

Background: In the brain, the type II nuclear receptors LXR and PPARγ control cholesterol efflux and inflammation, key processes in Alzheimer disease pathology.
Results: Combining the LXR and PPARγ agonists decreases the levels of Aβ and inflammation, resulting in improved cognition.
Conclusion: The LXR and PPARγ agonists complement each other, possibly by modulating microglial function.
Significance: Targeting multiple nuclear receptors expands the therapeutic opportunities for AD treatment.
Keywords: Alzheimer disease, amyloid β (Aβ), apolipoprotein E (apoE), microglia, peroxisome proliferator-activated receptor (PPAR), liver X receptor (LXR)
Abstract
Alzheimer disease (AD) is characterized by the extracellular accumulation of amyloid β (Aβ), which is accompanied by a robust inflammatory response in the brain. Both of these pathogenic processes are regulated by nuclear receptors, including the liver X receptors (LXRs) and peroxisome-proliferator receptor γ (PPARγ). Agonists of LXRs have been demonstrated previously to reduce Aβ levels and improve cognitive deficits in AD mouse models by inducing the transcription and lipidation of apolipoprotein E (apoE). Agonists targeting PPARγ reduce the microglial expression of proinflammatory genes and have also been shown to modulate apoE expression. Here we investigate whether a combination therapy with both LXR and PPARγ agonists results in increased benefits in an AD mouse model. We found that the LXR agonist GW3965 and the PPARγ agonist pioglitazone were individually able to increase the levels of apoE and related genes, decrease the expression of proinflammatory genes, and facilitate Aβ decreases in the hippocampus. Combined treatment with both agonists provoked a further increase in the expression of apoE and a decrease in the soluble and deposited forms of Aβ. The decrease in plaques was associated with increased colocalization between microglia and plaques. In addition, the PPARγ agonist in the combined treatment paradigm was able to counteract the elevation in plasma triglycerides that is a side effect of LXR agonist treatment. These results suggest that combined LXR/PPARγ agonist treatment merits further investigation for the treatment of AD.
Introduction
Alzheimer disease (AD)2 is a progressive neurodegenerative disorder characterized by the accumulation of amyloid β (Aβ) peptides in the brain. In cases of late-onset AD, which is the predominant form of the disease, the elevation of soluble forms of Aβ and their subsequent deposition into plaques arise principally from a deficiency in Aβ clearance (1, 2). Apolipoprotein E (apoE) modulates Aβ deposition and clearance in an isoform-dependent manner (3–5). Possession of an apoE4 allele, the strongest genetic risk factor for late-onset AD (6, 7), has been linked to decreased efficiency of Aβ clearance (3–5).
ApoE is the principal apolipoprotein in the brain, where it acts as a scaffold for HDL-like particles and facilitates the trafficking of lipids throughout the CNS. Cholesterol and phospholipids are loaded onto nascent apoE primarily by the lipid transporter ATP-binding cassette A1 (ABCA1) and, additionally, by ATP-binding cassette G1 (ABCG1) (8). Lipidated apoE is produced in the brain by astrocytes and, to a lesser extent, microglia. We have shown previously that lipidated apoE facilitates the proteolytic clearance of soluble forms of Aβ (9), which, in microglia is effected by increasing the rate of Aβ trafficking to lysosomes for degradation (10). The ability of apoE to facilitate soluble Aβ clearance is strongly dependent on the apoE isoform (3–5) and its lipidation state, with ABCA1-dependent lipidation playing an important role (11–13).
The production of apoE is transcriptionally regulated by liver X receptors (LXRs) α and β (14, 15). LXRs are type II nuclear receptors that are activated by oxysterol ligands and function as cholesterol sensors. Upon ligand binding, LXRs form obligate heterodimers with retinoid X receptors (RXRs) and promote the transcription of apoE, ABCA1, and ABCG1, among other genes involved in cholesterol metabolism (16). Treatment of murine models of AD with synthetic LXR ligands such as GW3965 or TO901317 has been shown previously to result in increased apoE expression and lipidation in the brain, memory improvements, and reduced levels of Aβ (9, 11, 13, 17–25). Ligands that target peroxisome proliferator-activated receptor γ (PPARγ) also produce increases in LXR target genes and the associated benefits, likely by inducing the expression of LXRα as well as interaction with enhancer elements (26–28). A large body of literature indicates that activation of LXR, PPARγ, or their heterodimeric partner RXR is able to ameliorate Aβ pathology and mediate behavioral improvements in mouse models of AD (29, 30).
Deposition of Aβ induces a robust inflammatory response in microglia, resulting in their migration and subsequent association with Aβ deposits and increased cytokine and chemokine production (31–33). PPARγ activates a program of gene expression that promotes phagocytosis and tissue repair in macrophages (34, 35) and has been shown to mediate a similar phenotypic conversion in microglia (27, 36, 37). LXR agonists also modulate microglial function and act as regulators of phagocytosis in AD models (23, 38), partially through the transcriptional control of phagocytic proteins such as MerTK and Axl (39). PPARγ and LXR agonists have been reported to mitigate neuroinflammation in AD models by exerting robust anti-inflammatory activity (37, 40). Additionally, ligand activation of PPARγ or LXR results in their sumoylation, and sumoylated PPARγ or LXR interacts with corepressor complexes at NFκB and AP1 promoters to prevent their clearance (37, 40), suppressing proinflammatory gene expression. LXR and PPARγ each act independently to transrepress an overlapping but distinct subset of proinflammatory genes (37, 40, 41).
In this study, we evaluated the therapeutic potential of combining LXR and PPAR agonists in treating APPswe/PSEN1dE9 (APP/PS1) mice, which express familial human mutations in both APP and presenilin. We reproduce the previous findings that LXR and PPAR agonists individually are able to decrease Aβ plaques in the hippocampi of AD mice, produce anti-inflammatory effects, and mediate behavioral improvements. Additionally, we demonstrate that a combination of LXR and PPARγ agonists elicits biochemical and behavioral improvements while ameliorating LXR-mediated plasma hypertriglyceridemia. Combination treatment was able to effect a further increase in LXR target gene production and effectively reduce inflammatory markers. Importantly, combination treatment was more effective than individual agonist treatments at stimulating the colocalization of microglia and plaques in APP/PS1 mice and promoting Aβ intracellular degradation. These data provide a rationale for the further investigation of combination therapies utilizing LXR and PPARγ agonists in AD.
Experimental Procedures
Animals and Treatment
APPswe/PS1Δe9 (APP/PS1) mice (The Jackson Laboratory) coexpress a chimeric human-mouse amyloid precursor protein containing the APPswe mutations (K595N/M596L) and human presenilin 1 with an exon 9 deletion (Δe9) from the mouse prion promoter. Male 6-month-old APP/PS1 mice or non-transgenic littermates were orally gavaged orally every day for 9 days with 50 mg/kg/day GW3965, 80 mg/kg/day pioglitazone, both 50 mg/kg/day GW3965 and 80 mg/kg/day pioglitazone, or a vehicle control (0.5% DMSO in sesame oil). Behavioral analysis was performed during the last 2 days of treatment. The animals were then sacrificed, and one hemisphere was fixed in 4% PFA and processed for immunohistochemistry. The hippocampus and cortex were removed from the other hemisphere and snap-frozen and subjected to RNA and protein extraction. For analysis of plasma triglycerides, male 2-month-old C57Bl/6 mice were treated with GW3965, pioglitazone, both, or vehicle as above, with plasma collected on day 9 of treatment within 3 h of the last dose. All experiments involving animals followed protocols approved by the Case Western Reserve University School of Medicine Institutional Animal Care and Use Committee.
Behavioral Testing
Contextual Fear Conditioning
Freezing behavior was monitored by an automated tracking system (Coulbourn Instruments). On day 8 of drug treatment, mice underwent training, which consisted of 2 min of free exploration in the shock chamber followed by 30 s of the conditioned stimulus (an 85-db sound at 2800 Hz). After a 2-s delay, the unconditioned stimulus (0.56 mA) was delivered, and the freezing response was measured for 30 s. The training paradigm was repeated four times. Twenty-four hours later, the retention test was performed, during which mice were returned to the same shock chamber for 5 min for contextual freezing measurements in the absence of conditioned and unconditioned stimuli. The percent of time frozen and number of freezes were recorded.
Protein Extraction
Cortices and hippocampi dissected from hemibrains were homogenized in 800 μl of tissue homogenization buffer (250 mm sucrose, 20 mm Tris (pH 7.4), 1 mm EDTA, and 1 mm EGTA in diethylpyrocarbonate-treated water). Homogenates were centrifuged at 5000 × g for 10 min at 4 °C, and supernatants were stored at −80 °C for Western blot analysis. For extraction of soluble Aβ species, 250 μl of homogenate was added to an equal volume of 0.4% diethylamine in 100 mm NaCl, and the samples were again homogenized mechanically. The samples were then centrifuged at 135,000 × g for 1 h at 4 °C. 0.5 m Tris-HCl (pH 6.8) was added to the supernatant, which was stored at −80 °C for analysis of soluble Aβ species by ELISA. The remaining pellet was sonicated in cold 70% formic acid and centrifuged at 109,000 × g for 1 h at 4 °C. The supernatant was neutralized, and the samples were stored at −80 °C for analysis of insoluble Aβ species by ELISA.
Cell Culture
Primary microglia and astrocytes were prepared from postnatal day 0–3 mice as described previously (9). Purified microglia and astrocytes were maintained in DMEM/F12 (Invitrogen) containing 5% heat-inactivated FBS and 1% penicillin/streptomycin for 3 days. Twenty-four hours before treatment, medium was changed to serum-free DMEM/F12 containing 1% penicillin/streptomycin. For Western blot and qPCR analyses, cells were plated in 6 well plates at 1 × 106 cells/well and treated for 24 h with GW3965, pioglitazone, or GW3965 and pioglitazone or vehicle (DMSO) at the indicated concentrations.
Intracellular Aβ Degradation Assay
Soluble Aβ was prepared by dissolving lyophilized Aβ1–42 in DMSO to a final concentration of 1 mg/ml to create a solution of mostly monomeric Aβ species with very few oligomers (42). Primary microglia were plated in 12-well plates at a density of 4 × 105 cells/well. Microglia were then pretreated for 24 h with GW3965, pioglitazone, or GW3965 and pioglitazone or vehicle (DMSO) at the indicated concentrations and then incubated with 2 μg/ml Aβ1–42 (American Peptide Co.) for 18 h. Plates were washed with PBS, and cells were lysed in 1% SDS with Protease Inhibitor Cocktail (Roche). The remaining intracellular Aβ was measured by ELISA.
Aβ ELISA
For the intracellular Aβ degradation assay, ELISAs were performed using 6E10 as the capture antibody and 4G8-HRP as the detection antibody (Covance). To analyze the levels of soluble and insoluble Aβ in brain homogenates, ELISAs were performed using 6E10 as the capture antibody and Aβ1–40-HRP or Aβ1–42-HRP (Covance) for detection. The results were read using a Spectramax colorimetric plate reader (Molecular Devices) and normalized to the total protein.
Western Blot Analysis
Cell lysates or brain homogenates were resolved on BisTris 4–12% gels (Invitrogen), transferred to PVDF membranes, and immunodetected using anti-ABCA1 (Novus Biologicals), anti-ABCG1 (Novus Biologicals), anti-apoE (Santa Cruz Biotechnology), and anti-β-actin (Santa Cruz Biotechnology). Band intensities were quantified using NIH ImageJ software.
Native PAGE
Cell lysates or brain homogenates were resolved on Tris-Glycine 4–12% gels (Invitrogen), transferred to PVDF membranes, and immunodetected using an anti-ApoE antibody (Santa Cruz Biotechnology). Native high molecular weight standards (GE Healthcare, high molecular weight native marker kit, catalog no. 17044501) were run on each gel and used to determine the Stokes diameter of samples. The intensity of the bands above 8 nm in size was quantified using NIH ImageJ software to determine the apoE lipidation index.
RNA Extraction, Reverse Transcription, and Quantitative PCR
Quantification of pro- or anti-inflammatory gene expression was performed as described previously (27). For qPCR analysis of cells, RNA was isolated using the RNeasy mini kit (Qiagen) according to the instructions of the manufacturer. For qPCR analysis of brain homogenate, 200 μl of homogenate was combined with an equal volume of RNA-Bee (Tel-Test). Chloroform was added, and the samples were shaken vigorously. The samples were incubated on ice for 15 min, followed by centrifugation at 13,000 × g for 15 min at 4 °C. The aqueous phase was collected, and RNA extraction was performed as above. RNA samples (0.5 μg) were reverse-transcribed using the QuantiTect reverse transcription kit (Quiagen). TaqMan PreAmp Master Mix (Life Technologies) was then used to preamplify cDNA for 14 cycles according to the instructions of the manufacturer. Preamplified cDNA was run in a 10-μl reaction for 40 cycles on the StepOne Plus real-time PCR system (Applied Biosystems) using TaqMan Gene Expression Master Mix (Life Technologies). Primers labeled with FAM probes were from Life Technologies and included Iba1 (Mm00479862_g1), CD45 (Mm01293575_m1), Tnfα (Mm99999068_m1), Il-1β (Mm01336189_m1), Nos2 (Mm01309902_m1), IL6 (Mm00446190_m1), and GAPDH (4352339E-0904021). The comparative CT method (ΔΔCT) was used to analyze gene expression.
Immunohistochemistry and Image Analysis
Coronal sections (30 μm) were taken from post-fixed hemispheres using a cryostat. Alternate sections were immunostained for analysis of the plaque area, microglial area, and microglia/plaque colocalization. Antigen retrieval was performed with 20 μl/ml proteinase K in TE buffer (50 mm Tris,1 mm EDTA, and 0.5% Triton X-100 (pH 8)), and slides were blocked in 5% normal goat serum in PBS and 0.1% Triton X-100. Primary antibodies (6E10, 1:1000, Covance; Iba1, 1:500, Wako) were incubated overnight at 4 °C. Analysis was performed on two sections per slide on three slides spaced evenly throughout the hippocampus. Images were analyzed by a blinded observer using Image Pro-Plus software (Media Cybernetics) for the percentage area of 6E10-positive plaques in the cortex or hippocampus and the percentage area occupied by Iba1-positive microglia. The percentage of 6E10-positive area also positive for Iba1 immunostaining was determined for every individual plaque in each section, and the results were normalized to Iba1 intensity in a non-plaque area in each image to determine Iba1 enrichment at plaques.
Triglyceride Assay
Plasma was collected using 3.8% sodium citrate in water as an anticoagulant. Blood was centrifuged at 1000 × g for 10 min at 4 °C. The plasma was removed and stored at −80 °C for further analysis. Levels of plasma triglycerides were determined using the triglyceride colorimetric assay kit (Cayman Chemical Company) according to the instructions of the manufacturer. The assays were read using a Spectramax colorimetric plate reader (Molecular Devices).
Statistics
All statistical analyses were performed using Prism software (GraphPad, San Diego, CA). Two-tailed Student's t test, one-way analysis of variance (ANOVA) with a Tukey post-test or two-way ANOVA was used to determine p values.
Results
Enhanced Effects of Combined Activation of PPARγ and LXR in Vitro
Because PPARγ and LXR agonists have been reported independently to increase expression of LXR target genes (16, 27), we analyzed whether a combined activation of PPARγ and LXR would lead to a greater increase in LXR target gene expression. We cultured primary astrocytes and quantified target gene expression after a 24-h treatment with an LXR agonist (GW3965), a PPARγ agonist (pioglitazone), or a combined treatment with both. Consistent with our previous findings, we observed that LXR and PPARγ agonists were able to increase protein expression of apoE and its lipidating proteins ABCA1 and ABCG1 in cultured astrocytes (Fig. 1A) (9, 27). Combined treatment with GW3965 and pioglitazone did not produce an additive effect in the expression of apoE protein but significantly increased the expression of its lipid transporter ABCA1 over pioglitazone alone (Fig. 1A). To determine how this up-regulation in ABCA1 affected apoE, we assessed the quantity of large-diameter apoE particles secreted by astrocytes into the extracellular medium by native PAGE (Fig. 1B). Detection of large-diameter apoE particles by native PAGE has been shown to correlate with increases in apoE lipidation (43), but we are unable to exclude the possibility that native PAGE detects some amount of apoE aggregation. As expected, GW3965 and pioglitazone were able to independently increase the amount of large-diameter apoE secreted, and combination treatment induced a further increase in large-diameter apoE particles (Fig. 1B).
FIGURE 1.

Combination therapy increases target gene expression and Aβ degradation and decreases inflammatory markers in vitro. A, cultured primary astrocytes were incubated for 24 h with DMSO (μm), 500 nm GW3965, 100 nm pioglitazone, or both doses combined, and then LXR target genes were quantified by immunoblotting and normalized to actin. Representative blots are shown in the right panel. B, conditioned medium from the same treated primary astrocytes was collected, separated by native PAGE, and immunoblotted for apoE. C, primary microglia were treated for 24 h with GW, pio, or GW + pio, followed by the addition of 2 μg/ml Aβ1–42 for 18 h. Remaining intracellular Aβ was quantified using ELISA and normalized to total protein. D, cultured primary microglia were incubated for 24 h with the indicated concentrations of drug or DMSO for controls, and then 100 ng/ml LPS was introduced to the medium for 12 h. RNA was extracted, and the expression of proinflammatory genes was examined by qPCR analysis. The dotted line indicates baseline transcript levels in the DMSO-only control. The LPS-treated control was preincubated with DMSO for 24 h and then with 100 ng/ml LPS for 12 h (n = 4). *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with the vehicle-treated control; ##, p < 0.01; ###, p < 0.001 compared with the LPS-treated control; Student's t test. a.u., arbitrary units.
Microglia internalize soluble Aβ from the medium and degrade it intracellularly, and this process is facilitated by agonists of LXR, PPARγ, and RXR (9, 10, 27, 29). Microglia were incubated with Aβ for 18 h, resulting in the uptake and degradation of Aβ. The amount of intracellular Aβ remaining after that time was measured as an indicator of Aβ degradation efficiency. Pretreatment with GW3965 or pioglitazone for 24 h enhanced the degradation efficiency in microglia, and pretreatment with a combination of both drugs further enhanced this effect (Fig. 1C).
LXR and PPARγ are also known to have anti-inflammatory effects in microglia because of transrepression of NFκB at the promoters of inflammatory genes (37). To determine whether combination therapy targeting both receptors could enhance these anti-inflammatory effects, we pretreated primary cultured microglia for 24 h with DMSO, GW3965, pioglitazone, or both nuclear receptor agonists and then induced an inflammatory response with LPS. Induction of transcription for the proinflammatory cytokines TNFα and IL1β was less in microglia pretreated with either GW3965 or pioglitazone (Fig. 1D). Microglia pretreated with a combination therapy exhibited enhanced suppression of these proinflammatory genes (Fig. 1D). Pretreatment with GW3965 or pioglitazone alone did not decrease transcript levels of inducible nitric oxide synthase (iNOS), but the combination pretreatment produced a significant decrease in iNOS transcripts (Fig. 1D).
Combination Therapy Increases apoE Particle Size in APP/PS1 Mice
Previously published data indicate that increasing apoE protein levels and lipidation were associated with a reversal of the behavioral deficits and some aspects of pathology in AD mouse models (30). On the basis of our in vitro data indicating that combination therapy enhances the transcriptional effects of LXR and PPARγ agonists and their facilitation of Aβ degradation (Fig. 1), we chose to treat APP/PS1 mice with the combination therapy. Four treatment groups of age-matched transgenic male 6-month-old animals were generated and treated for 9 days by oral gavage. The first group was treated with 50 mg/kg/day GW3965, the second with 80 mg/kg/day pioglitazone, the third with 50 mg/kg/day GW3965 and 80 mg/kg/day pioglitazone in the same volume, and the fourth with a vehicle of DMSO in sesame oil. Western blot analysis on hemibrain cortical/hippocampal homogenate indicated that GW3965 and pioglitazone increased transcription of the LXR target genes ABCA1, ABCG1, and apoE, with combination therapy providing a significant increase in apoE over GW3965 alone (Fig. 2, A and B). Non-denaturing gel electrophoresis was performed on cortical/hippocampal homogenates and detected with apoE antibody to determine the size distribution of apoE. GW3965 and pioglitazone were both able to increase the proportion of large-diameter apoE particles, and combination therapy further increased this effect (Fig. 2C).
FIGURE 2.
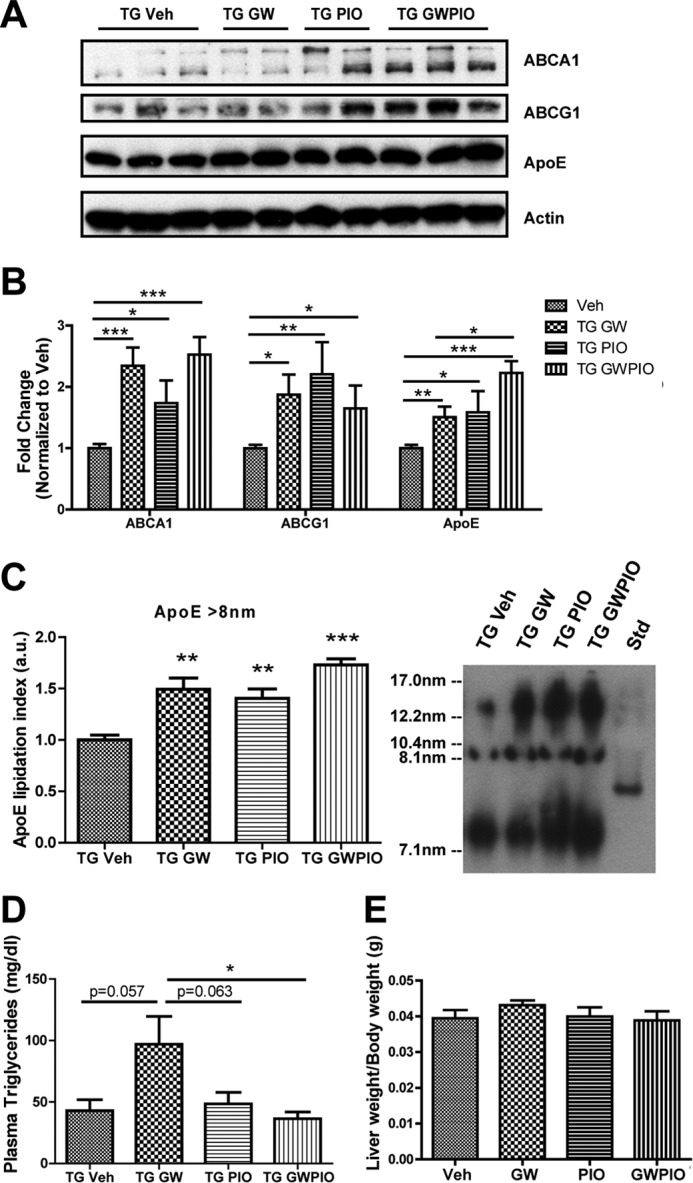
Nuclear receptor agonists stimulate transcription of LXR target genes in AD mice. A, 6-month-old transgenic (TG) APPswe/PSENΔE9 mice were orally gavaged with 50 mg/kg/day GW3965, 80 mg/kg/day pioglitazone, or both for 9 days. Cortical/hippocampal homogenate was immunoblotted for ABCA1, ABCG1, and apoE. Representative blots are shown. Veh, vehicle. B, each sample was normalized to actin, and results are expressed as fold difference compared with vehicle controls. C, equal volumes of whole-brain homogenate from each treatment group were analyzed by native PAGE and immunoblotted for apoE (n = 7–11 animals/group). D and E, 6-month-old transgenic APPswe/PSENΔE9 or non-transgenic littermate control animals were orally gavaged with the doses described above for 9 days. D, plasma was collected and analyzed for triglyceride content by colorimetric assay (n = 6 animals/group). E, complete livers were removed from each animal and weighed, and liver weight was normalized to total body weight for each animal. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with vehicle-treated control; #, p < 0.05 compared with GW-treated group; Student's t test. a.u., arbitrary units.
Pioglitazone Treatment Reduces the GW3965-mediated Elevation of Plasma Triglycerides
LXRs agonists have been reported to up-regulate the synthesis of fatty acids in the liver because of LXR-mediated stimulation of a gene expression program that includes SREBP-1c and fatty acid synthase, among other lipogenic enzymes (41). In a preliminary study in a cohort of WT mice, we observed a significant increase in plasma triglycerides after 9 days of treatment with GW3965 compared with vehicle, which was not observable after 9 days of treatment with pioglitazone or with both GW3965 and pioglitazone (data not shown). In 6-month-old APP/PS1 mice, we observed a strong trend for GW3965 to increase plasma triglycerides, which was reduced significantly by addition of pioglitazone in our combination treatment (Fig. 2D). Treatment with either GW3965, pioglitazone, or both agonists did not correspond to an increased liver weight (Fig. 2E).
Combination Therapy Reduces Inflammatory Markers in APP/PS1 Mice
APP/PS1 animals have increased expression of Iba1 and CD45, microglial markers that are reflective of a proinflammatory phenotype (Fig. 3, A–D). GW3965 and pioglitazone were able to reduce expression of Iba1, as assessed by qPCR on cortical/hippocampal homogenate (Fig. 3A) and immunostaining on hemicortical sections, significantly reducing Iba1 immunostaining compared with vehicle or GW3965 alone (Fig. 3, B and C). Interestingly, combination therapy effected the largest decrease in Iba1, reducing Iba1 levels to a point below baseline wild-type levels (Fig. 3A). The levels of CD45 were also decreased by combination therapy (Fig. 3D).
FIGURE 3.

Nuclear receptor agonists reduce the proinflammatory environment in APP/PS1 mice. A and B, transcript levels from RNA isolated from whole-brain homogenate (A) and quantification of immunofluorescence in the cortex and hippocampus (B) of the microglia marker Iba1 in 6-month-old APPswe/PSENΔE9 mice orally gavaged with the indicated drugs for 9 days. Nine hemicoronal sections per animal were immunostained for Iba1, and Iba1 area analysis was performed as described under “Experimental Procedures.” TG, transgenic; Veh, vehicle. C, representative images of Iba1 immunostaining in the cortex. D–G, transcript levels of the proinflammatory markers CD45 (D), IL6 (E), IL1β (F), and TNFα (G) were analyzed by qPCR on RNA isolated from whole-brain homogenate (n = 7–11 animals/group). *, p < 0.05; **, p < 0.01 compared with vehicle-treated APP/PS1 mice; #, p < 0.05; ##, p < 0.01; ###, p < 0.001 compared with non-transgenic littermate control mice. A and D–G, one-way ANOVA with Tukey post-test. B, Student's t test.
Our in vitro data indicate that treating microglia with GW3965 and pioglitazone together decreases the inflammatory response they exhibit upon challenge with LPS (Fig. 1D). In the APP/PS1 mouse model, mice treated with GW3965, pioglitazone, or both all exhibited different levels of proinflammatory gene expression, as measured by qPCR (Fig. 3, E–G). IL6 was the only cytokine increased significantly in APP/PS1 animals at 6 months of age, and pioglitazone and GW + pio were able to significantly decrease IL6 gene expression (Fig. 3E). IL1β was decreased significantly decreased by GW3965 and GW + pio (Fig. 3F), and TNFα was decreased by pioglitazone but increased by GW + pio treatment (Fig. 3G), suggesting that GW3965, pioglitazone, and GW + pio act on microglia to differently regulate their inflammatory state.
Combination Therapy Reduces Aβ Deposition by Increasing Colocalization between Microglia and Plaques
To evaluate the ability of nuclear receptor agonists to reduce Aβ species in 6-month-old APP/PS1 mice, we sequentially extracted soluble and insoluble Aβ species from cortical and hippocampal homogenates. Treatment with GW3965 alone did not significantly reduce soluble or insoluble Aβ levels, whereas pioglitazone treatment significantly decreased only levels of soluble Aβ40 by about 25% (Fig. 4, A and B). However, when we evaluated Aβ levels in treated mice by immunohistochemistry (Fig. 4, C–E), we were able to observe significant decreases in the amount of hippocampal (Fig. 4E) but not cortical (Fig. 4D) 6E10 staining, indicating that GW3965 and pioglitazone were able to facilitate the clearance of deposited Aβ in the hippocampus.
FIGURE 4.

Combination therapy significantly reduces the amyloid burden in AD mice. A and B, Aβ was sequentially extracted from whole brain homogenates using diethylamine for soluble Aβ (A) and formic acid for insoluble Aβ (B). Samples were analyzed by ELISA, and Aβ values were normalized for total protein loaded and to vehicle-treated animals. To quantify plaque load by immunohistochemistry, 9 hemicoronal sections/animal were immunostained for 6E10, and plaque area analysis was performed as described under “Experimental Procedures.” TG, transgenic; Veh, vehicle. C, representative images from the cortex. The Aβ plaque area was quantitated in the cortex (D) and hippocampus (E) (n = 7–11 animals/group). *, p < 0.05; **, p < 0.01 compared with vehicle-treated APP/PS1 mice; Student's t test.
In contrast, mice treated with combination therapy exhibited decreased levels of both soluble Aβ40 and Aβ42 as measured by ELISA (Fig. 4A). Insoluble Aβ40 levels were also decreased by about 50%, but the decreases in insoluble Aβ42 were not significant (Fig. 4B). These findings correlated with an approximate 60% decrease in cortical plaques and a 50% decrease in hippocampal plaques, as quantified by immunohistochemistry (Fig. 4, C–E).
Decreases in insoluble species of Aβ and plaque are likely mediated principally by microglia, which are capable of taking up and degrading fibrillar forms of Aβ. We found that the overall Iba1+ area in the brain is decreased following nuclear receptor agonist treatments (Fig. 3C) and, therefore, follows overall plaque burden. However, we observed that there are more microglia at sites of plaque deposition (Fig. 5). To determine the amount of colocalization between microglia and plaques, we quantified the amounts of 6E10/Iba1 double-positive areas in the cortex (Fig. 5B) and hippocampus (Fig. 5C) of treated and control mice by immunohistochemistry. The GW3965, pio, and GW + pio treatments significantly increased the amount of microglia/plaque colocalization in the cortex, and GW3965 and GW + pio were able to significantly increase colocalization in the hippocampus. These observations suggest that nuclear receptor agonists mediate reductions in insoluble Aβ by altering the interaction between microglia and plaques.
FIGURE 5.

Nuclear receptor agonists promote microglial colocalization with plaques. A, hemicoronal sections from each treatment group were coimmunostained with Iba1 (green) for microglia and 6E10 (red) for Aβ plaques. Representative images from the cortex are shown. TG, transgenic; Veh, vehicle. B and C, colocalization between 6E10 and Iba1 was analyzed as described under “Experimental Procedures” in the cortex (B) and hippocampus (C). n = 7–11 animals/group; *, p < 0.05; **, p < 0.01 compared with vehicle-treated APP/PS1 mice; Student's t test.
Treatment with Nuclear Receptor Agonists Reverses the Cognitive Deficits in APP/PS1 Mice
To determine whether treatment with nuclear receptor agonists is able to improve behavioral impairments in APP/PS1 animals, we evaluated hippocampus-dependent memory in a contextual fear conditioning test. APP/PS1 animals exhibited a decreased freezing response compared with WT animals during the training period of the task, but drug-treated animals did not exhibit this impairment in learning during the training period (Fig. 6A). As expected, WT age-matched littermate controls froze for about 25% of the testing period, whereas, at 6 months of age, APP/PS1 mice exhibited deficits in freezing behavior, freezing about 15% less than WT littermate animals (Fig. 6B). Consistent with studies published previously, 9-day treatments with GW3965 and pioglitazone were able to restore freezing behavior in APP/PS1 animals to WT levels. A 9-day treatment with GW + pio was able to achieve an equivalent cognitive improvement (Fig. 6B).
FIGURE 6.
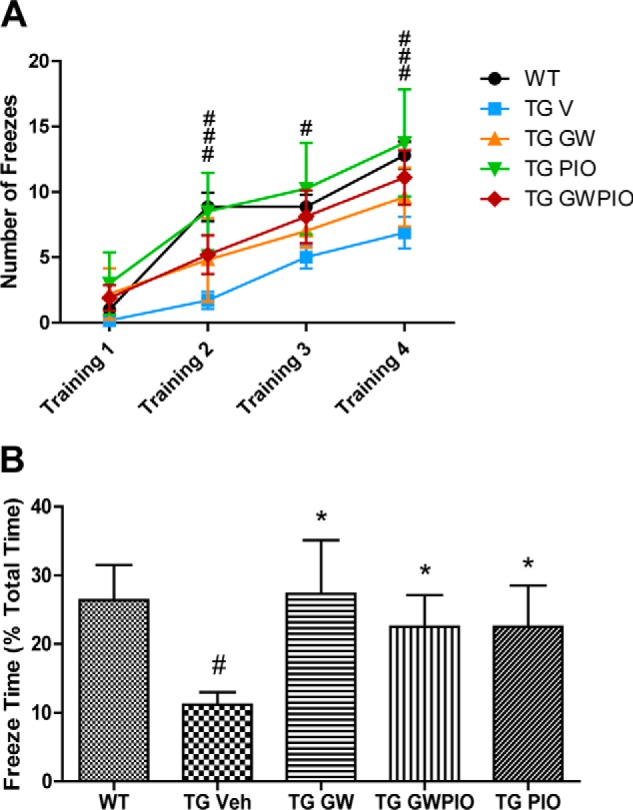
Nuclear receptor agonists ameliorate cognitive deficits in AD mice. Training for the fear conditioning assay was performed on day 8 of drug treatment, and mice were tested on day 9. A, the number of freezes by each treatment group during training is shown as a function of periods. #, p < 0.05; ###, p < 0.001 between non-transgenic (TG) control mice and transgenic vehicle (Veh) mice by two-way ANOVA. B, the percentage of time spent freezing by each treatment group during the contextual fear conditioning test period. *, p < 0.05 compared with vehicle-treated APP/PS1 mice; #, p < 0.05 compared with non-transgenic littermate control mice by one-way ANOVA with Tukey post-test; n = 7–11 animals/group.
Discussion
LXR and PPAR agonists have been demonstrated previously to have beneficial effects in AD mouse models. The first study utilizing nuclear receptor agonists in AD was published in 2003 by Yan et al. (44) and reported that long-term treatment of Tg2576 mice with pioglitazone was able to decrease levels of soluble Aβ. However, subsequent studies using PPAR agonists in several AD mouse models had variable success in observing changes in Aβ levels. Several studies using various treatment paradigms with PPARγ agonists observed decreases in soluble Aβ species (45, 46), plaques (47, 48) or both (27, 36, 49, 50), and others reported decreased intracellular Aβ (51, 52). However, there were also cases where PPARγ agonists exhibited no measurable effect on Aβ pathology (53–55). LXR agonists have had a similarly mixed success since 2005, when Koldamova et al. (11) reported that TO901317 was able to reduce soluble Aβ levels in APP23 mice. Since then, LXR agonists have been reported to decrease soluble Aβ (9, 18, 20, 22, 23, 25, 56), decrease insoluble Aβ or plaques (9, 19, 20, 22–24, 56), or, in one case, decrease plaques while increasing soluble Aβ species (13). One study observed no biochemical changes in Aβ pathology in aged animals (21) but reported behavioral improvements. Although the majority of studies report that PPARγ and LXR agonists are able to reduce Aβ pathology, the variability in the clearance of soluble Aβ species and plaques remains unexplained. The genotype, age of the mice, differences in diet, and drug formulation are all likely to influence drug efficacy, but these factors have not been explored systematically.
We found that in whole-brain homogenates, treatment with GW3965 did not significantly stimulate the clearance of soluble and insoluble species of Aβ, and pioglitazone was only able to stimulate the clearance of soluble Aβ40. We also visualized plaques using immunohistochemistry to evaluate regional effects on Aβ deposition. Interestingly, although plaques did not decrease significantly in the cortices of pioglitazone- or GW3965-treated animals, both drugs were able to induce significant decreases in hippocampal plaque load in the same animals. Our results are similar to those of Riddell et al. (18), who reported that an LXR agonist selectively induced apoE and ABCA1 and reduced Aβ42 in the hippocampi of Tg2576 mice. Donkin et al. (13) also reported that GW3965 mediated a hippocampus-specific reduction in dense-core plaques in APP/PS1 mice, although, when Aβ deposition across the whole brain was quantified, no significant reduction was observed.
Combination therapy with GW + pio significantly reduced the overall levels of soluble Aβ40 and Aβ42. It is likely that the reduction in soluble Aβ we observed in GW + pio treatment is in part due to the enhanced transcription of apoE and its lipid transporters because levels of lipidated apoE have been shown previously to increase the rate of degradation of Aβ by microglia (9, 10). However, we also observed an acute reduction of insoluble Aβ40 and deposited Aβ plaques in both the cortex and hippocampus over the course of our 9-day treatment with GW + pio, which is more likely due to active phagocytic clearance by microglia (57). We postulate that the reductions in Aβ we observed were mainly due to modulation of Aβ clearance rather than Aβ production because previous studies using GW3965 (9) and pioglitazone (27) have established that these drugs do not affect the production or processing of APP in transgenic mouse models of AD. This agrees with our in vitro observations that GW + pio treatment is able to enhance microglial degradation of Aβ to a greater degree than either agonist alone. The mechanistic basis of the enhanced effectiveness of the combined drug treatment may be the ability of the drugs to target independent enhancer sequences to which the nuclear receptors bind and act combinatorially to promote gene expression (7). Future studies will be able to determine whether the Aβ clearance resulting from engaging PPARγ and LXR together via a combination therapy will be more reproducible than the effects of individual PPARγ and LXR agonists.
It is possible that the facilitation of microglial phenotypic conversion to an anti-inflammatory, phagocytic phenotype contributes to the increased benefits of GW + pio treatment. APP/PS1 mice at 6 months of age already exhibit increased microgliosis and astrogliosis as well as proinflammatory alterations in their brain cytokine profiles (27). Importantly, PPARγ agonists have been shown to not only reduce inflammation but also to increase phagocytosis of Aβ in vitro and in vivo (27, 36). The effects of LXR agonists on inflammation in AD models have not been studied as thoroughly, but treatment with LXR agonists has been reported to be anti-inflammatory (19, 24), increase colocalization between glia and plaques (23), and regulate transcription of phagocytic genes (39). Together with evidence showing that, in macrophages, PPARγ and LXR are able to transrepress overlapping but distinct sets of proinflammatory genes (37, 38, 41, 58), this body of evidence led us to hypothesize that costimulation with LXR and PPARγ agonists would have a greater anti-inflammatory effect than either agonist alone. We found that GW + pio treatment effectively decreased proinflammatory markers in vitro and in vivo with an interesting exception. GW + pio reduced TNFα transcript levels in cultured primary microglia but not in whole brains in APP/PS1 mice. This could indicate that GW + pio treatment acts differently on non-microglial brain cells that produce TNFα, such as astrocytes, or imply that environment is an important factor for determining the effects of these drugs on microglia. Importantly, GW + pio also increased the colocalization between plaques and microglia. We propose that this enhanced colocalization facilitates improved phagocytosis and, together with the increased degradation of Aβ by microglia, accounts for the reduction of Aβ species we observed with GW + pio treatment.
Our investigation also reports behavioral improvements in APP/PS1 mice after LXR or PPARγ agonist treatment, which is consistent with the vast majority of previous studies. LXR (9, 13, 18, 20–24) and PPARγ (27, 36, 46, 48, 50, 52, 55, 59, 60) agonists have been reported to ameliorate memory deficits using a variety of tests in a range of mouse models. It should be noted, however, that Nicolakakis et al. (53), Masciopinto et al. (59), and Papadopoulos et al. (54) reported no behavioral improvements after treating mice with pioglitazone. We found that APP/PS1 mice at 6 months of age are impaired in the performance of the contextual fear conditioning task and that this impairment is rectified by treatment with GW, pio, or both agonists. Our dosage paradigm did not allow a closer examination of this effect because we utilized high doses of GW + pio that have been shown previously to restore freezing behavior to wild-type levels. It will be important in future studies to use lower doses of GW + pio that are less individually effective to determine whether combination treatment is able to further enhance memory improvements.
Importantly, although pioglitazone has entered phase III clinical trials for the treatment of AD, no LXR agonists have reached clinical trials because of an unfavorable side effect profile that includes hypertriglyceridemia (61, 62). This is likely due to LXR-dependent up-regulation of the sterol response element binding protein 1c (SREBP1C) pathway in the liver. PPARα agonists have been shown to decrease hypertriglyceridemia, possibly through stimulating β oxidation of fatty acids (42), and a PPARα agonist has been shown to counteract LXR agonist-induced hypertriglyceridemia in mice (63). There is evidence that PPARγ activation could also normalize LXR-mediated hypertriglyceridemia, possibly by mediating the redistribution of triglycerides from the plasma to storage in fatty tissue by inducing genes involved in lipoprotein uptake and hydrolysis (64, 65). Our finding that the addition of a PPARγ agonist is able to normalize hypertriglyceridemia observed in LXR agonist-treated mice renews the potential for LXR agonists to enter clinical trials for AD. Additionally, we did not observe changes in liver weight in mice treated with GW3965, pioglitazone, or GW + pio, which is consistent with work reported previously (66–70).
In this study, we provide support for the beneficial actions of LXR and PPARγ agonists in the treatment of amyloid pathology, inflammation, and behavioral impairments in AD mouse models. LXR and PPARγ activation, in combination, more effectively improves several biochemical markers and alleviates cognitive impairments in AD mice, with an additional reduction in side effects. Our results demonstrate the potential for therapies targeting multiple heterodimer partners of RXR for the treatment of AD.
Author Contributions
R. S. and G. E. L. designed the study and wrote the paper. M. P. P. performed and analyzed the experiments shown in Figs. 3 and 4 and contributed to the preparation of the figures. B. T. C designed, performed, and analyzed the serum triglyceride assays and provided technical assistance. L. T. provided technical assistance and contributed to the preparation of the figures. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank J. Colleen Karlo for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 AG030482 (to G. E. L.) and 5T32NS067431-13 (to R. R. S.). This work was also supported by the Gail and Elliott Schlang Philanthropic Fund and CAPES Foundation Grant 5758/12-2 (to M. P. P.). The authors declare that they have no conflicts of interest with the contents of this article.
- AD
- Alzheimer disease
- Aβ
- β-amyloid
- apoE
- apolipoprotein E
- LXR
- liver X receptor
- RXR
- retinoid X receptor
- PPARγ
- peroxisome proliferator-activated receptor γ
- qPCR
- quantitative PCR
- DMSO
- dimethyl sulfoxide
- ANOVA
- analysis of variance
- pio
- pioglitazone
- GW
- GW3965
- APP
- amyloid precursor protein.
References
- 1. Mawuenyega K. G., Sigurdson W., Ovod V., Munsell L., Kasten T., Morris J. C., Yarasheski K. E., Bateman R. J. (2010) Decreased clearance of CNS β-amyloid in Alzheimer's disease. Science 330, 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wildsmith K. R., Holley M., Savage J. C., Skerrett R., Landreth G. E. (2013) Evidence for impaired amyloid β clearance in Alzheimer's disease. Alzheimers. Res. Ther. 5, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Castellano J. M., Kim J., Stewart F. R., Jiang H., DeMattos R. B., Patterson B. W., Fagan A. M., Morris J. C., Mawuenyega K. G., Cruchaga C., Goate A. M., Bales K. R., Paul S. M., Bateman R. J., Holtzman D. M. (2011) Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 3, 89ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bales K. R., Liu F., Wu S., Lin S., Koger D., DeLong C., Hansen J. C., Sullivan P. M., Paul S. M. (2009) Human APOE isoform-dependent effects on brain β-amyloid levels in PDAPP transgenic mice. J. Neurosci. 29, 6771–6779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deane R., Sagare A., Hamm K., Parisi M., Lane S., Finn M. B., Holtzman D. M., Zlokovic B. V. (2008) apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Invest. 118, 4002–4013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Corder E. H., Saunders A. M., Strittmatter W. J., Schmechel D. E., Gaskell P. C., Small G. W., Roses A. D., Haines J. L., Pericak-Vance M. A. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923 [DOI] [PubMed] [Google Scholar]
- 7. Schmechel D. E., Saunders A. M., Strittmatter W. J., Crain B. J., Hulette C. M., Joo S. H., Pericak-Vance M. A., Goldgaber D., Roses A. D. (1993) Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 90, 9649–9653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holtzman D. M., Herz J., Bu G. (2012) Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiang Q., Lee C. Y., Mandrekar S., Wilkinson B., Cramer P., Zelcer N., Mann K., Lamb B., Willson T. M., Collins J. L., Richardson J. C., Smith J. D., Comery T. A., Riddell D., Holtzman D. M., Tontonoz P., Landreth G. E. (2008) ApoE promotes the proteolytic degradation of Aβ. Neuron 58, 681–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee C. Y., Tse W., Smith J. D., Landreth G. E. (2012) Apolipoprotein E promotes β-amyloid trafficking and degradation by modulating microglial cholesterol levels. J. Biol. Chem. 287, 2032–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koldamova R. P., Lefterov I. M., Staufenbiel M., Wolfe D., Huang S., Glorioso J. C., Walter M., Roth M. G., Lazo J. S. (2005) The liver X receptor ligand T0901317 decreases amyloid β production in vitro and in a mouse model of Alzheimer's disease. J. Biol. Chem. 280, 4079–4088 [DOI] [PubMed] [Google Scholar]
- 12. Wahrle S. E., Jiang H., Parsadanian M., Kim J., Li A., Knoten A., Jain S., Hirsch-Reinshagen V., Wellington C. L., Bales K. R., Paul S. M., Holtzman D. M. (2008) Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Invest. 118, 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Donkin J. J., Stukas S., Hirsch-Reinshagen V., Namjoshi D., Wilkinson A., May S., Chan J., Fan J., Collins J., Wellington C. L. (2010) ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J. Biol. Chem. 285, 34144–34154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lehmann J. M., Kliewer S. A., Moore L. B., Smith-Oliver T. A., Oliver B. B., Su J. L., Sundseth S. S., Winegar D. A., Blanchard D. E., Spencer T. A., Willson T. M. (1997) Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J. Biol. Chem. 272, 3137–3140 [DOI] [PubMed] [Google Scholar]
- 15. Tall A. R. (2008) Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J. Intern. Med. 263, 256–273 [DOI] [PubMed] [Google Scholar]
- 16. Hong C., Tontonoz P. (2014) Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat. Rev. Drug Discov. 13, 433–444 [DOI] [PubMed] [Google Scholar]
- 17. Burns M. P., Vardanian L., Pajoohesh-Ganji A., Wang L., Cooper M., Harris D. C., Duff K., Rebeck G. W. (2006) The effects of ABCA1 on cholesterol efflux and Aβ levels in vitro and in vivo. J. Neurochem. 98, 792–800 [DOI] [PubMed] [Google Scholar]
- 18. Riddell D. R., Zhou H., Comery T. A., Kouranova E., Lo C. F., Warwick H. K., Ring R. H., Kirksey Y., Aschmies S., Xu J., Kubek K., Hirst W. D., Gonzales C., Chen Y., Murphy E., Leonard S., Vasylyev D., Oganesian A., Martone R. L., Pangalos M. N., Reinhart P. H., Jacobsen J. S. (2007) The LXR agonist TO901317 selectively lowers hippocampal Aβ42 and improves memory in the Tg2576 mouse model of Alzheimer's disease. Mol. Cell. Neurosci. 34, 621–628 [DOI] [PubMed] [Google Scholar]
- 19. Lefterov I., Bookout A., Wang Z., Staufenbiel M., Mangelsdorf D., Koldamova R. (2007) Expression profiling in APP23 mouse brain: inhibition of Aβ amyloidosis and inflammation in response to LXR agonist treatment. Mol. Neurodegener. 2, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fitz N. F., Cronican A., Pham T., Fogg A., Fauq A. H., Chapman R., Lefterov I., Koldamova R. (2010) Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J. Neurosci. 30, 6862–6872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vanmierlo T., Rutten K., Dederen J., Bloks V. W., van Vark-van der Zee L. C., Kuipers F., Kiliaan A., Blokland A., Sijbrands E. J., Steinbusch H., Prickaerts J., Lütjohann D., Mulder M. (2011) Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol. Aging 32, 1262–1272 [DOI] [PubMed] [Google Scholar]
- 22. Wesson D. W., Borkowski A. H., Landreth G. E., Nixon R. A., Levy E., Wilson D. A. (2011) Sensory network dysfunction, behavioral impairments, and their reversibility in an Alzheimer's β-amyloidosis mouse model. J. Neurosci. 31, 15962–15971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Terwel D., Steffensen K. R., Verghese P. B., Kummer M. P., Gustafsson J.-Å., Holtzman D. M., Heneka M. T. (2011) Critical role of astroglial apolipoprotein E and liver X receptor-α expression for microglial Aβ phagocytosis. J. Neurosci. 31, 7049–7059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cui W., Sun Y., Wang Z., Xu C., Peng Y., Li R. (2012) Liver X receptor activation attenuates inflammatory response and protects cholinergic neurons in APP/PS1 transgenic mice. Neuroscience 210, 200–210 [DOI] [PubMed] [Google Scholar]
- 25. Fitz N. F., Castranio E. L., Carter A. Y., Kodali R., Lefterov I., Koldamova R. (2014) Improvement of memory deficits and amyloid-β clearance in aged APP23 mice treated with a combination of anti-amyloid-β antibody and LXR agonist. J. Alzheimers. Dis. 41, 535–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yue L., Mazzone T. (2009) Peroxisome proliferator-activated receptor γ stimulation of adipocyte ApoE gene transcription mediated by the Liver receptor X pathway. J. Biol. Chem. 284, 10453–10461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mandrekar-Colucci S., Karlo J. C., Landreth G. E. (2012) Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease. J. Neurosci. 32, 10117–10128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Daniel B., Nagy G., Hah N., Horvath A., Czimmerer Z., Poliska S., Gyuris T., Keirsse J., Gysemans C., Van Ginderachter J. A., Balint B. L., Evans R. M., Barta E., Nagy L. (2014) The active enhancer network operated by liganded RXR supports angiogenic activity in macrophages. Genes Dev. 28, 1562–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cramer P. E., Cirrito J. R., Wesson D. W., Lee C. Y., Karlo J. C., Zinn A. E., Casali B. T., Restivo J. L., Goebel W. D., James M. J., Brunden K. R., Wilson D. A., Landreth G. E. (2012) ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Skerrett R., Malm T., Landreth G. (2014) Nuclear receptors in neurodegenerative diseases. Neurobiol. Dis. 72, 104–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heneka M. T., O'Banion M. K., Terwel D., Kummer M. P. (2010) Neuroinflammatory processes in Alzheimer's disease. J. Neural Transm. 117, 919–947 [DOI] [PubMed] [Google Scholar]
- 32. Hickman S. E., Allison E. K., El Khoury J. (2008) Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer's disease mice. J. Neurosci. 28, 8354–8360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mandrekar-Colucci S., Landreth G. E. (2010) Microglia and inflammation in Alzheimer's disease. CNS Neurol. Disord. Drug Targets 9, 156–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chawla A. (2010) Control of macrophage activation and function by PPARs. Circ. Res. 106, 1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chinetti-Gbaguidi G., Staels B. (2011) Macrophage polarization in metabolic disorders: functions and regulation. Curr. Opin. Lipidol. 22, 365–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yamanaka M., Ishikawa T., Griep A., Axt D., Kummer M. P., Heneka M. T. (2012) PPARγ/RXRα-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J. Neurosci. 32, 17321–17331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saijo K., Crotti A., Glass C. K. (2013) Regulation of microglia activation and deactivation by nuclear receptors. Glia 61, 104–111 [DOI] [PubMed] [Google Scholar]
- 38. Zelcer N., Khanlou N., Clare R., Jiang Q., Reed-Geaghan E. G., Landreth G. E., Vinters H. V., Tontonoz P. (2007) Attenuation of neuroinflammation and Alzheimer's disease pathology by liver x receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 10601–10606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. A-Gonzalez N., Bensinger S. J., Hong C., Beceiro S., Bradley M. N., Zelcer N., Deniz J., Ramirez C., Díaz M., Gallardo G., de Galarreta C. R., Salazar J., Lopez F., Edwards P., Parks J., Andujar M., Tontonoz P., Castrillo A. (2009) Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 31, 245–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ghisletti S., Huang W., Ogawa S., Pascual G., Lin M.-E., Willson T. M., Rosenfeld M. G., Glass C. K. (2007) Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARγ. Mol. Cell 25, 57–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bensinger S. J., Tontonoz P. (2008) Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 454, 470–477 [DOI] [PubMed] [Google Scholar]
- 42. Shen C. L., Murphy R. M. (1995) Solvent effects on self-assembly of β-amyloid peptide. Biophys. J. 69, 640–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hu J., Liu C.-C., Chen X.-F., Zhang Y.-W., Xu H., Bu G. (2015) Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Aβ metabolism in apoE4-targeted replacement mice. Mol. Neurodegener. 10, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yan Q., Zhang J., Liu H., Babu-Khan S., Vassar R., Biere A. L., Citron M., Landreth G. (2003) Anti-inflammatory drug therapy alters β-amyloid processing and deposition in an animal model of Alzheimer's disease. J. Neurosci. 23, 7504–7509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lacombe P., Mathews P. M., Schmidt S. D., Breidert T., Heneka M. T., Landreth G. E., Feinstein D. L., Galea E. (2004) Effect of anti-inflammatory agents on transforming growth factor β over-expressing mouse brains: a model revised. J. Neuroinflammation 1, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pedersen W. A., McMillan P. J., Kulstad J. J., Leverenz J. B., Craft S., Haynatzki G. R. (2006) Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp. Neurol. 199, 265–273 [DOI] [PubMed] [Google Scholar]
- 47. Toledo E. M., Inestrosa N. C. (2010) Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1ΔE9 mouse model of Alzheimer's disease. Mol. Psychiatry 15, 272–285, 228 [DOI] [PubMed] [Google Scholar]
- 48. O'Reilly J.-A., Lynch M. (2011) Rosiglitazone improves spatial memory and decreases insoluble Aβ1–42 in APP/PS1 Mice. J. Neuroimmune Pharmacol. 7, 140–144 [DOI] [PubMed] [Google Scholar]
- 49. Heneka M. T., Sastre M., Dumitrescu-Ozimek L., Hanke A., Dewachter I., Kuiperi C., O'Banion K., Klockgether T., Van Leuven F., Landreth G. E. (2005) Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1–42 levels in APPV717I transgenic mice. Brain 128, 1442–1453 [DOI] [PubMed] [Google Scholar]
- 50. Escribano L., Simón A.-M., Gimeno E., Cuadrado-Tejedor M., López de Maturana R., García-Osta A., Ricobaraza A., Pérez-Mediavilla A., Del Río J., Frechilla D. (2010) Rosiglitazone rescues memory impairment in Alzheimer's transgenic mice: mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology 35, 1593–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sastre M., Dewachter I., Rossner S., Bogdanovic N., Rosen E., Borghgraef P., Evert B. O., Dumitrescu-Ozimek L., Thal D. R., Landreth G., Walter J., Klockgether T., van Leuven F., Heneka M. T. (2006) Nonsteroidal anti-inflammatory drugs repress β-secretase gene promoter activity by the activation of PPARγ. Proc. Natl. Acad. Sci. U.S.A. 103, 443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Searcy J. L., Phelps J. T., Pancani T., Kadish I., Popovic J., Anderson K. L., Beckett T. L., Murphy M. P., Chen K.-C., Blalock E. M., Landfield P. W., Porter N. M., Thibault O. (2012) Long-term pioglitazone treatment improves learning and attenuates pathological markers in a mouse model of Alzheimer's disease. J. Alzheimers. Dis. 30, 943–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nicolakakis N., Aboulkassim T., Ongali B., Lecrux C., Fernandes P., Rosa-Neto P., Tong X.-K., Hamel E. (2008) Complete rescue of cerebrovascular function in aged Alzheimer's disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor γ agonist. J. Neurosci. 28, 9287–9296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Papadopoulos P., Rosa-Neto P., Rochford J., Hamel E. (2013) Pioglitazone improves reversal learning and exerts mixed cerebrovascular effects in a mouse model of Alzheimer's disease with combined amyloid-β and cerebrovascular pathology. PLoS ONE 8, e68612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Denner L. A., Rodriguez-Rivera J., Haidacher S. J., Jahrling J. B., Carmical J. R., Hernandez C. M., Zhao Y., Sadygov R. G., Starkey J. M., Spratt H., Luxon B. A., Wood T. G., Dineley K. T. (2012) Cognitive enhancement with rosiglitazone links the hippocampal PPARγ and ERK MAPK signaling pathways. J. Neurosci. 32, 16725–35a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hu Y., Yang Y., Yu Y., Wen G., Shang N., Zhuang W., Lu D., Zhou B., Liang B., Yue X., Li F., Du J., Bu X. (2013) Synthesis and identification of new flavonoids targeting liver X receptor β involved pathway as potential facilitators of Aβ clearance with reduced lipid accumulation. J. Med. Chem. 56, 6033–6053 [DOI] [PubMed] [Google Scholar]
- 57. Savage J. C., Jay T., Goduni E., Quigley C., Mariani M. M., Malm T., Ransohoff R. M., Lamb B. T., Landreth G. E. (2015) Nuclear receptors license phagocytosis by trem2+ myeloid cells in mouse models of Alzheimer's disease. J. Neurosci. 35, 6532–6543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gosselin D., Glass C. K. (2014) Epigenomics of macrophages. Immunol. Rev. 262, 96–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Masciopinto F., Di Pietro N., Corona C., Bomba M., Pipino C., Curcio M., Di Castelnuovo A., Ciavardelli D., Silvestri E., Canzoniero L. M., Sekler I., Pandolfi A., Sensi S. L. (2012) Effects of long-term treatment with pioglitazone on cognition and glucose metabolism of PS1-KI, 3xTg-AD, and wild-type mice. Cell Death Dis. 3, e448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rodriguez-Rivera J., Denner L., Dineley K. T. (2011) Rosiglitazone reversal of Tg2576 cognitive deficits is independent of peripheral gluco-regulatory status. Behav. Brain Res. 216, 255–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Baranowski M. (2008) Biological role of liver X receptors. J. Physiol. Pharmacol. 59, 31–55 [PubMed] [Google Scholar]
- 62. Li A. C., Glass C. K. (2004) PPAR- and LXR-dependent pathways controlling lipid metabolism and the development of atherosclerosis. J. Lipid Res. 45, 2161–2173 [DOI] [PubMed] [Google Scholar]
- 63. Beyer T. P., Schmidt R. J., Foxworthy P., Zhang Y., Dai J., Bensch W. R., Kauffman R. F., Gao H., Ryan T. P., Jiang X.-C., Karathanasis S. K., Eacho P. I., Cao G. (2004) Coadministration of a liver X receptor agonist and a peroxisome proliferator activator receptor-α agonist in mice: effects of nuclear receptor interplay on high-density lipoprotein and triglyceride metabolism in vivo. J. Pharmacol. Exp. Ther. 309, 861–868 [DOI] [PubMed] [Google Scholar]
- 64. Schoonjans K., Peinado-Onsurbe J., Lefebvre A. M., Heyman R. A., Briggs M., Deeb S., Staels B., Auwerx J. (1996) PPARα and PPARγ activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J. 15, 5336–5348 [PMC free article] [PubMed] [Google Scholar]
- 65. Evans R. M., Barish G. D., Wang Y.-X. (2004) PPARs and the complex journey to obesity. Nat. Med. 10, 355–361 [DOI] [PubMed] [Google Scholar]
- 66. Miao B., Zondlo S., Gibbs S., Cromley D., Hosagrahara V. P., Kirchgessner T. G., Billheimer J., Mukherjee R. (2004) Raising HDL cholesterol without inducing hepatic steatosis and hypertriglyceridemia by a selective LXR modulator. J. Lipid Res. 45, 1410–1417 [DOI] [PubMed] [Google Scholar]
- 67. Quinet E. M., Savio D. A., Halpern A. R., Chen L., Schuster G. U., Gustafsson J. A., Basso M. D., Nambi P. (2006) Liver X receptor (LXR)-β regulation in LXRα-deficient mice: implications for therapeutic targeting. Mol. Pharmacol. 70, 1340–1349 [DOI] [PubMed] [Google Scholar]
- 68. Kruit J. K., Plösch T., Havinga R., Boverhof R., Groot P. H., Groen A. K., Kuipers F. (2005) Increased fecal neutral sterol loss upon liver X receptor activation is independent of biliary sterol secretion in mice. Gastroenterology 128, 147–156 [DOI] [PubMed] [Google Scholar]
- 69. van der Hoorn J., Lindén D., Lindahl U., Bekkers M., Voskuilen M., Nilsson R., Oscarsson J., Lindstedt E., Princen H. (2011) Low dose of the liver X receptor agonist, AZ876, reduces atherosclerosis in APOE*3Leiden mice without affecting liver or plasma triglyceride levels. Br. J. Pharmacol. 162, 1553–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ye J. M., Doyle P. J., Iglesias M. A., Watson D. G., Cooney G. J., Kraegen E. W. (2001) Peroxisome proliferator-activated receptor (PPAR)-α activation lowers muscle lipids and improves insulin sensitivity in high fat-fed rats: comparison with PPAR-γ activation. Diabetes 50, 411–417 [DOI] [PubMed] [Google Scholar]