

Background: Herpes simplex virus attachment protein gC comprises a glycosaminoglycan-binding site and a mucin-like region.
Results: Removal of the mucin-like region modifies gC interaction with glycosaminoglycans.
Conclusion: The mucin-like region balances the gC-glycosaminoglycan interaction during virus binding to and release from target cells.
Significance: The finding might be of relevance for similar proteins on other GAG-binding viruses.
Keywords: carbohydrate-binding protein, glycoprotein, glycosaminoglycan, glycosylation, herpesvirus, surface plasmon resonance (SPR), mucin-like region
Abstract
Glycoprotein C (gC) mediates the attachment of HSV-1 to susceptible host cells by interacting with glycosaminoglycans (GAGs) on the cell surface. gC contains a mucin-like region located near the GAG-binding site, which may affect the binding activity. Here, we address this issue by studying a HSV-1 mutant lacking the mucin-like domain in gC and the corresponding purified mutant protein (gCΔmuc) in cell culture and GAG-binding assays, respectively. The mutant virus exhibited two functional alterations as compared with native HSV-1 (i.e. decreased sensitivity to GAG-based inhibitors of virus attachment to cells and reduced release of viral particles from the surface of infected cells). Kinetic and equilibrium binding characteristics of purified gC were assessed using surface plasmon resonance-based sensing together with a surface platform consisting of end-on immobilized GAGs. Both native gC and gCΔmuc bound via the expected binding region to chondroitin sulfate and sulfated hyaluronan but not to the non-sulfated hyaluronan, confirming binding specificity. In contrast to native gC, gCΔmuc exhibited a decreased affinity for GAGs and a slower dissociation, indicating that once formed, the gCΔmuc-GAG complex is more stable. It was also found that a larger number of gCΔmuc bound to a single GAG chain, compared with native gC. Taken together, our data suggest that the mucin-like region of HSV-1 gC is involved in the modulation of the GAG-binding activity, a feature of importance both for unrestricted virus entry into the cells and release of newly produced viral particles from infected cells.
Introduction
Carbohydrate components at the cell surface, such as sialic acid residues and glycosaminoglycan (GAG)4 chains of proteoglycans, are frequently targeted by different viruses to assist their attachment to susceptible host cells. This interaction has to be neatly regulated to permit, on the one hand, attachment and entry of the virus into the cells and, on the other hand, detachment and liberation of trapped virions, in the case of non-productive, dead-end interactions. This is particularly evident during virus egress, where all biomolecular interactions between the host cell and the virion need to be overcome to ensure particle release and escape of the newly assembled virions to infect surrounding cells. Some sialic acid-binding viruses, such as influenza viruses, express the receptor-degrading enzyme sialidase to balance the virus interaction with sialic acid. This enzyme prevents both trapping of progeny virions at the cell surface during release and the formation of virus clusters due to cross-linking via sialic acid residues on viral glycoproteins (1). A detailed study of the activity of viral sialidase has led to the development of potent anti-influenza drugs (2).
Many viruses, including herpes simplex virus (HSV) (3, 4), respiratory syncytial virus (3, 5), Ebola virus (6), and HIV (7), utilize GAGs on the host cell surface for attachment purposes. For these GAG-binding viruses, the underlying mechanisms regulating binding and unbinding events between the viral attachment component and the GAG chains are far from being fully understood (8). A common structural feature of the GAG-binding proteins of HSV, respiratory syncytial virus, and Ebola virus is the presence of a region(s) comprising numerous, clustered O-linked glycans also known as the mucin-like region(s). It is believed that biological functions of such regions are to stretch the protein to enhance its availability for binding, to protect the protein against proteolytic degradation, and to modulate the host immune response (9–11). However, less is known about the influence of mucin-like regions on the interaction of viral glycoproteins with GAGs on the cell surface.
We have recently found that serial passages of HSV-1 in cultured cells in the presence of the GAG-mimetic oligosaccharide PI-88 (also known as muparfostat) resulted in the selection of viral variants that were resistant to this inhibitor, due to deletion of an entire mucin-like domain located at the N-terminal part of the viral attachment protein gC (12). Interestingly, similar selection experiments performed with HSV-2 resulted in viral variants lacking gG, a virus envelope protein also carrying a mucin-like region (13). These results, indicating that the mucin-like regions of viral glycoproteins could modify the virus sensitivity to GAG mimetics, encouraged us to further investigate the influence of these domains on the GAG-binding activity of viral attachment.
In this study, we investigate the GAG-binding properties of the HSV-1 glycoprotein gC, a protein known to mediate virus attachment to GAGs on the cell surface (14, 15). Specifically, we compare the binding behavior of native gC, carrying the mucin-like region, with that of a mutated gC variant, lacking the mucin-like region (gCΔmuc) (Fig. 1, a and b), to surface-immobilized GAGs using surface plasmon resonance (SPR)-based sensing. We take advantage of a previously established interaction platform consisting of GAGs immobilized to a streptavidin surface. The GAGs were biotinylated at their reducing end to allow for end-on surface immobilization in a brushlike arrangement, reflecting the grafting architecture of the GAG chains on proteoglycans (Fig. 1c) (16–18). Other advantages of this platform over the more common side-on immobilization using multiple biotin groups along the GAG chain (16, 19) include a better accessibility and a reduced risk of interference with protein binding caused by the chemical modification.
FIGURE 1.

Herpes simplex virus glycoprotein gC and the GAG interaction platform. a, schematic structure of gC. The mucin-like region found in the N-terminal region contains multiple sites for O-linked (green) and N-linked (red) glycosylation and lies close to the positively changed GAG-binding region. The mucin-like region is present in native gC but deleted in gCΔmuc. TMR, transmembrane region. b, electrophoretic profiles of native gC and gCΔmuc. c, schematic image representing the GAG immobilization strategy. d, streptavidin (SA) and GAG mass deposited on the sensor surface measured by SPR, the calculated chain density, and chain-to-chain distance. SAM, self-assembled monolayer.
Our study demonstrates that the deletion of the mucin-like region affects the gC interaction with GAGs by decreasing the binding affinity between gC and sulfated GAGs and also by making the dissociation from sulfated GAGs slower. This indicates that glycosylation can play an important role in modulating the interaction between virions and the surface of host cells. This is further confirmed by cell data showing a decrease in sensitivity of mutant virus to GAG mimetic inhibitors and a reduced release of these viruses from the surface of infected cells.
Experimental Procedures
Materials
All materials, unless otherwise stated, were purchased from commercial sources. Oligo(ethylene glycol) (OEG) disulfides with terminal hydroxyl (dS-OEG; structure, -(S-CH2-(CH2-O-CH2)7-CH2-OH)2; molecular weight, 771.0 Da) and biotin groups (dS-OEG-biotin; structure, -(S-C2H4-CO-NH-(CH2-O-CH2)9-NH-CO-C4H8-biotin)2; molecular weight, 1539.9 Da) were purchased from Polypure (Oslo, Norway). Streptavidin was purchased from Sigma. Chondroitin sulfate (CS) was purchased from Kraeber (Ellerbek, Germany), and hyaluronan (HA) was purchased from Aqua Biochem (Dessau, Germany). Neuraminidase type V from Clostridium perfringens (N2876) was purchased from Sigma. The GAG-mimetic oligosaccharide PI-88 was prepared as described previously (20) and obtained from Progen (Brisbane, Australia). Heparin was obtained from Medicarb (Stockholm, Sweden). Monoclonal antibodies B1C1, C2H12, and C4H11, specific for HSV-1 gC, were prepared as described previously (21). PKH26 red fluorescent cell linker was purchased from Sigma-Aldrich, and illustra MicrospinTM columns were from GE Healthcare. Lipids were obtained from Avanti Polar Lipids (Alabaster, AL). PBS buffer at pH 7.4 (137 mm NaCl, 2.7 mm KCl, 10 mm phosphate buffer) was purchased as tablets from Sigma. Water was deionized (resistivity >18.2 megaohms/cm) and filtered using a Milli-Q system (Millipore). All buffers were filtered and degassed before use.
Cells and Viruses
African green monkey kidney (GMK AH1) cells (22) were cultivated in Eagle's minimum essential medium supplemented with 2% fetal calf serum, 0.05% Primaton RL substance (Kraft Inc., Norwich, CT), 100 units/ml penicillin, and 100 μg/ml streptomycin. The virus strain used was HSV-1 KOS (ATCC, VR- 1493) (23). A variant of HSV-1 KOS strain deficient in expression of gC (KOS-gCdef) due to a frameshift-inducing mutation (deletion of cytosine at position 366) was also used.
Preparation of HSV-1 Variants Lacking the Mucin-like Domain in gC; Purification of Viruses and gC
HSV-1 KOS variants resistant to GAG-mimetic PI-88 due to deletion of amino acids 33–116 of gC (i.e. a fragment comprising an entire mucin-like region of this protein) were used. A full protocol of the selection of these variants has been described previously (12). Because these variants may, apart from a deletion in gC, possess mutations in other viral proteins, a PCR-amplified fragment encompassing nucleotides −152 to 1659 of gC of these mutant viruses was transfected, along with DNA purified from KOS-gCdef, into GMK AH1 cells, using the marker transfer procedure described previously (12). The resulting viral variant (KOS-gCΔmuc) possessed a designed deletion in the gC in a background similar to the native KOS strain. The reactivity of the two virus strains with the monoclonal anti-gC antibodies B1C1, C2H12, and C4H11 was studied by the ELISA-based method performed on the surface of infected cells as described (24). Methyl-[3H]thymidine-labeled extracellular HSV-1 particles were purified by centrifugation through a three-step discontinuous sucrose gradient as described previously (25).
Native gC and gC lacking the mucin-like domain (gCΔmuc) were isolated from lysates of extracellular virus particles and virus-infected cells by immunoaffinity chromatography as described previously (25). Glycoproteins were aliquoted in deionized water, stored at −80 °C, and dissolved in PBS prior to measurements. Treatment of gC with neuraminidase was performed by incubation of purified protein in acetate buffer, pH 6.5 (50 mm sodium acetate/acetic acid, 154 mm NaCl, 9 mm CaCl2), with broad-spectrum neuraminidase (1 milliunit/μg of protein) for 2 h at 37 °C.
Viral Assays
The effect of PI-88 and heparin on infectivity of HSV-1 was tested by the viral plaque number reduction assay as described previously (13).
The yield of infectious virus in extracellular medium and in infected cells was analyzed by the one-step growth-based assay as follows. GMK AH1 cells were infected with KOS or KOS-gCΔmuc at a multiplicity of infection (MOI) of 3. Following a virus adsorption period for 90 min at 37 °C, the cells were rinsed three times with Eagle's minimum essential medium and further incubated in the same medium at 37 °C. At specific time points counting from the end of the virus attachment period, infectious culture medium and infected cells were harvested to determine the amount of infectious virus by a plaque titration assay. Infected cells were harvested by scraping into fresh supernatant medium and then subjected to a rapid freeze-thaw cycle at −80 °C ethanol and a 37 °C water bath, respectively, to release infectious virus. To study the effect of PI-88 on liberation of viral particles from the cell surface, GMK AH1 cells were infected with virus at an MOI of 3, followed by incubation in PI-88-containing medium (20 μg/ml) for 20 h.
Treatment of HSV-1 with neuraminidase was performed as follows. HSV-1 virions that spontaneously released from infected cells into supernatant culture medium were pelleted by centrifugation at 20,000 × g for 90 min. The pelleted virions were gently resuspended in acetate buffer, pH 6.5, comprising 250 milliunits of neuraminidase. Following incubation for 1 h at 37 °C, the virions were pelleted by centrifugation 20,000 × g for 90 min and gently resuspended in PBS.
Electron microscopy of cells infected with HSV-1 was carried out as described previously (26). Briefly, GMK AH1 cells were propagated on a Melinex polyester insert (Agar Scientific Ltd., Stansted, UK) to avoid any scraping or pelleting of cells during the sample preparation. The cells were infected with native KOS or KOS-gCΔmuc at an MOI of 3, and at 20 h postinfection, they were fixed with 2.5% glutaraldehyde for 1 h at 37 °C. Subsequently, the cells were triple-rinsed with 0.05 m Tris-HCl buffer (pH 7.4) supplemented with 2 mm CaCl2 and processed for electron microscopy as described by Widéhn and Kindblom (27).
Quantification of Viral DNA
Extraction and quantification of HSV-1 DNA has been described previously (28). Briefly, viral DNA from infected cells and infectious supernatant medium was extracted with the MagNA Pure LC DNA Isolation Kit I (Roche Diagnostics, Mannheim, Germany). HSV-1 DNA quantification was performed through real-time quantitative PCR by detecting and amplifying a 118-nucleotide segment of the highly conserved region on HSV-1 glycoprotein B with a pair of primers and a gB-specific probe. A specific cycle threshold (Ct) value was obtained for each sample, and Ct values were then related to a standard curve with known concentrations of DNA copies.
Glycosaminoglycan Derivatives
End-on biotinylated GAGs hyaluronan (b-HA, 23 kDa), chondroitin sulfate (b-CS, 20 kDa), and sulfated hyaluronan (b-sHA, 30 kDa) were provided from INNOVENT e.V. (Jena, Germany). The molecular weight determination, synthetic sulfation, and biotinylation procedure of these derivatives have been described previously (16). Briefly, b-CS (mixture of 70% chondroitin-4-sulfate and 30% chondroitin-6-sulfate) has an average natural degree of sulfation (DSsulfate) of 0.9, and b-sHA has a synthetically induced DSsulfate of 3.1, indicating that on average, every repeating disaccharide unit carries one or three sulfate groups, respectively. b-HA carries no sulfate groups.
Sensor Functionalization
Biotinylation of SPR sensor chips was performed as described previously (16, 29). Briefly, gold-coated sensor chips (SIA Kit Au, GE Healthcare, Uppsala, Sweden) were first cleaned in a 5:1:1 solution of water, 25% ammonia, and 30% hydrogen peroxide for 10 min at 80 °C. After rinsing with water, the sensor chips were dried under a flow of nitrogen and placed in a 0.5 mm ethanol solution of dS-OEG (99 mol %) and dS-OEG-biotin (1 mol %) for incubation for >12 h to obtain a well ordered self-assembled monolayer. After incubation, the sensor chips were ultrasonicated for 3 min in ethanol to remove any loosely bound disulfides before mounting the sensor chips in the instrument.
SPR-based Interaction Experiments
Interactions between surface-bound GAGs and HSV-1 gCs were monitored in flow mode and real time in experiments based on surface plasmon resonance (SPR) detection using a Biacore 2000 instrument (GE Healthcare). All experiments were performed at 22 °C, using PBS as running buffer at a flow rate of 5 or 20 μl/min, for equilibrium and kinetic binding curves, respectively. Biotin-functionalized sensor chips were used, and a baseline was established in PBS. Streptavidin was introduced at 25 μg/ml, followed by rinsing in PBS. The surface was functionalized with end-on biotinylated GAGs: b-CS (0.5 mg/ml), b-HA, or b-sHA (0.1 mg/ml). b-CS and b-sHA were injected until saturation, whereas b-HA was injected to match the surface coverage of b-CS. If not stated otherwise, gC-binding experiments were performed using an injection volume of 325 μl. The measurement was carried out under a constant protein flow until the signal stabilized because equilibrium was reached. The signal was considered stable when the ratio of the slope at equilibrium was <5% of the initial slope (slopes were measured over a time period of 10 min). The protein was used directly after thawing and was not refrozen.
Inhibition experiments with IgG monoclonal antibody B1C1 were performed by mixing the antibody and the gC at a molar ratio MB1C1/MgC = 6 (assuming a molecular weight of 160 kDa for B1C1) for at least 5 min before injecting it over the GAG-presenting surface. To estimate the electrostatic contribution to the gC-GAG binding, experiments were also carried out in high ionic strength PBS buffer containing 262 mm NaCl, and in HEPES buffer containing 3 mm Ca2+. To account for changes in protein activity, these experiments were compared with binding under normal buffer conditions using the same protein aliquot. The adsorbed mass on the sensor surface was estimated from the SPR signal as described previously (16, 30). For b-HA and the sulfated b-CS and b-sHA, a refractive index increment, dn/dc, of 0.147 ml/g (31) and 0.160 ml/g (32–34) was used, respectively. dn/dc values for native gC and gCΔmuc were calculated using a linear combination of dn/dc values for the protein and glycan part of the glycoprotein, weighted according to the respective molecular weights (35), and were found to be 0.162 and 0.170 ml/g, respectively.
To obtain information on binding affinity, binding curves were obtained at low flow (5 μl/min) until equilibrium, at increasing concentrations with rinsing in between. Equilibrium binding levels were plotted against gC concentration, and the resulting curve was fitted to the Hill equation, which has previously been used for similar interaction systems (17, 36, 37),
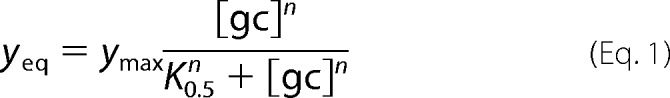 |
where yeq is the equilibrium binding at a certain gC concentration, [gC], ymax is the maximum binding (i.e. the saturation level), K0.5 equals [gC] at half-maximal binding, and n is an indicator for cooperativity (37). n > 1 indicates positive cooperativity, where the binding of one ligand increases the chance of a second ligand to bind. For n = 1, Langmuir conditions apply, and K0.5 = KD.
Total Internal Reflection Fluorescence (TIRF) Microscopy Experiments
Supported lipid bilayers made of 0.5 mg/ml 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine and 1 weight % 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) were formed on cleaned coverglasses by surface-induced vesicle rupture. The liposomes were obtained by the hydration and extrusion method as described previously (38). This was followed by the sequential additions of 25 μg/ml streptavidin and biotinylated GAGs (0.5 mg/ml for b-CS and 0.1 mg/ml for b-HA) for at least 20 min. KOS and KOS-gCΔmuc viruses were fluorescently labeled with PKH26 red fluorescent cell linker and filtered for 1 min at 2000 × g. The concentrations of the virus suspensions were adjusted prior to labeling, according to viral DNA quantification. Concentration in viral DNA during a measurement was 1.3·109 copies/ml. Measurements were made about 60 min after the addition of virus to reach equilibrium. All incubation steps were performed in PBS buffer.
Time lapse movies (15-s time interval, 250 frames) were recorded with a Nikon Eclipse Ti-E inverted microscope using a ×60 magnification (numerical aperture = 1.49) oil immersion objective (Nikon Corp.). The microscope was equipped with an X-Cite 120 lamp (Lumen Dynamics Group Inc.), a TRITC filter cube (Nikon Corp.), and an Andor DU879E-CSBV camera (Andor Technology). The movies were analyzed with a MatLab script (MathWorks, Inc.), written in-house. Briefly, only virus particles of an intensity above a set threshold were detected. They were considered bound if present for >5 consecutive frames. The number of newly arrived vesicles over time was plotted and fitted linearly. The first 30 frames were excluded from the fit.
Results
Biological Activities of HSV-1 Mutant Lacking the Mucin-like Region on gC
In this study, we sought to investigate whether the mucin-like region on gC of HSV-1 plays a role in the interaction between gC and GAGs. To this end, we prepared a mutant of the HSV-1 KOS strain that lacked an entire mucin-like domain but contained the GAG-binding site on gC (KOS-gCΔmuc) (Fig. 1a). To verify that the deletion of the mucin-like region did not induce gross conformational alterations to the rest of the protein, the reactivity of the mutant virus with three different anti-gC monoclonal antibodies targeting different sites (i.e. B1C1, C2H12, and C4H11) was tested in an ELISA-based assay. The reactivity of KOS-gCΔmuc with the antibodies was similar to that of native KOS strain (data not shown), thus excluding the possibility of major conformational alterations in the structure of truncated gC. Specific infectivity (i.e. the number of viral particles (DNA copy equivalents) per infectious unit in preparations of purified virus) was 9.9 for KOS and 16.5 for KOS-gCΔmuc. This indicates that the defect in expression of the mucin-like domain in gC modestly affected the virus infectivity because ∼1.7 times more viral particles of the mutant virus than native KOS strain was required to infect the cells.
The effects of heparin and sulfated oligosaccharide PI-88 (a GAG mimetic) on the infection of GMK-AH1 cells by KOS and KOS-gCΔmuc are shown in Fig. 2, a and b, respectively. The viruses were mixed with the inhibitors and incubated for 10 min at room temperature prior to the infection of cells. Approximately 3 times higher concentrations of heparin and PI-88 were needed to inhibit infectivity of KOS-gCΔmuc by 50%, as compared with the native KOS strain. Furthermore, ∼15–40% of KOS-gCΔmuc remained non-neutralized even at the highest heparin/PI-88 concentration tested. To investigate whether sialic acid, a terminal sugar residue on different glycoconjugates, including clustered O-linked glycans of the mucin-like domain of HSV-1 gC, could interfere with the virus-GAG interaction, we treated KOS and KOS-gCΔmuc virions with broad-spectrum neuraminidase prior to their assessment for heparin sensitivity. The neuraminidase-treated viruses exhibited a slightly altered pattern of sensitivity to heparin as compared with mock-treated HSV-1 (Fig. 2c). These data indicate that the HSV-1 mutant lacking the mucin-like domain in gC was less sensitive to GAG-based inhibitors of the virus attachment to cells and that removal of sialic acid from HSV-1 KOS strain slightly decreased its sensitivity to heparin. This possibly reflects a decreased or altered binding of these inhibitors to the virus particle.
FIGURE 2.
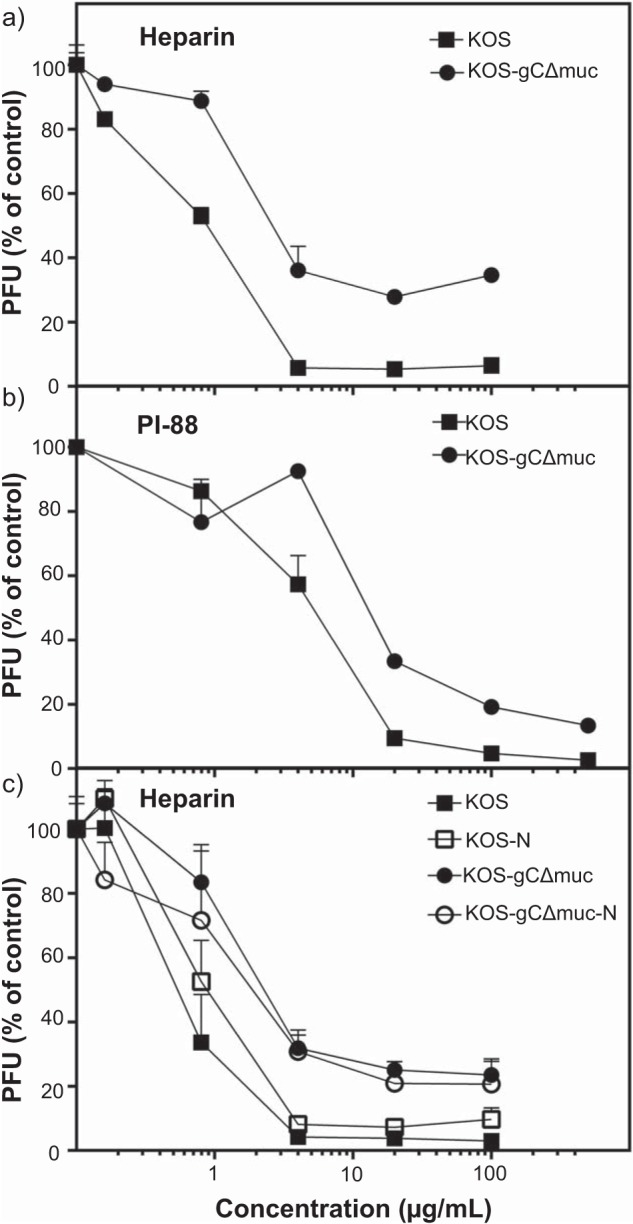
The mucin-like domain of gC of HSV-1 influences the virus sensitivity to sulfated oligosaccharides. The HSV-1 mutant lacking the mucin-like domain in gC (KOS-gCΔmuc) and native KOS strain were tested for sensitivity to the sulfated oligosaccharides of heparin (a) and PI-88 (b) by the viral plaque-forming unit (PFU) reduction assay. c, the neuraminidase-treated viruses (KOS-N and KOS-gCΔmuc-N) as well as mock-treated viruses were tested for their sensitivity to heparin. For each assay, two separate experiments were performed, and the data are expressed as the percentage of the number of viral plaques found in the presence of the test compounds, relative to those developed in mock-treated infected cells. Error bars, S.D.
We also assayed the yield of infectious virus produced by GMK AH1 cells following their infection with KOS and KOS-gCΔmuc. The cells were infected at an MOI of 3, and the amount of infectious virus retained by infected cells (cell-associated virus) and released from cells into culture medium (extracellular virus) was determined by a plaque assay. Because infected cells retain infectious virus in the cytoplasm and on the cell plasma membrane, these cells were subjected to a quick freeze-thaw cycle to liberate viral particles prior to the titer determination. Depending on the time after infection of cells, the KOS-gCΔmuc mutant produced ∼2–5 times less cell-associated virus than the native KOS strain (Fig. 3a). However, the amount of extracellular virus of KOS-gCΔmuc was significantly lower (∼20–600 times) than that of native KOS (Fig. 3b). One interpretation of these differences is that most of the newly produced virions of KOS-gCΔmuc virus are trapped by GAG chains of proteoglycans at the surface of infected cells. To verify this possibility, we investigated the virus release from infected cells in the presence of PI-88, which might help to liberate the viral particles from the cells surface. To this end, GMK AH1 cells were infected with the virus at an MOI of 3 and then incubated in a medium comprising PI-88 (20 μg/ml) for an ∼20-h period of virus production and release from infected cells. Despite the fact that PI-88 was suggested in a previous experiment to show higher affinity for the native KOS strain than for KOS-gCΔmuc (Fig. 2b), this compound enhanced the amount of extracellular virus of KOS strain by only ∼75%, whereas it enhanced the amount of KOS-gCΔmuc by ∼420% (Fig. 3c). To assess the amount of viral particles present on the cell surface, we analyzed the virus-infected cells by electron microscopy. GMK AH1 cells, growing on Melinex polyester inserts to avoid scraping and pelleting, were infected at an MOI of 3 and examined by electron microscopy 20 h after infection (Fig. 4). As expected, the viral particles were present on the surface of cells infected with both native KOS (Fig. 4a) and KOS-gCΔmuc (Fig. 4b). We counted the number of viral particles present on the surface of 15 cells. The average number of viral particles/cell was 68 ± 60 for native KOS and 118 ± 58 for KOS-gCΔmuc. This indicates that although the KOS-gCΔmuc produced ∼2 times less infectious virus in infected cells than native KOS strain after 20 h of infection (Fig. 3a), there were ∼2 times more viral particles of mutant virus retained on the cell surface. Altogether, these data suggest that the absence of the mucin-like region in the HSV-1 attachment component gC alters the sensitivity of viral particles to GAG-based inhibitors of virus attachment to cells and may lead to trapping of newly produced virions on the surface of infected cells.
FIGURE 3.
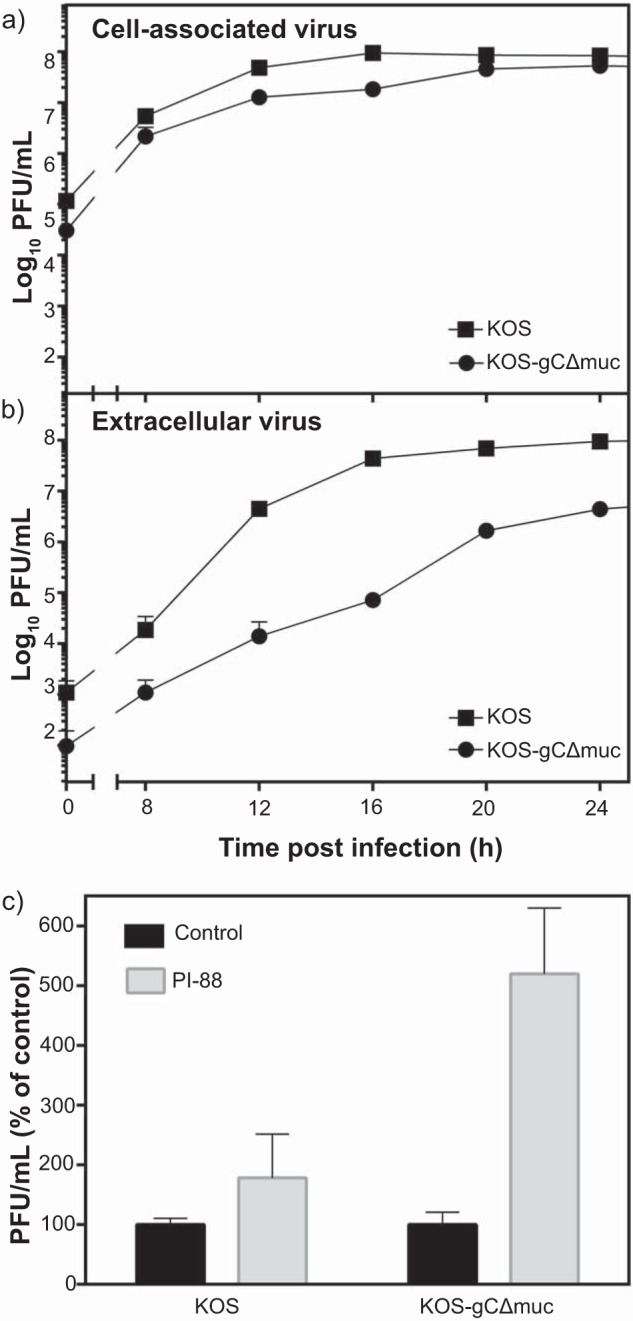
The mucin-like region of gC of HSV-1 promotes release of infectious virus from infected cells. GMK AH1 cells were inoculated with the native HSV-1 KOS strain or the mutant virus lacking the mucin-like region in gC (KOS-gCΔmuc) at a multiplicity of infection of 3. The amounts of virus in infected cells (a) and infectious culture medium (b) at specific time points after the end of the inoculation period were determined. The data are means of two separate determinations and are expressed as the titers of infectious virus determined by the viral plaque (PFU) assay. c, the GMK AH1 cells were inoculated with the virus for 3 h and then incubated with PI-88 (20 μg/ml) or without this compound for 18 h. The data are means of four determinations from two separate experiments and are expressed as a percentage of infectious titer found in PI-88-treated cells as related to mock-treated controls. Error bars, S.D.
FIGURE 4.
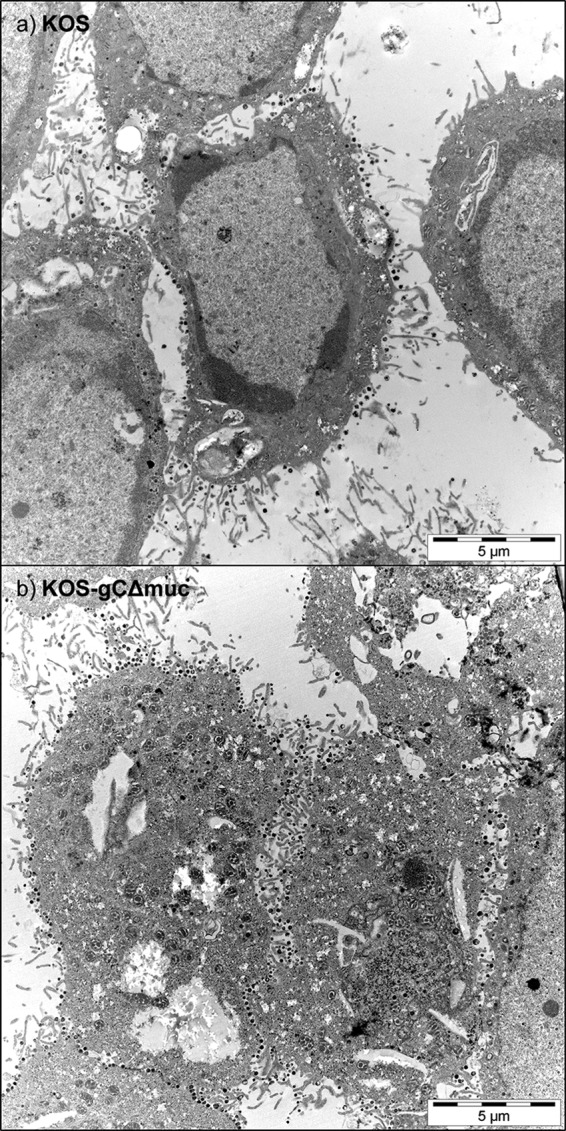
The mucin-like domain of HSV-1 gC promotes release of viral particles from the surface of infected cells. GMK-AH1 cells were infected with native KOS strain (a) or KOS-gCΔmuc (b) and processed for electron microscopy 20 h after infection. The viral particles at the cell plasma membrane are seen as black dots. For each virus, at least 15 images were captured and assessed independently by two investigators.
Surface-based Assay to Characterize gC-GAG Interactions
To further explain the data obtained with whole virus particles (Figs. 2–4), gC glycoproteins were isolated from KOS and KOS-gCΔmuc virions and characterized by electrophoresis on BisTris gels (Fig. 1b). As expected, native gC exhibited a higher apparent molecular mass (112 kDa) than KOS-gCΔmuc, where deletion of the mucin-like domain (gCΔmuc) decreased its mass to ∼64 kDa. gC-GAG binding studies were performed by SPR-based sensing, using GAGs that were biotinylated at the reducing end, enabling an end-on configuration when bound to streptavidin on the sensor surface (Fig. 1, c and d). In this study, non-sulfated hyaluronan (b-HA), synthetically sulfated hyaluronan (b-sHA), and naturally sulfated chondroitin sulfate (b-CS) were used. CS is naturally occurring on the surface of many cell types and known to be a receptor for HSV-1 attachment (21). Synthetically sulfated b-sHA is included to study the effect of sulfation as such, because protein binding to GAGs is predominately mediated through sulfate groups. b-sHA is evenly sulfated along the chain and carries ∼3 times more sulfate groups than b-CS (DSsulfate = 3.1 and DSsulfate = 0.9, respectively). The masses of b-sHA (18 ± 3 ng/cm2) and b-CS (7 ± 2 ng/cm2) immobilized on the sensor surface are in good agreement with the ones reported previously (16). The surface coverage of the non-sulfated b-HA was tuned to reach a GAG density comparable with that of b-CS (Fig. 1d). Our study revealed that both native gC and gCΔmuc bound to sulfated GAGs (i.e. b-sHA and b-CS) (Fig. 5), indicating that the presence of the mucin-like domain in viral gC is not a prerequisite for binding to GAGs. As expected, both these proteins bound poorly to the non-sulfated b-HA (<10 and <15% of the specific signal for native gC and gCΔmuc, respectively), thus confirming the requirement of sulfated groups for the gC-GAG interaction.
FIGURE 5.
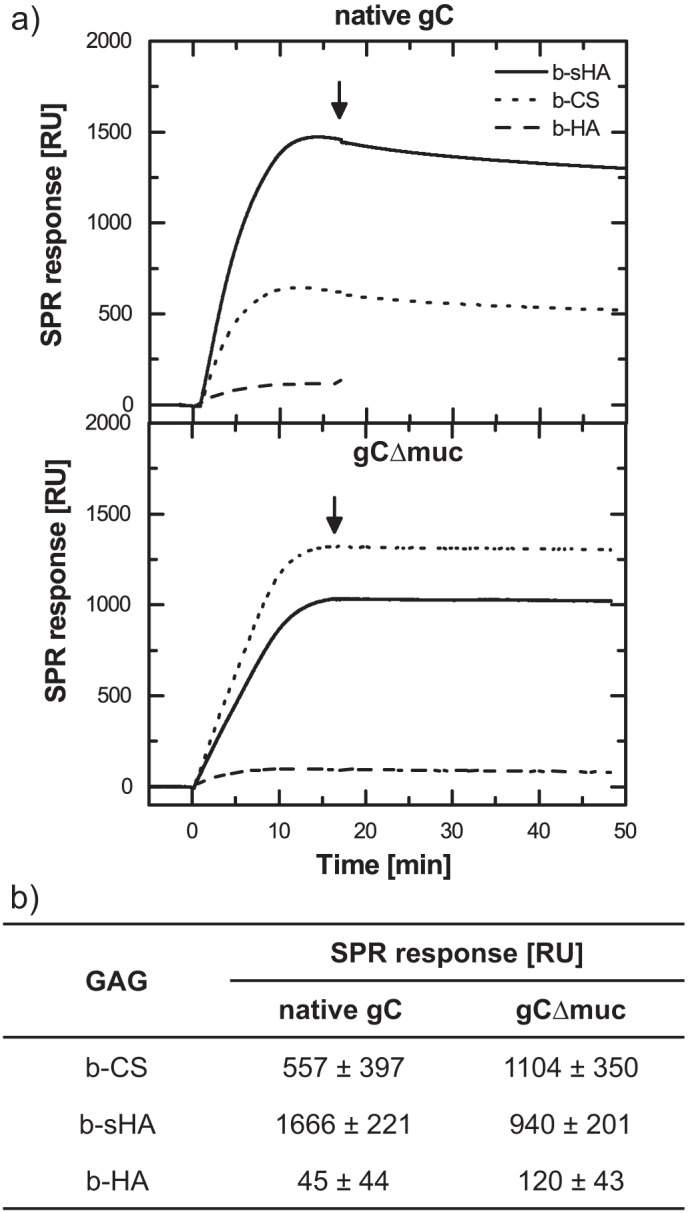
Binding of gC to surface-immobilized GAGs as measured with SPR. a, real-time SPR response showing binding of native gC and gC lacking the mucin-like region (gCΔmuc) to surface-immobilized b-CS (dotted line), b-HA (dashed line), and b-sHA (solid line). The protein concentration was 11 nm, and onset of rinsing is marked with an arrow. b, mean and S.D. values for at least three measurements.
Nature of the gC-GAG Interaction
To gain further insights into the nature of the gC-GAG interaction and into the contribution of electrostatic interactions, inhibition experiments were performed. A competitive assay was carried out using the gC-reactive antibody B1C1, previously shown to inhibit HSV-1 attachment to CS-expressing cells (21). This antibody recognizes a gC region that partly or entirely overlaps with the CS- and heparan sulfate (HS)-binding site of gC (Fig. 1a) (21, 24, 39, 40). The antibody B1C1 was premixed with native gC or with gCΔmuc, and the resulting interference with GAG binding was assessed by SPR-based sensing (Fig. 6, solid and dotted lines). Binding was efficiently reduced for both native gC (to b-CS, 89 ± 4%; to b-sHA, 88 ± 10%) and gCΔmuc (to b-CS, 97 ± 2%; to b-sHA, 91 ± 4%). Therefore, our results strongly suggest that the interaction of GAGs with both native gC and gCΔmuc is specific for the B1C1-/GAG-binding site. It is known that electrostatic interactions play an important role in the binding of proteins to GAGs. This is also true for gC, where the GAG-binding domain comprises clusters of positively charged amino acid residues (24, 39, 40) that can bind to negatively charged sulfate/carboxyl groups on the GAG chain. To investigate the contribution of electrostatic interactions, PBS in which the concentration of NaCl was increased to 262 mm was used, and the binding levels at saturation were compared with the ones measured in PBS with isotonic ionic strength (Fig. 6a, solid and dashed lines). Binding was reduced by 52 ± 10 and 52 ± 24% when native gC bound to b-CS and b-sHA, respectively, and by 51 ± 24 and 4 ± 8% for the equivalent binding of gCΔmuc. The presence of Ca2+ did not significantly affect the gC-GAG interaction, because no dependence on the presence of Ca2+ was found for native gC interacting with surface-immobilized GAGs (data not shown).
FIGURE 6.
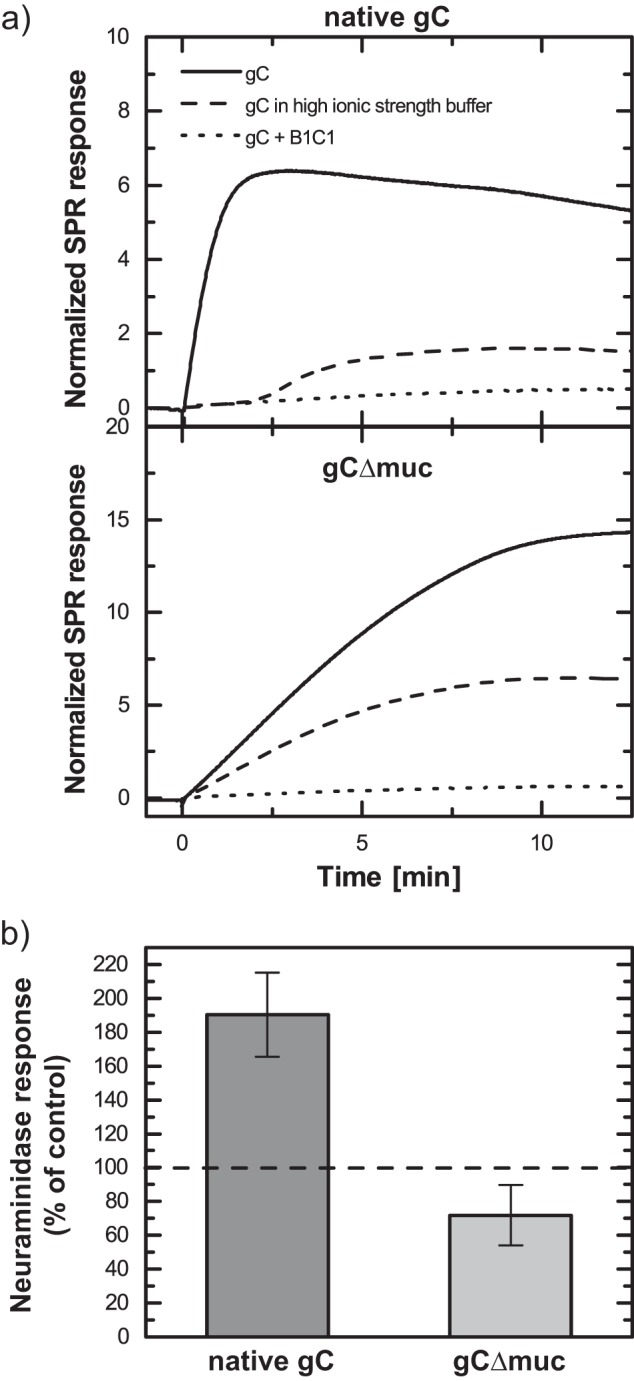
Binding of gC to surface-immobilized GAGs is affected by a specific anti-gC monoclonal antibody, by increased ionic strength, and by neuraminidase treatment. a, binding of native gC and gCΔmuc were compared with binding when the anti-gC monoclonal antibody B1C1 was included (dotted lines) and when the ionic strength was raised to 0.26 m NaCl (dashed lines). Here, binding is shown to b-CS, and SPR response is normalized to the amount of b-CS on the surface. b, binding to b-CS of gC treated with neuraminidase was compared with mock-treated gC. The graph represents mean and S.D. (error bars) values of three measurements, and the data are expressed as the percentage of binding for neuraminidase-treated versus mock-treated gC.
To gain further insights into the modulating factors, GAG-binding experiments were performed using gCs treated with neuraminidase. The binding properties were significantly affected by the desialylation process; for KOS-gC, binding was increased by a factor of ∼2 compared with mock-treated gCs. For KOS-gCΔmuc, on the other hand, treatment with neuraminidase led to a slight decrease (∼25%) in binding. (Fig. 6b).
Affinity Determination
To determine the binding affinity of native gC and gCΔmuc to GAGs, equilibrium-binding curves were generated by plotting the SPR signal at binding equilibrium against the gC concentration in solution. To account for differences in the amount of GAGs immobilized on the surface and hence to provide a more direct comparison of the binding to the different GAGs, the SPR response for the gC binding was normalized to the SPR response for the GAG immobilization. The curves were fitted to Equation 1 (Fig. 7, a and b), to gain both quantitative and qualitative information about the binding behavior of the different systems at equilibrium (Fig. 7c).
FIGURE 7.

Equilibrium binding characteristics for the binding of gC proteins to surface-immobilized GAGs. a, representative equilibrium binding curves (symbols) fitted to the Hill equation (line) for native gC (left) and gCΔmuc (right) to b-CS, b-sHA, and b-HA. The binding is normalized to the SPR response for each respective GAG on the surface. b, parameters obtained from fitting the equilibrium binding curves in a and b to the Hill equation. K0.5 = [gC] at half-maximal binding. For n = 1, K0.5 = KD. The maximal binding is normalized to the amount of GAG on the surface and used for calculating the number of gCs bound per GAG chain at saturation. Mean and S.D. values of 2–4 measurements are shown.
The factor n in Equation 1 can be used to estimate the extent of cooperativity. n > 1 indicates positive cooperativity, where the binding of one ligand will facilitate the binding of the next ligand. No cooperativity was found for binding of native gC to b-CS, as n = 0.9 ± 0.5. For gCΔmuc, the average n values were between 1.5 and 2, which is significantly less than the large stoichiometric value (∼15), indicating a weak positive cooperative behavior.
Moreover, quantitative insights into binding affinities between the molecules can be gained through the factor K0.5, given by the gC concentration at half-maximal binding in the equilibrium binding curves. For binding events that follow the Langmuir equation (n = 1), K0.5 = KD. For binding of native gC to b-CS, the measured KD (as n = 1 in this case) was 5.3 ± 0.6 nm. The apparent equilibrium dissociation constant (K0.5) for the binding of gCΔmuc to the same GAG was about 10 times higher (47.7 ± 11.2), indicating lower affinity. No significant differences between the naturally sulfated b-CS and the artificially sulfated b-sHA were seen for either of the gCs.
Equation 1 can further be used to estimate the saturation limit (i.e. the maximum binding (ymax) of gCs that can be reached). To account for the difference in molecular weight between native gC and gCΔmuc, the number of bound gCs per GAG chain was also calculated (Fig. 7c). Native gC bound to the sulfated GAGs in an approximate 1:1 stoichiometry, whereas ∼15 copies of gCΔmuc bound to each GAG chain. Again, for each gC, no significant difference in stoichiometry could be seen between the two sulfated GAGs.
Kinetic Behavior
To further investigate the binding behavior of the two gCs to sulfated GAGs, we also investigated binding kinetics by real-time monitoring of gC binding and subsequent dissociation upon rinsing (Fig. 5). The dissociation behavior of native gC from b-CS is further emphasized in Fig. 8. Dissociation was only detectable for native gC, whereas binding of gCΔmuc appeared essentially irreversible during the time frame of the experiments. Generally, the dissociation rate constant koff can be directly determined by fitting the rinsing curve with an exponentially decaying function. For data regarding native gC, the dissociation was best fitted to a double exponential equation according to Equation 2,
where R2 = 0.999. Corresponding values for fits to a single exponential equation and single exponential equation with an irreversible fraction were 0.919 and 0.988, respectively (fits not shown).
FIGURE 8.
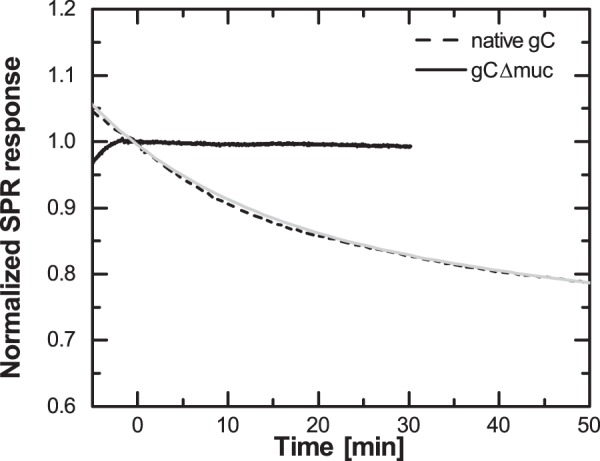
Native gC and gCΔmuc differ in their dissociation from surface-immobilized GAGs. Normalized dissociation of gCΔmuc (solid curve) and native gC (dashed curve) from b-CS. The dashed curve was fitted to Equation 2; y = 0.15·e−0.06t + 0.84·e−0.0015t (light gray).
Such a dissociation behavior indicates that the sample is heterogeneous and contains at least two, but most likely more, fractions with distinct dissociation rate constants. The fitted values of koff1 and koff2 from Equation 2 therefore have little meaning (38). Generally, for a Langmuir binding, the association rate constant kon can be determined using the relation KD = koff/kon. Although more complex binding models are needed in our case, the higher affinity of native gC, together with the apparent higher koff, suggests that kon is higher for native gC than for gCΔmuc. These results indicate that gCΔmuc has less of a propensity to bind to GAGs compared with native type gC but that once formed, the gCΔmuc-GAG complex is more stable.
Association of Virus Particles with Glycosaminoglycans
Although SPR experiments make it possible to quantify the dissociation rate constant of the protein, the determination of kon becomes cumbersome in view of the complex binding kinetics observed here. To gain further insights into the association behavior of whole virus particles with GAGs, we therefore complemented our data with TIRF microscopy experiments and analyzed the rate of arrival of individual fluorescently labeled viruses (Fig. 9a) under equilibrium conditions. As expected, both virus strains bound poorly to the non-sulfated b-HA (∼5 and ∼25% of the binding to b-CS for KOS and KOS-gCΔmuc, respectively). Provided that binding is reaction-limited, this technique allows for an independent determination of kon (as further detailed in Ref. 38). The slope of the association curve (Fig. 9b) is directly proportional to kon, the number of ligands on the surface, and to the number of particles in solution, making it possible to qualitatively compare the association behavior of KOS and KOS-gCΔmuc. In agreement with our data on solubilized protein, this experiment suggests that KOS-gCΔmuc has less of a propensity to bind compared to the wild type KOS: the association slope for KOS-gCΔmuc was indeed 15.3% ± 1.7% of the association slope of KOS.
FIGURE 9.
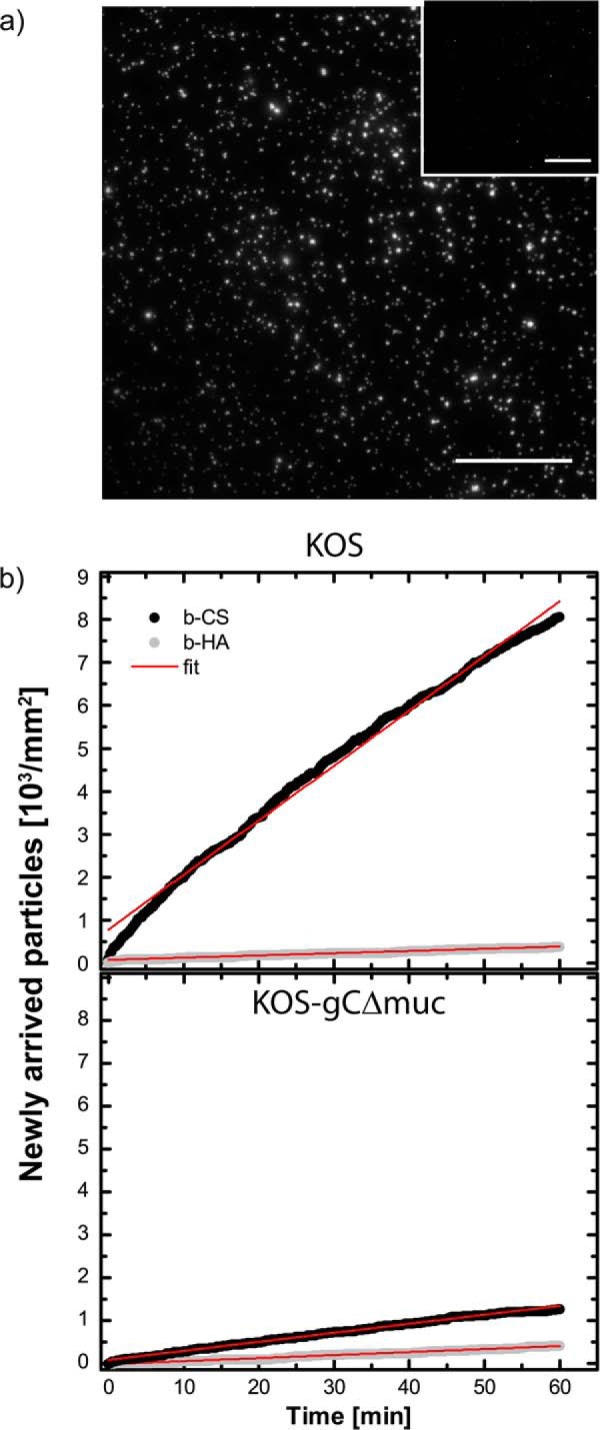
HSV-1 association with surface-bound GAGs investigated by total internal reflection fluorescence microscopy. a, TIRF images showing fluorescently labeled KOS viruses bound to b-CS and b-HA (inset). Scale bar, 50 μm. b, the number of newly arrived virus particles over time was assessed under equilibrium conditions for KOS and KOS-gCΔmuc to b-CS (black dots) and b-HA (gray dots). The data were fitted linearly (red lines).
Discussion
We have found that the HSV-1 mutant deficient in the expression of a mucin-like region of the attachment protein gC, exhibited two phenotypic traits: (i) a decreased virus sensitivity to GAG-based inhibitors of virus attachment to cells and (ii) a reduced release of newly produced viral particles from infected cells, most likely due to their trapping by GAGs at the cell surface. It has been recently reported (41) that HSV-1 enhances expression of cellular heparanase-1 to promote release of HSV-1 particles from the surface of infected cells. This finding was based inter alia on the observation that the knockdown of the heparanase-1 gene with shRNA decreased the release of HSV-1 particles from infected cells by ∼2–5-fold. We found that the deletion of the mucin-like domain from the viral attachment protein gC conferred a more pronounced defect in the virus release than the heparanase-1 knockdown. The quantity of KOS-gCΔmuc that spontaneously released from infected cells was usually reduced by >20-fold as compared with KOS.
To gain an in depth understanding of the molecular origin of the functional alterations conferred by the absence of a mucin domain, we purified gC from the native KOS and from the mutant variant KOS-gCΔmuc and investigated the interactions of the different gC variants with GAGs. To this end, SPR-based biosensing and a recently presented sensing platform designed to study interactions between GAGs and biomolecules were employed (16–18, 42). Our variant of the platform is based on a biotinylated self-assembled OEG monolayer with immobilized streptavidin. Such surfaces are commonly used to construct sensors for biomolecular interaction studies and have been shown to exhibit good non-fouling properties (29). The GAG chains were immobilized via their reducing end, a presentation strategy that mimics the attachment of GAGs to proteoglycans on the cell surface (42). In our study, the GAG chains were 40–60 nm long, which correlates well with those found on proteoglycans (43). They were immobilized with a chain-to-chain distance of roughly 20 nm, leaving enough space for the gC protein to bind (the length of gC has previously been estimated to be ∼24 nm (44), whereas our dynamic light scattering measurements suggest diameters on the order of 20 nm for gC and 12 nm for gCΔmuc (data not shown)). We found that both native gC and gCΔmuc bound to sulfated GAGs, indicating that the mucin-like region of gC is not critical for the binding to GAGs. Similar findings were reported by others (45, 46). GAG binding of gCΔmuc was reduced by preincubation with an anti-gC antibody, whose epitope overlaps with the GAG recognition site on the glycoprotein. This indicated that the binding to GAGs of gCΔmuc is specific to the GAG-binding domain. Interestingly, we found that for all gC-GAG combinations, except the binding of gCΔmuc to b-sHA, binding was reduced by increased ionic strength buffer. These results indicate that under the conditions of this experiment, electrostatic interactions play an important role in promoting association between the native gC and GAG molecules. The lack of the mucin-like domain could, however, potentially decrease the sensitivity of the gC to hypertonic NaCl when binding to some GAGs, suggesting an altered contribution of electrostatics versus other types of forces (e.g. hydrogen bonding and hydrophobic interactions). A previous study compared the effect of increased ionic strength on the binding of gC from HSV-1 and gC from HSV-2 to HS GAG chains. gC from HSV-2 naturally lacks the mucin-like region and was shown to be more resistant to increased ionic strength than the binding of gC from HSV-1 gC (25). This supports the hypothesis that mucin-like regions on viral proteins may promote electrostatic interactions. To gain further insights into the mechanisms modulating the gC-GAG interaction, the importance of divalent ions (Ca2+) and the presence of sialic acid on gC were assessed. Divalent ions are known to interact with GAGs and could influence the interaction by bridging between GAGs and proteins. Sialic acid could, on the other hand, increase the repulsion by adding negative charges to the glycoproteins. Interestingly, a combination of heavily sialylated mucin-like domain with the positively charged GAG-binding site as in gC of wild-type HSV-1 seems to prevent nonspecific interactions due to repulsion between sialylated mucins and negative charges at the cell surface and to sustain the specific affinity for GAG chains due to capability of the GAG-binding site to overcome the repulsive forces of sialylated mucins. Our results suggest that divalent ions did not significantly affect the gC-GAG interaction, whereas the removal of sialic acid from gC increased the interaction with the GAGs, suggesting that sialic acid can have a repulsive effect and hence play a role in modulating the interaction. However, desialylation of viral particles slightly decreased their GAG dependence, suggesting that these virions are more prone to nonspecific GAG-independent interaction. This effect was not observed with desialylated gC because this protein was tested in the binding assay where only GAG chains were present. The exact mechanisms leading to this modulation will be the subject of further investigation.
More importantly, we found that removing the mucin-like region from HSV-1 gC changed kinetic, affinity, and equilibrium characteristics of the gC-GAG interaction. Kinetics as well as equilibrium binding curves revealed that native gC exhibited a higher apparent affinity (K0.5 ≈ 5 nm) for the GAGs compared with gCΔmuc (K0.5 ≈ 50 nm). The dissociation rate of both gCs from the GAGs was low. In fact, for gCΔmuc, no apparent dissociation could be seen during the experiment. Characterization experiments performed with dynamic light scattering and nanoparticle tracking analysis further suggest that at the concentration used to acquire the data in Fig. 8, the percentage of protein likely to be found in an oligomerized state is well below 1% (data not shown). This excludes the possibility that the observed difference in dissociation behavior is an artifact due to protein aggregation.
Taken together, our data indicate that the mutant protein has less of a propensity to bind to GAGs compared with native gC but that the complex is more stable once formed. This interpretation is in good agreement with the additional observation that the binding of KOS virus particles to sensor-immobilized GAGs has a higher kon than the binding of KOS-gCΔmuc particles. These findings are also qualitatively in good agreement with observations by Rux et al. (46), who have studied an HSV-1 gC mutant protein carrying a similar deletion (Δ33–123) as the gCΔmuc (Δ33–116) studied here. Indeed, their mutant protein exhibited a lower affinity compared with native gC for surface-immobilized heparin and HS. Also, the dissociation rate of native gC was higher compared with gCΔ33–123, an observation in line with our findings. It should, however, be noted that the affinities determined in our study were ∼10 times higher than those reported by Rux et al. (46), most likely due to the different GAGs used and to the fact that a full-length gC comprising the transmembrane region was used here as opposed to the other study. The transmembrane region was kept in order to preserve the native structure of the protein. The decreased affinity of gCΔmuc measured with SPR is also in line with the observed reduction in sensitivity to heparin and PI-88 of the virus mutant carrying gCΔmuc. In addition to this, our data suggest that the mucin-like domain of gC may contribute to the reversibility of its binding to cell surface GAGs, a feature of importance in all dead-end virus-GAG interactions that lead to non-productive infection of cells and in the release of virus from infected cells. One consequence of a decreased dissociation of gCΔmuc from GAGs seems to be that a significant proportion of KOS-gCΔmuc virions remained trapped at the cell surface. Interestingly, other mutations leading to an overexpression of O-linked glycans in the mucin-like region have been shown to hinder the attachment of HSV-1 to CS-expressing gro2C cells, which further emphasizes the modulating function of a mucin-like region (47).
In addition to the affinity determination, the GAG-presenting platform together with the equilibrium binding curves enabled us to study the stoichiometry of the gC-GAG interaction. Here, a clear difference was seen between the two gCs, because the number of gC copies that bound to a single GAG chain was on average 1 for native gC, as suggested previously for gC binding to heparin/HS (46), whereas it was close to 15 for gCΔmuc. The CS chains used here were ∼43 nm long, consisting of 43 disaccharides (∼1 nm/disaccharide unit (48)), whereas the minimum gC-binding sequence on HS has been estimated to consist of 5–6 disaccharide units (49). Assuming a similar requirement as for HS, one CS chain in our study would contain ∼8 such binding sites for gC. The observation that only one copy of native gC bound to a single CS chain can most likely be related to increased steric hindrance due to the presence of the mucin-like region. Also, clusters of the O-linked glycans are thought to induce a straight, rodlike conformation of glycoproteins (44, 50). This is also the case for gC on HSV-1, where this region may extend the protein to ∼24 nm (44). On the other hand, a gC lacking this region is reduced in size, and more copies of such protein are likely to be accommodated on a single GAG chain. Using dynamic light scattering, we observed that both proteins had a small propensity to form aggregates with sizes between 60 and 100 nm in diameter. Our complementary nanoparticle tracking analysis data acquired at concentrations within the saturation plateau of Fig. 7 suggest, however, that aggregation in solution is negligible (data not shown). Nevertheless, GAG-induced protein-protein interactions cannot be excluded as a contributing factor for the high stoichiometric number measured here. In any case, the lack of a mucin-like region seems to increase protein binding of purified protein to GAG chains. Because viral particles contain numerous copies of gC, the presence of gCΔmuc may potentiate the multivalent attachment of viral particles to cells via multiple glycoproteins. Multivalent interactions are well known to play a key role in the context of virus binding and results in an increased avidity of the virus for the cell membrane. This feature could therefore further explain the reduced release of the KOS-gCΔmuc viral particles from the surface of infected cells.
The interaction of the protein with the surface-immobilized GAG is characterized by a complex binding behavior. This is in the first place reflected by the dissociation behavior, which could only be fitted with multiexponential functions. This is consistent with a heterogeneous binding behavior characterized by the presence of multiple protein fractions having different dissociation rate constants (koff). We speculate that the presence of a spectrum of dissociation rates directly reflect the presence of heterogeneities in the mucin-like region, a characteristic feature of enzyme-driven processes such as glycosylation. Accordingly, it is also highly likely that association is characterized by multiple kon values, although this is difficult to assess experimentally.
An additional observation further highlighting the complexity of the observed binding kinetics is that native gC, as opposed to the gCΔmuc, exhibits an overshoot kinetic behavior (Fig. 5a). Upon gC addition, binding to sulfated GAGs increased until a plateau value was reached and then decreased, although gC was continuously added. Overshoot phenomena have been reported in a variety of studies (51–54). In short, overshooting can occur for both single-protein and multiprotein samples. In samples containing multiple species that differ in affinity and diffusive behavior, the fast diffusing species binds first and is overcome by the slower, higher affinity species, present either in bulk or induced upon a structural change at the interface (52, 53). For this kind of overshoot to be apparent using label-free techniques, the species have to differ in molecular weight and/or the space they occupy. It is noteworthy that the mucin-like region of gC comprises numerous O-linked glycans that differ in the number of sugar residues. In most cases, they comprise a single GalNAc or GalNAc-Gal disaccharide that can be further modified with negatively charged sialic acid (8). One cannot exclude the possibility that these glycoforms of gC may differ somewhat in their affinity for GAGs, thus inducing an overshoot phenomenon.
By investigating the binding of gC to different GAGs (b-HA, b-CS, and b-sHA), not only the influence of the mucin-like region, but also the effect of GAG-sulfation on the binding process could be explored (55, 56). Very little binding of the gCs was seen to the non-sulfated GAG b-HA, confirming that sulfate groups are required for the interaction of viral attachment components with GAGs (49). However, for both gC variants, binding affinity and maximum binding level to b-CS and b-sHA were comparable, despite the fact that the b-sHA surface exhibited about an order of magnitude higher surface density of sulfate groups. This observation suggests that the mere increase of sulfate groups is not sufficient to promote a stronger attachment of the protein, but that the position of sulfation groups on the disaccharide or along the GAG chain is likely to play a critical role in promoting and modulating the gC binding behavior to sulfated GAGs. The investigation of the role of sulfation is, however, beyond the scope of the present work but will be the subject of further investigation.
In summary, our findings demonstrate that the mucin-like region of HSV-1 gC, although not critical for binding to GAGs per se, balances the gC-GAG interaction to, on the one hand, facilitate the attachment of viral particles to cells and, on the other hand, allow an efficient release of viral progeny from the surface of infected cells. This function of the mucin-like region, although non-enzymatic, recalls to some extent the balancing functions of sialidase in binding of influenza virus to sialic acid. It is not unlikely that the mucin-like region enhances the gC-GAG affinity by presenting the binding sites in a more outstretched and accessible conformation. Moreover, once the protein is bound, the presence of the negatively charged sialic acid residues on some O-glycans of the mucin-like region in close proximity to the GAG-binding site may, depending on the relative importance of hydrogen bond formation and entropic effects, induce repulsion of bound gC from the negative GAG chains, thus modulating the dissociation behavior of the GAG-gC complex.
Author Contributions
M. B., T. B., and E. T. conceived and coordinated this study. M. B. and N. A. designed the SPR study and performed all experiments on surface-immobilized glycosaminoglycans. C. E. and E. T. prepared and characterized virus mutants, designed and performed all cell-based experiments, and performed DNA quantification. N. P. and N. A. designed the TIRF experiments and evaluated the data. N. A. and S. S. developed the GAG surface modification strategy. S. S. assisted with interpretation of all data. S. M. and M. S. prepared the glycosaminoglycan derivatives. T. P.-X. assisted with acquisition and interpretation of the dynamic light scattering data. N. A., C. E., E. T., M. B., and T. B. wrote the manuscript. All authors read and approved the manuscript.
Acknowledgments
Fredrik Höök is acknowledged for valuable discussions and comments on the manuscript. We further thank Hanna Gawlitza for performing parts of the SPR measurements, Maria Johansson for support with protein isolation, and Karin Norling for acquiring supporting data (nanoparticle tracking analysis, protein quantification). Déborah Rupert and Nagma Parveen are acknowledged for help with the supporting data.
This work was supported by the European Union Seventh Framework Programme (FP7/2007–2013) under Grant Agreement NMP4-SL2009-229292 (“Find & Bind”), the Chalmers Area of Advance in Materials Science, and Vetenskapsrådet (through the Linneus program SUPRA). The authors declare that they have no conflicts of interest with the contents of this article.
- GAG
- glycosaminoglycan
- gC
- glycoprotein C
- SPR
- surface plasmon resonance
- OEG
- oligo(ethylene glycol)
- dS-OEG
- disulfide-OEG
- CS
- chondroitin sulfate
- b-CS
- biotinylated chondroitin sulfate
- MOI
- multiplicity of infection
- HA
- hyaluronan
- b-sHA
- biotinylated sulfated hyaluronan
- b-HA
- biotinylated hyaluronan
- TIRF
- total internal reflection fluorescence
- TRITC
- tetramethylrhodamine isothiocyanate
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- HS
- heparan sulfate
- DSsulfate
- degree of sulfation.
References
- 1. Palese P., Tobita K., Ueda M., Compans R. W. (1974) Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 61, 397–410 [DOI] [PubMed] [Google Scholar]
- 2. von Itzstein M., Wu W. Y., Kok G. B., Pegg M. S., Dyason J. C., Jin B., Van Phan T., Smythe M. L., White H. F., Oliver S. W., Colman P. M., Varghese J. N., Ryan D. M., Woods J. M., Bethell R. C., Hotham V. J., Cameronf J. M., Penn C. R. (1993) Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 363, 418–423 [DOI] [PubMed] [Google Scholar]
- 3. Vaheri A. (1964) Heparin and related polyionic substances as virus inhibitors. Acta Pathol. Microbiol. Scand. Suppl. 171, 171–198 [PubMed] [Google Scholar]
- 4. WuDunn D., Spear P. G. (1989) Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 63, 52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krusat T., Streckert H. J. (1997) Heparin-dependent attachment of respiratory syncytial virus (RSV) to host cells. Arch. Virol. 142, 1247–1254 [DOI] [PubMed] [Google Scholar]
- 6. Salvador B., Sexton N. R., Carrion R., Jr., Nunneley J., Patterson J. L., Steffen I., Lu K., Muench M. O., Lembo D., Simmons G. (2013) Filoviruses utilize glycosaminoglycans for their attachment to target cells. J. Virol. 87, 3295–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saphire A. C. S., Bobardt M. D., Zhang Z., David G., Gallay P. A. (2001) Syndecans serve as attachment receptors for human immunodeficiency virus type 1 on macrophages. J. Virol. 75, 9187–9200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Olofsson S., Bergström T. (2005) Glycoconjugate glycans as viral receptors. Ann. Med. 37, 154–172 [DOI] [PubMed] [Google Scholar]
- 9. Vigerust D. J., Shepherd V. L. (2007) Virus glycosylation: role in virulence and immune interactions. Trends Microbiol. 15, 211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Olofsson S., Hansen J. E. S. (1998) Host cell glycosylation of viral glycoproteins: a battlefield for host defence and viral resistance. Scand. J. Infect. Dis. 30, 435–440 [DOI] [PubMed] [Google Scholar]
- 11. Friedman H. M., Cohen G. H., Eisenberg R. J., Seidel C. A., Cines D. B. (1984) Glycoprotein C of herpes simplex virus 1 acts as a receptor for the C3b complement component on infected cells. Nature 309, 633–635 [DOI] [PubMed] [Google Scholar]
- 12. Ekblad M., Adamiak B., Bergefall K., Nenonen H., Roth A., Bergstrom T., Ferro V., Trybala E. (2007) Molecular basis for resistance of herpes simplex virus type 1 mutants to the sulfated oligosaccharide inhibitor PI-88. Virology 367, 244–252 [DOI] [PubMed] [Google Scholar]
- 13. Adamiak B., Ekblad M., Bergström T., Ferro V., Trybala E. (2007) Herpes simplex virus type 2 glycoprotein G is targeted by the sulfated oligo- and polysaccharide inhibitors of virus attachment to cells. J. Virol. 81, 13424–13434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Herold B. C., WuDunn D., Soltys N., Spear P. G. (1991) Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 65, 1090–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Spear P. G. (2004) Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol. 6, 401–410 [DOI] [PubMed] [Google Scholar]
- 16. Altgärde N., Nilebäck E., de Battice L., Pashkuleva I., Reis R. L., Becher J., Möller S., Schnabelrauch M., Svedhem S. (2013) Probing the biofunctionality of biotinylated hyaluronan and chondroitin sulfate by hyaluronidase degradation and aggrecan interaction. Acta Biomater. 9, 8158–8166 [DOI] [PubMed] [Google Scholar]
- 17. Baranova N. S., Nilebäck E., Haller F. M., Briggs D. C., Svedhem S., Day A. J., Richter R. P. (2011) The inflammation-associated protein TSG-6 cross-links hyaluronan via hyaluronan-induced TSG-6 oligomers. J. Biol. Chem. 286, 25675–25686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Richter R. P., Hock K. K., Burkhartsmeyer J., Boehm H., Bingen P., Wang G., Steinmetz N. F., Evans D. J., Spatz J. P. (2007) Membrane-grafted hyaluronan films: a well-defined model system of glycoconjugate cell coats. J. Am. Chem. Soc. 129, 5306–5307 [DOI] [PubMed] [Google Scholar]
- 19. Osmond R. I. W., Kett W. C., Skett S. E., Coombe D. R. (2002) Protein-heparin interactions measured by BIAcore 2000 are affected by the method of heparin immobilization. Anal. Biochem. 310, 199–207 [DOI] [PubMed] [Google Scholar]
- 20. Yu G., Gunay N. S., Linhardt R. J., Toida T., Fareed J., Hoppensteadt D. A., Shadid H., Ferro V., Li C., Fewings K., Palermo M. C., Podger D. (2002) Preparation and anticoagulant activity of the phosphosulfomannan PI-88. Eur. J. Med. Chem. 37, 783–791 [DOI] [PubMed] [Google Scholar]
- 21. Adamiak B., Trybala E., Mardberg K., Johansson M., Liljeqvist J. A., Olofsson S., Grabowska A., Bienkowska-Szewczyk K., Szewczyk B., Bergstrom T. (2010) Human antibodies to herpes simplex virus type 1 glycoprotein C are neutralizing and target the heparan sulfate-binding domain. Virology 400, 197–206 [DOI] [PubMed] [Google Scholar]
- 22. Guenalp A. (1965) Growth and cytophatic effect of rubella virus in a line of green monkey kidney cells. Proc. Soc. Exp. Biol. Med. 118, 85–90 [PubMed] [Google Scholar]
- 23. Holland T. C., Marlin S. D., Levine M., Glorioso J. (1983) Antigenic variants of herpes simplex virus selected with glycoprotein-specific monoclonal antibodies. J. Virol. 45, 672–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Trybala E., Bergström T., Svennerholm B., Jeansson S., Glorioso J. C., Olofsson S. (1994) Localization of a functional site on herpes simplex virus type 1 glycoprotein C involved in binding to cell surface heparan sulphate. J. Gen. Virol. 75, 743–752 [DOI] [PubMed] [Google Scholar]
- 25. Trybala E., Liljeqvist J. A., Svennerholm B., Bergström T. (2000) Herpes simplex virus types 1 and 2 differ in their interaction with heparan sulfate. J. Virol. 74, 9106–9114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ekblad M., Adamiak B., Bergstrom T., Johnstone K. D., Karoli T., Liu L., Ferro V., Trybala E. (2010) A highly lipophilic sulfated tetrasaccharide glycoside related to muparfostat (PI-88) exhibits virucidal activity against herpes simplex virus. Antiviral Res. 86, 196–203 [DOI] [PubMed] [Google Scholar]
- 27. Widéhn S., Kindblom L. G. (1990) Agarose embedding: a new method for the ultrastructural examination of the in-situ morphology of cell cultures. Ultrastructural Pathology 14, 81–85 [DOI] [PubMed] [Google Scholar]
- 28. Namvar L., Olofsson S., Bergström T., Lindh M. (2005) Detection and typing of herpes simplex virus (HSV) in mucocutaneous samples by TaqMan PCR targeting a gB segment homologous for HSV types 1 and 2. J. Clin. Microbiol. 43, 2058–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nilebäck E., Feuz L., Uddenberg H., Valiokas R., Svedhem S. (2011) Characterization and application of a surface modification designed for QCM-D studies of biotinylated biomolecules. Biosens. Bioelectron. 28, 407–413 [DOI] [PubMed] [Google Scholar]
- 30. Larsson C., Rodahl M., Höök F. (2003) Characterization of DNA immobilization and subsequent hybridization on a 2D arrangement of streptavidin on a biotin-modified lipid bilayer supported on SiO2. Anal. Chem. 75, 5080–5087 [DOI] [PubMed] [Google Scholar]
- 31. Hokputsa S., Jumel K., Alexander C., Harding S. E. (2003) Hydrodynamic characterisation of chemically degraded hyaluronic acid. Carbohydr. Polym. 52, 111–117 [Google Scholar]
- 32. Horkay F., Basser P. J., Hecht A. M., Geissler E. (2012) Chondroitin sulfate in solution: effects of mono- and divalent salts. Macromolecules 45, 2882–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. DeAngelis P. L., Gunay N. S., Toida T., Mao W. J., Linhardt R. J. (2002) Identification of the capsular polysaccharides of Type D and F Pasteurella multocida as unmodified heparin and chondroitin, respectively. Carbohydr. Res. 337, 1547–1552 [DOI] [PubMed] [Google Scholar]
- 34. Satoh A., Toida T., Yoshida K., Kojima K., Matsumoto I. (2000) New role of glycosaminoglycans on the plasma membrane proposed by their interaction with phosphatidylcholine. FEBS Lett. 477, 249–252 [DOI] [PubMed] [Google Scholar]
- 35. Arakawa T., Wen J. (2001) Determination of carbohydrate contents from excess light scattering. Anal. Biochem. 299, 158–161 [DOI] [PubMed] [Google Scholar]
- 36. Hill A. V. (1913) The Combinations of haemoglobin with oxygen and with carbon monoxide. I. Biochem. J. 7, 471–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weiss J. N. (1997) The Hill equation revisited: uses and misuses. FASEB J. 11, 835–841 [PubMed] [Google Scholar]
- 38. Bally M., Gunnarsson A., Svensson L., Larson G., Zhdanov V. P., Höök F. (2011) Interaction of single viruslike particles with vesicles containing glycosphingolipids. Phys. Rev. Lett. 107, 188103. [DOI] [PubMed] [Google Scholar]
- 39. Mårdberg K., Trybala E., Glorioso J. C., Bergström T. (2001) Mutational analysis of the major heparan sulfate-binding domain of herpes simplex virus type 1 glycoprotein C. J. Gen. Virol. 82, 1941–1950 [DOI] [PubMed] [Google Scholar]
- 40. Mårdberg K., Trybala E., Tufaro F., Bergström T. (2002) Herpes simplex virus type 1 glycoprotein C is necessary for efficient infection of chondroitin sulfate-expressing gro2C cells. J. Gen. Virol. 83, 291–300 [DOI] [PubMed] [Google Scholar]
- 41. Hadigal S. R., Agelidis A. M., Karasneh G. A., Antoine T. E., Yakoub A. M., Ramani V. C., Djalilian A. R., Sanderson R. D., Shukla D. (2015) Heparanase is a host enzyme required for herpes simplex virus-1 release from cells. Nat. Commun. 6, 6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hoffman J., Larm O., Scholander E. (1983) A new method for covalent coupling of heparin and other glycosaminoglycans to substances containing primary amino groups. Carbohydr. Res. 117, 328–331 [DOI] [PubMed] [Google Scholar]
- 43. Tkachenko E., Rhodes J. M., Simons M. (2005) Syndecans: new kids on the signaling block. Circ. Res. 96, 488–500 [DOI] [PubMed] [Google Scholar]
- 44. Stannard L. M., Fuller A. O., Spear P. G. (1987) Herpes simplex virus glycoproteins associated with different morphological entities projecting from the virion envelope. J. Gen. Virol. 68, 715–725 [DOI] [PubMed] [Google Scholar]
- 45. Tal-Singer R., Peng C., Ponce De Leon M., Abrams W. R., Banfield B. W., Tufaro F., Cohen G. H., Eisenberg R. J. (1995) Interaction of herpes simplex virus glycoprotein gC with mammalian cell surface molecules. J. Virol. 69, 4471–4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rux A. H., Lou H., Lambris J. D., Friedman H. M., Eisenberg R. J., Cohen G. H. (2002) Kinetic analysis of glycoprotein C of herpes simplex virus types 1 and 2 binding to heparin, heparan sulfate, and complement component C3b. Virology 294, 324–332 [DOI] [PubMed] [Google Scholar]
- 47. Mårdberg K., Nyström K., Tarp M. A., Trybala E., Clausen H., Bergström T., Olofsson S. (2004) Basic amino acids as modulators of an O-linked glycosylation signal of the herpes simplex virus type 1 glycoprotein gC: functional roles in viral infectivity. Glycobiology 14, 571–581 [DOI] [PubMed] [Google Scholar]
- 48. Carlström D. (1957) The crystal structure of α-chitin (poly-N- acetyl-d-glucosamin). J. Biophys. Biochem. Cytol. 3, 669–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Feyzi E., Trybala E., Bergström T., Lindahl U., Spillmann D. (1997) Structural requirement of heparan sulfate for interaction with herpes simplex virus type 1 virions and isolated glycoprotein C. J. Biol. Chem. 272, 24850–24857 [DOI] [PubMed] [Google Scholar]
- 50. Jentoft N. (1990) Why are proteins O-glycosylated? Trends Biochem. Sci. 15, 291–294 [DOI] [PubMed] [Google Scholar]
- 51. Pearson J. T., Hill J. J., Swank J., Isoherranen N., Kunze K. L., Atkins W. M. (2006) Surface plasmon resonance analysis of antifungal azoles binding to CYP3A4 with kinetic resolution of multiple binding orientations. Biochemistry 45, 6341–6353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lebedev K., Mafé S., Stroeve P. (2006) Convection, diffusion and reaction in a surface-based biosensor: modeling of cooperativity and binding site competition on the surface and in the hydrogel. J. Colloid Interface Sci. 296, 527–537 [DOI] [PubMed] [Google Scholar]
- 53. Tao Y., Carta G., Ferreira G., Robbins D. (2011) Adsorption of deamidated antibody variants on macroporous and dextran-grafted cation exchangers: II. adsorption kinetics. J. Chromatogr. A 1218, 1530–1537 [DOI] [PubMed] [Google Scholar]
- 54. Rabe M., Verdes D., Seeger S. (2011) Understanding protein adsorption phenomena at solid surfaces. Adv. Colloid Interface Sci. 162, 87–106 [DOI] [PubMed] [Google Scholar]
- 55. Gama C. I., Hsieh-Wilson L. C. (2005) Chemical approaches to deciphering the glycosaminoglycan code. Curr. Opin. Chem. Biol. 9, 609–619 [DOI] [PubMed] [Google Scholar]
- 56. Raman R., Sasisekharan V., Sasisekharan R. (2005) Structural insights into biological roles of protein-glycosaminoglycan interactions. Chem. Biol. 12, 267–277 [DOI] [PubMed] [Google Scholar]