

Background: Vaccination against Tau reduces pathology in vivo; however, the mechanism of action remains unclear.
Results: Antibodies promote uptake of Tau fibrils in microglia or block uptake in neurons in a size- and epitope-dependent manner.
Conclusion: Antibodies have multiple potential mechanisms.
Significance: Establishing specific mechanisms of antibody activity may help in design and optimization of more effective agents.
Keywords: immunotherapy, microglia, neuron, Tau protein (Tau), tauopathy, clearance
Abstract
Tauopathies are neurodegenerative diseases characterized by accumulation of Tau amyloids, and include Alzheimer disease and certain frontotemporal dementias. Trans-neuronal propagation of amyloid mediated by extracellular Tau may underlie disease progression. Consistent with this, active and passive vaccination studies in mouse models reduce pathology, although by unknown mechanisms. We previously reported that intracerebroventricular administration of three anti-Tau monoclonal antibodies (HJ8.5, HJ9.3, and HJ9.4) reduces pathology in a model overexpressing full-length mutant (P301S) human Tau. We now study effects of these three antibodies and a negative control antibody (HJ3.4) on Tau aggregate uptake into BV2 microglial-like cells and primary neurons. Antibody-independent Tau uptake into BV2 cells was blocked by heparin, consistent with a previously described role for heparan sulfate proteoglycans. Two therapeutic antibodies (HJ8.5 and HJ9.4) promoted uptake of full-length Tau fibrils into microglia via Fc receptors. Surprisingly, HJ9.3 promoted uptake of fibrils composed of the Tau repeat domain or Alzheimer disease-derived Tau aggregates, but failed to influence full-length recombinant Tau fibrils. Size fractionation of aggregates showed that antibodies preferentially promote uptake of larger oligomers (n ≥∼20-mer) versus smaller oligomers (n ∼10-mer) or monomer. No antibody inhibited uptake of full-length recombinant fibrils into primary neurons, but HJ9.3 blocked neuronal uptake of Tau repeat domain fibrils and Alzheimer disease-derived Tau. Antibodies thus have multiple potential mechanisms, including clearance via microglia and blockade of neuronal uptake. However these effects are epitope- and aggregate size-dependent. Establishing specific mechanisms of antibody activity in vitro may help in design and optimization of agents that are more effective in vivo.
Introduction
Tauopathies are neurodegenerative diseases characterized by the accumulation within neurons of the microtubule associated protein Tau (1). These include Alzheimer disease (AD),3 which also features amyloid-β (Aβ) accumulation, and myriad dementia syndromes associated only with Tau accumulation. Tau pathology correlates with other measures of disease progression including neuronal dysfunction, synaptic loss, and functional decline (2–5), and progresses along neuroanatomical pathways in disease-specific patterns (6–8). The mechanism of progression is incompletely understood, but we and others have posited that Tau aggregates escape from diseased neurons into the extracellular space prior to entering adjacent, or synaptically connected cells. Aggregates then convert monomeric Tau to an amyloid by acting as a conformational template. This may mediate the relentless spread of Tau pathology in humans (9). Importantly, therapeutic interventions that prevent the uptake of aggregate seeds into naive neurons or promote microglial clearance might inhibit disease progression.
Microglia, the brain's resident immune cells, might clear antibody-bound Tau from the extracellular space. A recent study reported that microglia display modest levels of phagocytic capacity for Tau oligomers that are increased by LPS activation (10). It is not clear how microglia might clear antibody-bound Tau. Considering that pro-inflammatory cytokines are chronically heightened in the aging brain (11) as well as neurodegenerative diseases (12–14), the typical disease state may be sufficient to induce phagocytic activity in microglia.
The model of transcellular propagation suggests that extracellular Tau might be vulnerable to antibody-mediated therapies. Importantly, we and others have observed that passive immunization with anti-Tau antibodies reduces pathology in several mouse models of tauopathy (15–19), but the mechanisms are unknown. Several theories have been proposed, including inhibition or reversal of fibril formation (20, 21), clearance of intraneuronal fibrils via the lysosome (22), and blocking neuronal uptake of Tau seeds (18). We hypothesized that therapeutic antibodies might promote uptake of Tau aggregates into microglia. We have tested this idea using two forms of recombinant Tau protein fibrillized in vitro, as well as AD-derived Tau assemblies, evaluating the effects of antibodies in cell culture systems.
Experimental Procedures
Tau Expression, Purification, Fibrillization, and Labeling
Full-length 2N4R or repeat domain (RD) Tau, comprising amino acids 243–375 with a C-terminal HA tag (YPYDVPDYA) (23), was subcloned into pRK172. Recombinant 2N4R and RD Tau were prepared as described previously and stored lyophilized at −80 °C (24). Tau fibrillization was induced with either heparin or octadecyl sulfate (ODS). For heparin-induced fibrillization, a final concentration of 8 μm lyophilized Tau was reconstituted in 2 mm DTT for 1 h at room temperature followed by incubation without agitation at 37 °C in 10 mm HEPES, pH 7.4, 100 mm NaCl, and 8 μm heparin. RD was incubated for 24 h, whereas 2N4R was incubated for 1 week for fibrillization. Alternatively, 2N4R Tau was fibrillized with ODS by reconstituting a final concentration of 3 μm lyophilized Tau in 5 μm DTT for 1 h at room temperature followed by incubation without agitation at 37 °C in 10 mm HEPES, pH 7.4, 100 mm NaCl, and 50 μm ODS. To fluorescently label Tau protein, 1.6 nmol of Tau protein or equivalent volume of buffer alone was incubated with 25 μg of Alexa Fluor succinimidyl ester dye for 1 h at room temperature and then overnight at 4 °C with end-over-end rotation. Unconjugated dye was removed by dialysis in PBS and then quenched with 100 mm glycine for 1 h. Immediately before use, fibrils were sonicated using the Qsonica Q700 at amplitude 50 for 30 s.
Antibodies
HJ9.3 and HJ9.4 monoclonal antibodies recognize mouse and human Tau, whereas HJ8.5 recognizes only human Tau. Both HJ8.5 and HJ9.4 bind the N terminus (epitope residues 25–30 and 7–13 of National Center for Biotechnology Information (NCBI) reference sequence NP_005901, respectively). HJ9.3 antibody recognizes the third repeat domain (epitope residues 306–320 of NCBI reference sequence NP_005901). As a negative control, we used HJ3.4 mouse monoclonal antibody, which recognizes the N-terminal region of human Aβ peptide (epitope residues 1–16). All HJ antibodies investigated here are monoclonal and of the IgG2b isotype except for HJ8.5, which is of the IgG2ab subtype. Iba1 is a polyclonal rabbit antibody commercially available from Wako Chemicals (code number 019-19741). Polyclonal rabbit anti-Tau antibody used for Western blot is commercially available from Abcam (ab64139).
Cell Culture
BV2 cells were cultured in DMEM supplemented with 2% FBS, 100 μg/ml penicillin, and 100 μg/ml streptomycin. Cultures were maintained in a humidified atmosphere of 5% CO2 at 37 °C. Primary neuron cultures were prepared from the cortex of embryonic day 18.5 mouse embryos collected from CD1 timed-pregnant females purchased from Charles River. Cortical tissue was isolated and digested with 2 mg/ml papain and 20 units/ml DNase I. Neurons were seeded at 500,000 cells/ml in a 96-well plate precoated with 10 μg/ml poly-d-lysine and maintained in Neurobasal medium containing serum-free B-27, GlutaMAX, and penicillin/streptomycin. Neurons were used for experiments on DIV 4.
Tau Extraction from Brain and Size Exclusion Chromatography (SEC)
This study used only archival, de-identified postmortem brain tissue samples from autopsies performed with informed consent of each patient or relative via procedures approved by the relevant institutional committees at Washington University in St. Louis. Frontal lobe of normal control or AD brain tissue was homogenized in TBS buffer containing protease and phosphatase inhibitor cocktail (Roche Applied Science) using a probe sonicator (Omni Sonic Ruptor 250) at 4 °C. Samples were centrifuged at 6000 × g for 10 min at 4 °C to remove cellular debris. Supernatant was aliquoted, snap-frozen, and stored at −80 °C until use. Immunopurification of Tau was performed with monoclonal antibodies HJ9.3 and HJ8.5 at a ratio of 1 μg of antibody per 50 μg of total protein, incubating overnight at 4 °C rotating end-over-end. 150 μl of 50% slurry protein G agarose beads (Pierce) were washed three times with TBS buffer and then added to each 1 ml of antibody/brain homogenate. Antibody/brain/bead slurry was incubated overnight at 4 °C with end-over-end rotation and then centrifuged at 1000 × g for 3 min, and the supernatant was discarded. Beads were washed with cold Ag/Ab Binding Buffer, pH 8.0 (Thermo Scientific) four times. Tau bound to beads was eluted in 100 μl of low pH elution buffer (Thermo Scientific) and neutralized with 10 μl of Tris base, pH 8.5. To maximize recovery, Tau was eluted a second time with 50 μl of elution buffer and neutralized with 5 μl of Tris base, pH 8.5, for a total recovery volume of 165 μl. Eluted Tau was then labeled with 20 μg of Alexa Fluor 647 by incubating overnight at 4 °C with end-over-end rotation. The next day, unbound dye was quenched with 0.1 m glycine and centrifuged at 40,000 × g for 10 min, and the supernatant was loaded onto a HiPrep 16/60 Sephacryl S-500 HR column (GE Healthcare). SEC fractions were evaluated for their protein content by Alexa Fluor 647 fluorescence as measured by a Tecan M1000 fluorescence plate reader and by Micro BCA assay (Thermo Scientific).
Immunofluorescence and Microscopy
For microscopy imaging experiments, BV2 cells were seeded at 80,000 cells/ml. The next day, 50 nm 2N4R Tau fibrils conjugated to Alexa Fluor 488 were incubated with 5 μg/ml antibodies, as indicated, for 2 h at room temperature with gentle agitation. Tau-antibody complex was applied to BV2 cells for 1 h. Following uptake, cells were trypsinized to remove Tau bound to the cell membrane and replated on acid-etched poly-d-lysine-coated glass coverslips for microscopy imaging. BV2 cells were allowed to recover for 2 h and then washed once with PBS and fixed with 4% paraformaldehyde. BV2 cells were demarcated by immunostaining with rabbit anti-Iba1 (1 μg/ml; Wako Chemicals) primary antibody and Alexa Fluor 647-conjugated goat anti-rabbit IgG secondary antibody (4 μg/ml; Invitrogen). Treatment antibodies were identified by Alexa Fluor 546-conjugated goat anti-mouse IgG secondary antibody (4 μg/ml; Invitrogen). Following immunostaining, coverslips were mounted using Fluoromount-G (SouthernBiotech), sealed with clear nail enamel, and imaged using Zeiss LSM 5 PASCAL Vario Two RGB confocal system coupled to a Zeiss Axiovert 200 microscope.
Recombinant Tau Uptake Assays
BV2 cells were plated at 80,000 cells/ml in a 96-well plate. The next day, 50 nm Tau fibrils conjugated to Alexa Fluor 647 were sonicated and incubated with antibodies, as indicated, for 2 h at room temperature with gentle agitation. Following preincubation, Tau-antibody complexes were applied to BV2 cells for 30 min at 37 °C. Cells were harvested with 0.25% trypsin for 5 min and then centrifuged at 1000 × g for 5 min, the media were removed, and the pellets were resuspended in flow cytometry buffer (10× Hanks' balanced salt solution diluted to 1× with PBS plus 1% FBS and 1 mm EDTA). Cells were counted with a MACSQuant VYB flow cytometer (Miltenyi Biotec) gating for the single cell population, as identified by forward and side scatter profiles. Each experiment was conducted in triplicate, counting 10,000 single cells per experiment. Uptake experiments involving primary neurons were conducted similarly, except that neurons were plated at 500,000 cells/ml in a 96-well plate. Following preincubation, Tau-antibody complexes were applied to neurons for 3 h. Cells were harvested and analyzed identical to BV2 cells.
Immunoprecipitation and Western Blot of Recombinant Tau Protein
Immunoprecipitations were performed using components of a Pierce Classic IP kit. 2N4R Tau was fibrillized in vitro using ODS and then conjugated to Alexa Fluor 647 as described above. 1 μg of protein was diluted in 300 μl of IP Lysis/Wash Buffer (Input) and incubated with 15 μg of antibody as indicated for 2 h at room temperature with end-over-end rotation. Meanwhile, protein A/G UltraLink resin (Pierce 53132) was washed with IP Lysis/Wash Buffer and then incubated with 3% BSA diluted in IP Lysis/Wash Buffer for 1 h at room temperature with end-over-end rotation to block nonspecific interactions with the resin. Excess BSA solution was removed from IP columns by centrifugation, and the Tau-antibody immune complex was incubated with resin for 1 h at room temperature with end-over-end rotation. Following immune complex capture, unbound Tau was removed from the IP column by centrifugation and identified as flow-through. Tau captured by immune complex was eluted from resin by heating with reducing sample buffer and identified as elution. Equivalent volumes of input, flow-through, and elution were separated by SDS-PAGE (NuPAGE 4–12% Bis-Tris gel) and transferred to nitrocellulose membrane. Membrane was probed with rabbit anti-Tau antibody (0.04 μg/ml) and imaged using the Odyssey infrared imaging system (LI-COR).
Statistical Analysis
Tests for statistical analysis were performed using GraphPad Prism 6 software. Data are represented as mean ± S.D. Group mean values were analyzed by two-way analysis of variance to test for significance with Dunnett's test to correct for multiple comparisons.
Results
Anti-Tau Antibodies Promote Fibril Uptake by BV2 Cells
Intracerebroventricular infusion (18) or peripheral administration (19) of three monoclonal anti-Tau antibodies reduces pathology in mice that overexpress full-length human Tau containing the disease-associated P301S mutation (18). The mechanisms of this reduction are not well understood, but may involve antibody-Tau complex formation in the extracellular space. This is predicted by the hypothesis that transcellular movement of Tau aggregates propagates pathology among neurons. This involves the release of Tau assemblies into the extracellular space and uptake into a neighboring neuron, where they serve as templates for amyloid formation by native Tau (9). It has been unknown whether the therapeutic antibodies we previously described function by promoting microglial uptake, blocking neuronal uptake, or both (18). To test these ideas, we developed models based on BV2 cells and primary cortical neurons. BV2 are immortalized murine microglial cells that mimic many behaviors of primary microglia and exhibit a robust phagocytic response (25–27). Although they do not recapitulate all the activities of primary microglial cells, their highly reproducible phagocytic behavior in vitro made them preferable for these studies, which required minimal experimental variability.
We formed full-length 2N4R Tau fibrils in vitro with heparin and conjugated them to Alexa Fluor 488. We sonicated the fibrils in a water bath, diluted them to 50 nm, and incubated them with 5 μg/ml antibody as indicated. Next we applied the Tau-antibody complexes to the media of the BV2 cells. After 3 h, we removed Tau bound to the cell surface via trypsin treatment, and replated the cells on poly-d-lysine-coated glass coverslips. After a 2-h recovery, we washed the cells once with PBS and fixed them in 4% paraformaldehyde/PBS. We then stained them with rabbit anti-Iba1 primary antibody followed by Alexa Fluor 647-labeled anti-rabbit IgG secondary antibody to detect the cell surface and Alexa Fluor 546-labeled anti-mouse IgG secondary antibody to detect the treatment antibody. Weinitially used confocal microscopy to monitor uptake. We observed more uptake of Tau fibrils after preincubation with Tau-specific antibodies (Fig. 1). We quantified uptake using flow cytometry. In this case we formed full-length 2N4R Tau fibrils using ODS as an inducer (so as not to impact fibril binding to the cell surface) and conjugated them to Alexa Fluor 647. After sonication as before, we incubated fibrils with 0.01–10.0 μg/ml antibody prior to their application to BV2 cells. Following uptake, cells were washed once with PBS and trypsinized. Trypsinization removes any Tau bound to the plasma membrane and also dissociates the cells for flow cytometry analysis. Therefore, fluorescence signal measured by flow cytometry is indicative of uptake rather than binding to the cellular surface (28). We gated for single cell analysis. An Aβ-specific antibody, HJ3.4, served as a negative control. Preincubation with antibodies HJ8.5 and HJ9.4, but not HJ9.3, increased the median fluorescence intensity of the gated cells (Fig. 2A). To determine the effect of anti-Tau antibodies on neurons, we used a similar approach. We applied Tau-antibody complexes for 3 h to primary cortical neurons after 4 days in vitro. No antibody changed uptake of 2N4R Tau into neurons (Fig. 2B).
FIGURE 1.

Tau antibodies promote fibril uptake into BV2 cells. Full-length recombinant 2N4R Tau was fibrillized in vitro with heparin and then covalently conjugated to Alexa Fluor 488 (green). A–P, Tau was incubated with HJ3.4 (A–D), HJ8.5 (E–H), HJ9.3 (I–L), or HJ9.4 (M–P) for 2 h at room temperature with gentle agitation and then applied to BV2 cells for 1 h. Following uptake, cells were trypsinized to remove Tau bound to extracellular membrane and replated on poly-d-lysine-coated glass coverslips. Cells were allowed to recover for 2 h and then washed once with PBS, fixed with 4% paraformaldehyde, and immunostained with rabbit anti-Iba1 to identify the BV2 cell boundary (blue) and 546-conjugated goat anti-mouse IgG to detect the therapeutic antibody (red). Images were captured using confocal microscopy. Tau-directed antibodies promoted uptake of Tau fibrils. Arrowheads denote Tau aggregates and bound antibody.
FIGURE 2.
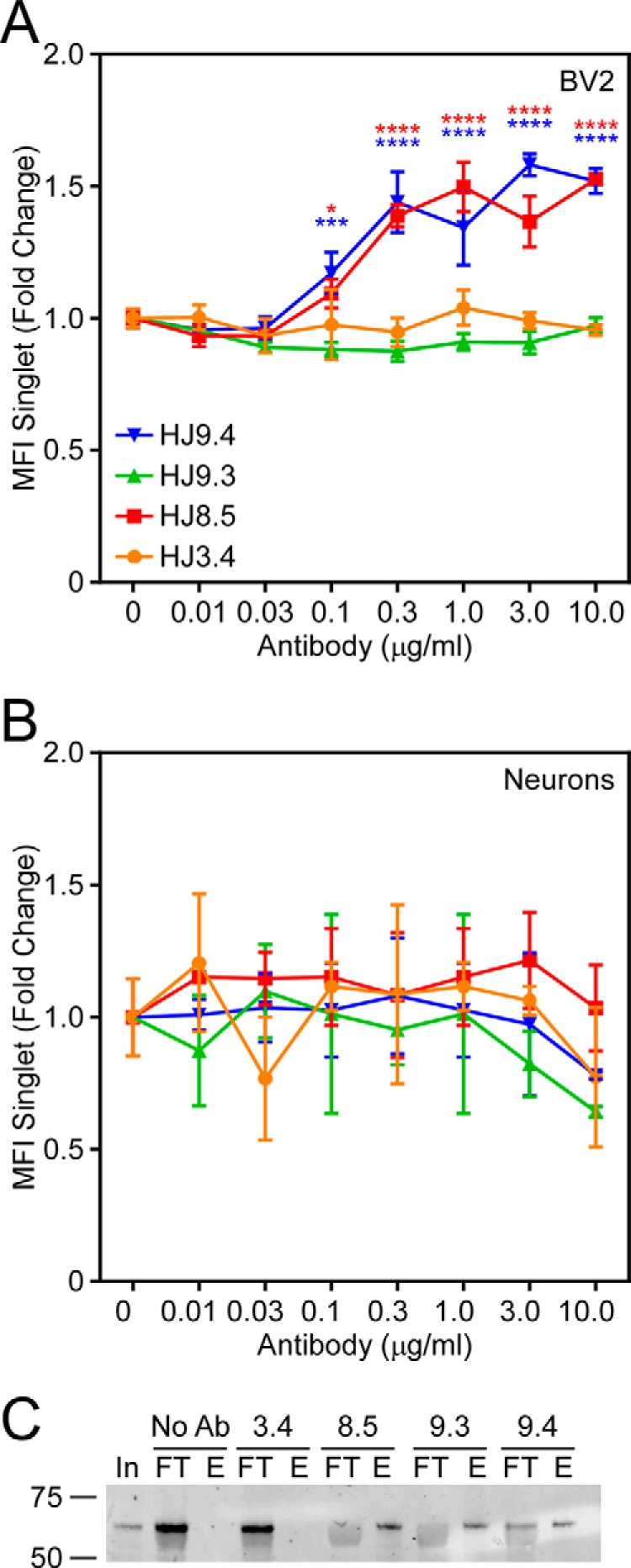
HJ8.5 and HJ9. 4, but not HJ9.3, promote uptake of 2N4R Tau in BV2 cells. 2N4R Tau was fibrillized in vitro with ODS and then covalently conjugated to Alexa Fluor 647. A and B, Tau was incubated with HJ3.4 (orange), HJ8.5 (red), HJ9.3 (green), or HJ9.4 (blue) and then applied to BV2 cells (A) or primary cortical neurons (B) at 37 °C. Cells were trypsinized and analyzed for Tau uptake by flow cytometry. HJ8.5 and HJ9.4, but not HJ3.4 or HJ9.3, promoted uptake of 2N4R Tau in BV2 cells. No antibodies had any effect on neuronal uptake of Tau aggregates. MFI, median fluorescence intensity. C, 2N4R Tau fibrils were immunoprecipitated using indicated antibodies. No Ab, no antibody; In, input; FT, flow-through; E, elution. Data are represented as mean ± S.D. Significance relative to HJ3.4 treatment, *, p < 0.05, ***, p < 0.001, ****, p < 0.0001.
The failure of HJ9.3 to promote BV2 uptake was surprising. Upon intracerebroventricular administration, HJ9.3 reduces Tau pathology in vivo, as do HJ8.5 and HJ9.4 (18). Although HJ8.5 and HJ9.4 bind Tau in the projection domain near the N terminus, HJ9.3 binds Tau in the third repeat domain of the aggregation-competent core of the protein. We hypothesized that the amino acids flanking the repeat domain might hinder the interactions necessary to promote Tau uptake into BV2 cells, due either to low avidity of the antibody to the full-length fibril or to reduced binding of the fibril-bound antibody to its cell surface receptors. To discriminate these two possibilities, we first tested the ability of HJ9.3 to bind fibrillized recombinant full-length Tau using immunoprecipitation. We conjugated fibrillized full-length Tau to Alexa Fluor 647 as described previously, incubating it alone or with each of the four antibodies described above. We captured the immune complexes using protein A/G UltraLink resin and analyzed the eluted protein and the flow-through fractions by Western blot. Upon SDS-PAGE, immunoprecipitated Tau fibrils resolved as monomer. All Tau-specific antibodies, but not negative control HJ3.4 or resin lacking antibody, equivalently immunoprecipitated full-length Tau fibrils (Fig. 2C). We concluded that HJ9.3 binds full-length Tau fibrils. We next tested whether the flanking amino acids of full-length Tau might somehow prevent fibril uptake in response to HJ9.3. We used the aggregation-competent RD of Tau, which nonetheless contains the epitope for HJ9.3. We formed RD fibrils in vitro, conjugated them to Alexa Fluor 647, and preincubated them with HJ3.4 or HJ9.3. Because RD lacks epitopes for HJ8.5 and HJ9.4, we did not study these antibodies. HJ9.3 promoted uptake of RD-647 fibrils in BV2 cells, whereas HJ3.4 had no effect (Fig. 3). Although the three anti-Tau antibodies had no effect on full-length 2N4R Tau fibril uptake in primary cultured neurons, surprisingly HJ9.3 blocked RD fibril uptake in these cells (Fig. 3). These results emphasize that despite the presence of an available epitope within a fibril, the same antibody can have distinct activities depending on the size of the fibrillar protein, and possibly its conformation. In this case, HJ9.3 had no effect on full-length Tau fibrils, whereas it potently blocked RD fibril uptake into neurons and promoted uptake into BV2 cells.
FIGURE 3.

HJ9.3 promotes uptake of RD fibrils in BV2 cells and inhibits uptake in primary cortical neurons. Peptide comprising the repeat domain of Tau was fibrillized in vitro with heparin and then covalently conjugated to Alexa Fluor 647. A–D, Tau was incubated with HJ3.4 (A and B) or HJ9.3 (C and D) and then applied to BV2 cells (A and C) or primary cortical neurons (B and D). HJ3.4 had no effect on Tau uptake in either cell type, but HJ9.3 promoted uptake of 4RD Tau in BV2 cells and inhibited uptake in primary cortical neurons. MFI, median fluorescence intensity. Data are represented as mean ± S.D. ***, p < 0.001, ****, p < 0.0001.
Antibody-dependent versus Antibody-independent Uptake into BV-2 Cells
We have previously described the role of heparan sulfate proteoglycans as mediators of spontaneous, antibody-independent uptake of full-length Tau fibrils into HEK293 cells and neurons (28). Fc receptors are expressed on the surface of immune cells and bind the antibody Fc region. Fc γ receptor (FcγR) specifically binds IgG, initiating a signaling cascade that triggers phagocytosis of antibody-bound antigens (29). We hypothesized that FcγR might facilitate uptake of antibody-Tau complexes in BV2 cells, as has been described for α-synuclein aggregates and Aβ aggregates (30, 31). To block the FcγR, we used an antibody specific to FcγII/III receptors, which bind antibodies with low affinity but high valency (29), as might be expected in an antibody-aggregate complex. We treated BV2 cells overnight with 60 ng/ml FcγR blocking antibody, and the next day we added 50 nm 2N4R-647 Tau fibrils preincubated with 5 μg/ml antibodies. HJ8.5 and HJ9.4 promoted uptake of 2N4R-647 fibrils, and treatment with FcγR blocking antibody eliminated this effect. FcγR blockade did not inhibit antibody-independent uptake of 2N4R Tau fibrils (Fig. 4). Thus although antibody-dependent uptake of Tau fibrils relieson FcγR, antibody-independent uptake uses a distinct mechanism.
FIGURE 4.
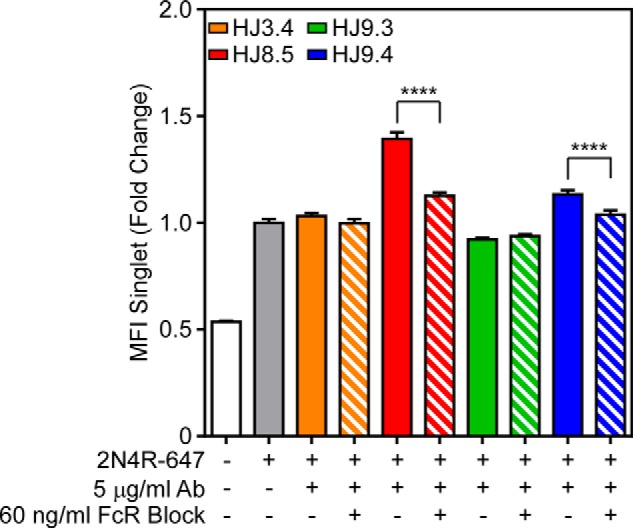
FcγRs facilitate antibody-mediated uptake of 2N4R Tau in BV2 cells. 2N4R Tau was fibrillized in vitro with ODS and then conjugated to Alexa Fluor 647. Tau was incubated with HJ3.4 (orange), HJ8.5 (red), HJ9.3 (green), or HJ9.4 (blue) and applied to BV2 cells that had been preincubated overnight with 60 ng/ml antibody specific to FcγR II/III. HJ8.5 and HJ9.4 promoted uptake of 2N4R Tau fibrils. This effect was abolished by preincubating cells with FcγR II/III-specific antibody. MFI, median fluorescence intensity. FcR, Fc receptor. Data are represented as mean ± S.D. ****, p < 0.0001.
To test whether antibody-independent uptake of Tau is mediated by HSPGs in BV2 cells, we incubated 2N4R-647 fibrils with heparin, F6, or polymeric dextran (PD). Although heparin binds Tau monomer to promote its fibrillization, it has no effect on state or stability of fibrils once they have formed, and blocks their interaction with HSPGs. This prevents fibril uptake into neurons, which is HSPG-dependent (28). F6 is a heparin mimetic that similarly binds Tau and prevents interaction with and uptake by HSPGs. PD is a negative control heparin mimetic that lacks the sulfation necessary for Tau interaction. HJ8.5 and HJ9.4 promoted uptake of 2N4R-647 in BV2 cells (Fig. 5A). Treatment of BV2 cells with heparin (Fig. 5C) and F6 (Fig. 5D), but not PD (Fig. 5B), inhibited uptake of Tau fibrils in the absence of antibody treatment (dark gray bars). Preincubation of Tau fibrils with HJ8.5 (red bars) and HJ9.4 (blue bars), but not HJ3.4 (orange bars) or HJ9.3 (green bars), overcame this inhibition (Fig. 5). Together, these data suggest that FcγR mediates antibody-dependent uptake of Tau fibrils, whereas HSPGs mediate antibody-independent uptake.
FIGURE 5.

HSPG-mediated uptake of 2N4R Tau is antibody-independent. 2N4R Tau was fibrillized in vitro with ODS and then conjugated to Alexa Fluor 647. A–D, Tau was incubated with HJ3.4 (orange), HJ8.5 (red), HJ9.3 (green), or HJ9.4 (blue) alone (A) or plus 1 μg/ml PD (B), heparin (C), or F6 (D). HJ8.5 and HJ9.4 alone promoted uptake of 2N4R Tau fibrils. PD had no effect on baseline uptake or antibody-mediated uptake of Tau fibrils. Heparin and F6 inhibited baseline uptake of Tau fibrils, but HJ8.5 and HJ9.4 rescued this decrease. MFI, median fluorescence intensity. Data are represented as mean ± S.D. *, p < 0.05, ***, p < 0.001, ****, p < 0.0001.
Antibodies Promote Uptake of AD-derived Large Fibrils, but Not Small Fibrils or Monomer
Fc receptor requires receptor cross-linking by multimeric antibody binding to trigger endocytosis. Therefore, we hypothesized that large aggregates might be more efficiently internalized by antibody-mediated uptake in BV2 cells. To test this, we fibrillized 2N4R Tau in vitro, conjugated the fibrils to Alexa Fluor 647, and then resolved them by SEC. We used molecular weight standards to estimate aggregate sizes as we have previously described (32). We estimated that we isolated monomer, ∼10-mer, and >20-mer (Fig. 6A). We normalized protein concentrations by Alexa Fluor 647 fluorescence and confirmed these measurements by Micro BCA analysis. We incubated equal amounts of Tau-enriched fractions (40 nm protein) with 5 μg/ml antibodies prior to application to BV2 cells. We then trypsinized cells and analyzed them by flow cytometry. HJ8.5 and HJ9.4 promoted uptake of aggregates, but not monomer (Fig. 6B). To test these effects on primary cultured neurons, we applied Tau-antibody complexes to cortical mouse neurons. HJ9.3 had a small inhibitory effect on uptake in neurons (Fig. 6C).
FIGURE 6.

Antibody effects on 2N4R and AD-derived Tau fibrils are size- and antibody-dependent. A, 2N4R Tau was fibrillized in vitro, conjugated to Alexa Fluor 647, and resolved by SEC (black). Additionally, Tau aggregates were immunopurified from normal control (blue) or AD brain (red) and then conjugated to Alexa Fluor 647 and resolved by SEC. Aggregate size was approximated using known protein standards (gray). AU, arbitrary units; Elution Vol, elution volume. B–E, recombinant (B and C) or AD-derived (D and E) Tau monomer, ∼10-mer, or >20-mer were incubated with HJ3.4, HJ8.5, HJ9.3, or HJ9.4 and applied to either BV2 cells (B and D) or primary cortical neurons (C and E). B, HJ8.5 and HJ9.3 promoted uptake of 10-mer and >20-mer Tau aggregates but not monomer in BV2 cells. HJ9.3 modestly inhibited uptake of 10-mer and >20-mer Tau aggregates in primary neurons. HJ8.5 and HJ9.3 promoted uptake of >20-mer Tau aggregates but not monomer or 10-mer in BV2 cells. E, HJ9.3 inhibited uptake of 10-mer and >20-mer Tau aggregates in primary neurons. MFI, median fluorescence intensity. Data are represented as mean ± S.D. *, p < 0.05, **, p < 0.01, ****, p < 0.0001.
Tau has six splice isoforms and multiple potential patterns of post-translational modification, and forms aggregates of distinct sizes. Because it is impossible to replicate all of these variables using recombinant Tau, we tested the effect of antibody treatment on Tau fibrils immunopurified from AD brain, covalently labeled with Alexa Fluor 647, and resolved by SEC. To confirm isolation of Tau aggregates, we also resolved Tau immunoprecipitated from normal control brain tissue, which elutes from the SEC column predominantly as monomer (Fig. 6A). Therapeutic antibodies promoted uptake of aggregates >20-mer but not ∼10-mers or monomers (Fig. 6D). To test these effects on primary cultured neurons, we applied Tau-antibody complexes to cortical mouse neurons. HJ8.5 and HJ9.4 did not alter neuronal uptake of AD-derived Tau aggregates, whereas we observed consistent inhibition by HJ9.3 on the uptake of AD-derived Tau aggregates (Fig. 6E). Aggregate size did not affect the activity of HJ9.3 in this case (Fig. 6E). In summary, HJ8.5 and HJ9.4 strongly promoted uptake of larger AD-derived Tau aggregates into BV2 cells and failed to affect smaller assemblies. Conversely, although HJ9.3 only modestly increased uptake of AD-derived Tau aggregates into BV2 cells, it alone prevented uptake into primary cultured neurons. These findings highlight the role of epitope and aggregate size in the efficacy of therapeutic antibodies.
Discussion
Summary
It is currently unknown how therapeutic antibodies reduce pathology in animal models, or how they might function in patients. This could be of great importance in the development of more effective therapies. Congdon et al. (22) suggested that antibodies act on intraneuronal Tau aggregates following clathrin-mediated endocytosis mediated by low affinity FcγRs. However, it is generally accepted that low affinity FcγRs differ from other receptors in that their activation requires cross-linking of polyvalent immune complexes, such as when multiple antibodies are bound to a protein aggregate (29, 33). The mechanism by which the antibodies could escape the endocytic vesicle to act on cytosolic Tau aggregates is unknown. We previously hypothesized that antibodies bind extracellular Tau and prevent its uptake by adjacent or post-synaptic neurons (18). However, here we find that only HJ9.3 can prevent uptake into neurons, whereas other therapeutic antibodies HJ8.5 and HJ9.4 have no effect on neuronal uptake. On the other hand, multiple prior studies have suggested a role for microglia-mediated clearance of α-synuclein (30) and Aβ aggregates (31, 34). Our studies are consistent with this mechanism, but suggest a more nuanced interpretation. HJ8.5 and HJ9.4 had a robust effect in this regard, whereas HJ9.3 had differential effects, suggesting a role for both epitope and aggregate characteristic in determining uptake patterns.
We have evaluated three therapeutic antibodies we previously studied in a mouse model of tauopathy (18). We began by studying their effects on full-length recombinant Tau fibrils. Two antibodies (HJ8.5 and HJ9.4) promoted uptake of full-length Tau fibrils into microglia, whereas another (HJ9.3) had no effect on full-length fibrils, but worked with RD and AD-derived Tau assemblies. Antibody-mediated uptake into BV2 cells required FcγR receptors, as it could be blocked by an anti-FcγR antibody. In neurons, HJ8.5 and HJ9.4 did not inhibit aggregate uptake of any form of Tau we tested. Conversely, despite having little effect on recombinant full-length Tau fibrils, HJ9.3 inhibited uptake of recombinant RD and AD-derived Tau assemblies, suggesting that this may occur in vivo. Finally, we observed a clear size dependence of the microglial clearance pathway. All antibodies selectively promoted uptake of larger but not smaller assemblies into BV2 cells. Taken together, this work emphasizes that therapeutic antibodies have multiple potential modes of action that might be optimized to improve efficacy in vivo. Further, the effects of aggregate type (recombinant full-length, RD, AD-derived Tau), size, and epitope targeted by each antibody will potentially specify unique therapeutic mechanisms.
Promoting Microglial Uptake
HJ8.5 and HJ9.4 readily promoted FcγR-mediated uptake of full-length recombinant and AD-derived Tau aggregates into BV2 cells. HJ9.3 also clearly promoted uptake of Tau RD into BV2 cells and modestly (but significantly) increased uptake of AD-derived aggregates. However, HJ9.3 did not increase BV2 uptake of recombinant, full-length Tau fibrils. Microglia highly express FcγRs, which bind the constant region of IgG. Activation results in phagocytosis (35), which would be required for the aggregate uptake observed. Three classes of FcγR are known: FcγRI, FcγRII, and FcγRIII (36). FcγRI, a high affinity receptor, binds monomeric IgG, whereas low affinity receptors FcγRII and FcγRIII bind only multimeric IgG (29). We have concluded that Tau-directed antibodies likely promote uptake in microglia via FcγRII and FcγRIII because an antibody specific to these receptors inhibited the antibody-mediated uptake of Tau fibrils in BV2 cells. However, it is likely that not all antibodies will need to promote microglial clearance to be effective. Studies in FcγR-deficient mice and application of antibodies with reduced or no effector function have suggested mechanisms of amyloid removal independent of FcγR binding (20, 37).
Our observation that antibodies specifically promote uptake of AD-derived Tau fibrils of ≥20-mer, but not monomer or ∼10-mer, is consistent with a role for lower affinity FcγRs, which depend on multimerized opsonization. BV2 cells also utilize HSPGs for antibody-independent uptake, identical to the antibody-independent uptake mechanism we previously reported for neurons (28). However, in BV2 cells, only partial blockade by heparin and F6 (which completely block uptake into neurons) suggests that additional antibody-independent phagocytic mechanisms likely exist. We now conclude that at least part of the antibodies' therapeutic effect in vivo could be to promote microglial clearance, although this effect varies by aggregate size and epitope.
Blocking Neuronal Uptake
HJ9.3 uniquely inhibited uptake of RD and AD brain-derived Tau fibrils into primary neurons. Neurons utilize HSPGs to internalize Tau aggregates via macropinocytosis (28). Heparin binding motifs on target proteins bind HSPGs to trigger internalization, and Tau contains several such motifs. HSPG binding sequences generally consist of a stretch of positively charged lysines or arginines. In Tau, these exist in the R2 and R3 repeat domains, in regions flanking the proline-rich region between the N-terminal inserts and the repeat domain, and in the lysine-rich region downstream of R4 (38, 39). Titration curves with short heparin fragments have defined HSPG binding motifs in R2 and R3 (residues 274–335). The PHF6 peptide (306VQIVYK311) within R3 has a particularly strong interaction (39). Because the HJ9.3 binding epitope maps to residues 306–320 (18), it is very plausible that HJ9.3 blocks HSPG-mediated uptake via steric hindrance.
It is unclear why HJ9.3 weakly blocks uptake of recombinant, full-length 2N4R Tau, whereas it effectively blocks RD and AD-derived Tau oligomers. This may be explained by the presence of multiple heparin binding domains on full-length Tau, which may be available to facilitate uptake although one is blocked via HJ9.3 binding. In contrast, the RD peptide only contains a single putative HSPG binding domain. Post-translational modifications such as cleavage events or other modifications that mask the binding domain (40) in AD-derived Tau may be responsible for reducing the number of available heparin binding domains, and thus could enable HJ9.3 to block uptake in AD-derived Tau assemblies.
Effects of Aggregate Size on Antibody Activity
We observed that therapeutic antibodies do not promote the uptake of monomer or small aggregates into BV2 cells. This implies an important role for polyvalency in antibody-mediated microglial clearance. Coupled with our observation that low affinity FcγRs likely mediate uptake (and could thus participate in clearance in vivo), this predicts that the size of extracellular aggregates that mediate propagation in vivo will influence the efficacy of therapeutic antibodies. Although relatively small Tau oligomers may be the most toxic form of aggregated protein (41–43), these might occur in a variety of sizes. Because the size and characteristics of toxic Tau assemblies could vary among the tauopathies, especially given the diversity of Tau prion strains present (44), this implies that antibody efficacy would similarly vary.
Mechanisms of Antibody Therapeutics
There are no disease-modifying treatments for AD or other tauopathies; however, numerous studies indicate that vaccines are a potential strategy, especially given the putative role for extracellular Tau (reviewed in Ref. 45). We propose that antibodies can promote uptake of Tau aggregates into microglia, and under certain conditions, inhibit uptake into neurons. However, these activities depend on antibody epitopes and the size of the aggregates, and clearly different mechanisms are possible for these and other antibodies. The recently described “glymphatic” system suggests that therapeutic interventions that promote bulk flow of aggregates into the periphery, which might be achieved with antibodies in vivo, may be an additional therapeutic antibody mechanism (46). Also anti-Tau antibodies may exert their effects on extracellular Tau by 1) neutralization of toxicity; 2) clearance via interstitial fluid to cerebrospinal fluid to plasma and 3) cellular degradation independent of the antibody Fc domain.
We found that HJ8.5 and HJ9.4 had similar activities to promote uptake into BV2 cells, without any appreciable effect on neuronal uptake. Importantly, both antibodies have epitopes within the N-terminal region of Tau. Conversely, HJ9.3, which targets an internal region of Tau, possibly encompassing an HSPG binding motif, had less consistent effects in triggering fibril uptake into BV2 cells, but additionally blocked neuronal uptake of some Tau species. As Tau antibodies make their way into the clinic, we suggest that elucidation of mechanisms of action of therapeutic antibodies will help predict complementary or synergistic functions and speed the development of more effective treatments.
Future Directions
These data identify microglia as potentially important effectors of therapeutic antibodies, whereas blockade of neuronal uptake may enhance benefit for some others. In BV2 cells, antibodies promote uptake via FcγRs. Our data are most consistent with a model in which therapeutic antibodies facilitate clearance of extracellular Tau, thereby reducing its potential for transcellular propagation. Future studies will be necessary to clarify the importance of these mechanisms in the therapeutic effects of different anti-Tau antibodies in vivo.
Author Contributions
K. E. F. and M. I. D. designed the study and wrote the paper. H. M. prepared the SEC-separated Tau aggregates. H. J. and D. M. H. supplied the experimental antibodies. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to Nigel Cairns, Erin Householder, the technical support of the Betty Martz Laboratory for Neurodegenerative Research, and the Knight Alzheimer's Disease Research Center at Washington University in St. Louis for brain tissue, supported by the following National Institutes of Health grants: P50AG005681 (Knight Alzheimer's Disease Research Center) and P01AG003991 (Healthy Aging and Senile Dementia).
This work was supported by the Alzheimer's Disease Research Program of the BrightFocus Foundation (to K. E. F.) and the Tau Consortium and National Institutes of Health Grant 1R01NS071835 (to M. I. D.). M. I. D., D. M. H., and H. J. are inventors on a submitted patent “Antibodies to Tau” that is licensed by Washington University. M. I. D. is a cofounder of ARTA Biosciences. D. M. H. cofounded and is on the scientific advisory board C2N Diagnostics. D. M. H. consults for Genentech, AstraZeneca, Neurophage, and Eli Lily. H. J., D. M. H., and M. I. D. are inventors of the antibodies described here, all of which have been licensed to C2N Diagnostics, LLC.
- AD
- Alzheimer disease
- Aβ
- amyloid-β
- RD
- repeat domain
- ODS
- octadecyl sulfate
- SEC
- size exclusion chromatography
- IP
- immunoprecipitation
- FcγR
- Fc γ receptor
- HSPG
- heparan sulfate proteoglycan
- PD
- polymeric dextran
- Bis-Tris
- 2-(bis(2-hydroxyethyl)amino)-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Mandelkow E.-M., Mandelkow E. (2012) Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2, a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arriagada P. V., Growdon J. H., Hedley-Whyte E. T., Hyman B. T. (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42, 631–639 [DOI] [PubMed] [Google Scholar]
- 3. Bancher C., Braak H., Fischer P., Jellinger K. A. (1993) Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer's and Parkinson's disease patients. Neurosci. Lett. 162, 179–182 [DOI] [PubMed] [Google Scholar]
- 4. Polydoro M., Acker C. M., Duff K., Castillo P. E., Davies P. (2009) Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J. Neurosci. 29, 10741–10749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Small S. A., Duff K. (2008) Linking Aβ and tau in late-onset Alzheimer's disease: a dual pathway hypothesis. Neuron 60, 534–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Braak H., Braak E. (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. (Berl.). 82, 239–259, 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- 7. Braak H., Braak E. (1995) Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol. Aging 16, 271–278; discussion 278–284 [DOI] [PubMed] [Google Scholar]
- 8. Braak H., Thal D. R., Ghebremedhin E., Del Tredici K. (2011) Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969 [DOI] [PubMed] [Google Scholar]
- 9. Holmes B. B., Diamond M. I. (2014) Prion-like properties of Tau protein: the importance of extracellular Tau as a therapeutic target. J. Biol. Chem. 289, 19855–19861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Majerova P., Zilkova M., Kazmerova Z., Kovac A., Paholikova K., Kovacech B., Zilka N., Novak M. (2014) Microglia display modest phagocytic capacity for extracellular tau oligomers. J. Neuroinflammation 11, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Godbout J. P., Chen J., Abraham J., Richwine A. F., Berg B. M., Kelley K. W., Johnson R. W. (2005) Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 19, 1329–1331 [DOI] [PubMed] [Google Scholar]
- 12. Griffin W. S. T. (2006) Inflammation and neurodegenerative diseases. Am. J. Clin. Nutr. 83, 470S–474S [DOI] [PubMed] [Google Scholar]
- 13. Mrak R. E., Griffin W. S. T. (2005) Potential inflammatory biomarkers in Alzheimer's disease. J. Alzheimers Dis. 8, 369–375 [DOI] [PubMed] [Google Scholar]
- 14. Streit W. J., Conde J. R., Fendrick S. E., Flanary B. E., Mariani C. L. (2005) Role of microglia in the central nervous system's immune response. Neurol. Res. 27, 685–691 [DOI] [PubMed] [Google Scholar]
- 15. Asuni A. A., Boutajangout A., Quartermain D., Sigurdsson E. M. (2007) Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J. Neurosci. 27, 9115–9129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boutajangout A., Ingadottir J., Davies P., Sigurdsson E. M. (2011) Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J. Neurochem. 118, 658–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chai X., Wu S., Murray T. K., Kinley R., Cella C. V., Sims H., Buckner N., Hanmer J., Davies P., O'Neill M. J., Hutton M. L., Citron M. (2011) Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J. Biol. Chem. 286, 34457–34467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yanamandra K., Kfoury N., Jiang H., Mahan T. E., Ma S., Maloney S. E., Wozniak D. F., Diamond M. I., Holtzman D. M. (2013) Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80, 402–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yanamandra K., Jiang H., Mahan T. E., Maloney S. E., Wozniak D. F., Diamond M. I., Holtzman D. M. (2015) Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann. Clin. Transl. Neurol. 2, 278–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bacskai B. J., Kajdasz S. T., McLellan M. E., Games D., Seubert P., Schenk D., Hyman B. T. (2002) Non-Fc-mediated mechanisms are involved in clearance of amyloid-β in vivo by immunotherapy. J. Neurosci. 22, 7873–7878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Solomon B., Koppel R., Frankel D., Hanan-Aharon E. (1997) Disaggregation of Alzheimer β-amyloid by site-directed mAb. Proc. Natl. Acad. Sci. U.S.A. 94, 4109–4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Congdon E. E., Gu J., Sait H. B. R., Sigurdsson E. M. (2013) Antibody uptake into neurons occurs primarily via clathrin-dependent Fcγ receptor endocytosis and is a prerequisite for acute tau protein clearance. J. Biol. Chem. 288, 35452–35465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wille H., Drewes G., Biernat J., Mandelkow E. M., Mandelkow E. (1992) Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J. Cell Biol. 118, 573–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goedert M., Jakes R. (1990) Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 9, 4225–4230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bocchini V., Mazzolla R., Barluzzi R., Blasi E., Sick P., Kettenmann H. (1992) An immortalized cell line expresses properties of activated microglial cells. J. Neurosci. Res. 31, 616–621 [DOI] [PubMed] [Google Scholar]
- 26. Koenigsknecht J., Landreth G. (2004) Microglial phagocytosis of fibrillar β-amyloid through a β1 integrin-dependent mechanism. J. Neurosci. 24, 9838–9846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kopec K. K., Carroll R. T. (1998) Alzheimer's β-amyloid peptide 1–42 induces a phagocytic response in murine microglia. J. Neurochem. 71, 2123–2131 [DOI] [PubMed] [Google Scholar]
- 28. Holmes B. B., DeVos S. L., Kfoury N., Li M., Jacks R., Yanamandra K., Ouidja M. O., Brodsky F. M., Marasa J., Bagchi D. P., Kotzbauer P. T., Miller T. M., Papy-Garcia D., Diamond M. I. (2013) Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U.S.A. 110, E3138–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. García-García E., Rosales C. (2002) Signal transduction during Fc receptor-mediated phagocytosis. J. Leukoc. Biol. 72, 1092–1108 [PubMed] [Google Scholar]
- 30. Bae E.-J., Lee H.-J., Rockenstein E., Ho D.-H., Park E.-B., Yang N.-Y., Desplats P., Masliah E., Lee S.-J. (2012) Antibody-aided clearance of extracellular α-synuclein prevents cell-to-cell aggregate transmission. J. Neurosci. 32, 13454–13469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bard F., Cannon C., Barbour R., Burke R. L., Games D., Grajeda H., Guido T., Hu K., Huang J., Johnson-Wood K., Khan K., Kholodenko D., Lee M., Lieberburg I., Motter R., Nguyen M., Soriano F., Vasquez N., Weiss K., Welch B., Seubert P., Schenk D., Yednock T. (2000) Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 6, 916–919 [DOI] [PubMed] [Google Scholar]
- 32. Mirbaha H., Holmes B. B., Sanders D. W., Bieschke J., Diamond M. I. (2015) Tau trimers are the minimal propagation unit spontaneously internalized to seed intracellular aggregation. J. Biol. Chem. 290, 14893–14903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Metzger H., Kinet J. P. (1988) How antibodies work: focus on Fc receptors. FASEB J. 2, 3–11 [DOI] [PubMed] [Google Scholar]
- 34. Bard F., Barbour R., Cannon C., Carretto R., Fox M., Games D., Guido T., Hoenow K., Hu K., Johnson-Wood K., Khan K., Kholodenko D., Lee C., Lee M., Motter R., Nguyen M., Reed A., Schenk D., Tang P., Vasquez N., Seubert P., Yednock T. (2003) Epitope and isotype specificities of antibodies to β-amyloid peptide for protection against Alzheimer's disease-like neuropathology. Proc. Natl. Acad. Sci. U.S.A. 100, 2023–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takai T., Li M., Sylvestre D., Clynes R., Ravetch J. V. (1994) FcRγ chain deletion results in pleiotrophic effector cell defects. Cell 76, 519–529 [DOI] [PubMed] [Google Scholar]
- 36. Ravetch J. V., Bolland S. (2001) IgG Fc receptors. Annu. Rev. Immunol. 19, 275–290 [DOI] [PubMed] [Google Scholar]
- 37. Das P., Howard V., Loosbrock N., Dickson D., Murphy M. P., Golde T. E. (2003) Amyloid-β immunization effectively reduces amyloid deposition in FcRγ−/− knock-out mice. J. Neurosci. 23, 8532–8538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Elbaum-Garfinkle S., Rhoades E. (2012) Identification of an aggregation-prone structure of tau. J. Am. Chem. Soc. 134, 16607–16613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sibille N., Sillen A., Leroy A., Wieruszeski J.-M., Mulloy B., Landrieu I., Lippens G. (2006) Structural impact of heparin binding to full-length Tau as studied by NMR spectroscopy. Biochemistry (Mosc.) 45, 12560–12572, 10.1021/bi060964o [DOI] [PubMed] [Google Scholar]
- 40. Gong C.-X., Liu F., Grundke-Iqbal I., Iqbal K. (2005) Post-translational modifications of tau protein in Alzheimer's disease. J. Neural Transm. 112, 813–838 [DOI] [PubMed] [Google Scholar]
- 41. Gómez-Ramos A., Díaz-Hernández M., Cuadros R., Hernández F., Avila J. (2006) Extracellular tau is toxic to neuronal cells. FEBS Lett. 580, 4842–4850 [DOI] [PubMed] [Google Scholar]
- 42. Gómez-Ramos A., Díaz-Hernández M., Rubio A., Miras-Portugal M. T., Avila J. (2008) Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell. Neurosci. 37, 673–681 [DOI] [PubMed] [Google Scholar]
- 43. Lasagna-Reeves C. A., Castillo-Carranza D. L., Guerrero-Muoz M. J., Jackson G. R., Kayed R. (2010) Preparation and characterization of neurotoxic tau oligomers. Biochemistry (Mosc.) 49, 10039–10041, 10.1021/bi1016233 [DOI] [PubMed] [Google Scholar]
- 44. Sanders D. W., Kaufman S. K., DeVos S. L., Sharma A. M., Mirbaha H., Li A., Barker S. J., Foley A. C., Thorpe J. R., Serpell L. C., Miller T. M., Grinberg L. T., Seeley W. W., Diamond M. I. (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82, 1271–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wisniewski T., Goñi F. (2014) Immunotherapy for Alzheimer's disease. Biochem. Pharmacol. 88, 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Iliff J. J., Wang M., Liao Y., Plogg B. A., Peng W., Gundersen G. A., Benveniste H., Vates G. E., Deane R., Goldman S. A., Nagelhus E. A., Nedergaard M. (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 4, 147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]