA encoding wild-type FimA (lanes 5 and 6). Strikingly, whereas the C116A and C157A mutations did not affect pilus production, the mutants targeting the C-terminal Cys residues (C394A and C445A) produced no visible pilus polymers (last 8 lanes). Instead, a low molecular mass form of FimA (less than 55 kDa) was abundantly secreted into the extracellular media of these mutants (Fig. 1C, M lanes for C394A and C445A). Pilus assembly was further examined using immunogold electron microscopy (IEM). Similar to the Western blotting results, no fimbriae were detected on the surface of pC394A or pC445A mutants (Fig. 2).
FIGURE 2.
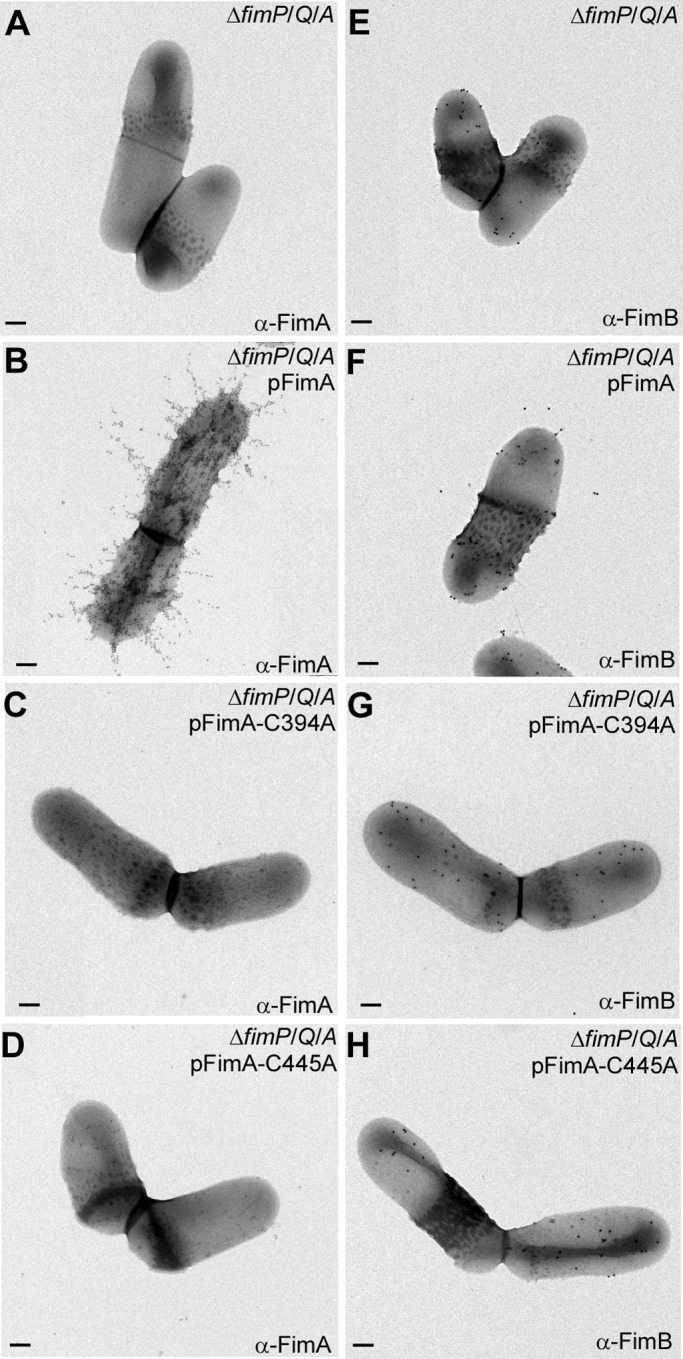
Assembly of A. oris type 2 fimbriae requires the formation of disulfide bonds. A. oris cells of various strains immobilized on nickel grids were stained with α-FimA (A–D) or α-FimB (E–H) followed by anti-rabbit IgG conjugated to 12- or 18-nm gold particles, respectively. The samples were stained with 1% uranyl acetate and viewed by a transmission electron microscope. Scale bars indicate 0.2 μm. Note that the experiments were performed with A. oris strains lacking type 1 fimbriae, i.e. ΔfimP/Q/A, to eliminate background.
To assess the bacterial phenotypes further, we examined if the Cys mutations affected A. oris biofilm formation and co-aggregation with S. oralis, two key processes known to require type 2 fimbriae (5). To cultivate biofilm, A. oris were grown under conditions of biofilm development (see “Experimental Procedures”) and after 48 h, the resulting biofilms were stained with crystal violet. Wild-type A. oris cells formed a robust biofilm, whereas the ΔfimA mutant failed to produce any as expected (Fig. 1D). Complementation with pFimA as well as the pC116A or pC157A mutants restored biofilm growth, but pC394A and pC445A did not form biofilm. These strains were also analyzed for interspecies interactions using S. oralis co-aggregation assays (5). Coaggregation between A. oris MG-1 and S. oralis So34 was visible, but undetectable when A. oris MG-1 was combined with S. oralis OC1, a mutant that lacks the receptor for type 2 fimbriae (Fig. 1E). The ΔfimA mutant did not coaggregate with So34, but complementation with pFimA, pC116A, or p157A restored the interaction. Identical to the ΔfimA mutant, A. oris containing pC394A or pC445A also failed to co-aggregate. Altogether, these data show that the C-terminal Cys394-Cys445 disulfide bond is essential for type 2 fimbrial assembly, whereas the N-terminal Cys116-Cys157 disulfide bond is dispensable.
The Disulfide Bond-forming Machine in A. oris
Because bacterial coaggregation is linked to FimA assembly, we aimed to identify disulfide bond-forming factors by screening a Tn5 transposon library of ∼3,000 clones for A. oris mutants that failed to co-aggregate with S. oralis. Mapping of Tn5 inserts in the four mutants by DNA sequencing revealed the expected insertions disrupting fimA and srtC2, the type 2 fimbria-specific sortase. One insertion, however, disrupted a vkor homolog (Fig. 3A). The A. oris VKOR homolog is a 27-kDa protein predicted to contain five predicted transmembrane helices and an N-terminal CXXC motif located in an exoplasmic loop. To confirm that the phenotype of the vkor::Tn5 mutant was not due to a polar effect, we created an unmarked vkor deletion mutant targeted by allelic exchange. This mutant also failed to coaggregate with S. oralis, and the defect was rescued by plasmid complementation (Fig. 3A).
FIGURE 3.
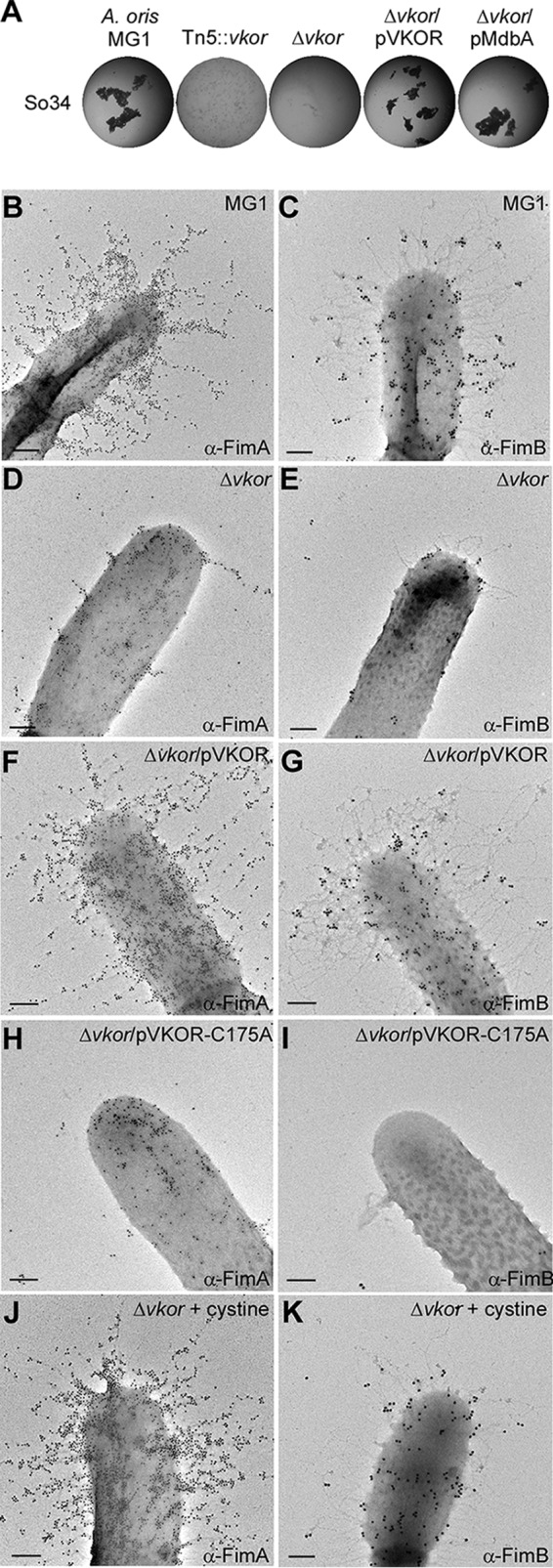
A. oris VKOR is required for type 2 fimbrial assembly. A, coaggregation was performed as described in the legend to Fig. 1E. B–K, A. oris cells of various strains grown in the absence or presence of cystine were immobilized on nickel grids and stained with α-FimA (B, D, F, H, and J) or α-FimB (C, E, G, I, and K), followed by anti-rabbit IgG conjugated to 18-nm gold particles. The samples were stained with 1% uranyl acetate and viewed by a transmission electron microscope. Scale bars indicate 0.5 μm.
To elucidate the coaggregation defect of the Δvkor mutant, we examined type 2 fimbriae by IEM using specific antibodies against the pilus shaft FimA and tip fimbrillin FimB, i.e. α-FimA and α-FimB, respectively. Compared with MG-1, pilus formation was severely diminished in the Δvkor mutant, but restored upon ectopic expression of vkor (Fig. 3, panels B–G). Importantly, mutating the first Cys of the CXXC motif of VKOR to Ala (C175A) abolished complementation of the Δvkor mutant indicating the functional importance of the motif (Fig. 3, H and I). Moreover, media supplemented with the oxidizing agent cystine restored type 2 fimbrial assembly in the Δvkor mutant (Fig. 3, J and K). We conclude that A. oris VKOR is required for fimbrial assembly through disulfide bond formation.
Incidentally, the tip fimbrillin FimB harbors 12 Cys residues. Although the cell surface of the ΔfimA mutant contains FimB (Fig. 2E), it was rarely detected on the Δvkor mutant (Fig. 3E). This suggested that FimB folding requires disulfide bond formation, consistent with a similar requirement we reported for CafA, another pilus tip protein of A. oris (6). Logically, we hypothesized that VKOR targets multiple fimbrial substrates. To explore this, we probed the assembly of type 1 fimbriae made of FimP, whose structure predicts the presence of two disulfide bonds in its N- and C-terminal IgG-like domains (12). Similar to type 2 fimbriae, type 1 fimbrial structures were barely detected in the Tn5::vkor mutant (Fig. 4, panels A–F) as well as the non-polar Δvkor mutant (Fig. 4H); instead, some FimP was secreted into the culture medium (Fig. 4G). These defects were rescued by VKOR complementation or the addition of cystine to the culture (Fig. 4, G and H). Collectively, these data establish the oxidoreductase activity of VKOR as a key common factor for pilus assembly.
FIGURE 4.
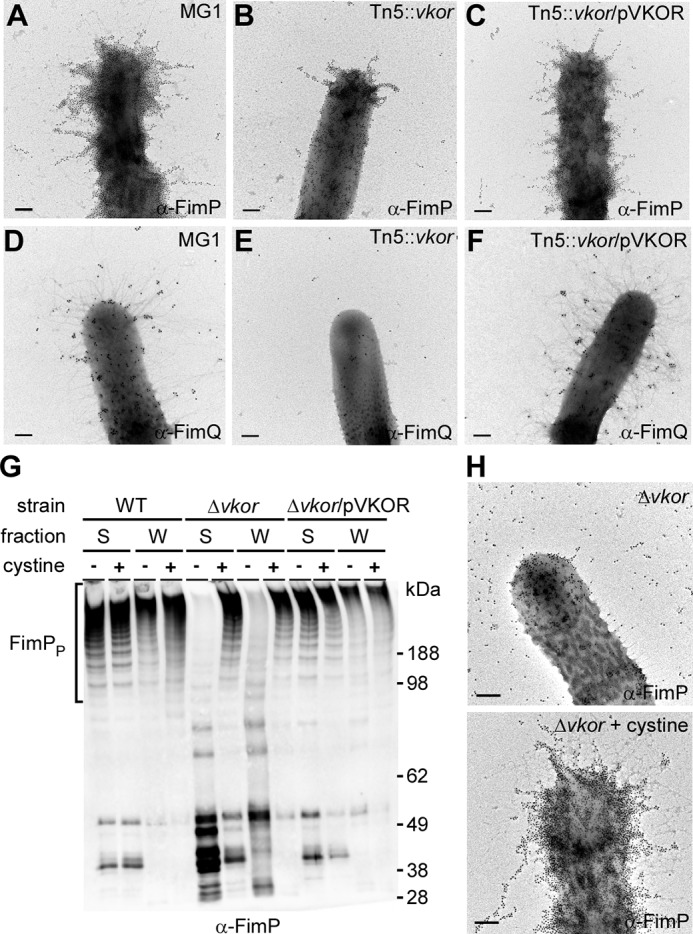
A. oris VKOR is required for type 1 fimbrial assembly. A–F and H, A. oris cells of various strains grown in the absence or presence of cystine were subject to IEM as described in the legend to Fig. 2 with α-FimP (A–C and H) or α-FimQ (D–F). Scale bars indicate 0.5 μm. G, the medium (S) and cell wall (W) fractions were collected from A. oris and its isogenic derivatives grown with (100 μg/ml) or without cystine. Protein samples were analyzed by immunoblotting with α-FimP.
The Primary Thiol-disulfide Oxidoreductase MdbA
Because cystine can rescue pilus assembly in the Δvkor mutant, we posited that VKOR may act to reoxidize a primary oxidoreductase enzyme in A. oris. A search for oxidoreductases in the A. oris MG-1 genome revealed ANA_1994. Similar to VKOR, ANA_1994, predicted to be a 32-kDa transmembrane protein (confirmed in Fig. 4A), harbors a CXXC motif. Based on the membrane-associated status of ANA_1994 in A. oris, as well as its thioredoxin-like fold and in vitro oxidoreductase activities (see below), we named ANA_1994 as MdbA for monoderm disulfide bond forming protein A. Multiple attempts to delete mdbA by conventional methods were unsuccessful, suggesting that this gene is essential. We then generated a conditional mdbA deletion mutant, whereby mdbA was removed from the bacterial chromosome in the presence of MdbA expressed from an arabinose-inducible plasmid (pAraC-MdbAAo). In the absence of arabinose (Fig. 5C, 0%), although a small amount of MdbA was produced due to promoter leakage (Fig. 5A), no type 2 fimbriae were detected on the cell surface by IEM; concomitantly, cell morphology was altered. In contrast, induction of MdbA restored the wild-type phenotypes (Fig. 5D, 2%). Remarkably, overproduction of MdbA from a multicopy plasmid restored both type 2 fimbrial assembly (Fig. 5F) and bacterial coaggregation with S. oralis (Fig. 3A) in the Δvkor mutant. However, an MdbA variant, in which the Cys136 of the CXXC motif was mutated to Ala, failed to restore pilus assembly (Fig. 5G). The data suggest that MdbA is a thiol-disulfide oxidoreductase that functions upstream of VKOR.
FIGURE 5.
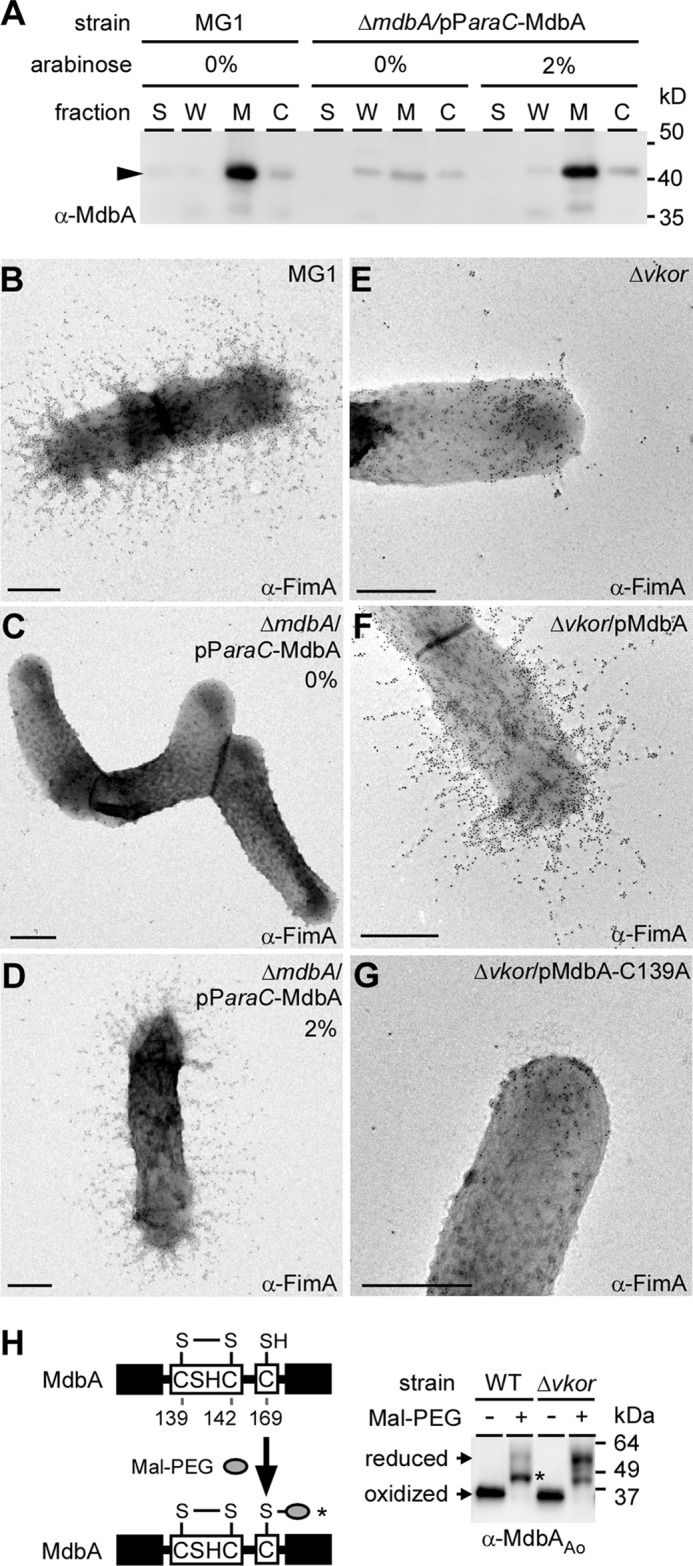
Assembly of A. oris type 2 fimbriae requires putative thiol-oxidoreductases VKOR and MdbA. A, cell fractionation on the parental strain MG-1 and its conditional deletion mdbA strain grown in the presence or absence of arabinose were performed. Protein samples collected from culture (S), cell wall (W), membrane (M), and cytoplasmic (C) fractions were immunoblotted with α-MdbA. B-G, A. oris strains were subject to IEM as previously described with α-FimA. Scale bars indicate 0.5 μm. H, whole cell lysates of the wild-type A. oris and Δvkor strains were treated with Mal-PEG prior to immunoblotting with α-MdbA. Molecular mass markers and reduced and oxidized forms of MdbA are indicated.
We hypothesized that Δvkor cells fail to produce fimbriae because they cannot “reoxidize” the reduced form of MdbA following catalysis. If this is true, the redox status of MdbA should be altered in the Δvkor mutant. To investigate this, whole cell lysates of wild-type and Δvkor strains were prepared, and samples collected by TCA precipitation were reacted with Mal-PEG, followed by SDS-PAGE analysis and immunoblotting using α-MdbA. In wild-type samples, alkylation with Mal-PEG resulted in a slight up-shift in MdbA migration compared with the untreated protein band. This is consistent with the modification of solo Cys169, which is not part of the CXXC motif; however, Mal-PEG treatment of the Δvkor lysates caused a higher up-shift, consistent with alkylation of Cys residues in the CXXC motif along with Cys169 (Fig. 5H). These data support that MdbA is reduced in the Δvkor mutant, suggesting that they form a redox pair required for disulfide bond formation in pilin precursors.
Crystal Structure of MdbA
The fact that A. oris MdbA has little sequence similarity with known DsbA proteins (supplemental Fig. S1 A), but is required for pilus assembly prompted us to attempt structural determination by x-ray crystallization using selenomethionine single-wavelength anomalous diffraction. An MdbA protein (residues 76–310) lacking its transmembrane domain was produced in E. coli. A structure for the reduced form of MdbA was refined to 1.55-Å resolution with R-work and R-free factors equal to 12.4 and 18.0%, respectively (Tables 1 and 2). The overall structure represents a typical DsbA protein family fold (42, 43), which incorporates a thioredoxin-like domain and an extended α-helical domain (Fig. 6A). The thioredoxin-like domain consists of 5-stranded β-sheet in an order of β1↑ − β3↓ − β2↓ − β4↑ − β5↓ and 5 flanking helices (2 α- and three 310-helices); the α-helical domain comprises 5 α-helices (H2–H6) and two 310 helices (Fig. 6A and supplemental Fig. S1B). The CSHC active-site motif, residues 139–142, is located on the N-terminal end of helix H1 (Fig. 6B). Significantly, the MdbA structure also harbors a cis-Pro loop (residues Thr285 and Pro286), a conserved feature of the TRX-protein family (44). The MdbA cis-Pro element is located on the loop between H6 α helix and β4 strand, and interacts with the CXXC motif via a hydrogen bond forming between Thr285 and Cys139 (Fig. 6B).
TABLE 1.
Crystal data collection statistics
| Statistics | |
|---|---|
| X-ray wavelength (Å) | 0.9792 |
| Space group | C 2221 |
| Unit cell dimensions | a = 46.9 Å, b = 66.9 Å, c = 256.2 Å, α = β = γ = 90° |
| Resolutiona (Å) | 38.4−1.55 (1.58−1.55) |
| No. of unique reflections | 59,458 (2,938) |
| Completeness | 99.9% (98.4%) |
| Rmerge | 0.10 (0.61) |
| CC1/2 (Å2) | −(0.71) |
| I/σ | 9.1 (2.16) |
| Redundancy | 6.3 (4.3) |
| Wilson plot B-factor (Å2) | 25.2 |
| Molecules per asymmetric unit | 2 |
| No. of protein residues | 400 |
a Numbers in parentheses are shown for the highest resolution shell.
TABLE 2.
Structure refinement statistics
| Statistics | |
|---|---|
| Resolution range (Å) | 38.4−1.55 (1.59−1.55) |
| Reflections | 56,424 (4,241) |
| σ cutoff | None |
| R-value (all) (%) | 12.67 |
| R-value (R-work) (%) | 12.39 (18.1) |
| Free R-value (%) | 17.99 (24.9) |
| Root mean square deviations from ideal geometry | |
| Bond length (Å) | 0.013 |
| Angle (degrees) | 1.61 |
| Chiral (Å) | 0.102 |
| No. of atoms | |
| Protein | 3,175 |
| CAPSO | 30 |
| Water | 451 |
| Mean B-factor (Å2) | |
| All atoms | 22.2 |
| Protein atoms | 20.6 |
| Protein main chain | 18.2 |
| protein side chain | 23.1 |
| CAPSO | 30.4 |
| Water | 33.8 |
| Molprobity Ramachandran plot statistics | |
| Residues in favored regions (%) | 99.6 |
| Residues in allowed regions (%) | 100.0 |
| Residues in disallowed region (%) | 0.0 |
FIGURE 6.

Structural analysis of A. oris MdbA. A, the A. oris MdbA crystal structure (residues 86–296) solved to a 1.55-Å resolution has two domains, a thioredoxin-like domain (rainbow colors) and an α-helical domain (purple). B, the MdbA active site is made of Cys139, Ser140, His141, and Cys142. The Sγ group in the two cysteine residues forms a hydrogen bond with Thr268 of the Cis-Pro element. C–F, the MdbA structure (purple) was aligned with B. subtilis BdbB (C; Protein Data Bank code 3EU3), M. tuberculosis DsbA (D; Protein Data Bank code 4JR6), S. aureus DsbA (E; Protein Data Bank code 3BCI), and E. coli DsbA (F; Protein Data Bank code 1FVK).
According to the DALI server (45), the closest MdbA structural homolog is Bacillus subtilis BdbD (46) with Z-score (strength of structural similarity in standard deviations above the mean) and root mean square deviations (root mean square deviation of superimposed atoms in Å) equal to 19 and of 2.7, respectively, for 186 residues. These proteins share only limited amino acid homology, mainly at the C-terminal fragments (22% identities for 147 residue alignment, based on NCBI Blastp server). Despite significant structural similarity (Fig. 6C), MdbA significantly differs from that of B. subtilis BdbD by two distinct features. First, the MdbA CXXC active site sequence is CSHC, whereas BdbD harbors a CPSC motif. Second, MdbA lacks a metal-binding site present in BdbD formed by residues Gln49, Glu115, and Asp180 (46). The two last residues are replaced by glycine residues in MdbA. The subsequent most similar structures to MdbA are DsbA-like proteins expressed by M. tuberculosis and Staphylococcus aureus (22, 48) (Fig. 6, D and E). By contrast, the E. coli DsbA structure (42) (Fig. 5F) is fairly distant on the DALI homolog list with Z-score and root meant square deviation values equal to 11.4 and 3.7, respectively. All DsbA-like proteins from Gram-positive bacteria, including A. oris MdbA, differ from the E. coli DsbA and others expressed by Gram-negative bacteria by β-sheet topology (i.e. the β-strand order of 3-2-4-5-1 in E. coli). Notably, the Gram-positive proteins were proposed to have highly charged electrostatic surface potential located on the α-helical domain adjacent to the catalytic site in place of a neutral hydrophobic patch in Gram-negative enzymes (42, 49). The A. oris MdbA structure shows a nearly neutral surface potential in that region (supplemental Fig. S1 C). Altogether, the data support the role of A. oris MdbA as a disulfide bond-forming enzyme that shares some common characteristics with DsbA proteins, but possesses novel features as well.
Reconstitution of Disulfide Bond Formation in Vitro
To unequivocally establish that MdbA enzymes directly oxidize proteins, we sought to reconstitute the disulfide bond-forming machine in vitro using A. oris recombinant MdbA enzymes and FimA as its physiological substrate. The reduced form of FimA was generated in vitro by DTT treatment. Upon removing DTT, FimA was incubated with MdbA or a catalytically inactive mutant in glutathione redox buffer at 37 °C for 30 min. At specific time points, reactions were quenched by the addition of Mal-PEG, and analyzed by SDS-PAGE and Coomassie staining. When reduced FimA was incubated with wild-type A. oris MdbA, a faster migrating band of FimA (i.e. not modified by Mal-PEG because Cys residues became oxidized) was visible after only 5 min of incubation (Fig. 6A). After 30 min, the majority of reduced FimA formed disulfide bonds (Fig. 7A, lane 30). FimA remained reduced in the reactions that contained no enzyme (mock-treated lanes) or an inactive enzyme with the catalytic residue Cys216 mutated to Ala, signifying that the substrate remained accessible to Mal-PEG (Fig. 7A, next 8 lanes).
FIGURE 7.
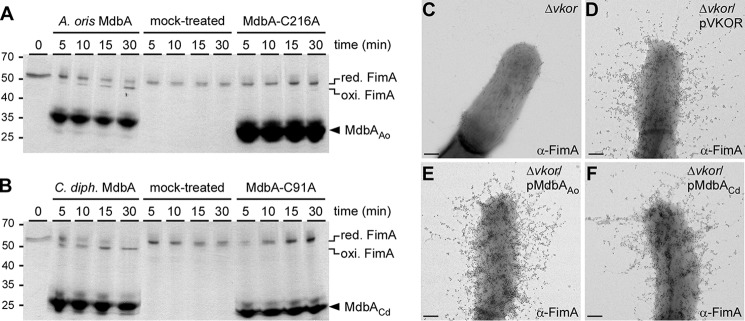
Reconstitution of a disulfide bond forming machine. A and B, reduced recombinant FimA was left untreated or treated with recombinant wild-type or catalytically inactive MdbA proteins from A. oris (A; MdbAAo) or C. diphtheriae (B; MdbACd). At timed intervals, the reactions were stopped by addition of Mal-PEG, and the protein samples were analyzed by SDS-PAGE, followed by Coomassie staining. Oxidized and reduced forms of FimA and MdbA proteins are indicated. C–F, the presence of FimA fimbriae was analyzed by IEM; scale bars of 0.2 μm.
The Gram-positive actinobacterium C. diphtheriae also encodes a predicted MdbA homolog (DIP_1880), and a plasmid expressing this protein in the A. oris Δvkor mutant restored pilus assembly similar to plasmid-borne VKOR or A. oris MdbA (Fig. 7, compare E and F). Indeed, the recombinant MdbA enzyme of C. diphtheriae also displayed oxidoreductase activity upon the FimA substrate; when reduced FimA was incubated with wild-type C. diphtheriae MdbA, all of the substrate became oxidized by 30 min. Substituting the catalytic Cys residue to Ala (MdbA-C91A) abolished C. diphtheriae MdbA oxidative folding activity (Fig. 7B). Altogether, we conclude that MdbA is necessary and sufficient for the oxidative folding of secreted disulfide containing pilus proteins and suggest that oxidative protein folding may be a common pathway for maturation of secreted proteins in Actinobacteria.
Discussion
Bacteria secrete a wide variety of proteins whose proper folding and function depends on disulfide bond formation. Gram-negative bacteria possess an oxidative periplasmic compartment with Dsb proteins that are required for disulfide bond formation in many secreted proteins. Although these pathways have been well characterized in these bacteria, little is known about how oxidative protein folding occurs in Gram-positive monoderms that lack periplasms (50). Here we report an oxidative folding pathway for pilus proteins in A. oris (Fig. 8) that likely represents a conserved mechanism to fold secreted proteins in other Actinobacteria, including two major pathogens C. diphtheriae and M. tuberculosis.
FIGURE 8.
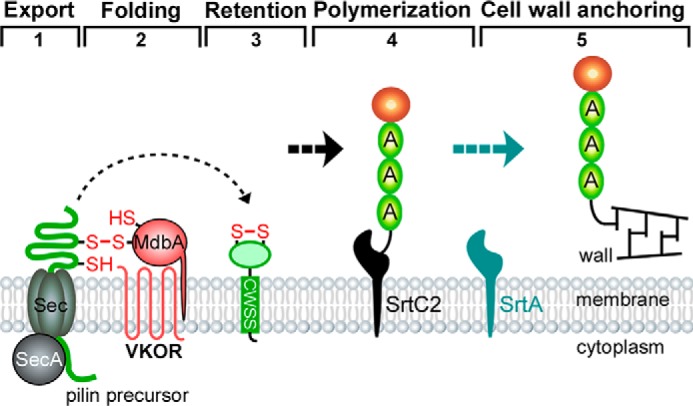
A revised model of pilus assembly in A. oris. Pilin precursors are synthesized in the cytoplasm. The transmembrane MbdA enzyme forms a mixed disulfide bond with nascent precursors emerging from the Sec translocon (step 1), allowing the precursors to fold into a near-native state before MdbA catalysis of disulfide bond formation, resulting in fully folded proteins that are subsequently imbedded in the membrane (steps 2 and 3). Pilin subunits are assembled into covalently linked pilus polymers by a pilus-specific sortase (step 4). The resulting polymers are anchored to the cell wall by another sortase (step 5). MdbA may serve as a “placeholder” to guide the pairing of cysteine residues into native disulfide bonds (47).
Our studies began with the Actinomyces FimA pilin constituting the type 2 fimbriae important for establishing biofilms within the oral cavity (5). FimA contains two disulfide bonds, a common feature of actinobacterial pilins (4, 11, 12). We have shown here that the C-terminal disulfide bond is essential for fimbrial assembly and function in biofilm formation and polymicrobial interactions (Figs. 1 and 2). Importantly, this disulfide bond (Cys394 and Cys445) stabilizes a loop proximal to the cell wall sorting signal, a feature required for sortase-mediated processing of the pilin precursor and pilus polymerization (51). We postulate that this linkage is important for maintaining the proper conformation of this region as it is required for sortase processing (Fig. 8). The folding may also protect the protein from proteolysis because a loss of oxidative folding causes the degradation of FimA in addition to a defect in pilus assembly (Fig. 1).
Although disulfide bonds can form spontaneously, we suspected that disulfide bond formation in FimA might be facilitated by an extra cytoplasmic factor. To uncover it, we screened a random transposon library of A. oris for mutants defective in pilus assembly and identified vkor (Fig. 3A). VKOR is believed to function as a DsbB analogue, because expression of the M. tuberculosis vkor rescues an E. coli dsbB mutant (52). An M. tuberculosis vkor mutant is available, but its physiological function has not been assessed (21). Independently, we surveyed the A. oris genome for genes encoding extracellular CXXC-containing proteins and found MdbA. Unlike E. coli dsbA and dsbB, which are non-essential genes, deletion of A. oris mdbA is lethal, suggesting that MdbA substrates are involved in essential processes. We then provided several lines of evidence that MdbA is a thiol-disulfide oxidoreductase and that MbdA and VKOR function as a redox pair to catalyze protein oxidation. First, whereas depletion of mdbA abolished pilus assembly, ectopic expression of mdbA restored fimbrial assembly in the mdbA and vkor mutants, and the functions of MdbA required its catalytic CXXC motif (Figs. 5 and 7). Second, MdbA possesses a thioredoxin-like domain (Fig. 6 and supplemental Fig. S1). Third, we were able to demonstrate that recombinant MdbA protein catalyzes disulfide bonds within FimA in vitro and its enzymatic activity requires the conserved CXXC motif (Fig. 7). Finally, we showed that in the absence of VKOR, the CXXC motif of MdbA was reduced suggesting that VKOR was required for its recycling (Fig. 5).
Although VKOR is evidently required to maintain a functional MdbA, which is essential, we were able to construct a Δvkor strain. At first glance, this result seems contradictory. It is possible that MdbA may have an essential function that is independent of VKOR and future studies will address why A. oris mdbA is essential. Alternatively, a VKOR-like factor could be present in the exoplasm that can partially compensate for the loss of vkor. However, it is also possible that laboratory conditions used in this study artificially supported the survival of the vkor mutant. For this study, A. oris were grown in rich media in the presence of oxygen. Thus, oxidizing agents present in this environment might suffice to activate enough MdbA to keep cells viable. In support of this, a small portion of MdbA detected in Δvkor was not alkylated by Mal-PEG, suggesting that they harbored an oxidized CXXC motif (Fig. 5). Additional support for this notion comes from a recent study of Dsb proteins expressed by the Gram-negative bacterium Francisella tulerancis that observed mixed populations of reduced and oxidized DsbA in a dsbB mutant (53). As a primary colonizer of the oral cavity, A. oris resides within the lower, anaerobic layers of biofilm, which do not contain oxidizing agents like oxygen. Thus, within its natural environment, A. oris vkor may be essential.
It is noteworthy that that although many Gram-positive bacteria seemingly exclude Cys residues from their secretomes (25), Actinobacteria, like A. oris and possibly C. diphtheriae, utilize disulfide bond formation as a key folding pathway. Are they exceptions, and if so, why? The cell envelope of corynebacteria and mycobacteria contains an outer layer of mycolic acids that is proposed to be equivalent to the outer membrane of Gram-negative bacteria (54, 55). However, A. oris is not known to produce mycolic acid. Its ultrastructure needs a closer inspection to examine whether it harbors any distinct compartments. It is likely that its peri-membranous environment contributes to disulfide bond formation within secreted polypeptides. Notably, because A. oris resides within the innermost, anaerobic layers of oral biofilms, its niche may shield it from aberrant protein oxidation (56).
Finally, specialized zones of secretion have been identified in Gram-positive bacteria including Streptococcus pyogenes, Enterococcus faecalis, and C. diphtheriae (57–59). It is possible that protein secretion, folding, and processing are closely coupled and coordinated to avoid damage that may be caused by extracellular oxygen. Unlike E. coli DsbA, A. oris MdbA is a membrane-bound protein. Because pilins may complete folding prior to interacting with sortase, it is possible that the disulfide bond forming machinery is both physically and functionally coupled to secretion (Fig. 8). This would ensure that proper disulfide bond formation is catalyzed rapidly before any aberrant oxidation can occur.
Author Contributions
M. E. R.-R., A. J., and H. T.-T. designed research; M. E. R.-R., J. O., C. W., C. C., and N. J. performed research; M. E. R.-R., J. O., A. D., and H. T.-T. analyzed data; and M. E. R.-R., J. O., A. D., and H. T.-T. wrote the paper.
Supplementary Material
Acknowledgments
We thank members of the Structural Biology Center at Argonne National Laboratory for their help in conducting x-ray diffraction data collection and our lab members for critical review and discussion of the manuscript. Argonne is operated by the University of Chicago Argonne, LLC, for the United States Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
This work was supported, in whole or in part, by National Institutes of Health Grant GM094585 from the NIGMS (to J. O. and A. J.) and NIDCR Grants F31DE024004 (to M. E. R.-R.) and DE017382 and DE025015 (to H. T.-T.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 4Z7X) have been deposited in the Protein Data Bank (http://wwpdb.org/).

This article contains supplemental Tables S1 and S2 and Fig. S1.
- VKOR
- vitamin K epoxide reductase
- CAPSO
- 3-(cyclohexylamino)-2-hydroxy-1-propanesulfonic acid
- IEM
- immunogold electron microscopy.
References
- 1. Girard A. E., Jacius B. H. (1974) Ultrastructure of Actinomyces viscosus and Actinomyces naeslundii. Arch. Oral Biol. 19, 71–79 [DOI] [PubMed] [Google Scholar]
- 2. Ton-That H., Das A., Mishra A. (2011) Actinomyces oris Fimbriae: an Adhesive Principle in Bacterial Biofilms and Tissue Tropism. in Actinomyces oris Fimbriae: an Adhesive Principle in Bacterial Biofilms and Tissue Tropism (Kolenbrander P. E., ed) pp. 63–77, ASM Press, Washington, D. C. [Google Scholar]
- 3. Wu C., Mishra A., Yang J., Cisar J. O., Das A., Ton-That H. (2011) Dual function of a tip fimbrillin of Actinomyces in fimbrial assembly and receptor binding. J. Bacteriol. 193, 3197–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mishra A., Devarajan B., Reardon M. E., Dwivedi P., Krishnan V., Cisar J. O., Das A., Narayana S. V., Ton-That H. (2011) Two autonomous structural modules in the fimbrial shaft adhesin FimA mediate Actinomyces interactions with streptococci and host cells during oral biofilm development. Mol. Microbiol. 81, 1205–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mishra A., Wu C., Yang J., Cisar J. O., Das A., Ton-That H. (2010) The Actinomyces oris type 2 fimbrial shaft FimA mediates co-aggregation with oral streptococci, adherence to red blood cells and biofilm development. Mol. Microbiol. 77, 841–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reardon-Robinson M. E., Wu C., Mishra A., Chang C., Bier N., Das A., Ton-That H. (2014) Pilus hijacking by a bacterial coaggregation factor critical for oral biofilm development. Proc. Natl. Acad. Sci. U.S.A. 111, 3835–3840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ton-That H., Schneewind O. (2003) Assembly of pili on the surface of Corynebacterium diphtheriae. Mol. Microbiol. 50, 1429–1438 [DOI] [PubMed] [Google Scholar]
- 8. Marraffini L. A., Dedent A. C., Schneewind O. (2006) Sortases and the art of anchoring proteins to the envelopes of Gram-positive bacteria. Microbiol. Mol. Biol. Rev. 70, 192–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mandlik A., Swierczynski A., Das A., Ton-That H. (2008) Pili in Gram-positive bacteria: assembly, involvement in colonization and biofilm development. Trends Microbiol. 16, 33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Swaminathan A., Mandlik A., Swierczynski A., Gaspar A., Das A., Ton-That H. (2007) Housekeeping sortase facilitates the cell wall anchoring of pilus polymers in Corynebacterium diphtheriae. Mol. Microbiol. 66, 961–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kang H. J., Paterson N. G., Gaspar A. H., Ton-That H., Baker E. N. (2009) The Corynebacterium diphtheriae shaft pilin SpaA is built of tandem Ig-like modules with stabilizing isopeptide and disulfide bonds. Proc. Natl. Acad. Sci. U.S.A. 106, 16967–16971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Persson K., Esberg A., Claesson R., Strömberg N. (2012) The Pilin Protein FimP from Actinomyces oris: crystal structure and sequence analyses. PLoS ONE 7, e48364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pedone E., Limauro D., Bartolucci S. (2008) The machinery for oxidative protein folding in thermophiles. Antioxid. Redox Signal. 10, 157–169 [DOI] [PubMed] [Google Scholar]
- 14. Goldberger R. F., Epstein C. J., Anfinsen C. B. (1963) Acceleration of reactivation of reduced bovine pancreatic ribonuclease by a microsomal system from rat liver. J. Biol. Chem. 238, 628–635 [PubMed] [Google Scholar]
- 15. Goldberger R. F., Epstein C. J., Anfinsen C. B. (1964) Purification and properties of a microsomal enzyme system catalyzing the reactivation of reduced ribonuclease and lysozyme. J. Biol. Chem. 239, 1406–1410 [PubMed] [Google Scholar]
- 16. Bardwell J. C., McGovern K., Beckwith J. (1991) Identification of a protein required for disulfide bond formation in vivo. Cell 67, 581–589 [DOI] [PubMed] [Google Scholar]
- 17. Kadokura H., Beckwith J. (2009) Detecting folding intermediates of a protein as it passes through the bacterial translocation channel. Cell 138, 1164–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bardwell J. C., Lee J. O., Jander G., Martin N., Belin D., Beckwith J. (1993) A pathway for disulfide bond formation in vivo. Proc. Natl. Acad. Sci. U.S.A. 90, 1038–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Missiakas D., Georgopoulos C., Raina S. (1993) Identification and characterization of the Escherichia coli gene dsbB, whose product is involved in the formation of disulfide bonds in vivo. Proc. Natl. Acad. Sci. U.S.A. 90, 7084–7088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kobayashi T., Ito K. (1999) Respiratory chain strongly oxidizes the CXXC motif of DsbB in the Escherichia coli disulfide bond formation pathway. EMBO J. 18, 1192–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dutton R. J., Boyd D., Berkmen M., Beckwith J. (2008) Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc. Natl. Acad. Sci. U.S.A. 105, 11933–11938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang L., Li J., Wang X., Liu W., Zhang X. C., Li X., Rao Z. (2013) Structure analysis of the extracellular domain reveals disulfide bond forming-protein properties of Mycobacterium tuberculosis Rv2969c. Protein Cell 4, 628–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Premkumar L., Heras B., Duprez W., Walden P., Halili M., Kurth F., Fairlie D. P., Martin J. L. (2013) Rv2969c, essential for optimal growth in Mycobacterium tuberculosis, is a DsbA-like enzyme that interacts with VKOR-derived peptides and has atypical features of DsbA-like disulfide oxidases. Acta Crystallogr. D Biol. Crystallogr. 69, 1981–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chim N., Harmston C. A., Guzman D. J., Goulding C. W. (2013) Structural and biochemical characterization of the essential DsbA-like disulfide bond forming protein from Mycobacterium tuberculosis. BMC Struct. Biol. 13, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Daniels R., Mellroth P., Bernsel A., Neiers F., Normark S., von Heijne G., Henriques-Normark B. (2010) Disulfide bond formation and cysteine exclusion in Gram-positive bacteria. J. Biol. Chem. 285, 3300–3309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stols L., Gu M., Dieckman L., Raffen R., Collart F. R., Donnelly M. I. (2002) A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 25, 8–15 [DOI] [PubMed] [Google Scholar]
- 27. Wu C., Huang I. H., Chang C., Reardon-Robinson M. E., Das A., Ton-That H. (2014) Lethality of sortase depletion in Actinomyces oris caused by excessive membrane accumulation of a surface glycoprotein. Mol. Microbiol. 94, 1227–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu Y. G., Whittier R. F. (1995) Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics 25, 674–681 [DOI] [PubMed] [Google Scholar]
- 29. Walsh M. A., Dementieva I., Evans G., Sanishvili R., Joachimiak A. (1999) Taking MAD to the extreme: ultrafast protein structure determination. Acta Crystallogr. D Biol. Crystallogr. 55, 1168–1173 [DOI] [PubMed] [Google Scholar]
- 30. Gorrec F., Palmer C. M., Lebon G., Warne T. (2011) Pi sampling: a methodical and flexible approach to initial macromolecular crystallization screening. Acta Crystallogr. D Biol. Crystallogr. 67, 463–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rosenbaum G., Alkire R. W., Evans G., Rotella F. J., Lazarski K., Zhang R. G., Ginell S. L., Duke N., Naday I., Lazarz J., Molitsky M. J., Keefe L., Gonczy J., Rock L., Sanishvili R., Walsh M. A., Westbrook E., Joachimiak A. (2006) The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. J. Synchrotron Radiat. 13, 30–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Minor W., Cymborowski M., Otwinowski Z., Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution: from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 33. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 34. Sheldrick G. M. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Terwilliger T. C. (2003) SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol. 374, 22–37 [DOI] [PubMed] [Google Scholar]
- 36. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 37. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 38. Collaborative Computational Project, Number 4. (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 39. Davis I. W., Murray L. W., Richardson J. S., Richardson D. C. (2004) MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 32, W615–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chang C., Huang I. H., Hendrickx A. P., Ton-That H. (2013) Visualization of Gram-positive bacterial pili. Methods Mol. Biol. 966, 77–95 [DOI] [PubMed] [Google Scholar]
- 41. Makmura L., Hamann M., Areopagita A., Furuta S., Muñoz A., Momand J. (2001) Development of a sensitive assay to detect reversibly oxidized protein cysteine sulfhydryl groups. Antioxid. Redox Signal. 3, 1105–1118 [DOI] [PubMed] [Google Scholar]
- 42. Martin J. L., Bardwell J. C., Kuriyan J. (1993) Crystal structure of the DsbA protein required for disulphide bond formation in vivo. Nature 365, 464–468 [DOI] [PubMed] [Google Scholar]
- 43. Shouldice S. R., Heras B., Walden P. M., Totsika M., Schembri M. A., Martin J. L. (2011) Structure and function of DsbA, a key bacterial oxidative folding catalyst. Antioxid. Redox Signal. 14, 1729–1760 [DOI] [PubMed] [Google Scholar]
- 44. Ren G., Stephan D., Xu Z., Zheng Y., Tang D., Harrison R. S., Kurz M., Jarrott R., Shouldice S. R., Hiniker A., Martin J. L., Heras B., Bardwell J. C. (2009) Properties of the thioredoxin fold superfamily are modulated by a single amino acid residue. J. Biol. Chem. 284, 10150–10159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Crow A., Lewin A., Hecht O., Carlsson Möller M., Moore G. R., Hederstedt L., Le Brun N. E. (2009) Crystal structure and biophysical properties of Bacillus subtilis BdbD: An oxidizing thiol:disulfide oxidoreductase containing a novel metal site. J. Biol. Chem. 284, 23719–23733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kosuri P., Alegre-Cebollada J., Feng J., Kaplan A., Inglés-Prieto A., Badilla C. L., Stockwell B. R., Sanchez-Ruiz J. M., Holmgren A., Fernández J. M. (2012) Protein folding drives disulfide formation. Cell 151, 794–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Heras B., Kurz M., Jarrott R., Shouldice S. R., Frei P., Robin G., Cemazar M., Thöny-Meyer L., Glockshuber R., Martin J. L. (2008) Staphylococcus aureus DsbA does not have a destabilizing disulfide: a new paradigm for bacterial oxidative folding. J. Biol. Chem. 283, 4261–4271 [DOI] [PubMed] [Google Scholar]
- 49. McMahon R. M., Premkumar L., Martin J. L. (2014) Four structural subclasses of the antivirulence drug target disulfide oxidoreductase DsbA provide a platform for design of subclass-specific inhibitors. Biochim. Biophys. Acta 1844, 1391–1401 [DOI] [PubMed] [Google Scholar]
- 50. Sarvas M., Harwood C. R., Bron S., van Dijl J. M. (2004) Post-translocational folding of secretory proteins in Gram-positive bacteria. Biochim. Biophys. Acta 1694, 311–327 [DOI] [PubMed] [Google Scholar]
- 51. Ton-That H., Marraffini L. A., Schneewind O. (2004) Sortases and pilin elements involved in pilus assembly of Corynebacterium diphtheriae. Mol. Microbiol. 53, 251–261 [DOI] [PubMed] [Google Scholar]
- 52. Wang X., Dutton R. J., Beckwith J., Boyd D. (2011) Membrane topology and mutational analysis of Mycobacterium tuberculosis VKOR, a protein involved in disulfide bond formation and a homologue of human vitamin K epoxide reductase. Antioxid. Redox Signal. 14, 1413–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ren G., Champion M. M., Huntley J. F. (2014) Identification of disulfide bond isomerase substrates reveals bacterial virulence factors. Mol. Microbiol. 94, 926–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Burkovski A. (2013) Cell envelope of corynebacteria: structure and influence on pathogenicity. ISRN Microbiol. 2013, 935736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bayan N., Houssin C., Chami M., Leblon G. (2003) Mycomembrane and S-layer: two important structures of Corynebacterium glutamicum cell envelope with promising biotechnology applications. J. Biotechnol. 104, 55–67 [DOI] [PubMed] [Google Scholar]
- 56. Kolenbrander P. E., Palmer R. J., Jr., Periasamy S., Jakubovics N. S. (2010) Oral multispecies biofilm development and the key role of cell-cell distance. Nat. Rev. Microbiol. 8, 471–480 [DOI] [PubMed] [Google Scholar]
- 57. Guttilla I. K., Gaspar A. H., Swierczynski A., Swaminathan A., Dwivedi P., Das A., Ton-That H. (2009) Acyl enzyme intermediates in sortase-catalyzed pilus morphogenesis in Gram-positive bacteria. J. Bacteriol. 191, 5603–5612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kline K. A., Kau A. L., Chen S. L., Lim A., Pinkner J. S., Rosch J., Nallapareddy S. R., Murray B. E., Henriques-Normark B., Beatty W., Caparon M. G., Hultgren S. J. (2009) Mechanism for sortase localization and the role of sortase localization in efficient pilus assembly in Enterococcus faecalis. J. Bacteriol. 191, 3237–3247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rosch J. W., Caparon M. G. (2005) The ExPortal: an organelle dedicated to the biogenesis of secreted proteins in Streptococcus pyogenes. Mol. Microbiol. 58, 959–968 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.