

Background: Engineering Fabs with high affinity toward two distinct antigens is challenged by the competing constraints of a shared binding surface.
Results: Deep mutational scan unveiled the sequences of Fabs with sub-nanomolar affinity for two angiogenic targets.
Conclusion: Fabs potent against two structurally unrelated targets were discovered.
Significance: Efficacious generation of dual action Fab expands the therapeutic potential of Fab molecules in ocular indications and beyond.
Keywords: antibody engineering, cooperativity, crystal structure, drug discovery, vascular endothelial growth factor (VEGF), angiopoietin2 (Ang2), deep mutational scanning, deep sequencing, dual specificity, neovascular age-related macular degeneration
Abstract
The development of dual targeting antibodies promises therapies with improved efficacy over mono-specific antibodies. Here, we engineered a Two-in-One VEGF/angiopoietin 2 antibody with dual action Fab (DAF) as a potential therapeutic for neovascular age-related macular degeneration. Crystal structures of the VEGF/angiopoietin 2 DAF in complex with its two antigens showed highly overlapping binding sites. To achieve sufficient affinity of the DAF to block both angiogenic factors, we turned to deep mutational scanning in the complementarity determining regions (CDRs). By mutating all three CDRs of each antibody chain simultaneously, we were able not only to identify affinity improving single mutations but also mutation pairs from different CDRs that synergistically improve both binding functions. Furthermore, insights into the cooperativity between mutations allowed us to identify fold-stabilizing mutations in the CDRs. The data obtained from deep mutational scanning reveal that the majority of the 52 CDR residues are utilized differently for the two antigen binding function and permit, for the first time, the engineering of several DAF variants with sub-nanomolar affinity against two structurally unrelated antigens. The improved variants show similar blocking activity of receptor binding as the high affinity mono-specific antibodies against these two proteins, demonstrating the feasibility of generating a dual specificity binding surface with comparable properties to individual high affinity mono-specific antibodies.
Introduction
In neovascular age-related macular degeneration (NV AMD),2 a condition affecting more than 1 million persons in the United States (1), choroidal neovascularization and vascular permeability lead to the death of photoreceptor cells and subsequent vision loss (2). Vascular endothelial growth factor-A (VEGF) is a clinically validated driver of choroidal neovascularization, and neutralization of VEGF, for example by the Food and Drug Administration-approved anti-VEGF Fab ranibizumab (Genentech), leads to stable or improved vision for the majority of treated patients (3). However, there exists a demand for therapies with broader and improved efficacy than what is currently available because a subset of patients does not significantly respond to anti-VEGF therapy. Targeting additional angiogenic signaling pathways may improve the therapeutic efficacy of VEGF inhibitors (4, 5). One pathway of interest is the receptor tyrosine kinase Tie2 and its ligand angiopoietin1/2, deregulation of which is believed to be involved in ocular vascular instability. The signaling of Ang1 through Tie2 can stabilize vascular structures, and the binding of Ang2 to Tie2 in many cases counteracts these stabilizing signals (6). An inhibitory effect on choroidal neo-vascularization by Ang2 blockade has been demonstrated in mouse models (7), and dual targeting of Ang2 and VEGF may result in improved efficacy in NV AMD treatment compared with anti-VEGF treatment alone.
One dual specificity antibody format that may be suitable to serve as a next generation therapeutic for NV AMD is a dual action Fab (DAF) (8), which harbors dual specificity in a single Fab. A Fab is small in size and in general is highly stable and soluble, and thus it is well suited for formulation for intravitreal injection, the established delivery option for AMD therapy. DAFs contain an engineered antigen-binding site that is able to bind two structurally unrelated epitopes. Achieving high monovalent affinity toward two antigens in a DAF has been challenging, as reflected in the three published DAF molecules where none has reached the sub-nanomolar range of the dissociation constant (Kd) for both antigens (8–10). It is an open question as to whether a single antibody-combining site can in fact bind two distinct antigens with such high affinity.
Strategies for in vitro affinity maturation typically involve generating large libraries of variants (106 to 1010) of the antibody of interest using site-directed or random mutagenesis of the variable domains or CDRs. This is followed by affinity-based selection and screening a limited number of clones by affinity assay and sequencing (11). The positions to include for combinatorial mutation and the extent of randomization in the libraries are quite limited as the library size that can be generated and screened is finite. Traditional mutagenesis methods such as alanine scanning can determine critical regions for ligand interaction, termed “hot spots,” and can rationally guide the affinity maturation library design to avoid alteration of sites that are unlikely to tolerate mutation (12, 13). To identify favorable mutations for affinity maturation directly from mutational scans, recent strategies implemented libraries of variants with single positions randomized to all 20 (14) or a subset of amino acids (15). Affinity-based selection and sequencing identified the variants with improved binding function or fitness. Individual mutations can then be combined to create a higher affinity clone. Next generation sequencing further expands this approach by examining the sequences of whole libraries with millions of reads, allowing calculation of the enrichment or depletion of each mutation during the selection process, thereby generating a comprehensive overview of the potential effect of each mutation (16). The expanded approach called deep mutational scanning has been applied to affinity improvement of protein inhibitors (17) and antibodies (18, 19). However, these studies have focused on identification of single mutations that are significantly enriched during the selection for binding function for affinity improvement.
Here, we generated an initial Two-in-One antibody with DAF, called 5A12, which has a Kd of 5 nm for both hVEGF and hAng2. To match the blocking activity observed for high affinity mono-specific antibodies against hAng2 and hVEGF, we aimed to improve the 5A12 DAF Kd value to subnanomolar values for both antigens. We employed the deep mutational scanning approach to map the CDRs for dual affinity maturation by combining phage library selection with deep sequencing using MiSeq. The phage libraries with single-site saturated mutation in each of the 5A12 CDRs were sorted separately for Ang2 and VEGF binding function prior to sequencing. Information on enrichment as well as depletion of all mutations at all CDR positions during antigen-binding selection was used to identify favored or disfavored mutation for each binding function,. We further extend the analysis of deep mutational scanning from single mutations to double mutations, which allowed us identify fold-stabilizing single mutations as well as mutation pairs that enhance the binding function of the DAF. This comprehensive data set was used to generate the first examples of DAFs with sub-nanomolar dual specificity affinities. An optimized 5A12 variant shows in vitro blocking efficacy comparable with mono-specific anti-Ang2 and anti-VEGF antibodies.
Experimental Procedures
DAF Isolation through Phage Library Selection
To recruit hAng2 binding to an anti-VEGF G6, phage-displayed libraries with randomized light chain CDRs were created using oligonucleotide-directed Kunkel mutagenesis of G6 Fab using methods described previously (8, 20). All three LC CDRs were randomized as described (8). The phage libraries (1010 transformants) were subjected to four rounds of binding selection using Fc C-terminally fused with the receptor binding domain (RBD) of hAng2 protein (Fc.hAng2). Fc-fused RBD of hAng1 protein (Fc.hAng1) was added to phage in the third round before incubating the phage with Fc.hAng2-coated wells to remove hAng1 binders and the fifth round phage libraries additionally being panned against hVEGF 8–109 (hVEGF). Random clones were picked and screened for hAng2 and hVEGF binding by ELISA (21). The positive clones were sequenced as described (22). Unique Ang2-binding clones that do not bind Ang1 and can block Tie2 binding were selected for affinity improvement as described (23). Phage competition ELISAs to derive the phage IC50 values for relative affinity measurement were performed as described (11). Briefly, Fab-displaying phage were first incubated with serial dilutions of antigens for 1 h. The unbound phage were then captured on antigen-coated ELISA wells and measured by incubating with anti-M13 antibody horseradish peroxidase (HRP) conjugate followed by HRP substrate and read spectrophotometrically at 450 nm. Phage IC50 is the antigen concentration that inhibits ∼50% of Fab-displaying phage from binding the antigen-coated wells.
DAF Affinity Maturation
For initial affinity improvement, phage libraries were constructed from a chosen DAF clone with selected LC or HC CDR residues mutated using either limited or soft randomization to allow either limited diversity based on natural amino acids or ∼50% of wild type with ∼50% all other amino acids, respectively. Resultant libraries were sorted with hVEGF or human His-tagged Ang2 RBD (hAng2his) with increasing stringency and screened for improved affinity as described (23). 5A12 was selected based on DNA sequencing, phage IC50, and then BIAcore-measured affinity as purified Fab.
For further affinity improvement by deep mutational scanning, phage libraries were constructed from 5A12 Fab format with either LC or HC CDR residues mutated using a mixture of degenerate oligonucleotides for each CDR loop. Each oligonucleotide randomizes one of the CDR sites with the NNK codon that encodes for all 20 amino acids with 32 codons, including a stop codon (N = G, A, T, and C and K = G and T in equal portion). Libraries were designed to allow either one NNK mutation in each of the three LC or HC CDRs called 3NNK libraries or one NNK mutation in a single CDR called 1NNK. Resultant library DNA was electroporated into Escherichia coli XL1 cells yielding ∼109 transformants. Libraries were sorted for either hVEGF109 or hAng2his binding separately with increasing stringencies by incubating with decreasing concentrations of antigens as above. For deep sequencing, phagemid double-stranded DNA was isolated from the selected rounds. The VH and the VL segments from each sample were amplified by an 18-cycle PCR amplification using Phusion DNA polymerase (New England Biolabs). The amplicon was purified on a 2% agarose gel.
Illumina Sequencing and Data Analysis
Amplicons were prepared with standard Illumina library preparation methods, using TruSeq DNA Sample Prep (Illumina). Adapter-ligated libraries were subjected to a single cycle of PCR and sequenced on the Illumina MiSeq, using paired end 200- or 300-bp reads as appropriate to cover the entire length of the amplicon. Sequencing data were analyzed using the statistical programming language R (24) and ShortRead (25). Quality control was performed on identified CDR sequences, where each CDR sequence was checked for the correct length and was allowed to carry only up to one NNK mutation and no non-NNK mutations. Calculating the frequency of all mutations, of every randomized position, generated position weight matrices. Enrichment ratios (ER) for all mutations were calculated by dividing the frequency of a given mutation at a given position in the sorted sample by the frequency of the very same mutation in the unsorted sample, as described previously (16). To test the performance of the ER as a classifier for identifying affinity-improving mutations, receiver operator characteristics were used. The performance of single mutation variants in a phage competition ELISA was used as a gold standard (IC50 (mutant)/IC50 (wild type) >1.5 for affinity improving mutations and IC50 (mutant)/IC50 (wild type) <1.5 for mutations that do not improve the affinity).
The ER of double mutations was calculated from the 3NNK library by calculating the ER of all clones that carry two NNK mutations at two given CDR positions while ignoring the third NNK mutation in those clones. To filter out sampling effects, mutation pairs that had less than 10 sequence counts either in the sorted or unsorted samples were removed from the analysis. Of 122,598 possible mutation pairs in the heavy chain, the ER of 43,224 pairs in the hAng2 panned dataset and of 32,120 pairs in the hVEGF panned dataset could be calculated. From the light chain library, which contains 77,616 possible mutation pairs, the ER of 37,006 pairs from hAng2 panning and 57,854 pairs from hVEGF panning could be determined. Four different models describing cooperativity were tested (26) to determine which model is the best representative for the data. A multiplicative model is used to calculate the expected ER (ERexp) of mutation pairs from the measured ER of each single mutation (ERmeas) as shown in Equation 1,
The cooperativity used here is thus defined as given in Equation 2,
 |
The partner potentiation scores to identify fold-stabilizing mutations are calculated as described previously (27) by calculating first the partner normalized cooperativity score for all available mutation pairs as shown in Equation 3,
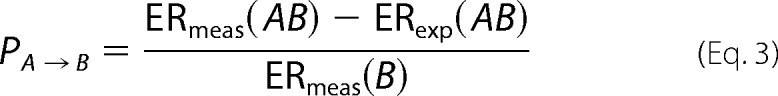 |
The partner potentiation score is the mean of the partner normalized cooperativity scores of a given mutation A is shown in Equation 4,
 |
Data were plotted using ggplot2 (28) and Circos (29). Crystal structures were visualized using PyMOL (30).
Antibody Characterization
The VL and VH of selected phage clones were cloned into vectors previously designed for transient human IgG or Fab expression in mammalian cells (21). IgGs were purified with protein A affinity chromatography and screened for specificity by ELISA using their respective antigen(s) and by baculovirus ELISA (31). To confirm Tie2 blocking activity, a competitive ELISA format was used where Tie2 Fc fusion was immobilized on Maxisorp ELISA plates, and biotinylated hAng2 was incubated with serial dilutions of purified Fab prior to capturing unbound biotin-hAng2 with the immobilized Tie2 and detected with streptavidin-conjugated HRP. The above format was also used to test for blocking of the hVEGF-VEGFR1(Flt1) interaction using immobilized VEGFR1 to capture free VEGF after solution equilibration of biotinylated hVEGF with serial dilutions of purified Fab (8). For Kd determination of Fab, surface plasmon resonance (SPR) measurements using a BIAcore T200 and CM5 sensor chip was used and dilutions of Fab as analyte were injected over the immobilized RBD of hVEGF or hAng2 at 25 °C to determine monovalent affinities. Association rates (kon) and dissociation rates (koff) were calculated using a simple one-to-one Langmuir binding model. The equilibrium dissociation constant (Kd) was calculated as the ratio koff/kon.
The melting temperature (Tm) was determined using differential scanning fluorimetry. 100 μg/ml purified Fab solution was mixed 1:500 with SYPRO Orange dye. The sample was gradually heated in a quantitative PCR machine from 20 to 100 °C, and the fluorescence was measured. The melting temperature (Tm) is the inflection point of the transition curve (32).
Protein Purification and Crystallization
C-terminal His-tagged RBD of Ang2 (residues 277–496, hAng2his) was expressed extracellularly in Trichoplusia ni insect cells. The supernatant was loaded onto a nickel-nitrilotriacetic acid Superflow column, washed with 50 mm Tris-HCl, pH 8.0, and 300 mm NaCl, and eluted with 250 mm imidazole in the same buffer. Fractions containing hAng2 were further purified over a Superdex S200 size exclusion column in 20 mm Tris-Cl, pH 7.5, and 300 mm NaCl. hVEGF (residues 8–109) was expressed, refolded, and purified as described previously (33). Fab 5A12 was expressed in E. coli and purified first with a protein G column followed by an SP-Sepharose column equilibrated with 20 mm MES, pH 5.5. The protein was eluted with a salt gradient of 0 to 0.25 m NaCl and peak fractions passed over a S200 column in 20 mm Tris-Cl, pH 7.5, and 250 mm NaCl (buffer A).
The hAng2his-5A12 complex was prepared by mixing at a 1.2:1 molar ratio and purified on an S200 column in buffer A. Fractions containing the complex were pooled and concentrated to 31.6 mg/ml for crystallization trials. Crystals were grown at 19 °C using vapor diffusion by mixing protein with an equal volume of a well solution containing 0.1 m Tris, pH 8.5, 0.2 m lithium sulfate, 25% PEG3350 and 3% d(+)-sucrose. The hVEGF-5A12 complex was prepared using the same protocol as for the hAng2his-5A12 complex, concentrated to 13.3 mg·ml, and crystallized under 0.1 m Tris, pH 8.0, 2 m ammonium sulfate, and 0.3 m NDSB-195. Both crystals were cryo-protected in artificial mother liquor containing 20% glycerol and flash-frozen in liquid nitrogen.
Structure Determination, Refinement, and Analysis
Data were collected at the Advanced Photon Source beamlines 21-IDG and 21-IDF. The hAng2–5A12 structure was determined by molecular replacement using the published Fab structure of G6 (Protein Data Bank code 2FJF) split into variable and constant regions and a published hAng2 structure (Protein Data Bank code 1Z3S) as search models. The asymmetric unit contains a 1:1 stoichiometry of Ang2-Fab. The hVEGF-5A12 structure was determined similarly but using the 5A12 model from the hAng2 complex structure and a published hVEGF structure (Protein Data Bank code 2FJG) as search models. The asymmetric unit contains a VEGF dimer with a Fab bound at each end (2:2) creating an elongated complex ∼200 Å in length. One of the Fab copies has significantly higher crystallographic B factors and retains a significant feature in the Fo − Fc difference density near its light chain (distant from the CDRs) that could not be unambiguously fit by any of the buffer or solvent components in the crystallization experiment. Structures were refined by iterative manual rebuilding PyMOL (30), COOT (34), and automated refinement BUSTER (35) and Phenix (36). Statistics are reported in Table 1. Coordinates and structure factors have been deposited in the Protein Data Bank under codes 4ZFG (hAng2–5A12) and 4ZFF (hVEGF-5A12).
TABLE 1.
Summary of crystallographic information
| VEGF-Fab5A12 (4ZFF) | Ang2-Fab5A12 (4ZFG) | |
|---|---|---|
| Wavelength (Å) | 0.97856 | 0.97856 |
| Resolution range (Å) | 50–2.75 (2.85–2.75) | 50–2.27 (2.35–2.27) |
| Space group | P 21212 | C 2 |
| Unit cell | 87.41, 313.89, 51.13 | 181.67, 109.29, 43.95 |
| 90, 90, 90 | 90, 100.47, 90 | |
| Total reflections | 57,584 (2426) | 149,581 (14263) |
| Unique reflections | 37,721 (370) | 38,887 (3794) |
| Multiplicity | 6.4 (6.6) | 3.8 (3.8) |
| Completeness (%) | 99.90 (100.00) | 99.29 (97.13) |
| Mean I/σ(I) | 16.49 (3.61) | 10.12 (1.77) |
| Wilson B-factor | 51.03 | 47.5 |
| R-merge | 0.115 (0.584) | 0.067 (0.655) |
| R-meas | 0.1185 | 0.079 |
| R-work | 0.2038 (0.2514) | 0.1773 (0.2861) |
| R-free | 0.2452 (0.3225) | 0.2201 (0.3111) |
| No. of atoms | 8374 | 5445 |
| Macromolecules | 7940 | 5027 |
| Ligands | 5 | 21 |
| Water | 429 | 397 |
| Protein residues | 1035 | 648 |
| Root mean square (bonds) | 0.027 | 0.019 |
| Root mean square (angles) | 1.84 | 1.81 |
| Ramachandran favored (%) | 96 | 96 |
| Ramachandran outliers (%) | 0.79 | 0.31 |
| Clash score | 5.61 | 3.54 |
| Average B-factor | 68.6 | 59.9 |
| Macromolecules | 69.6 | 59.5 |
| Ligands | 50.7 | 87.3 |
| Solvent | 49.9 | 64 |
The structural epitope and paratope were identified using the program “Contact” in the CCP4 suite and a cutoff value of 5Å (37). To classify Fab CDR positions as buried or solvent-exposed, the crystal structures of the Fab molecules in the hAng2- and hVEGF-bound state without the antigen present were analyzed using the program Areaimol of the CCP4 suite (37). The resulting solvent-accessible area (SAA) was compared with the maximal solvent accessible area (mSAA) (38). Residues that have an SAA/mSAA of <0.1 were classified as buried; residues that have an SAA/mSAA of >0.5 were classified as solvent-exposed. To calculate the functional paratope, for every CDR position, the enrichment of mutations was calculated by Equation 5
where FrqMutSort = 1 − FrqWTSort, and FrqMutNoSort = 1 − FrqWTNoSort, FrqWTSort, and FrqWTNoSort is the frequency of wild type amino acid at a given position in the sorted or unsorted library sample, respectively.
Results and Discussion
Generation of an Initial Ang2/VEGF DAF 5A12
We used the VEGF-blocking antibody G6 (39) as a starting template to recruit the second specificity toward Ang2 by mutation in its light chain (LC) CDRs using the engineering strategy previously described (8). G6 has been demonstrated to block human VEGF (hVEGF) binding to its receptors, VEGFR1 and VEGFR2, and its affinity-improved clones such as G6.31 are highly potent in vitro and in vivo as blockers of VEGF (39, 40). The initial DAF clone had weak affinity toward the receptor-binding domain (RBD) of human Ang2 (hAng2, Kd = 100 nm) and hVEGF (Kd = 50 nm). Affinity maturation using a “soft randomization” strategy (23) yielded 5A12, which exhibited a dissociation constant (Kd) of 5 nm toward both hAng2 and hVEGF as determined by BIAcore. 5A12 retains the VEGF blocking function of G6 and blocks the binding of hAng2 to its receptor Tie2 (data not shown). However, the affinity of 5A12 for hVEGF is 25-fold weaker than that of the well described hVEGF blocking antibodies ranibizumab (Kd < = 0.2 nm) (41) and G6.31 (Kd = 0.2 nm) (39). Similarly, the affinity of 5A12 for hAng2 does not match the affinity of an in-house generated anti-Ang2 Fab (G5.5 (Kd = 0.2 nm), which shows potent in vitro and in vivo hAng2 blocking activity.3 Thus, further affinity maturation is required to convert 5A12 to a similarly potent blocker as the high affinity mono-specific antibodies against hAng2 and hVEGF.
Crystal Structure of 5A12 in Complex with hAng2 and hVEGF
To examine the molecular details of the dual interaction, we generated the crystal structures of the 5A12 Fab in complex with the RBD of hAng2 or hVEGF with resolutions of 2.27 and 2.75 Å, respectively (Fig. 1, A and B, and Table 1, electron density in Fig. 4). The two antigens in the complex structures are highly similar to previously reported structures of hAng2 in complex with Tie2 (root mean square deviation 0.567 Å over 1405 atoms (42)) and hVEGF in complex with the G6 Fab (root mean square deviation 0.43 Å over 1110 atoms (40)), demonstrating that 5A12 does not induce any conformational changes in the antigens. The structural epitope recognized by 5A12 largely overlaps with the Tie2-binding site explaining the mechanism of hAng2 blockade (Fig. 1C). The epitope of 5A12 on hVEGF is nearly identical to the epitope of G6 (Fig. 1D) (39).
FIGURE 1.

Structural characterization of angiopoietin 2 and VEGF binding by DAF 5A12. Crystal structures of 5A12 Fab in complex with RBD of human Ang2 (A) or VEGF (B). A top down view of 5A12 antigen-binding sites for Ang2 or VEGF. The surface representation of the heavy chain is colored blue and the light chain is green. Structural paratopes or residues that are 5 Å or less away from Ang2 or VEGF are colored orange or red, respectively, and show an extensive overlap. C, comparison of the epitope of 5A12 (circled with black line) and Tie2 (red lines) on hAng2. D, comparison of the epitope of 5A12 (black line) and the parental antibody G6 (red line) on hVEGF. E, top view onto the binding site of 5A12 in the hVEGF and hAng2 bound state in comparison with the loop conformations of G6 in the antigen-free form and the hVEGF-bound form. Plasticity in the Cα backbone can be observed in the CDR-H2 and CDR-H3. F, CDR-H2 loop conformations of 5A12 in the hVEGF- and hAng2-bound state in comparison with G6 in the antigen-free form and the hVEGF-bound form. The CDR-H2 loop adopts the same conformation in hVEGF-bound state of G6 and 5A12, whereas the CDR-H2 loop conformation in the hAng2-bound state of 5A12 is the same as observed for the antigen-free form of G6.
FIGURE 4.

Deep mutational scan of 5A12 reflects the different role of CDR-H2 for binding its two antigens. A, 5A12 (schematic representation, green and blue) in binding hAng2 (surface representation, light blue) does not involve the CDR-H2 loop (cartoon with electron density) while using CDR-H2 for hVEGF binding (surface representation, gray) (B). Detailed view of the CDR-H2 loop of 5A12 with the respective electron density (carved at 2 Å for better visibility) in the Ang2-bound (C) and VEGF-bound (D) conformation is shown. Discernible residues are labeled. Heat map representation of the log2 enrichment ratios of CDR-H2 residues scanned by selection of heavy chain 3NNK library for Ang2 (E) or VEGF (F) binding. Asterisks mark the wild type residues.
The structures shows that seven CDR positions of 5A12 are involved in binding both hVEGF and hAng2: HC-F97, HC-F98, HC-L99, HC-P100, LC-I29, LC-D31, and LC-L92 (the Chothia numbering scheme will be used in this work (43), contact distance cutoff <5 Å). Moreover, when mapping the two structural paratopes on the surface of the antigen-binding region, it becomes evident that the spatial organization of the two paratopes is highly intertwined (Fig. 1, A and B). To accommodate the binding of two structurally unrelated epitopes with essentially the same binding site, 5A12 utilizes a large degree of structural plasticity similar to that described for a Her2/VEGF DAF (44). CDR-H2 in particular undergoes drastic conformational change in the hVEGF-bound state compared with the hAng2-bound state. HC-A53, for example, moves by 14 Å (Fig. 1, E and F). When overlaying the structures of 5A12VEGF and 5A12Ang2 with the parental G6 in the hVEGF-bound state (G6VEGF) or in the unbound state (G6unbound), it is evident that the conformations of CDR-H2 and CDR-H3 in 5A12VEGF and G6VEGF are identical, whereas the conformations of the two loops in 5A12Ang2 resemble G6unbound. Thus, the plasticity of the 5A12 CDRs is inherited from the parental antibody G6 and is not added when recruiting dual specificity. In addition to the large conformational changes of the CDR-H2 and CDR-H3 region, some side chains in other CDR loops use different rotamers during binding of the two antigens thereby providing further plasticity without the requirement for rearrangement of the main chain backbone. The intertwined paratopes and the high structural plasticity of the 5A12-binding site required for dual affinity underscore the challenges to identify mutations that are compatible with improving the binding toward both antigens.
Deep Mutational Scanning for Beneficial Single and Double Mutations
To further affinity-mature 5A12, we turned to deep mutational scanning that combines the use of combinatorial library selection and deep sequencing to assess the effect of all possible single mutations and many double mutations of 5A12 CDRs in binding both antigens. The application of deep mutational scanning for antibody affinity maturation has so far focused on mutating each CDR site one at a time (18, 19). This single site approach limits the assessment to the effect of single mutations and does not allow examination of the impact of mutation pairs on the binding function. Synergistic interactions between mutations can be an important driver for the fitness of a protein as they might overcome local fitness minima and/or promote stability (45). A recent study described the identification of fold-stabilizing mutations by analyzing cooperativity between mutation pairs in their deep mutational scanning of a WW domain (27). We wanted to assess here whether large scale cooperativity analysis of mutation pairs can also provide binding-enhancing mutations. We therefore used a library design in which all three CDRs of each chain of 5A12 were mutated simultaneously (Table 2) (46). Each clone in this library carried three NNK-encoded mutations (n = A or T or G or C; K = G or T), one in each of the three CDRs. We call this library design triple-NNK (3NNK). We speculated that this library design strategy would allow us to estimate the effect of single as well as double mutations on antibody fitness and therefore enable the analysis of cooperativity. As a control to check whether the 3NNK library design allows for the correct fitness assessment of single mutations, we generated a library where each CDR of the 5A12 heavy chain was mutated separately (1NNK) (Table 2). Each library was sorted against hAng2 and hVEGF separately, and both the sorted and unsorted libraries were sequenced with MiSeq (Table 3).
TABLE 2.
Library design overview
| Library name | Randomization strategy | Residues randomized (Chothia numbering) | Theoretical library size (on DNA level) | Library size (no. of transformants) |
|---|---|---|---|---|
| HC-1NNK | Separate single site mutagenesis of HC-CDRs by NNK | CDR1, 28–35; CDR2, 49–58; CDR3, 93–100b | 928 | 1 × 107 |
| HC-3NNK | Simultaneous single site mutagenesis of all 3 HC-CDRs by NNK | CDR1, 28–35; CDR2, 49–58; CDR3, 93–100b | 2.9 × 107 | 5 × 108 |
| LC-3NNK | Simultaneous single site mutagenesis of all 3 LC-CDRs by NNK | CDR1, 28–35; CDR2, 50–58; CDR3, 89–96 | 1.5 × 107 | 1 × 109 |
TABLE 3.
Deep sequencing statistics
| Panning | Library | No. of raw reads | No. of reads with full CDRs after quality control | Observed (counts >10)/expected single amino acid mutations |
|---|---|---|---|---|
| Unsorted | LC-3NNK | 5,835,436 | 1,522,999 | 483/483 |
| Ang2 | LC-3NNK | 4,291,707 | 3,121,241 | |
| VEGF | LC-3NNK | 4,758,105 | 3,589,324 | |
| Unsorted | HC-3NNK | 6,670,070 | 1,381,130 | 609/609 |
| Ang2 | HC-3NNK | 4,838,445 | 2,740,029 | |
| VEGF | HC-3NNK | 4,721,529 | 2,398,256 | |
| Unsorted | HC-1NNK CDR1 | 279,535 | 83,312 | 168/168 |
| Ang2 | HC-1NNK CDR1 | 144,103 | 92,573 | |
| VEGF | HC-1NNK CDR1 | 250,058 | 115,401 | |
| Unsorted | HC-1NNK CDR2 | 552,189 | 168,783 | 231/231 |
| Ang2 | HC-1NNK CDR2 | 287,387 | 188,498 | |
| VEGF | HC-1NNK CDR2 | 491,958 | 185,947 | |
| Unsorted | HC-1NNK CDR3 | 158,186 | 66,989 | 210/210 |
| Ang2 | HC-1NNK CDR3 | 390,433 | 256,998 | |
| VEGF | HC-1NNK CDR3 | 467,303 | 229,836 |
The ER, the ratio of the frequency of each mutation in the sorted library over the frequency of the same mutation in the unsorted library, were calculated to represent the effect or fitness of each mutation on antigen binding. To first determine whether our assumption is true, i.e. it is possible to estimate the fitness score of single mutations from the 3NNK library design, we compared the ER obtained from the 3NNK library design with that obtained from the 1NNK library design. ER obtained with the two different library designs correlated well (by using a linear model, the r2 for the hAng2 sorted samples is 0.73, and for the hVEGF-sorted samples it is 0.82, see Fig. 2), demonstrating the validity of our approach.
FIGURE 2.

Enrichments for single mutations from selection of the heavy chain 3NNK and 1NNK library are similar. The log2 enrichment ratios obtained from the 1NNK and 3NNK heavy chain library designs panned against Ang2 (A) and VEGF (B) are compared in a scatter plot. The mutations are colored according to the CDR in which they are located.
Enrichment Ratios Reflect Differential Binding Modes of 5A12 to the Two Antigens
The ER of 1092 mutations for the two binding functions of 5A12 are plotted as a heatmap (Fig. 3A). Interestingly, most of the 52 CDR positions exhibit distinct profiles of enrichment (Fig. 3A, red) and depletion (blue) of the mutations with VEGF- or Ang2 -binding selection indicating that the two binding functions involve differential usage of these CDR positions. The highly conserved positions from Ang2 panning that tolerate few if any single substitutions are mostly located on CDR-L1, CDR-L2, and CDR-L3 as well as on CDR-H3, whereas most of the conserved positions from VEGF panning are located in the three heavy chain CDR loops. The distinct binding pattern for the two antigens is even more evident when the conservation (ER average per position) is mapped onto the surface of the 5A12 structure yielding the functional paratope (Fig. 3B). In contrast to the extensively overlapping structural paratopes (Fig. 1, A and B), the two functional paratopes have a degree of spatial separation. Hot spot positions (surface-exposed positions that are highly conserved) for hAng2 binding mutations are located mostly on the light chain with the exception of HC-Y100a, while hVEGF hot spot positions are located primarily on the heavy chain.
FIGURE 3.

A, single mutation ER for all randomized CDR positions from Ang2 and VEGF panning. The heat maps show the ER for 1040 mutations in the heavy (left) and light chain (right) CDRs obtained from Ang2 (upper panel) and VEGF panning (lower panels). The line plot shows the solvent accessibility for each scanned position of 5A12 Fab as an unbound Fab based on the crystal structure of the 5A12 Fab in the Ang2-bound (orange line) or the VEGF-bound form (magenta line). B, 5A12 functional paratopes for Ang2 or VEGF binding as mapped by a deep mutational scan are viewed at the same orientation as the 5A12 structural paratopes in Fig. 1, C and D. The means of ER of all mutations at every mutated CDR position were calculated and mapped on the structures color-coded similarly as the heat map.
Furthermore, the structural plasticity of the antigen-binding site is reflected in the mutational data. Structurally, the CDR-H2 loop of 5A12 is engaged with hVEGF, although it is not involved in binding hAng2 (Fig. 4). The heat maps consistently show that CDR-H2 tolerates mutations at most positions for hAng2 binding, while being highly conserved at several positions for hVEGF binding, for example HC-G50, HC-G54, and HC-G55 (Figs. 3 and 4). Consistent with previous structural analyses of G6, these glycine residues are implicated to provide the necessary flexibility for CDR-H2 to adopt the unusual conformation for VEGF binding (40). The distinct mutational profile reflects the strategy for generating 5A12. 5A12 is based on the anti-VEGF antibody G6, which was isolated from a single framework library with randomized heavy chain CDR positions. Ang2 binding was subsequently recruited from a G6-based library with light chain residues randomized.
However, there are also similarities in the observed enrichment patterns for a few CDR positions comparing the Ang2 and VEGF-sorted samples, suggesting antigen-independent effects at these positions. Positions where both samples show mutation-sensitive results likely play a common structural role. For example, HC-R94 and HC-A49 are structurally buried at the base of CDR loops not making contact with either antigen, yet both tolerate few if any mutations (Fig. 3A). HC-R94 forms a salt bridge with D101, which is important for CDR-H3 conformation (47). Positions where both samples show similar tolerance to most mutations tend to be surface-exposed and structurally distant from the antigen-binding area as exemplified by HC-28 and LC-56. Their tolerance to mutations indicates a lack of involvement in the antigen binding and fewer structural constraints.
The enrichment pattern further provides insight into the mechanism of affinity maturation toward the single antigens. Several contact residues are selected for either single mutation (e.g. LC-S30aT and LC-A53D for Ang2 or VEGF binding, respectively) or a series of mutations (e.g. LC-L93V/I/R/K or LC-F31H/S/T/N for Ang2 or VEGF binding, respectively) with high enrichment, suggesting that these mutations optimize the existing contact between the antigen and the antibody (Fig. 3A), which is a well known mechanism for driving affinity maturation of antibodies (48, 49). Furthermore, some buried positions that are not in direct contact with the antigens contain selected substitutions that are highly enriched (e.g. LC-A34M, LC-Q89HW for Ang2 binding, and HC-F95Y, A100bV for VEGF binding), demonstrating the important and antigen-specific role of some buried, noncontact sites in antibody affinity maturation.
Deep Mutational Scanning Identifies Potential Mutations for Dual-specific Affinity Maturation
To determine how well the enrichment data correlate with experimental antigen binding affinity, we selected 25 clones that showed increased enrichment to at least one of the antigens. To obtain an affinity estimate, the IC50 for both antigens of the single point mutants of 5A12 were determined in a phage competition ELISA (Fig. 5A). The results showed that the ER metric serves as an excellent classifier to distinguish affinity-improving mutations from mutations that reduce or have no effect on affinity (receiver operator characteristics, area under the curve 0.93).
FIGURE 5.

Potential mutations for improving dual binding function. A, correlation between the enrichment (as log2ER, y axis) for 25 selected single mutations (15 in HC and 10 in LC) based on the deep sequencing data set and the improvement of Ang2- (red) or VEGF- (blue) relative affinity determined with phage competition ELISA (as log2(IC50 (mutant)/IC50 (wild type), x axis). Many mutants were selected because they may improve binding affinity based on the positive ERs. The enrichment derived from HC 3NNK and HC 1NNK libraries or LC 3NNK libraries were compared against relative affinity change. The Pearson's correlation (ρ) is shown. A comparison of the enrichment (log2ER) derived from the hVEGF-sorted versus hAng2-sorted HC-3NNK library (B) or LC-3NNK library (C) is shown.
For the dual affinity maturation of 5A12, we looked for mutations that improve the affinity toward both antigens or improve the binding toward one antigen without reducing the affinity toward the other antigen. An example of the latter is mutation HC-A53D, which is enriched in the hVEGF-sorted sample, but it is not enriched nor depleted in the hAng2 sorting (Fig. 4, E and F). A comparison of the enrichment of all HC mutations (Fig. 5B) or LC mutations (Fig. 5C) for hVEGF binding versus hAng2 binding indicates that most mutations have a negative impact on one interaction or another, but mutations that may be beneficial for potentially improving dual affinity do exist.
Combining Enrichment and Cooperativity Analyses Identifies Synergistic Mutation Pairs
Using the 3NNK dataset, we were able to calculate ER for 170,204 out of 400,428 possible mutation pairs (see under “Experimental Procedures” for details). One surprising pattern we found when analyzing the most enriched mutation pairs is that a few mutations show up repeatedly in several highly enriched mutation pairs as visualized in a Circos plot (Fig. 6, A and B). For example, in the dataset obtained from hAng2 panning, the CDR-H3 mutation F97P forms strongly enriched mutation pairs with several distinct mutations located in the CDR-H1 and CDR-H2 loop (pink ribbons in Fig. 6A). Interestingly, the majority of the “mutation partners” of HC-F97P are not in close spatial proximity to HC-F97P as shown in the structure of 5A12Ang2 (Fig. 6A, bar graph). A similar pattern can be observed for mutations LC-L93K and HC-F98P.
FIGURE 6.

Identification of affinity-improving, synergistic mutation pairs using a combination of enrichment, epistasis, and structural analysis. The Circos plot visualizes the highest enriched position pairs as calculated from sequencing data obtained from the hAng2-sorted HC 3NNK libraries (A) and LC 3NNK libraries (B). For this visualization, the enrichment of mutation pairs at the same CDR positions were summed up. The circular segments represent CDR positions, and the ribbons, connecting two positions, represent a position pair. The width of ribbon represents the sum of enrichment at this position. At the end of each ribbon, the amino acids are listed that form the mutation pairs at the positions connected by the ribbon. The histogram at the outer layer of the Circos plot shows the Cα-Cα distance between two residues in pairs. The ribbons of the position pairs are highlighted in pink when they contain fold-stabilizing mutations (see C and D). Positions are colored orange when they contain affinity improving mutation pairs (see main text for details). The correlation between the log2 ER of a given mutation in heavy chain (C) or light chain (D) libraries and the partner potentiation score from Ang2 panning is shown in a scatterplot. The melting temperature of selected Fab variants is shown relative to the parental 5A12. Triplet repeats of Tm measurement are highly reproducible with differences less than 0.2 °C. E, fold change in affinity (Kd) for hAng2 and hVEGF as measured with BIAcore of selected double mutation pairs and the comprising single mutations. Error bars represent standard error of the mean from three independent measurements. F, locations of synergistic mutation pairs identified using double mutation enrichment analysis are mapped on the structure of 5A12Ang2. The heavy chain is colored red, and the light chain is colored blue. Pairs are marked by spheres and connected by an orange line.
Two scenarios may explain the frequent presence of a particular mutation in enriched mutation pairs as follows: one is that the mutation provides a specific improvement of the antigen-binding interface, and the resulting higher affinity is further improved or not diminished by various mutation partners. Another scenario is that the mutation exhibits an advantage in fitness other than affinity during phage selection. For HC-F97P, we suspect the latter because its mutation partners are mostly spatially distant. Indeed, when 5A12HC-F97P affinity was measured in phage competition ELISA, we detected no improvement in affinity (data not shown). A reason for the strong selection of HC-F97P may thus be an improved functional folding in E. coli for clones carrying HC-F97P. To confirm this, we used a modified analysis of cooperativity as introduced by Araya et al. (27) to predict whether HC-F97P and other similar mutations would improve the thermostability of the 5A12 fold. This analysis yields for each mutation a so-called “partner potentiation score.” Large partner potentiation scores are typical for fold-improving mutations. We calculated partner potentiation scores using a simple multiplicative cooperativity model for a large subset of all possible mutation pairs from the data sets obtained from hAng2 panning. Indeed, as Fig. 6, C and D, shows, HC-F97P, HC-F98P, and LC-L93K have the highest partner potentiation score (>20). Differential scanning fluorometry confirmed that all three single mutation variants show an improved thermostability compared with 5A12 (Fig. 6, C and D).
To select mutation pairs for true affinity improvement of 5A12, we avoided those pairs containing mutation with a high partner potentiation score. In addition to enrichment and partner potentiation score, we applied a third measure to identify affinity improving mutation pairs, the spatial distance between those mutations. The rationale is that strong synergy would require some type of change in conformation and/or dynamics, and this is more likely to occur between adjacent residues (50). Seven mutation pairs were identified from the hAng2-sorted 3NNK heavy and light chain dataset, and their respective single mutation variants were generated for kinetic binding analysis by SPR measurement using a BIAcore instrument. Mapping of the mutation pairs onto the 5A12Ang2 structure shows that all mutation pairs selected, except one (HC-D30M/I51W), are located at the base of the different CDR loops suggesting conformational roles (Fig. 6F). Five selected double mutations show an increase in hAng2 affinity over 5A12 (Fig. 6E). Interestingly, among the selected mutation pairs, two pairs, HC-H35D/G50K and HC-W33P/I51G, show a strong increase in hAng2 binding in the context of a double mutant, although the single mutations by themselves have a strong negative impact on the hAng2 binding function. Based on the single mutation enrichment data, none of the mutation pairs would have been selected for affinity maturation (e.g. log2 ER H35D, −1.231; G50K, −1.93). Three of the selected pairs also showed an improvement of hVEGF binding as follows: LC-V33L/Q89Y, LC-A34G/Q89W, and LC-A34S/Q89H. Our extended deep sequencing and library approach was able to identify mutation pairs that improve dual affinity.
Combination of Mutations Allows Generation of Several High Affinity Variants
As a starting point to generate high dual affinity variants, we chose the structurally buried double mutation pair LC-A34G/Q89W from our cooperativity analysis, as well as the single mutations LC-Q89H and LC-Q89W, all of which exhibited increased dual affinity. We then combined those mutations with favorable single mutations that are mostly surface-exposed positions and located in different CDRs so that the individual contributions of the mutations are likely additive (Fig. 7). Several 5A12 variants exhibit sub-nanomolar affinity for hAng2 as well as hVEGF (Fig. 7B). The improved variants were then tested for their ability to block ligand receptor interaction using an in vitro receptor blocking assay. Although the ranking of these variants based on the receptor blocking potency or based on Kd value is not identical, all the affinity-improved DAF variants indeed improved blocking potency over the parental antibody 5A12 with some exhibiting a similar blocking activity as the high affinity anti-VEGF antibody G6.31 and anti-Ang2 antibody G5.5 (Fig. 7C).
FIGURE 7.

Affinity improved variants with sub-nanomolar dual affinity show similar blocking activity as mono-specific antibodies. A, positions in the heavy (red) or light chain (blue) of 5A12, which have been mutated to generate higher affinity variants, are shown as spheres. B, dual-specific affinity matured variants of 5A12 with various mutation combination indicated and their affinity as the average Kd, kon, and koff values from three independent BIAcore SPR experiments (with errors less than 50%). C, in vitro receptor blocking assay to compare the blocking of hAng2 binding to Tie2 as well as of hVEGF to VEGFR1 by 5A12, the high affinity 5A12 variants, and the mono-specific hAng2 and hVEGF antibody G5.5 and G6.31, respectively.
Conclusion
We demonstrate the use of deep mutational scanning to optimize a dual specificity binding site of a Two-in-One antibody, and to achieve, to our knowledge for the first time, sub-nanomolar monovalent affinity against two structurally unrelated antigens. This demonstrated the capacity of a single antigen-binding site to evolve high dual affinity. The process of deep mutational scanning of the 52 CDR residues for VEGF/Ang2 dual affinity provided a high resolution functional map for the two distinct interactions within a highly shared binding site. The map elucidates the subtly or drastically different roles played by most of the antibody CDR residues, which are either in direct contact or part of the structural scaffold for the two interactions. The dual specificity with sub-nanomolar affinity is achieved by intricately balancing the needs of the two interactions and differentially utilizing most of the 52 CDR residues.
Compared with previous work on deep mutational scanning mapping antibody or protein interactions, we modified the strategy of library design and sequence analysis to take advantage of the increased capability of the next generation sequencing of MiSeq to efficiently read through all three CDRs of the heavy or light chain variable domain. As demonstrated, the library design facilitates the identification of affinity-improving mutation pairs from two different CDRs as well as fold-stabilizing mutations that are compatible with antigen binding. The protocol introduced in this work may prove beneficial for achieving high affinity for dual-specific and species cross-reactive as well as mono-specific antibodies especially in cases where single mutation space has already been extensively explored without success.
Dual targeting strategies for ocular diseases such as NV AMD promise treatments with improved efficacy. However, designing such dual targeting therapies holds several challenges. Among those are the requirement of sufficiently high affinity to counter the action of both targets, finding the optimal antibody format to ease the formulation and manufacturing hurdle of targeting two activities and determining the most efficacious ratio needed for the two activities. Although VEGF/Ang2 DAF likely provides similar efficacy as two mono-specific Fabs in combination or a bi-specific antibody, it offers several advantages. The DAF format is a dual targeting single Fab, which has been shown to readily achieve high concentration formulation for intra-vitreal injection. Fab as the smallest natural antigen binding fragment allows packing more binding sites into the small volume one can inject into the eye than other therapeutic options of larger molecular size. Another potential advantage of the high affinity DAF format for dual targeting in ocular diseases is when one target is present at a higher concentration than the other. DAF is an “either/or” Fab that binds either one or the other antigens at one time, and it is therefore not confined to a fixed ratio of two targeting functions. Each DAF molecule is at all times capable of blocking either one of its two targets as needed. The ability to engineer dual-specificity of desired affinity expands the therapeutic potential of Fab molecules and Two-in-One antibody in ocular indications and beyond.
Author Contributions
P. K. and G. F. conceived the manuscript. All authors contributed to the design of the experiments. P. K., C. V. L., S. S., J. S., P. W., and S. F. H. performed the experiments. P. K., S. F. H., and G. F, are the main writers of the manuscript.
Acknowledgments
We thank Minhong Yan, Gu Zhang, and John Ridgeway for providing angiopoietin 2 for initial DAF generation; Isidro Hötzel for helpful discussions on NNK library design; Maryelise Cieslewicz for technical support in library generation and panning; and Catherine Foo and Somasekar Seshagiri for MiSeq sequencing. We also thank Shamrock Structures, LLC, for crystallographic data collection.
All authors are paid employees of Genentech, Inc., a member of the Roche Group, and are inventors of a patent application based on the work described herein.
This article was selected as a Paper of the Week.
The atomic coordinates and structure factors (codes 4ZFG and 4ZFF) have been deposited in the Protein Data Bank (http://wwpdb.org/).
S. Sanowar, C. V. Lee, G. Zhang, J. Brady, M. Yan, J. Cheng, J. Le Couter, and G. Fuh, unpublished data.
- NV AMD
- neovascular age-related macular degeneration
- Ang
- angiopoietin
- Fab
- antigen-binding fragment
- DAF
- dual action Fab
- CDR
- complementarity determining region
- HC
- heavy chain
- LC
- light chain
- ER
- enrichment ratio
- RBD
- receptor binding domain
- h
- human
- SAA
- solvent-accessible area
- mSAA
- maximal solvent accessible area
- SPR
- surface plasmon resonance.
References
- 1. Klein R., Klein B. E. (2013) The prevalence of age-related eye diseases and visual impairment in aging: current estimates. Invest. Ophthalmol. Vis. Sci. 54, ORSF5–ORSF13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Lookeren Campagne M., LeCouter J., Yaspan B. L., Ye W. (2014) Mechanisms of age-related macular degeneration and therapeutic opportunities. J. Pathol. 232, 151–164 [DOI] [PubMed] [Google Scholar]
- 3. Rosenfeld P. J., Rich R. M., Lalwani G. A. (2006) Ranibizumab: phase III clinical trial results. Ophthalmol. Clin. North Am. 19, 361–372 [DOI] [PubMed] [Google Scholar]
- 4. Ferrara N. (2010) Vascular endothelial growth factor and age-related macular degeneration: from basic science to therapy. Nat. Med. 16, 1107–1111 [DOI] [PubMed] [Google Scholar]
- 5. Campochiaro P. A. (2013) Ocular neovascularization. J. Mol. Med. 91, 311–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thurston G., Daly C. (2012) The complex role of angiopoietin-2 in the angiopoietin-tie signaling pathway. Cold Spring Harb. Perspect. Med. 2, a006550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rennel E. S., Regula J. T., Harper S. J., Thomas M., Klein C., Bates D. O. (2011) A human neutralizing antibody specific to Ang-2 inhibits ocular angiogenesis. Microcirculation 18, 598–607 [DOI] [PubMed] [Google Scholar]
- 8. Bostrom J., Yu S. F., Kan D., Appleton B. A., Lee C. V., Billeci K., Man W., Peale F., Ross S., Wiesmann C., Fuh G. (2009) Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen-binding site. Science 323, 1610–1614 [DOI] [PubMed] [Google Scholar]
- 9. Schaefer G., Haber L., Crocker L. M., Shia S., Shao L., Dowbenko D., Totpal K., Wong A., Lee C. V., Stawicki S., Clark R., Fields C., Lewis Phillips G. D., Prell R. A., Danilenko D. M., et al. (2011) A Two-in-One antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell 20, 472–486 [DOI] [PubMed] [Google Scholar]
- 10. Lee C. V., Koenig P., Fuh G. (2014) A Two-in-One antibody engineered from a humanized interleukin 4 antibody through mutation in heavy chain complementarity-determining regions. mAbs 6, 622–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bostrom J., Lee C. V., Haber L., Fuh G. (2009) Improving antibody binding affinity and specificity for therapeutic development. Methods Mol. Biol. 525, 353–376 [DOI] [PubMed] [Google Scholar]
- 12. Cunningham B. C., Wells J. A. (1989) High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244, 1081–1085 [DOI] [PubMed] [Google Scholar]
- 13. Weiss G. A., Watanabe C. K., Zhong A., Goddard A., Sidhu S. S. (2000) Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. U.S.A. 97, 8950–8954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu H., Beuerlein G., Nie Y., Smith H., Lee B. A., Hensler M., Huse W. D., Watkins J. D. (1998) Stepwise in vitro affinity maturation of Vitaxin, an αvβ3-specific humanized mAb. Proc. Natl. Acad. Sci. U.S.A. 95, 6037–6042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rajpal A., Beyaz N., Haber L., Cappuccilli G., Yee H., Bhatt R. R., Takeuchi T., Lerner R. A., Crea R. (2005) A general method for greatly improving the affinity of antibodies by using combinatorial libraries. Proc. Natl. Acad. Sci. U.S.A. 102, 8466–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fowler D. M., Araya C. L., Fleishman S. J., Kellogg E. H., Stephany J. J., Baker D., Fields S. (2010) High-resolution mapping of protein sequence-function relationships. Nat. Methods 7, 741–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Whitehead T. A., Chevalier A., Song Y., Dreyfus C., Fleishman S. J., De Mattos C., Myers C. A., Kamisetty H., Blair P., Wilson I. A., Baker D. (2012) Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat. Biotechnol. 30, 543–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fujino Y., Fujita R., Wada K., Fujishige K., Kanamori T., Hunt L., Shimizu Y., Ueda T. (2012) Robust in vitro affinity maturation strategy based on interface-focused high-throughput mutational scanning. Biochem. Biophys. Res. Commun. 428, 395–400 [DOI] [PubMed] [Google Scholar]
- 19. Forsyth C. M., Juan V., Akamatsu Y., DuBridge R. B., Doan M., Ivanov A. V., Ma Z., Polakoff D., Razo J., Wilson K., Powers D. B. (2013) Deep mutational scanning of an antibody against epidermal growth factor receptor using mammalian cell display and massively parallel pyrosequencing. mAbs 5, 523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee C. V., Sidhu S. S., Fuh G. (2004) Bivalent antibody phage display mimics natural immunoglobulin. J. Immunol. Methods 284, 119–132 [DOI] [PubMed] [Google Scholar]
- 21. Lee C. V., Liang W. C., Dennis M. S., Eigenbrot C., Sidhu S. S., Fuh G. (2004) High-affinity human antibodies from phage-displayed synthetic Fab libraries with a single framework scaffold. J. Mol. Biol. 340, 1073–1093 [DOI] [PubMed] [Google Scholar]
- 22. Sidhu S. S., Li B., Chen Y., Fellouse F. A., Eigenbrot C., Fuh G. (2004) Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J. Mol. Biol. 338, 299–310 [DOI] [PubMed] [Google Scholar]
- 23. Lee C. V., Hymowitz S. G., Wallweber H. J., Gordon N. C., Billeci K. L., Tsai S. P., Compaan D. M., Yin J., Gong Q., Kelley R. F., DeForge L. E., Martin F., Starovasnik M. A., Fuh G. (2006) Synthetic anti-BR3 antibodies that mimic BAFF binding and target both human and murine B cells. Blood 108, 3103–3111 [DOI] [PubMed] [Google Scholar]
- 24. R Core Team (2013) R: A Language and Environment for Statistical Computing, Lucent Technologies, Murray Hill, NJ [Google Scholar]
- 25. Morgan M., Anders S., Lawrence M., Aboyoun P., Pagès H., Gentleman R. (2009) ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 25, 2607–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mani R., St Onge R. P., Hartman J. L. 4th, Giaever G., Roth F. P. (2008) Defining genetic interaction. Proc. Natl. Acad. Sci. U.S.A. 105, 3461–3466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Araya C. L., Fowler D. M., Chen W., Muniez I., Kelly J. W., Fields S. (2012) A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proc. Natl. Acad. Sci. U.S.A. 109, 16858–16863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wickam H. (2009) ggplot2: Elegant Graphics for Data Analysis, Springer, New York [Google Scholar]
- 29. Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S. J., Marra M. A. (2009) Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3r1 Schrodinger, LLC, New York [Google Scholar]
- 31. Hötzel I., Theil F. P., Bernstein L. J., Prabhu S., Deng R., Quintana L., Lutman J., Sibia R., Chan P., Bumbaca D., Fielder P., Carter P. J., Kelley R. F. (2012) A strategy for risk mitigation of antibodies with fast clearance. mAbs 4, 753–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Niesen F. H., Berglund H., Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 [DOI] [PubMed] [Google Scholar]
- 33. Christinger H. W., Muller Y. A., Berleau L. T., Keyt B. A., Cunningham B. C., Ferrara N., de Vos A. M. (1996) Crystallization of the receptor binding domain of vascular endothelial growth factor. Proteins 26, 353–357 [DOI] [PubMed] [Google Scholar]
- 34. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 35. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O. S., Vonrhein C., Womack T. O. (2011) BUSTER, Version 2.11.4, Global Phasing Ltd, Cambridge, U.K. [Google Scholar]
- 36. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miller S., Janin J., Lesk A. M., Chothia C. (1987) Interior and surface of monomeric proteins. J. Mol. Biol. 196, 641–656 [DOI] [PubMed] [Google Scholar]
- 39. Liang W. C., Wu X., Peale F. V., Lee C. V., Meng Y. G., Gutierrez J., Fu L., Malik A. K., Gerber H. P., Ferrara N., Fuh G. (2006) Cross-species vascular endothelial growth factor (VEGF)-blocking antibodies completely inhibit the growth of human tumor xenografts and measure the contribution of stromal VEGF. J. Biol. Chem. 281, 951–961 [DOI] [PubMed] [Google Scholar]
- 40. Fuh G., Wu P., Liang W. C., Ultsch M., Lee C. V., Moffat B., Wiesmann C. (2006) Structure-function studies of two synthetic anti-vascular endothelial growth factor Fabs and comparison with the Avastin Fab. J. Biol. Chem. 281, 6625–6631 [DOI] [PubMed] [Google Scholar]
- 41. Chen Y., Wiesmann C., Fuh G., Li B., Christinger H. W., McKay P., de Vos A. M., Lowman H. B. (1999) Selection and analysis of an optimized anti-VEGF antibody: crystal structure of an affinity-matured Fab in complex with antigen. J. Mol. Biol. 293, 865–881 [DOI] [PubMed] [Google Scholar]
- 42. Barton W. A., Tzvetkova-Robev D., Miranda E. P., Kolev M. V., Rajashankar K. R., Himanen J. P., Nikolov D. B. (2006) Crystal structures of the Tie2 receptor ectodomain and the angiopoietin-2-Tie2 complex. Nat. Struct. Mol. Biol. 13, 524–532 [DOI] [PubMed] [Google Scholar]
- 43. Al-Lazikani B., Lesk A. M., Chothia C. (1997) Standard conformations for the canonical structures of immunoglobulins. J. Mol. Biol. 273, 927–948 [DOI] [PubMed] [Google Scholar]
- 44. Bostrom J., Haber L., Koenig P., Kelley R. F., Fuh G. (2011) High affinity antigen recognition of the dual specificity variants of herceptin is entropy-driven in spite of structural plasticity. PLoS One 6, e17887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Romero P. A., Arnold F. H. (2009) Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 10, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li B., Fouts A. E., Stengel K., Luan P., Dillon M., Liang W. C., Feierbach B., Kelley R. F., Hötzel I. (2014) In vitro affinity maturation of a natural human antibody overcomes a barrier to in vivo affinity maturation. mAbs 6, 437–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Morea V., Tramontano A., Rustici M., Chothia C., Lesk A. M. (1998) Conformations of the third hypervariable region in the VH domain of immunoglobulins. J. Mol. Biol. 275, 269–294 [DOI] [PubMed] [Google Scholar]
- 48. Cauerhff A., Goldbaum F. A., Braden B. C. (2004) Structural mechanism for affinity maturation of an anti-lysozyme antibody. Proc. Natl. Acad. Sci. U.S.A. 101, 3539–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li Y., Li H., Yang F., Smith-Gill S. J., Mariuzza R. A. (2003) X-ray snapshots of the maturation of an antibody response to a protein antigen. Nat. Struct. Biol. 10, 482–488 [DOI] [PubMed] [Google Scholar]
- 50. Skinner M. M., Terwilliger T. C. (1996) Potential use of additivity of mutational effects in simplifying protein engineering. Proc. Natl. Acad. Sci. U.S.A. 93, 10753–10757 [DOI] [PMC free article] [PubMed] [Google Scholar]