

Background: Mutations in fibulin-4 cause autosomal recessive cutis laxa 1B, characterized by loose skin with vascular, lung, and skeletal abnormalities.
Results: A mouse strain carrying a recurrent fibulin-4 missense mutation was generated and characterized.
Conclusion: Mutant mice recapitulate the complete clinical features of the disease.
Significance: The study provides the first evidence that fibulin-4 regulates collagen fibrillogenesis.
Keywords: collagen, connective tissue, elastin, extracellular matrix, lysyl oxidase, skin, tendon, aneurysm, aorta, forelimb
Abstract
Fibulin-4 is an extracellular matrix protein essential for elastic fiber formation. Frameshift and missense mutations in the fibulin-4 gene (EFEMP2/FBLN4) cause autosomal recessive cutis laxa (ARCL) 1B, characterized by loose skin, aortic aneurysm, arterial tortuosity, lung emphysema, and skeletal abnormalities. Homozygous missense mutations in FBLN4 are a prevalent cause of ARCL 1B. Here we generated a knock-in mouse strain bearing a recurrent fibulin-4 E57K homozygous missense mutation. The mutant mice survived into adulthood and displayed abnormalities in multiple organ systems, including loose skin, bent forelimb, aortic aneurysm, tortuous artery, and pulmonary emphysema. Biochemical studies of dermal fibroblasts showed that fibulin-4 E57K mutant protein was produced but was prone to dimer formation and inefficiently secreted, thereby triggering an endoplasmic reticulum stress response. Immunohistochemistry detected a low level of fibulin-4 E57K protein in the knock-in skin along with altered expression of selected elastic fiber components. Processing of a precursor to mature lysyl oxidase, an enzyme involved in cross-linking of elastin and collagen, was compromised. The knock-in skin had a reduced level of desmosine, an elastin-specific cross-link compound, and ultrastructurally abnormal elastic fibers. Surprisingly, structurally aberrant collagen fibrils and altered organization into fibers were characteristics of the knock-in dermis and forelimb tendons. Type I collagen extracted from the knock-in skin had decreased amounts of covalent intermolecular cross-links, which could contribute to the collagen fibril abnormalities. Our studies provide the first evidence that fibulin-4 plays a role in regulating collagen fibril assembly and offer a preclinical platform for developing treatments for ARCL 1B.
Introduction
Cutis laxa is a heterogeneous group of disorders characterized by redundant and inelastic skin with variable internal organ involvement (1, 2). Fibulin-4, fibulin-5, and LTBP4 (latent TGF-β binding protein 4) are three elastic fiber components that underlie autosomal recessive cutis laxa (ARCL)2 type 1B (OMIM #614437), 1A (OMIM #219100), and 1C (OMIM #613177), respectively. Absence of any of the three proteins impairs elastic fiber formation during organogenesis. ARCL 1B, caused by mutations in the gene encoding fibulin-4 (EFEMP2/FBLN4, hereafter referred to as FBLN4), is a very severe and often lethal disease caused by cardiopulmonary failure (1, 2). The typical clinical manifestations are loose skin, aortic aneurysm, arterial tortuosity, and pulmonary emphysema. In addition, the patients display features of generalized connective tissue abnormalities, including bone fragility, kyphoscoliosis, arachnodactyly, pectus excavatum, joint laxity, and diaphragmatic and inguinal hernia. Some patients bearing FBLN4 mutations display severe cardiovascular and skeletal abnormalities but have mild or no apparent skin involvement (3, 4).
Fibulin-4 is a ∼50-kDa extracellular matrix protein consisting of a signature fibulin C-terminal domain, five tandem repeats of calcium-binding epidermal growth factor-like (cbEGF) modules, and an N-terminal modified cbEGF module (see Fig. 1A) (5). Its essential role in elastic fiber assembly has been uncovered through studies of mice lacking fibulin-4 (6). The Fbln4 null mice die perinatally because of cardiopulmonary failure resulting from an absence of intact elastic fibers. Further insights into the critical role of fibulin-4 in the vascular system have been provided by the investigation of a hypomorphic Fbln4 mutant mouse and two independent vascular smooth muscle-specific conditional Fbln4 null mice (7–9). These Fbln4 mutant mice live into adulthood and develop ascending aortic aneurysm and arterial tortuosity.
FIGURE 1.

Generation of a mouse model with fibulin-4 E57K mutation. A, modular structure of fibulin-4. FC, fibulin-type C terminus. Modified cbEGF domain contains an insertion of 28 amino acids. The E57K mutation is located in the N terminus of the modified-cbEGF domain. B, nucleotide and amino acid sequences of the human and mouse fibulin-4 genes surrounding the E57K mutation, which is located in the beginning of exon 4. Intron sequences are shown in lowercase letters. A G → A point mutation results in E57K substitution in both humans and mice. C, targeting strategy. The gene targeted region corresponds to exon 1–10 (exons shown as black boxes) of the wild type Fbln4 allele; amino acid residue Glu-57 (E57) is encoded by exon 4. The external and internal probes used for Southern blotting of electroporated mouse embryonic stem cells and key restriction enzymes sites are shown. In the targeting vector, a G → A mutation is introduced in exon 4 (shown as an asterisk), resulting in E57K substitution. A loxP/FRT flanked-pGK-gb2-Neo cassette is inserted in intron 4 in the opposite direction of Fbln4 transcription. Homologous recombination generates the targeted allele containing the pGK-gb2-Neo cassette in the intron. Crossing mice harboring the targeted allele with transgenic mice bearing FLP1 recombinase gene results in the knock-in allele, Fbln4E57K. D, screening of electroporated ES cells by Southern blotting using external and internal DNA probes. E, genotyping by PCR to identify Fbln4+/+, Fbln4+/E57K, and Fbln4E57K/E57K mice.
Elastic fiber formation during development is a multistep process requiring tropoelastin, as well as several other extracellular matrix molecules (10, 11). Newly secreted tropoelastin first self-associates into small aggregates (coacervation) on the cell surface, and then the tropoelastin molecules are cross-linked by lysyl oxidase (LOX). The small aggregates are next transferred to fibrillin microfibrils and undergo further coacervation and cross-linking, resulting in mature elastic fibers composed of an elastin core surrounded by fibrillin-rich microfibrils. Fibulin-4, fibulin-5, and LTBP4 participate in different steps of the assembly process (12–14). Several lines of evidence indicate that fibulin-4 facilitates cross-linking of tropoelastin by LOX. In Fbln4 null mice, elastin cross-links, measured by desmosine content, are ∼10% of the level found in wild type mice (6). Biochemical analyses show that fibulin-4 binds LOX and that the interaction is mediated through the N-terminal region of fibulin-4 and the propeptide of LOX (8, 14).
A total of 12 distinct FBLN4 pathogenic mutations have been reported thus far (3, 4, 15–22). The mutations fall into two groups: nine missense and three frameshift mutations. Three patients carrying frameshift mutations, either in homozygous or compound heterozygous state, have very severe clinical manifestations, dying at birth to age 18 months. The severe clinical presentation of patients with frameshift mutations is associated with a marked reduction or complete absence of normal fibulin-4 protein, consistent with the perinatal lethal phenotype of the Fbln4 null mice (6). On the other hand, patients carrying homozygous missense mutations have variable clinical severity. Previous studies demonstrated that mutant fibulin-4 proteins are produced by fibroblasts from some patients with missense mutations (4, 21, 22). However, the functional consequences of the missense mutations in vivo remain unclear. To better understand the pathophysiological mechanisms underlying FBLN4 missense mutations in vivo and to develop effective treatments for this life-threatening disorder, we generated a knock-in mouse strain carrying a recurrent homozygous missense mutation found in ARCL 1B patients, the E57K substitution (18, 22). We show that the knock-in mice survive into adulthood and display cutaneous, vascular, pulmonary, and skeletal defects reminiscent of the patients with the same missense change. We also show that collagen fibrils in skin and tendon of the mutant mice are abnormal, providing the first evidence that fibulin-4 is required not only for the formation of elastic fibers but also regulates collagen fibrillogenesis.
Experimental Procedures
Antibodies
Antibodies specific for fibulins were raised in rabbits using recombinant proteins expressed in HEK293 cells as described previously (23). Antibody for fibrillin-1 (pAb 9543) was kindly provided by Dr. Lynn Sakai. Antibody for tropoelastin (#387, against the C terminus of mouse tropoelastin) was purchased from Elastin Products Company (Owensville, MO); antibodies for LOX were from Sigma-Aldrich (L4669, against C terminus) and Santa Cruz Biotechnology (sc-66948, M-140, against amino acids 66–205). Rabbit monoclonal antibodies for calnexin (C5C9) and BiP (C50B12) were obtained from Cell Signaling Technology (Beverly, MA).
Generation of Fibulin-4 E57K Knock-in Mice
A knock-in mouse strain bearing the fibulin-4 E57K substitution (see Fig. 1B) was generated using a custom gene-targeting service (inGenious Targeting Laboratory, Inc., Ronkonkoma, NY). Briefly, an 8.7-kb subclone from a mouse C57BL/6 strain genomic BAC clone was used to construct the targeting vector, which contained a 4-kb 5′ homology arm including exons 1–4 and a 4.25-kb 3′ homology arm encompassing exons 5–10 (see Fig. 1C). A loxP/FRT flanked-pGK-gb2-Neo cassette was inserted in the 3′-5′ orientation into intron 4. The E57K (GAG > AAG) mutation in exon 4 was engineered by overlap extension PCR. The targeting vector was electroporated into mouse C57BL/6 embryonic stem cells and neomycin-resistant cell clones were screened by Southern blotting using internal and external probes (see Fig. 1, C and D). Correctly targeted ES clones were injected into mouse C57BL/6 blastocysts to generate chimeric mice, which were crossed with wild type C57BL/6 mice to obtain germ line transmission of the targeted allele, which contained the neomycin resistance gene (see Fig. 1C). Mice harboring the gene targeted allele were crossed with Flp transgenic mice that express a variant of the Saccharomyces cerevisiae FLP1 recombinase gene under the direction of the human β-actin promoter (B6.Cg-Tg(ACTFLPe)9205Dym/J; Jackson Laboratory) to remove the neomycin-resistant gene (see Fig. 1C). Genotyping of the mice was performed by PCR using forward primer 5′-CTCTGCTGCCGTCATCAGTGATCT-3′ located in exon 4 and reverse primer 5′-GCTTGGAACCCACAGAGATCCTC-3′ located in intron 4. Wild type and knock-in allele generated PCR products of 566 and 746 bp, respectively (see Fig. 1E). Animal care and experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University.
Dermal Fibroblasts and Immunoblot Analysis
Back skin tissues were dissected from newborn and 4-month-old littermates of all three genotypes. Primary fibroblasts were allowed to grow out from the skin, released by trypsin digestion and expanded in DMEM with 10% fetal bovine serum (Life Technologies, Inc.). For immunoblotting, confluent fibroblasts were cultured in serum-free DMEM for 24 h. Cells were lysed with radioimmune precipitation assay buffer (Sigma-Aldrich), and protein concentration was determined by protein assay reagent from Bio-Rad. Serum-free medium was concentrated using Centricon centrifugal filter devices (EMD Millipore, Billerica, MA). Cell lysates (20 μg of total protein) and culture medium (100 μl) were run on 4–12% NuPAGE Bis-Tris polyacrylamide gels (Life Technologies, Inc.) under nonreducing or reducing conditions (5% 2-mercaptoethanol), transferred to Immobilon PVDF membrane (EMD Millipore), and then incubated with antibodies. The bound antibodies were detected using SuperSignal WestFemto substrates (Pierce).
Histology and Immunostaining
Skin tissues were dissected from the upper backs of mice and frozen in optimal cutting temperature compound (VWR, Pittsburgh, PA). Cryosections of 10-μm thickness were stained with hematoxylin-eosin and Masson's trichrome reagents by standard protocols. For immunostaining, cryosections were incubated with primary antibodies at room temperature for 1 h and detected with Alexa Fluor 488-labeled secondary antibody (Life Technologies, Inc.). To visualize nuclei, sections were counterstained with DAPI (1:1000 dilution) for 2 min at room temperature. Slides were then covered with Fluorosafe reagent, and immunofluorescent images were obtained using a Nikon TS100F fluorescent microscope. Fluorescence intensity was quantified in 8–10 randomly selected interfollicular dermal regions from three mice using National Institutes of Health ImageJ software. Aorta and lung tissues were fixed with 10% buffered formalin and embedded in paraffin. Sections 5 μm thick were stained with hematoxylin-eosin and pentachrome reagents. For each genotype, three to five adult mice at age 3–4 months and two newborn mice were analyzed.
Elastin and Collagen Analysis
Back skin tissues were dissected from three adult mice (4 months old) of each genotype and hydrolyzed in 6 n HCl for 48 h at 110 °C. After removal of HCl under vacuum, the dried samples were resuspended in amino acid sample buffer and filtered using Ultrafree-MC (0.45 μm) centrifugal filters (EMD Millipore) to remove insoluble material. Aliquots of filtered samples were used to quantify levels of elastin and collagen by desmosine and hydroxyproline assays as described previously (24).
Assessment of Type I Collagen Cross-linking
Depilated back skin tissue from two 3-month-old mice of each genotype was extracted sequentially with 1.0 m NaCl and 0.5 m acetic acid (100 mg of wet tissue/ml extraction solution) for 5 days each at 4 °C. Extracts were collected by centrifugation at 14,000 × g for 30 min. Ten μl of salt extract, and 6 μl of acid extract were analyzed by electrophoresis on 4–20% Tris-glycine SDS-polyacrylamide gels under reduced conditions, which were stained with Coomassie blue (Bio-Rad).
Transmission Electron Microscopy
Back skin and forelimb tendon tissues were dissected from three 1-month-old mice of each genotype and fixed in 0.1 m cacodylate buffer (pH 7.4) containing 4% paraformaldehyde, 2.5% glutaraldehyde, and 8 mm CaCl2 and then post-fixed with 1% osmium tetroxide as described previously (25, 26). Thin sections were poststained with 2% aqueous uranyl acetate and 1% phosphotungstic acid (pH 3.2) and examined at 80 kV using a Tecnai 12 or JEOL 1400 transmission electron microscope equipped with a Gatan Ultrascan US100 2K digital camera or Gatan Orius wide field side mount CC digital camera (Gatan Inc., Pleasanton, CA).
Collagen Fibril Diameter Analysis
Collagen fibril diameters in skin and tendon from three mice of each genotype were measured using RM Biometrics-Bioquant Image Analysis System (Nashville, TN) as described previously (25). Diameters were determined along the minor axis of the fibril cross-section. Ten digital images from each animal taken at a magnification of 60,000 were analyzed. In each image, an area of interest containing a minimum of 80 fibrils was measured. Mean fibril diameter was calculated using 30 regions of interests from three animals of each genotype and presented as mean ± standard deviation. The dermal data approximated a normal distribution, and a Student's t test was performed with significance at p < 0.05. Data analysis and histograms were performed using Microsoft Excel 2010 (Redmond, WA).
X-Ray Analysis
Four-month-old littermates were euthanized, and x-ray radiographs were taken using IVIS Lumina XR whole animal scanner (Caliper LifeSciences, Hopkinton, MA).
Statistical Analysis
Data from at least three mice per group were analyzed using unpaired, two-tailed Student's t test (Excel 2010) and are expressed as means ± standard deviation. p < 0.05 was considered statistically significant.
Results
Generation of the Fbln4 E57K Knock-in Mouse
Fibulin-4 E57K substitution is located in the beginning of the modified cbEGF domain (Fig. 1A). The nucleotide and amino acid sequences surrounding the Glu-57 residue of fibulin-4 are highly conserved between humans and mice (Fig. 1B). A G → A mutation in the gene resulted in E57K amino acid substitution in both species. The targeting vector for the knock-in mouse contained a G → A point mutation in exon 4 of the Fbln4 gene and a loxP/FRT flanked-pGK-gb2-Neo cassette in intron 4 (Fig. 1C). Correctly targeted embryonic stem cells (on C57BL/6 background) and germ line transmission of the targeted allele were identified and characterized by Southern blotting using both external and internal probes (Fig. 1, C and D). The resulting heterozygous mice were crossed with transgenic mice expressing FLP1 recombinase under the direction of the human β-actin gene promoter to remove the neomycin-resistant gene. This yielded the Fbln4E57K knock-in allele, in which a copy of the loxP/FRT sequence remained in intron 4 (Fig. 1C). Heterozygous Fbln4+/E57K mice were intercrossed, resulting in offspring of all three genotypes on the C57BL/6 background (Fig. 1E).
Fbln4E57K/E57K Mice Are Viable and Display Gross Morphological Abnormalities
The Fbln4E57K/E57K mice were viable, unlike the perinatal lethality of the fibulin-4 null mice (6). Body sizes were comparable among littermates of three genotypes at all ages examined (newborn to 1 year). However, male Fbln4E57K/E57K mice could be readily distinguished from their littermates by the abnormal gross appearance, including loose skin, droopy eyelids, and bent forelimbs (Fig. 2, A–D). The abnormalities were detected in 50% of the homozygous male animals as early as the neonatal stage, and the severity progressed with age. In addition, patchy hair loss was noted in the back skin starting at approximately age 3–4 months, with 80% of the male homozygous mice developing partial alopecia by 1 year of age (Fig. 2E). Surprisingly, the gross morphological abnormalities were not apparent in female Fbln4E57K/E57K mice at the neonatal and young adult stages, although some female homozygous mice developed the abnormal phenotypes as they aged. The Fbln4E57K/E57K mice can survive to 1 year of age, but some animals developed respiratory distress with audible wheezing. Mice presented with severe signs of respiratory distress were euthanized, and these mice had enlarged hearts on autopsy. Male mice were used for further experiments in this study.
FIGURE 2.

Gross morphological abnormalities of the fibulin-4 E57K knock-in mice. A, 4-month-old Fbln4E57K/E57K mouse with droopy eyelid and abnormally bent forelimb (arrows). B, 4-month-old Fbln4+/+ mouse with normal eye and forelimb (arrows). C, loose and inelastic skin in 12-month-old Fbln4E57K/E57K mouse shown by picking up the skin with fingers (left panel). After the fingers were released, the skin did not recoil back but remained extended as a flat skin fold (right panel). D, bent forelimb in Fbln4E57K/E57K mouse. Littermates of three genotypes at age 4 months are shown. E, patchy hair loss in Fbln4E57K/E57K mice. Three 12-month-old mice of each genotype are shown. Note that body sizes of three genotypes were comparable.
Fibulin-4 E57K Protein Is Synthesized, Prone to Form Dimers, and Inefficiently Secreted
To determine whether the fibulin-4 E57K protein was produced by the mutant mice, dermal fibroblasts were established from littermates of all three genotypes. Cell lysates and culture media from the fibroblasts were analyzed by immunoblotting. Normal fibulin-4 protein produced by the Fbln4+/+ fibroblasts migrated at ∼50 kDa under reducing conditions (Fig. 3A) and at ∼45 kDa under nonreducing conditions (Fig. 3B). The majority of the fibulin-4 protein was secreted into the culture medium, and only a small fraction was present in the cell lysate. Mutant fibulin-4 E57K protein was detected in the Fbln4E57K/E57K fibroblasts (Fig. 3A). However, the majority of the mutant protein was found in the cell lysate, and only a small amount was secreted into the medium. Moreover, under nonreducing conditions, a new band at ∼100 kDa, corresponding to a dimer of fibulin-4, was detected in both cell lysate and culture medium (Fig. 3B). The 100-kDa band also was found, to a lesser extent, in the cell lysate of the Fbln4+/E57K fibroblasts. A higher amount of fibulin-4 protein was present in the Fbln4+/E57K cell lysate than the Fbln4+/+ counterpart. Together, the results suggested that the fibulin-4 E57K protein was synthesized in amounts comparable with the normal fibulin-4 protein and that the mutant protein was prone to form dimers and mostly retained intracellularly.
FIGURE 3.

Analyses of dermal fibroblasts from Fbln4+/+, Fbln4+/E57K, and Fbln4E57K/E57K mice. A and B, immunoblot analysis of fibulin-4. Confluent dermal fibroblasts from 4-month-old littermates of three genotypes were grown in serum-free medium for 24 h. Culture medium (100 μl) and cell lysates (20 μg total protein) were separated on 4–12% polyacrylamide gels under reducing (A) and nonreducing (B) conditions. Immunoblot analysis of β-actin in cell lysates (20 μg of total protein/lane) is shown in the bottom of A. C, immunostaining of dermal fibroblasts grown on chamber slides with antibodies against fibulin-4 (FBLN4) and tropoelastin (ELA). Graded increases in intracellular immunoreactivity (green color) of fibulin-4 and tropoelastin were seen in Fbln4+/E57K and Fbln4E57K/E57K fibroblasts, compared with Fbln4+/+ cells. Slides were counterstained with DAPI (blue) to visualize nuclei. D, immunostaining of dermal fibroblasts grown on chamber slides with antibodies against calnexin and BiP. Slides were counterstained with DAPI (blue) to visualize nuclei. Graded increases in immunoreactivity for both ER stress markers were seen in Fbln4+/E57K and Fbln4E57K/E57K fibroblasts, compared with the Fbln4+/+ fibroblasts. Note the punctate staining patterns for both markers in the Fbln4E57K/E57K fibroblasts.
Immunostaining was carried out using fibroblasts fixed and permeabilized with methanol to visualize intracellularly located fibulin-4. Cytoplasmic fibulin-4 immunoreactivity was increased in the Fbln4E57K/E57K fibroblasts, and to a lesser extent in the Fbln4+/E57K fibroblasts, compared with the Fbln4+/+ cells (Fig. 3C). The results are in good agreement with the immunoblotting experiments. Immunoreactivity of tropoelastin also was found to be increased inside the Fbln4E57K/E57K and Fbln4+/E57K cells relative to the Fbln4+/+ control. This suggested that secretion of tropoelastin was impaired in the presence of the fibulin-4 E57K mutant protein. The presence of excessive mutant fibulin-4 protein intracellularly may trigger an endoplasmic reticulum (ER) stress response. Therefore, fibroblasts were immunostained with markers for ER stress. Increased immunoreactivity with a punctate cytoplasmic pattern for calnexin and BiP, two ER-resident chaperones (27), was observed in the Fbln4E57K/E57K fibroblasts and to a lesser extent in the Fbln4+/E57K fibroblasts (Fig. 3D).
Lysyl Oxidase Processing from Its Precursor in Fbln4E57K/E57K Fibroblasts and Skin
Previous studies showed that cross-linking of tropoelastin by LOX was reduced in the absence of fibulin-4 (6). Therefore, LOX levels in fibroblasts of the three genotypes were assessed by immunoblotting. LOX is synthesized and secreted as a 50-kDa proenzyme (pro-LOX) and then cleaved extracellularly by procollagen C-proteinase/BMP-1 into an N-terminal 18-kDa propeptide and a C-terminal 32-kDa mature enzyme (28). Using an antibody recognizing the proenzyme and mature enzyme, but not the cleaved propeptide, similar amounts of pro-LOX were found in the cell lysates of all three genotypes (Fig. 4A). In the culture medium, most of the pro-LOX was processed, and the amounts of mature LOX in the three genotypes appeared comparable (Fig. 4B). However, there was relatively more pro-LOX in the Fbln4E57K/E57K and Fbln4+/E57K media than the Fbln4+/+ control, suggesting that the extracellular conversion of the pro-LOX to mature LOX in Fbln4E57K/E57K fibroblasts was compromised. To assess the extent of pro-LOX processing in vivo, cryosections of mouse back skin were immunostained using an antibody that recognizes predominantly the N-terminal propeptide region (amino acids 66–168) and also a small segment of the mature enzyme (amino acids 169–205) (Fig. 4C). The Fbln4E57K/E57K dermis showed a substantial increase in immunoreactivity compared with the Fbln4+/+ control. The result was consistent with the immunoblotting analysis, suggesting that there was more unprocessed LOX proenzyme in the Fbln4E57K/E57K mutant mice.
FIGURE 4.

Analysis of LOX expression in dermal fibroblasts and skin tissue. A and B, cell lysates (A, 20 μg of total protein) and culture medium (B, 100 μl) from dermal fibroblasts of newborn littermates were separated on 4–12% polyacrylamide gels under reducing conditions and immunoblotted with a polyclonal antibody (L4669, against LOX C terminus; Sigma) recognizing both LOX precursor (pro-LOX) and mature enzyme (LOX). Short exposure of LOX in culture medium is shown (B, right panel). C, immunostaining of back skin from 4-month-old littermates with an antibody recognizing predominantly the N-terminal propeptide region and also a small segment of the mature enzyme (sc-66948, against N-terminal residues 66–205; Santa Cruz Biotechnology). Fbln4E57K/E57K skin showed increased immunoreactivity. Tissue sections were counterstained with DAPI (blue) to visualize nuclei. Magnification bar, 50 μm. a.a., amino acids.
Fibulin-4 E57K Protein Is Expressed at a Low Level in Skin and Affects Elastic Fiber Assembly
To determine whether the fibulin-4 E57K protein is produced in vivo and its effect on elastic fiber assembly, cryosections of back skin from 4-month-old mice were immunostained with antibodies specific for fibulin-4 and several key elastic fiber components. In the wild type skin, long fibulin-4 containing elastic fibers were found near the dermal-epidermal junction, surrounding the hair follicles and throughout the dermis (Fig. 5). By contrast, fibulin-4 immunoreactivity in the Fbln4E57K/E57K skin was markedly reduced and found primarily in the pericellular regions of the hair follicles and dermis; long fibulin-4 fibers were rarely seen. The Fbln4+/E57K skin also had significantly fewer long fibulin-4 fibers and showed decreased immunoreactivity compared with the Fbln4+/+ skin. Quantification of the fibulin-4 immunoreactivity confirmed the graded decrease in fibulin-4 expression in heterozygous and homozygous mutant mice (Fig. 5, right column). Together, the results indicated that the fibulin-4 E57K protein was present in the tissue matrix at a relatively low level and suggested that the mutant fibulin-4 protein was not incorporated into long elastic fibers. Immunostaining with antibodies for tropoelastin showed that elastic fibers in the Fbln4E57K/E57K skin were shorter and less abundant compared with the other two genotypes, particularly in dermal region between hair follicles (Fig. 5). Immunoreactivity of elastic fiber-associated proteins, including fibulin-2, -3, and -5, in the Fbln4E57K/E57K skin also appeared reduced, and their staining patterns were similar to that of tropoelastin. Immunoreactivity for fibrillin-1 and LTBP4 was comparable among the three genotypes.
FIGURE 5.

Immunostaining of mouse skin with antibodies for fibulin-4 and selected elastic fiber components. Cryosections of back skin tissues from 4-month-old Fbln4+/+, Fbln4+/E57K, and Fbln4E57K/E57K male mice were immunostained with rabbit polyclonal antibodies specific for fibulin-4 (FBLN4), tropoelastin (ELN), latent TGF-β binding protein-4 (LTBP4), fibulin-5 (FBLN5), fibrillin-1 (FBN1), fibulin-2 (FBLN2), and fibulin-3 (FBLN3). Tissue sections were counterstained with DAPI (blue) to visualize nuclei. Magnification bar, 20 μm. Fluorescence intensity was quantified and shown in the right panels. The values are expressed as -fold over Fbln4+/+ and means ± S.D. (error bars). *, p < 0.05 between Fbln4E57K/E57K and Fbln4+/+. A low level of fibulin-4 E57K mutant protein is present in the Fbln4E57K/E57K skin. Immunoreactivity of tropoelastin, FBLN5, FBLN2, and FBLN3 was reduced, and elastic fibers were shorter in the Fbln4E57K/E57K skin. LTBP4 and FBN1 immunoreactivity was not significantly different.
Histological and Biochemical Analyses of the Fbln4E57K/E57K Skin
Histological examination of skin from 5-day-old mice showed that the dermis and panniculus carnosus muscle of the Fbln4E57K/E57K skin was notably thinner than the wild type counterparts (Fig. 6A). Dermis from the 3-month-old Fbln4E57K/E57K mice also appeared thinner and exhibited a darker purple color by hematoxylin and eosin staining, compared with the Fbln4+/+ skin. The darker purple staining of the Fbln4E57K/E57K skin by hematoxylin suggested the presence of more extracellular basophilic material, such as acidic proteoglycans. Collagen fibers, stained blue by Masson's trichrome reagent, were reduced in the 3-month-old Fbln4E57K/E57K dermis compared with the Fbln4+/E57K and Fbln4+/+ controls.
FIGURE 6.

Histological and biochemical analyses of skin from Fbln4+/+, Fbln4+/E57K, and Fbln4E57K/E57K mice. A, top row, cryosections of skin from newborn mice stained with hematoxylin-eosin (H&E), showing thinner dermis and panniculus carnosus muscle (PCM). Middle row, cryosections of skin from 3-month-old mice stained with hematoxylin-eosin, showing a darker purple color in the Fbln4E57K/E57K dermis. Bottom row, cryosections of skin from 3-month-old mice stained with Masson's trichrome reagent showing fewer collagen fibers (blue color) in the Fbln4E57K/E57K dermis. B, desmosine and hydroxylproline analyses of skin from 4-month-old mice (three mice of each genotype). Desmosine content of the Fbln4E57K/E57K skin is significantly reduced compared with the other two genotypes (left panel). The asterisk indicates statistically significant changes (p < 0.05, two-tailed distribution) between knock-in and wild type mice by unpaired Student's t test, assuming equal variances. Hydroxylproline contents of skin from different genotypes are comparable (right panel).
Biochemical analyses were performed to quantify the amounts of collagen and elastic fibers in skin of the three genotypes. Desmosine cross-links of elastic fibers were significantly reduced in the Fbln4E57K/E57K skin compared with the Fbln4+/E57K and Fbln4+/+ skin (Fig. 6B). The data were consistent with the reduced and fragmented elastic fibers observed by immunostaining described above (Fig. 5). Collagen content, determined using a hydroxylproline assay, was similar in the skin of all three genotypes (Fig. 6B).
Ultrastructural Abnormalities of Collagen and Elastic Fibers in the Fbln4E57K/E57K Skin
Dermis from 1-month-old Fbln4E57K/E57K and Fbln4+/+ mice was assessed by transmission electron microscopy. In the Fbln4E57K/E57K dermis, collagen fibers (fibril bundles) were smaller and less densely packed with fibrils (Fig. 7A, panels a and b). High magnification images showed many abnormally large and irregular-shaped collagen fibrils in the Fbln4E57K/E57K dermis, in contrast to circular collagen fibrils with variable diameter in the Fbln4+/+ dermis (Fig. 7A, panels c and d). The fibril diameter distribution was unimodal in both genotypes. However, the Fbln4E57K/E57K distribution was broader and shifted toward larger diameters (median, 86.6 nm versus 91.9 nm). Analysis of the mean diameters indicated that the means were significantly larger in the Fbln4E57K/E57K mice compared with wild type controls; 96.8 ± 10.3 nm versus 89.9 ± 8.7 nm (p = 0.006) for the Fbln4E57K/E57K and Fbln4+/+ dermal fibrils, respectively (Fig. 7B).
FIGURE 7.
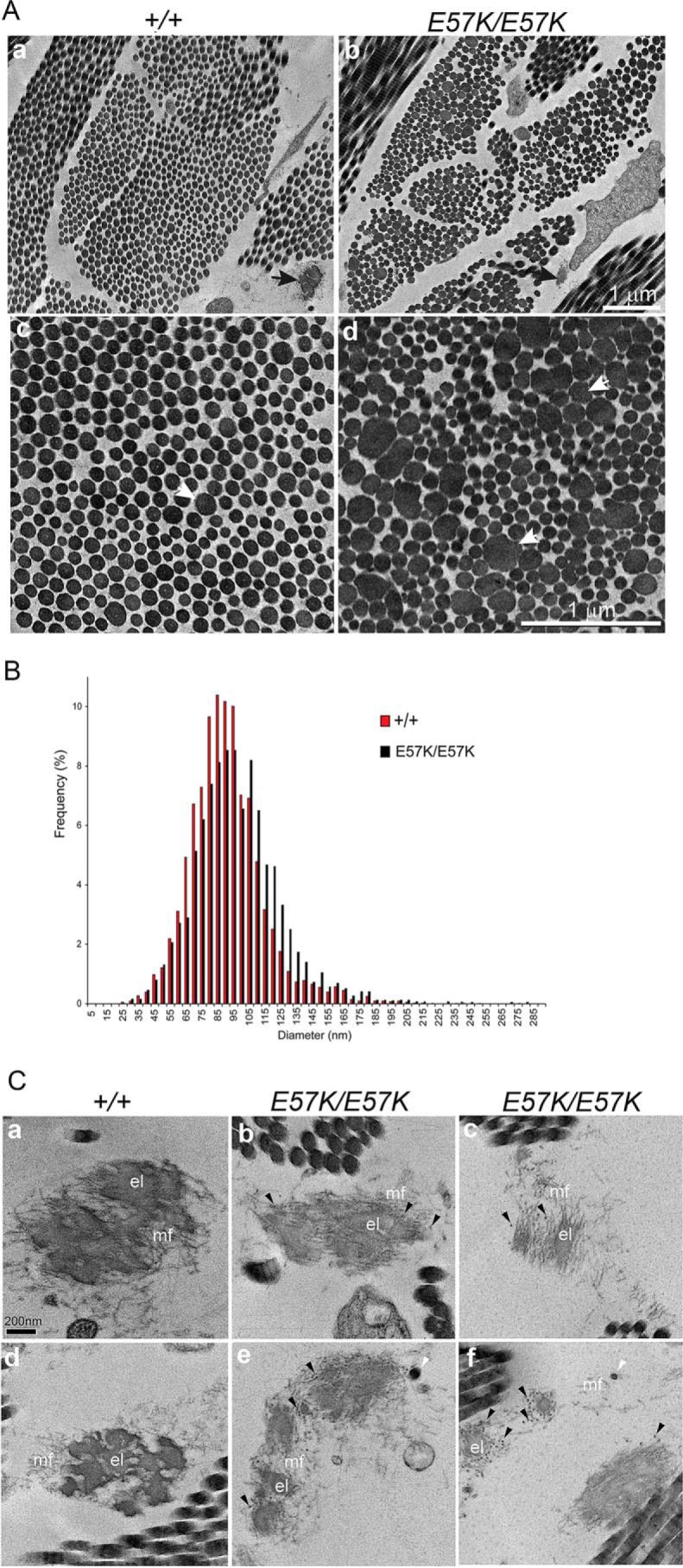
Transmission electron micrographs of skin from 1-month-old Fbln4+/+ and Fbln4E57K/E57K mice. A, collagen fibers. Panels a and b, low magnification micrographs show that in the Fbln4E57K/E57K dermis collagen fibers (fibril bundles) are smaller in size and less densely packed with collagen fibrils. Elastic fiber (black arrow) in the Fbln4E57K/E57K dermis is smaller than that in the Fbln4+/+ counterpart. Magnification bar, 1 μm. Panels c and d, high magnification images show many unusually large and abnormally shaped collagen fibrils (white arrows) in the Fbln4E57K/E57K dermis. Magnification bar, 1 μm. B, collagen fibril diameter distribution of the Fbln4+/+ (red) and Fbln4E57K/E57K (black) dermis. The Fbln4E57K/E57K distribution was broader and shifted toward larger diameters (median, 86.6 nm versus 91.9 nm). The mean diameters were significantly larger in the Fbln4E57K/E57K mice compared with wild type controls; 96.8 ± 10.3 nm versus 89.9 ± 8.7 nm (p = 0.006) for the Fbln4E57K/E57K and Fbln4+/+ dermal fibrils respectively. C, elastic fibers. Panels a and d, elastic fibers in the Fbln4+/+ skin are composed of dense elastin core (el) surrounded by microfibrils (mf). Panels b, c, e, and f, elastic fibers in the Fbln4E57K/E57K skin are structurally abnormal. The elastin cores are smaller with relatively low electron density or composed of separate, smaller globules with excess microfibrils. Some elastin aggregates are deposited outside of the central elastin core (white arrowheads). Many distinct, dark small particles (black arrowheads) are present in the periphery of the elastin cores and associated with microfibrils. Magnification bar, 200 nm.
Elastic fibers in the Fbln4E57K/E57K dermis were smaller in size and structurally abnormal (Fig. 7A, panels a and b, black arrows). In the Fbln4+/+ skin, elastic fibers were composed of large, electron-dense aggregates of elastin core surrounded by a mantle of fibrillin microfibrils (Fig. 7C, panels a and d). By contrast, the Fbln4E57K/E57K elastic fibers were deficient in polymerized elastin, appearing as small and less electron-dense globular deposits associated with excessive microfibrils (Fig. 7C, panels b, c, e, and f). Some polymerized elastin aggregates were deposited outside of the microfibrils (Fig. 7C, panels e and f, white arrowheads). In addition, many distinct, dark small particles, which might represent aberrantly assembled elastin aggregates, were associated with the elastic fibers (Fig. 7C, panels b, c, e, and f, black arrowheads).
Forelimb Defect and Abnormal Tendon Collagen Fibrils in Fbln4E57K/E57K Mice
Patients with fibulin-4 mutations show significant skeletal abnormalities, such as kyphoscoliosis, joint laxity, and multiple bone fractures. Therefore, 4-month-old littermates of three genotypes were analyzed by x-ray imaging for signs of skeletal defects. Apart from abnormally bent forelimbs noted by morphological analyses described above, the Fbln4E57K/E57K mice did not show other apparent skeletal abnormalities (Fig. 8). The forelimb abnormality was more often bilateral than unilateral. Histological examination of the forelimbs did not reveal significant skeletal muscle abnormalities in the Fbln4E57K/E57K mice (data not shown).
FIGURE 8.

X-ray radiographs of 4-month-old Fbln4+/+, Fbln4+/E57K, and Fbln4E57K/E57K littermates. A–C, lateral view; D–F, ventral view; G–I, forelimbs. Apart from the bent forelimb of the Fbln4E57K/E57K mouse (white arrows in F and I), there is no apparent difference in body size and skeletal patterning among three genotypes.
To determine whether the bent forelimbs resulted from a soft connective tissue defect, forelimb tendons from 1-month old Fbln4E57K/E57K and Fbln4+/+ mice were examined by transmission electron microscopy. Low magnification micrographs showed that collagen fibers in the Fbln4E57K/E57K tendon were smaller, and the interspace between collagen fibers was wider relative to the wild type control (Fig. 9A, top panels). High magnification micrographs revealed that the Fbln4E57K/E57K tendon had a high abundance of small diameter collagen fibrils. In addition, many large diameter fibrils showed somewhat irregular contours. Analysis of fibril diameter distributions demonstrated broad, multimodal distributions with substantial differences in both Fbln4E57K/E57K and Fbln4+/+ tendons. The Fbln4E57K/E57K tendon fibrils demonstrated a distinct fibril subpopulation at ∼50 nm that was not present in the Fbln4+/+ distribution. In addition, the Fbln4E57K/E57K distribution was broader and shifted toward larger diameters in the Fbln4E57K/E57K distribution (Fig. 9B). The mean diameter was 91.5 ± 14.5 nm for the Fbln4E57K/E57K fibrils and 114.2 ± 9.9 nm for the Fbln4+/+ fibrils.
FIGURE 9.

Transmission electron micrographs of forelimb tendons in 1-month-old Fbln4+/+ and Fbln4E57K/E57K mice. A, top panels, low magnification micrographs show that the Fbln4E57K/E57K tendon consists of smaller collagen fibers with wider space between collagen fibers than the wild type control. Magnification bars, 2 μm. Bottom panels, high magnification micrographs show that the Fbln4E57K/E57K tendon has a large number of small diameter fibrils; many large diameter collagen fibrils have irregular contours (white arrows). Magnification bars, 1 μm. B, collagen fibril diameter distribution of the Fbln4+/+ (red) and Fbln4E57K/E57K (black) tendon. The tendon fibril diameter distributions demonstrated broad, multimodal distributions in both genotypes. The Fbln4E57K/E57K distribution demonstrated a distinct fibril subpopulation at ∼50 nm that was not present in the Fbln4+/+ distribution. In addition, the Fbln4E57K/E57K distribution was broader and shifted toward larger diameters in the Fbln4E57K/E57K distribution. The mean diameter was 91.5 ± 14.5 nm for the Fbln4E57K/E57K fibrils and 114.2 ± 9.9 nm for the Fbln4+/+ fibrils.
Type I Collagen Cross-linking Is Reduced in the Fbln4E57K/E57K Skin
The finding that both collagen and elastic fibers were abnormal in the Fbln4E57K/E57K mice suggested the possible involvement of lysyl oxidase-mediated cross-linking of both fiber systems (29). Biochemical analysis of desmosine content described above suggested that cross-linking of elastic fibers was reduced in the Fbln4E57K/E57K skin. Lysyl oxidase stabilizes newly deposited collagen fibrils in extracellular matrix by forming divalent and then trivalent cross-links between lysine/hydroxylysine residues in the telopeptide and triple-helical domain of adjacent collagen molecules. The covalent cross-linking of collagen fibrils was therefore assessed by sequential extraction of skin tissue from two 3-month-old mice of each genotype with 1.0 m NaCl and 0.5 m acetic acid, using a constant ratio of 100 mg wet tissue/ml of extraction solution. In the NaCl extracts (Fig. 10A), almost all the collagen molecules in the Fbln4E57K/E57K extract were not cross-linked (α1, α2 chains), whereas the Fbln4+/E57K and Fbln4+/+ extracts contained approximately equal amounts of un-cross-linked and divalent cross-linked (β11, β12) collagen molecules and some trivalent cross-linked molecules (γ). Similarly, in the acetic acid extracts (Fig. 10B), substantially higher amounts of un-cross-linked collagen α chains were present in the Fbln4E57K/E57K sample compared with the other two genotypes. The amounts of divalent and trivalent cross-linked collagen molecules were comparable among the three genotypes. The results indicated that the overall covalent cross-linking of type I collagen was significantly reduced in the Fbln4E57K/E57K skin.
FIGURE 10.
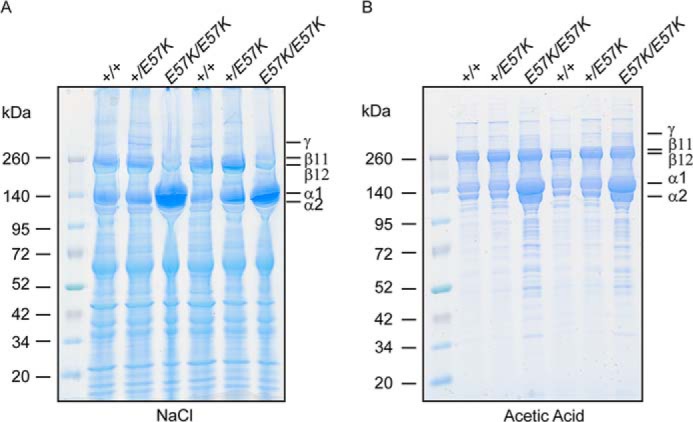
Analysis of collagen cross-linking in skin. Back skin from 3-month-old Fbln4+/+, Fbln4+/E57K, and Fbln4E57K/E57K mice (two of each genotype) was sequentially extracted in 1 m NaCl and 0.5 m acetic acid at a fixed ratio of 100 mg wet tissue/1 ml of extraction solution. Extracts were run on reduced 4–20% SDS-polyacrylamide gradient gels and stained with Coomassie Blue. A, 1.0 m NaCl extract. Each lane was loaded with 10 μl of extract from a separate mouse. B, 0.5 m acetic acid extract. Each lane was loaded with 6 μl of extract from a separate mouse. α1 and α2, un-cross-linked type I collagen chains; β11 and β12, divalently cross-linked collagen; γ, trivalently cross-linked collagen.
Aortic Aneurysm, Tortuous Artery, Enlarged Heart, and Pulmonary Emphysema in Fbln4E57K/E57K Mice
To determine whether the mutant mice had vascular and pulmonary abnormalities as patients with fibulin-4 gene mutations, morphological analyses of internal organs were performed. Dramatic aortic root dilatation and/or ascending aortic aneurysm were observed in ∼50% of the Fbln4 E57K/E57K mice by age 7 months (Fig. 11A). The increase in aortic diameter in Fbln4 E57K/E57K mice was at least 2-fold compared with age and sex-matched Fbln4+/+ and Fbln4+/E57K mice. All Fbln4E57K/E57K mice showed arterial tortuosity and elongation (Fig. 11B). Markedly enlarged hearts were observed in 12-month-old male Fbln4E57K/E57K mice, compared with the age- and sex-matched Fbln4+/+ and Fbln4+/E57K mice (Fig. 11C).
FIGURE 11.

Large vessels, hearts, and lungs of the Fbln4+/+, Fbln4+/E57K, and Fbln4E57K/E57K mice. A, ascending aortae. Note the two different types of dilations in the Fbln4E57K/E57K ascending aortae. B, left common carotid arteries. The Fbln4E57K/E57K artery is tortuous. C, hearts from Fbln4+/+, Fbln4+/E57K, and Fbln4E57K/E57K mice at age 12 months. The Fbln4E57K/E57K hearts are enlarged. D, histology of large vessels and lungs. Paraffin-embedded sections of ascending aortae (panels a–f), descending thoracic aortae (panels g–l), and lungs (panels m–o) stained with hematoxylin-eosin (HE; panels a–c, g–i, and m–o) and pentachrome reagents (PC; panels d–f and j–l). White arrows indicate discontinuities in elastic laminae of large vessels.
Histological analyses revealed thickening of the aortic wall in both ascending and descending aortae of the Fbln4E57K/E57K and Fbln4+/E57K mice (Fig. 11D, panels a–c and g–i). Smooth muscle cells in the ascending and descending aortae of the Fbln4+/+ mice had elongated nuclei, but round nuclei were predominantly seen in the Fbln4E57K/E57K smooth muscle cells, indicating an immature cellular phenotype. Pentachrome staining showed that elastic laminae of the Fbln4E57K/E57K ascending aorta were thin and discontinuous (Fig. 11D, panels d–f). Elastic laminae in Fbln4E57K/E57K descending aorta were relatively normal, although some discontinuities were observed (Fig. 11D, panels j–l). Histological examination of lungs from the three genotypes showed that the Fbln4E57K/E57K lung had enlarged air spaces, a feature consistent with human emphysema (Fig. 11D, panels m–o).
Discussion
Recessive missense mutations in fibulin-4 are a prevalent cause of ARCL 1B. Here, we generated and characterized a novel knock-in mouse model corresponding to fibulin-4 E57K homozygous missense mutation found in two unrelated ARCL 1B patients (18, 22). Both patients have cutis laxa, ascending aortic aneurysm, vascular tortuosity, and pulmonary emphysema/pneumothorax. They also display significant skeletal defects, one with multiple bone fractures at birth and the other with scoliosis, joint laxity, arachnodactyly, pectus excavatum, flexion contractures of the wrists, and feet anomalies. We show that the fibulin-4 E57K knock-in mice display the cutaneous, cardiovascular, pulmonary, and skeletal abnormalities of ARCL 1B. Previous studies of fibulin-4 null, conditional null, and hypomorphic mice have been limited to the vascular and pulmonary systems (6–9, 30). The fibulin-4 E57K knock-in mouse model allows the investigation of fibulin-4 function in the cutaneous and skeletal systems for the first time and future studies of other organ systems where fibulin-4 is expressed, such as eye and cartilage (31, 32).
Missense mutations in fibulin-4 typically affect the N-terminal calcium-binding consensus sequence, conserved cysteines or amino acid adjacent to the cysteines in any of the cbEGF domains (Fig. 12). The FBLN4 E57K missense mutation is located in the N terminus of FBLN4 protein in the beginning of the modified cbEGF domain and alters a conserved residue implicated in calcium-binding (Fig. 12). The E57K fibulin-4 mutant protein is efficiently synthesized and stable, because the total amounts of fibulin-4 protein (intracellular and secreted combined) produced by dermal fibroblasts from Fbln4+/+, Fbln4+/E57K, and Fbln4E57K/E57K mice are comparable (Fig. 3A). The E57K alteration is situated adjacent to the first cysteine residue (C1) of the modified cbEGF domain (Fig. 12). Substitution of a conserved acidic Glu-57 with an oppositely charged basic Lys residue is expected to significantly alter the local electrostatic property and thereby would interfere with the disulfide bonding of the adjacent C1 residue with C3 of the modified cbEGF domain. This likely would prevent proper folding of the mutant fibulin-4 protein, resulting in intracellular retention. In addition, some of the C1 and C3 residues would be free and thereby could form intermolecular disulfide bridges, leading to dimer formation. Indeed, biochemical analysis demonstrates that the mutant fibulin-4 protein is preferentially retained intracellularly, and some mutant fibulin-4 proteins form dimers (Fig. 3A). This leads to significant increases in the expression of ER-resident chaperone proteins, which can promote folding of the mutant fibulin-4 protein and/or target misfolded mutant fibulin-4 protein for degradation (Fig. 3D). Activation of the ER stress pathway and unfolded protein response may contribute to the abnormal phenotype of the knock-in mice.
FIGURE 12.
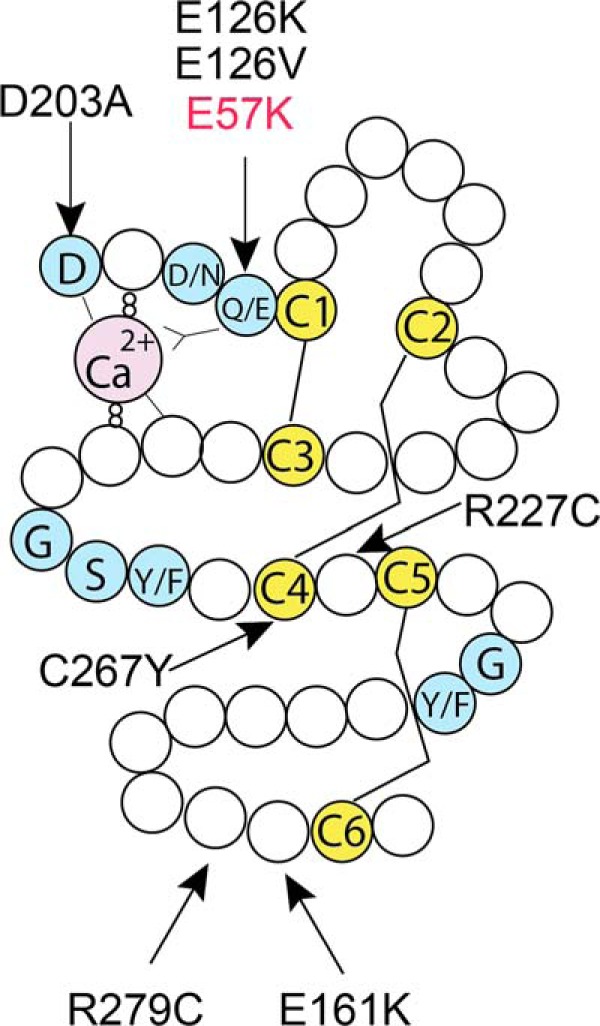
Fibulin-4 missense mutations found in patients. Schematic drawing of a typical cbEGF domain, showing the consensus sequences for calcium binding at the N terminus (blue), six conserved cysteine residues (C1–C6, yellow), and other conserved amino acids (blue). Three pairs of disulfide bridges are formed between C1 and C3, C2 and C4, and C5 and C6. The modified cbEGF domain contains a 28-amino acid insertion between C4 and C5. Arrows show the locations of known fibulin-4 missense mutations within the respective cbEGF domain.
The knock-in mice display marked skin abnormality, a hallmark of cutis laxa syndrome that has not been investigated in previously reported fibulin-4 mouse models. Our studies show that fibulin-4 is widely distributed in skin of normal mice, concentrated near the dermal-epidermal junction and surrounding hair follicles in a manner similar to the other elastic fiber components (Fig. 5). In the Fbln4E57K/E57K skin, immunoreactivity of elastin and other elastic fiber-associated proteins shows relatively smaller decreases compared with the significant reduction of fibulin-4 immunoreactivity. This suggests that the presence of the mutant fibulin-4 protein does not exert a dominant negative effect in elastic fiber formation. However, the elastic fibers in the mutant skin are notably shorter, particularly in the dermis between hair follicles. Electron microscopic analyses indicate that ultrastructural abnormalities of the elastic fibers in Fbln4E57K/E57K mice resemble those previously seen in the fibulin-5 null mouse skin (33). These include an elastin core consisting of small globules with low electron density, disconnection between small elastin aggregates and microfibrils, and an excess of microfibrils without an elastin core. These results are consistent with the unique and nonoverlapping roles of fibulin-4 and fibulin-5 in elastic fiber assembly.
A striking finding revealed by the ultrastructural analysis of skin is that the dermal collagen fibrils in the Fbln4E57K/E57K dermis are abnormal. This is in sharp contrast to a previous finding for the fibulin-5 null skin, in which no dermal collagen fibril defect was observed (33). The specific role of fibulin-4 in collagen fibrillogenesis is further supported by the irregular fibril contours and abnormal diameter distribution of collagen fibrils in the forelimb tendon, as well as altered organization into fibers, i.e. fibril bundles. Defective collagen fibrillogenesis may contribute to the forelimb abnormality seen in the Fbln4E57K/E57K mice and to the significantly more severe phenotype of the fibulin-4 null than fibulin-5 null mice (34, 35). The collagen fibril abnormalities found in the skin and tendon of the mutant mice are not the same. In the skin where collagen fibrils are randomly arranged, there is a pronounced increase in large, aberrantly shaped collagen fibrils, and the mean fibril diameter is higher in the knock-in skin compared with the wild type skin. On the other hand, in tendons where collagen fibrils are highly organized in parallel, there is a significant increase in small diameter collagen fibrils, as reflected by the smaller mean fibril diameter in the knock-in tendon compared with the wild type tendon.
Reduced collagen cross-links mediated by LOX might contribute to the abnormal collagen fibrillogenesis observed in the E57K fibulin-4 mutant mice. Fibulin-4 has been previously shown to interact with the LOX propeptide. Our immunoblotting and immunostaining experiments suggest that conversion of pro-LOX to mature LOX is less complete in the Fbln4E57K/E57K mice than wild type animals (Fig. 4). Although the amount of mature LOX detected by immunoblotting in culture media from the Fbln4E57K/E57K fibroblasts was not significantly reduced compared with the other two genotypes (Fig. 4B), the reduction of desmosine cross-links of elastin, as well as decreased amounts of divalent and trivalentcollagen cross-links in the Fbln4E57K/E57K skin, provide biochemical support that the LOX activity is reduced in vivo (Figs. 6B and 10). The LOX binding site on fibulin-4 is located in the N-terminal segment, which contains the E57K substitution (8). Recent in vitro binding assays using recombinant proteins produced in HEK293 cells indicate that human E57K fibulin-4 protein shows reduced binding to pro-LOX and tropoelastin.3 Together, the results imply that the fibulin-4 E57K missense mutation compromises LOX activation and thereby affects the assembly of both elastic fibers and collagen fibrils. Consistent with this proposition are the findings that 1) fibulin-5 does not bind LOX, and the fibulin-5 null mice do not display collagen abnormality (8, 14, 33); 2) LOX activity is required for ordered collagen fibrillogenesis by tendon cells (36); 3) abnormal collagen cross-linking affects collagen fibril structure and organization (37); and 4) LOX null mice are perinatal lethal and display aortic aneurysm, cardiopulmonary dysfunction, and diaphragmatic rupture, similar to the mice deficient in fibulin-4 (38, 39).
Several ARCL 1B patients, including one of the patients with a recessive E57K substitution, display multiple bone fractures at birth, a cardinal feature of osteogenesis imperfecta. Congenital bone fragility results from mutations in genes encoding collagen I and proteins responsible for prolyl 3-hydroxylation of collagens, among others (40). Our finding that fibulin-4 influences collagen fibril and fiber assembly resulting in altered structure and organization suggests an explanation of the bone abnormality observed in some ARCL 1B patients. However, unlike osteogenesis imperfecta syndrome, the ARCL 1B patients do not have short stature, and the Fbln4E57K/E57K mice are similar in size to the wild type mice. This might be related to increased TGF-β activity associated with fibulin-4 deficiency, as demonstrated in the aorta and lung tissue of fibulin-4 hypomorphic mice and an ARCL 1B patient (4, 9). Fibulin-4 has been shown to bind fibrillin-1 and is localized on fibrillin microfibrils (23, 41). The fibrillin microfibrils play a specific role in the extracellular sequestration of large latent TGF-β complex, comprising latent TGF-β binding proteins and TGF-β with its latency-associated propeptide (42). Fibulin-4 interacts with the N-terminal region of fibrillin-1 that also contains the binding site for LTBP1 (43). Biochemical analysis showed that fibulin-4 and LTBP1 compete for fibrillin-1 binding (43). Fibulin-4 also binds strongly to LTBP1; fibrillin-1 and LTBP1 can bind fibulin-4 simultaneously, forming a ternary complex (44). Indeed, there is significant overlap between clinical features of ARCL 1B and Marfan syndrome caused by fibrillin-1 mutations, such as thoracic aortic aneurysm/dilatation, emphysematous lung, joint laxity, pectus deformity, arachnodactyly, scoliosis, and kyphosis. It is noteworthy that excessive TGF-β signaling has recently been shown to be a common mechanism in the pathogenesis of osteogenesis imperfecta (45).
Fibulin-4 also has been shown to interact with the homeobox transcription factor Pitx2, which plays a role in ocular and forelimb development (32, 46). The possibility of an intracellular form of fibulin-4 generated from an alternative translational initiation site has been suggested by in vitro translation experiments (47). Whether or not intracellular interaction of fibulin-4 and Pitx2 contributes to the bent forelimb abnormality remains an open question.
Our studies show that the Fbln4E57K/E57K mice exhibit ascending aortic aneurysm and tortuous arteries. The vascular phenotypes closely parallel those in the smooth muscle cell-specific conditional fibulin-4 knock-out mice and the hypomorphic fibulin-4 mice (7–9). Comprehensive studies of the vascular abnormalities of the Fbln4E57K/E57K mice are currently underway and will be reported elsewhere.
It is unclear why female Fbln4E57K/E57K mice display few gross morphological abnormalities at neonatal and young adult stages. Skin structure has a clear gender difference, which is mediated by a complex interplay between estrogens and androgens (48). The dermis in male mice is 190% thicker than the female counterparts, whereas the epidermis and hypodermis are thicker in the female than male mice (49). Androgens are the main regulators of dermal thickness and hair follicle growth. Collagen and elastin are major components of the dermis and responsible for maintaining the dermal architecture. Abnormal collagen and elastic fibers associated with mutant fibulin-4 thus may have differential effects on male and female skin.
In conclusion, we have generated a fibulin-4 knock-in mouse strain that harbors a recurrent fibulin-4 missense mutation in ARCL 1B. We have demonstrated that the homozygous mutant mice exhibit cutaneous, vascular, pulmonary, and skeletal defects seen in ARCL 1B patients. Our studies show that missense fibulin-4 mutations give rise to abnormal collagen fibrils and altered assembly of fibrils into fibers, in addition to paucity of elastic fibers associated with fibulin-4 deficiency known previously. We provide evidence that fibulin-4 affects intermolecular collagen cross-linking mediated by LOX and regulates collagen fibril assembly. The knock-in mouse model thus provides a valuable tool to better understand the pathophysiological mechanisms underlying missense fibulin-4 mutations and to develop treatment for this life-threatening disease.
Author Contributions
M.-L. C. conceived and coordinated the study. O. I. and V. A. designed, performed, and analyzed the experiments shown in Figs. 2–6A, 8, and 10. C. M. H., I. S., and R. P. M. designed, performed, and analyzed the experiments shown in Figs. 6B and 11. S. M. A. and D. E. B. designed, performed, and analyzed the experiments shown in Figs. 7 and 9. T. S. contributed new reagents. M. A. and A. D. performed breeding, genotyping, and morphological analyses of mice. M.-L. C., O. I., D. E. B., and R. P. M. wrote the paper.
Acknowledgments
We thank Dr. Masahiro Iwamoto (Children's Hospital of Philadelphia) for help in the analysis of forelimb phenotype and Dr. Lynn Sakai (Shriners Hospital for Children, Portland, OR) for providing fibrillin-1 antibody.
This work was supported, in whole or in part, by National Institutes of Health Grants AR056523 (M.-L. C.), HL53325 (R. P. M.), HL105314 (R. P. M.), and AR044745 (D. E. B.). The authors declare that they have no conflicts of interest with the contents of this article.
T. Sasaki, unpublished observations.
- ARCL
- autosomal recessive cutis laxa
- LOX
- lysyl oxidase
- cbEGF
- calcium-binding epidermal growth factor-like
- ER
- endoplasmic reticulum.
References
- 1. Urban Z., Davis E. C. (2014) Cutis laxa: Intersection of elastic fiber biogenesis, TGFβ signaling, the secretory pathway and metabolism. Matrix Biol. 33, 16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Uitto J., Li Q., Urban Z. (2013) The complexity of elastic fibre biogenesis in the skin: a perspective to the clinical heterogeneity of cutis laxa. Exp. Dermatol. 22, 88–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Al-Hassnan Z. N., Almesned A. R., Tulbah S., Hakami A., Al-Omrani A., Al Sehly A., Mohammed S., Majid S., Meyer B., Al-Fayyadh M. (2012) Recessively inherited severe aortic aneurysm caused by mutated EFEMP2. Am. J. Cardiol. 109, 1677–1680 [DOI] [PubMed] [Google Scholar]
- 4. Renard M., Holm T., Veith R., Callewaert B. L., Adès L. C., Baspinar O., Pickart A., Dasouki M., Hoyer J., Rauch A., Trapane P., Earing M. G., Coucke P. J., Sakai L. Y., Dietz H. C., De Paepe A. M., Loeys B. L. (2010) Altered TGFβ signaling and cardiovascular manifestations in patients with autosomal recessive cutis laxa type I caused by fibulin-4 deficiency. Eur. J. Hum. Genet. 18, 895–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Giltay R., Timpl R., Kostka G. (1999) Sequence, recombinant expression and tissue localization of two novel extracellular matrix proteins, fibulin-3 and fibulin-4. Matrix Biol. 18, 469–480 [DOI] [PubMed] [Google Scholar]
- 6. McLaughlin P. J., Chen Q., Horiguchi M., Starcher B. C., Stanton J. B., Broekelmann T. J., Marmorstein A. D., McKay B., Mecham R., Nakamura T., Marmorstein L. Y. (2006) Targeted disruption of fibulin-4 abolishes elastogenesis and causes perinatal lethality in mice. Mol. Cell. Biol. 26, 1700–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang J., Davis E. C., Chapman S. L., Budatha M., Marmorstein L. Y., Word R. A., Yanagisawa H. (2010) Fibulin-4 deficiency results in ascending aortic aneurysms: a potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ. Res. 106, 583–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Horiguchi M., Inoue T., Ohbayashi T., Hirai M., Noda K., Marmorstein L. Y., Yabe D., Takagi K., Akama T. O., Kita T., Kimura T., Nakamura T. (2009) Fibulin-4 conducts proper elastogenesis via interaction with cross-linking enzyme lysyl oxidase. Proc. Natl. Acad. Sci. U.S.A. 106, 19029–19034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hanada K., Vermeij M., Garinis G. A., de Waard M. C., Kunen M. G., Myers L., Maas A., Duncker D. J., Meijers C., Dietz H. C., Kanaar R., Essers J. (2007) Perturbations of vascular homeostasis and aortic valve abnormalities in fibulin-4 deficient mice. Circ. Res. 100, 738–746 [DOI] [PubMed] [Google Scholar]
- 10. Yanagisawa H., Davis E. C. (2010) Unraveling the mechanism of elastic fiber assembly: The roles of short fibulins. Int. J. Biochem. Cell Biol. 42, 1084–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wagenseil J. E., Mecham R. P. (2007) New insights into elastic fiber assembly. Birth Defects Res. C Embryo Today 81, 229–240 [DOI] [PubMed] [Google Scholar]
- 12. Papke C. L., Yanagisawa H. (2014) Fibulin-4 and Fibulin-5 in elastogenesis and beyond: Insights from mouse and human studies. Matrix Biol. 37, 142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Noda K., Dabovic B., Takagi K., Inoue T., Horiguchi M., Hirai M., Fujikawa Y., Akama T. O., Kusumoto K., Zilberberg L., Sakai L. Y., Koli K., Naitoh M., von Melchner H., Suzuki S., Rifkin D. B., Nakamura T. (2013) Latent TGF-β binding protein 4 promotes elastic fiber assembly by interacting with fibulin-5. Proc. Natl. Acad. Sci. U.S.A. 110, 2852–2857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Choudhury R., McGovern A., Ridley C., Cain S. A., Baldwin A., Wang M. C., Guo C., Mironov A., Jr., Drymoussi Z., Trump D., Shuttleworth A., Baldock C., Kielty C. M. (2009) Differential regulation of elastic fiber formation by fibulin-4 and -5. J. Biol. Chem. 284, 24553–24567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sawyer S. L., Dicke F., Kirton A., Rajapkse T., Rebeyka I. M., McInnes B., Parboosingh J. S., Bernier F. P. (2013) Longer term survival of a child with autosomal recessive cutis laxa due to a mutation in FBLN4. Am. J. Med. Genet. A 161A, 1148–1153 [DOI] [PubMed] [Google Scholar]
- 16. Hebson C., Coleman K., Clabby M., Sallee D., Shankar S., Loeys B., Van Laer L., Kogon B. (2014) Severe aortopathy due to fibulin-4 deficiency: molecular insights, surgical strategy, and a review of the literature. Eur. J. Pediatr. 173, 671–675 [DOI] [PubMed] [Google Scholar]
- 17. Kappanayil M., Nampoothiri S., Kannan R., Renard M., Coucke P., Malfait F., Menon S., Ravindran H. K., Kurup R., Faiyaz-Ul-Haque M., Kumar K., De Paepe A. (2012) Characterization of a distinct lethal arteriopathy syndrome in twenty-two infants associated with an identical, novel mutation in FBLN4 gene, confirms fibulin-4 as a critical determinant of human vascular elastogenesis. Orphanet J. Rare Dis. 7, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Iascone M., Sana M. E., Pezzoli L., Bianchi P., Marchetti D., Fasolini G., Sadou Y., Locatelli A., Fabiani F., Mangili G., Ferrazzi P. (2012) Extensive arterial tortuosity and severe aortic dilation in a newborn with an EFEMP2 mutation. Circulation 126, 2764–2768 [DOI] [PubMed] [Google Scholar]
- 19. Erickson L. K., Opitz J. M., Zhou H. (2012) Lethal osteogenesis imperfecta-like condition with cutis laxa and arterial tortuosity in MZ twins due to a homozygous fibulin-4 mutation. Pediatr. Dev. Pathol. 15, 137–141 [DOI] [PubMed] [Google Scholar]
- 20. Hoyer J., Kraus C., Hammersen G., Geppert J. P., Rauch A. (2009) Lethal cutis laxa with contractural arachnodactyly, overgrowth and soft tissue bleeding due to a novel homozygous fibulin-4 gene mutation. Clin. Genet. 76, 276–281 [DOI] [PubMed] [Google Scholar]
- 21. Dasouki M., Markova D., Garola R., Sasaki T., Charbonneau N. L., Sakai L. Y., Chu M. L. (2007) Compound heterozygous mutations in fibulin-4 causing neonatal lethal pulmonary artery occlusion, aortic aneurysm, arachnodactyly, and mild cutis laxa. Am. J. Med. Genet. A 143A, 2635–2641 [DOI] [PubMed] [Google Scholar]
- 22. Hucthagowder V., Sausgruber N., Kim K. H., Angle B., Marmorstein L. Y., Urban Z. (2006) Fibulin-4: a novel gene for an autosomal recessive cutis laxa syndrome. Am. J. Hum. Genet. 78, 1075–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kobayashi N., Kostka G., Garbe J. H., Keene D. R., Bächinger H. P., Hanisch F. G., Markova D., Tsuda T., Timpl R., Chu M. L., Sasaki T. (2007) A comparative analysis of the fibulin protein family. Biochemical characterization, binding interactions, and tissue localization. J. Biol. Chem. 282, 11805–11816 [DOI] [PubMed] [Google Scholar]
- 24. Cheng J. K., Stoilov I., Mecham R. P., Wagenseil J. E. (2013) A fiber-based constitutive model predicts changes in amount and organization of matrix proteins with development and disease in the mouse aorta. Biomech. Model. Mechanobiol. 12, 497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Izu Y., Ansorge H. L., Zhang G., Soslowsky L. J., Bonaldo P., Chu M. L., Birk D. E. (2011) Dysfunctional tendon collagen fibrillogenesis in collagen VI null mice. Matrix Biol. 30, 53–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sicot F. X., Tsuda T., Markova D., Klement J. F., Arita M., Zhang R. Z., Pan T. C., Mecham R. P., Birk D. E., Chu M. L. (2008) Fibulin-2 is dispensable for mouse development and elastic fiber formation. Mol. Cell. Biol. 28, 1061–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ni M., Lee A. S. (2007) ER chaperones in mammalian development and human diseases. FEBS Lett. 581, 3641–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Panchenko M. V., Stetler-Stevenson W. G., Trubetskoy O. V., Gacheru S. N., Kagan H. M. (1996) Metalloproteinase activity secreted by fibrogenic cells in the processing of prolysyl oxidase. Potential role of procollagen C-proteinase. J. Biol. Chem. 271, 7113–7119 [DOI] [PubMed] [Google Scholar]
- 29. Lucero H. A., Kagan H. M. (2006) Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell Mol. Life Sci. 63, 2304–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ramnath N. W., van de Luijtgaarden K. M., van der Pluijm I., van Nimwegen M., van Heijningen P. M., Swagemakers S. M., van Thiel B. S., Ridwan R. Y., van Vliet N., Vermeij M., Hawinkels L. J., de Munck A., Dzyubachyk O., Meijering E., van der Spek P., Rottier R., Yanagisawa H., Hendriks R. W., Kanaar R., Rouwet E. V., Kleinjan A., Essers J. (2014) Extracellular matrix defects in aneurysmal fibulin-4 mice predispose to lung emphysema. PLoS One 9, e106054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xiang Y., Sekine T., Nakamura H., Imajoh-Ohmi S., Fukuda H., Yudoh K., Masuko-Hongo K., Nishioka K., Kato T. (2006) Fibulin-4 is a target of autoimmunity predominantly in patients with osteoarthritis. J. Immunol. 176, 3196–3204 [DOI] [PubMed] [Google Scholar]
- 32. Acharya M., Sharp M. W., Mirzayans F., Footz T., Huang L., Birdi C., Walter M. A. (2012) Yeast two-hybrid analysis of a human trabecular meshwork cDNA library identified EFEMP2 as a novel PITX2 interacting protein. Mol. Vis. 18, 2182–2189 [PMC free article] [PubMed] [Google Scholar]
- 33. Choi J., Bergdahl A., Zheng Q., Starcher B., Yanagisawa H., Davis E. C. (2009) Analysis of dermal elastic fibers in the absence of fibulin-5 reveals potential roles for fibulin-5 in elastic fiber assembly. Matrix Biol. 28, 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yanagisawa H., Davis E. C., Starcher B. C., Ouchi T., Yanagisawa M., Richardson J. A., Olson E. N. (2002) Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature 415, 168–171 [DOI] [PubMed] [Google Scholar]
- 35. Nakamura T., Lozano P. R., Ikeda Y., Iwanaga Y., Hinek A., Minamisawa S., Cheng C. F., Kobuke K., Dalton N., Takada Y., Tashiro K., Ross J., Jr., Honjo T., Chien K. R. (2002) Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature 415, 171–175 [DOI] [PubMed] [Google Scholar]
- 36. Herchenhan A., Uhlenbrock F., Eliasson P., Weis M., Eyre D., Kadler K. E., Magnusson S. P., Kjaer M. (2015) Lysyl oxidase activity is required for ordered collagen fibrillogenesis by tendon cells. J. Biol. Chem. 290, 16440–16450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cabral W. A., Perdivara I., Weis M., Terajima M., Blissett A. R., Chang W., Perosky J. E., Makareeva E. N., Mertz E. L., Leikin S., Tomer K. B., Kozloff K. M., Eyre D. R., Yamauchi M., Marini J. C. (2014) Abnormal type I collagen post-translational modification and crosslinking in a cyclophilin B KO mouse model of recessive osteogenesis imperfecta. PLoS Genet. 10, e1004465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hornstra I. K., Birge S., Starcher B., Bailey A. J., Mecham R. P., Shapiro S. D. (2003) Lysyl oxidase is required for vascular and diaphragmatic development in mice. J. Biol. Chem. 278, 14387–14393 [DOI] [PubMed] [Google Scholar]
- 39. Mäki J. M., Sormunen R., Lippo S., Kaarteenaho-Wiik R., Soininen R., Myllyharju J. (2005) Lysyl oxidase is essential for normal development and function of the respiratory system and for the integrity of elastic and collagen fibers in various tissues. Am. J. Pathol. 167, 927–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marini J. C., Blissett A. R. (2013) New genes in bone development: what's new in osteogenesis imperfecta. J. Clin. Endocrinol. Metab. 98, 3095–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. El-Hallous E., Sasaki T., Hubmacher D., Getie M., Tiedemann K., Brinckmann J., Bätge B., Davis E. C., Reinhardt D. P. (2007) Fibrillin-1 interactions with fibulins depend on the first hybrid domain and provide an adaptor function to tropoelastin. J. Biol. Chem. 282, 8935–8946 [DOI] [PubMed] [Google Scholar]
- 42. Ramirez F., Rifkin D. B. (2009) Extracellular microfibrils: contextual platforms for TGFβ and BMP signaling. Curr. Opin. Cell Biol. 21, 616–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ono R. N., Sengle G., Charbonneau N. L., Carlberg V., Bächinger H. P., Sasaki T., Lee-Arteaga S., Zilberberg L., Rifkin D. B., Ramirez F., Chu M. L., Sakai L. Y. (2009) Latent transforming growth factor β-binding proteins and fibulins compete for fibrillin-1 and exhibit exquisite specificities in binding sites. J. Biol. Chem. 284, 16872–16881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Massam-Wu T., Chiu M., Choudhury R., Chaudhry S. S., Baldwin A. K., McGovern A., Baldock C., Shuttleworth C. A., Kielty C. M. (2010) Assembly of fibrillin microfibrils governs extracellular deposition of latent TGF β. J. Cell Sci. 123, 3006–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grafe I., Yang T., Alexander S., Homan E. P., Lietman C., Jiang M. M., Bertin T., Munivez E., Chen Y., Dawson B., Ishikawa Y., Weis M. A., Sampath T. K., Ambrose C., Eyre D., Bächinger H. P., Lee B. (2014) Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nat. Med. 20, 670–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Holmberg J., Ingner G., Johansson C., Leander P., Hjalt T. A. (2008) PITX2 gain-of-function induced defects in mouse forelimb development. BMC Dev. Biol. 8, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gallagher W. M., Greene L. M., Ryan M. P., Sierra V., Berger A., Laurent-Puig P., Conseiller E. (2001) Human fibulin-4: analysis of its biosynthetic processing and mRNA expression in normal and tumour tissues. FEBS Lett. 489, 59–66 [DOI] [PubMed] [Google Scholar]
- 48. Dao H., Jr., Kazin R. A. (2007) Gender differences in skin: a review of the literature. Gend. Med. 4, 308–328 [DOI] [PubMed] [Google Scholar]
- 49. Azzi L., El-Alfy M., Martel C., Labrie F. (2005) Gender differences in mouse skin morphology and specific effects of sex steroids and dehydroepiandrosterone. J. Invest. Dermatol. 124, 22–27 [DOI] [PubMed] [Google Scholar]