

Background: How atypical PKCs are maintained in an active conformation is unknown.
Results: We identify an acidic surface on the aPKC scaffold, p62, that tethers the kinase's autoinhibitory pseudosubstrate to allow activity. The biologically active basic peptide, ZIP, competes for binding to this surface, resulting in localized aPKC autoinhibition.
Conclusion: p62 tethers aPKCs in an active conformation.
Significance: p62 is a molecular target for ZIP.
Keywords: enzyme mechanism, p62 (sequestosome 1(SQSTM1)), protein-protein interaction, scaffold protein, serine/threonine protein kinase, atypical protein kinase C (aPKC)
Abstract
Atypical protein kinase C (aPKC) enzymes signal on protein scaffolds, yet how they are maintained in an active conformation on scaffolds is unclear. A myristoylated peptide based on the autoinhibitory pseudosubstrate fragment of the atypical PKCζ, zeta inhibitory peptide (ZIP), has been extensively used to inhibit aPKC activity; however, we have previously shown that ZIP does not inhibit the catalytic activity of aPKC isozymes in cells (Wu-Zhang, A. X., Schramm, C. L., Nabavi, S., Malinow, R., and Newton, A. C. (2012) J. Biol. Chem. 287, 12879–12885). Here we sought to identify a bona fide target of ZIP and, in so doing, unveiled a novel mechanism by which aPKCs are maintained in an active conformation on a protein scaffold. Specifically, we used protein-protein interaction network analysis, structural modeling, and protein-protein docking to predict that ZIP binds an acidic surface on the Phox and Bem1 (PB1) domain of p62, an interaction validated by peptide array analysis. Using a genetically encoded reporter for PKC activity fused to the p62 scaffold, we show that ZIP inhibits the activity of wild-type aPKC, but not a construct lacking the pseudosubstrate. These data support a model in which the pseudosubstrate of aPKCs is tethered to the acidic surface on p62, locking aPKC in an open, signaling-competent conformation. ZIP competes for binding to the acidic surface, resulting in displacement of the pseudosubstrate of aPKC and re-engagement in the substrate-binding cavity. This study not only identifies a cellular target for ZIP, but also unveils a novel mechanism by which scaffolded aPKC is maintained in an active conformation.
Introduction
Coordination on protein scaffolds plays a key role in controlling specificity and fidelity in cell signaling (2). Binding to protein scaffolds is particularly important in controlling the function of atypical protein kinase C (aPKC)4 isozymes λ/ι (mouse/human) and ζ, a class of the Ser/Thr PKC family whose distinguishing feature is the lack of regulation by the lipid second messenger, diacylglycerol (3). Rather, protein interactions are central to the biological function of these isozymes, and a number of scaffolds have been identified that bind aPKCs to position them near substrates (4).
Atypical PKCs and the canonical diacylglycerol-regulated PKCs share the same architecture of a C-terminal kinase domain that is autoinhibited by intramolecular interactions with modules in the N-terminal moiety (5–8). Specifically, a pseudosubstrate segment immediately preceding a C1 domain occupies the substrate-binding cavity in the inactive conformation, and its release from the substrate-binding cavity is required for activation. However, atypical PKCs differ in how this activation is achieved. For conventional and novel PKC isozymes, binding of diacylglycerol to one of the tandem C1 domains induces a conformational change that expels the pseudosubstrate from the substrate-binding cavity (9). For conventional PKCs, binding of diacylglycerol is facilitated by Ca2+-dependent recruitment of the kinase via the plasma membrane-sensing Ca2+-regulated C2 domain (10–12). However, atypical PKCs do not have a diacylglycerol sensor. Although they do possess a C1 domain immediately following the pseudosubstrate segment, the ligand-binding pocket is compromised and neither the physiological ligand diacylglycerol nor the potent functional analogues, phorbol esters, bind atypical C1 domains (13, 14). Thus, it is unclear what the physiological mechanism is for release of the pseudosubstrate of atypical PKCs.
Atypical PKCs differ from conventional and novel PKCs by having an N-terminal Phox and Bem1 (PB1) domain, a protein-protein interaction module that homodimerizes with the PB1 domain of proteins such as the scaffold p62, also known as sequestosome 1 (SQSTM1) and zeta-interacting protein (15–17). In addition, they contain a Type III PDZ ligand that is predicted to target aPKCs to PDZ domain proteins that bind Type III ligands (18, 19). This tethering to protein scaffolds is likely the key determinant in specificity of signaling by aPKCs, particularly given their exceptionally low catalytic rate (1–2 orders of magnitude lower than that of conventional PKC isozymes, depending on the substrate (20)).5 However, how the pseudosubstrate is released from the substrate-binding cavity to achieve efficient signaling at these scaffolds remains to be resolved.
A myristoylated peptide based on the pseudosubstrate segment of PKCζ, zeta inhibitory peptide (ZIP), has been extensively used to inhibit aPKC activity. Although the peptide (and one in which the sequence is scrambled) effectively inhibits aPKC in vitro (1, 21), we have previously shown that ZIP does not inhibit the catalytic activity of endogenous aPKC or a construct of the unconstrained catalytic domain, PKMζ, in cells (1). This is not surprising, as the affinity of the pseudosubstrate for the active site of PKCs is 2 orders of magnitude lower than the nanomolar affinity required for peptides to effectively bind substrates in cells (22). However, ZIP has profound effects on cellular function. For example, it slows the assembly of tight junctions in Madin-Darby canine kidney epithelial cells (23), inhibits insulin-induced glucose uptake in human adipocytes (24), and inhibits insulin-dependent Rab4-GTP binding in 3T3-L1 adipocytes (25). Most strikingly, treatment of brain slices with ZIP inhibits long-term potentiation (LTP), the cellular correlate of memory (21, 26–29). Although a transcript variant of PKCζ that only encodes the kinase domain, PKMζ, is frequently invoked as the target of ZIP, mice lacking PKCζ/PKMζ retain full sensitivity to ZIP (30, 31) or scrambled ZIP (30, 31). This continued sensitivity to ZIP upon loss of PKCζ/PKMζ validates the finding that ZIP does not directly inhibit the intrinsic catalytic activity of the aPKC kinase domain in cells. Given the ability of the peptide to inhibit cellular function, it is likely that it has a bona fide target in cells unrelated to direct binding to the active site of aPKCs.
Here we sought to identify a bona fide target of ZIP and, in so doing, unveiled a novel mechanism by which aPKCs are maintained in an active conformation on a protein scaffold. Specifically, we identify a previously undescribed interaction between the pseudosubstrate of aPKCs and an acidic surface on p62 that stabilizes the open, active conformation of aPKC. Using a genetically encoded reporter for PKC activity fused to the p62 scaffold, we show that ZIP inhibits the activity of wild-type PKCλ but not a construct lacking the pseudosubstrate. These data support a model in which ZIP competes for binding tothe acidic surface, resulting in displacement of the pseudosubstrate of aPKC and re-engagement in the active site and autoinhibition of the kinase. This study not only identifies a cellular target for ZIP, but also unveils a novel mechanism by which a protein scaffold locks a kinase in an active conformation.
Experimental Procedures
Materials
Myristoylated PKC zeta, pseudosubstrate (ZIP), Cy5-conjugated ZIP, and the non-conjugated Cy5 control (HiLyteTM Fluor 647 amine) were purchased from AnaSpec (Fremont, CA). For co-immunoprecipitation experiments, monoclonal HA.11 (16B12) antibody (Covance, MMS-101P-599), along with protein A/G UltraLink resin (Thermo Scientific, 53145), was used to capture HA-tagged proteins, and normal mouse IgG (Santa Cruz Biotechnology, sc-2025) was used as control. Rat HA antibody (Sigma-Aldrich, 3F10), rabbit polyclonal GFP antibody (Cell Signaling, 2555S), phospho-PKCζ (Thr410) (Santa Cruz Biotechnology, sc-12894-R), phospho-PKCζ [EP2037AY] (Thr560) (Abcam, ab62372), and rabbit PKCλ (BD Transduction Laboratories, 610397) were used for Western blotting or visualization of peptide array. Auranofin (ANF, CAS 34031-32-8) was purchased from TOCRIS (catalogue number 4600). Gö6983 (CAS 133053-19-7) was purchased from Calbiochem (365251), and calyculin A (CAS 101932-71-2) was purchased from Cell Signaling (9902). DMEM (10-013-CV), Dulbecco's PBS (21-031-CV), and Hanks' balanced salt solution (21-022-CV) were obtained from Cellgro. PZ09 was synthesized as described (32).
Plasmids
The protein kinase C kinase activity reporter (CKAR) was described previously (33, 34). CKAR was fused to the N terminus of human p62 (a gift from Jorge Moscat, BC017222.1). Mouse PKCλ (NM_008857.3) and human PKCι (a gift from Alan Fields, NM_002740.5) constructs were N-terminally tagged with monomeric yellow or cherry fluorescent proteins. The truncation mutants are: PKCλ ΔPB1 missing residues 26–107, PKCλ ΔPS missing residues 114–131, and PKCι PB1-PS containing amino acid sequence 16–131. p62 constructs contain either a monomeric cyan or a monomeric yellow fluorescent protein fused to its N terminus, and the HA-p62 PB1 has an HA tag fused to the amino acid sequence 2–102 of p62. The ZIP-insensitive construct of p62, p62E32/81Q, was created by site-directed mutagenesis. To generate the GluR1-GFP and GluR2-GFP constructs, GFP (flanked by GGSGTRGGS and SGGSGGGG linkers) was inserted in the intracellular C-terminal portion of GluR1 (at Ser870) and GluR2 (at Ser862) using PCR cloning.
Cell Culture and Transfection
COS-7 and HEK293T cells were maintained in DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C in 5% CO2. Transient transfection was carried out using FuGENE 6 (Promega).
Immunoprecipitation
HEK293T cells were transfected with YFP-PKCι PB1PS and HA-p62 PB1 and treated with 5 μm myristoylated ZIP for 20 min or 10 μm Auranofin for 60 min at room temperature. Cells were rinsed with PBS and then lysed in lysis buffer (50 mm Tris, pH 7.4, 100 mm NaCl, 1% Triton X-100, 10 mm Na4P2O7, 50 mm NaF, 5 mm EDTA, 1 mm Na3VO4) with fresh protease inhibitors (50 μg/ml leupeptin, 2 mm benzamidine, and 100 μm PMSF) and 1 mm DTT. The insoluble pellet of the lysate was removed by >16,000 × g spin for 10 min at 4 °C. The soluble portion was incubated with 1 μl of monoclonal HA.11 (16B12) antibody for 2 h at 4 °C while constantly agitated. The antibody-bound proteins were captured by the addition of 10 μl of protein A/G UltraLink resin, rocked for 1 h at 4 °C, and pelleted by a 1-min 6,000 × g spin. The immunopellets were washed three times with 1 ml of lysis buffer and then denatured in 4× sample buffer for Western blot analysis.
Protein Structure Prediction
No homologous structure template is detected for the pseudosubstrate region of PKCζ. Ab initio structure prediction software QUARK (35) was used to model the three-dimensional structure of the pseudosubstrate region.
Protein-Protein Docking
The protein-protein docking was carried out in three steps. First, 20 initial binding poses between the p62 PB1 dimer (rat; Protein Data Bank ID 2KTR) and the PKCζ pseudosubstrate region were generated using ZDock (36) and HEX (37); these putative complex structures were refined using RosettaDock (38). The charge distributions in the binding interface were visualized and analyzed. The binding pose with the complementary charge distributions in the binding interface was selected.
Live Cell FRET Imaging
To access protein-protein interactions using FRET, COS-7 cells were plated onto sterilized glass coverslips in 35-mm dishes and transfected with CFP-tagged p62 and various YFP-tagged constructs. Approximately 24 h after transfection, cells were washed and imaged in Hanks' balanced salt solution with 1 mm CaCl2 in the dark at room temperature. 5 μm ZIP, 10 μm Auranofin, 5 μm PZ09, or 50 nm calyculin A was added as indicated. Images from CFP, YFP, and FRET channels were acquired and analyzed as described previously (34, 39). The FRET for the protein-protein interaction experiments FRET/CFP ratio was plotted. In CKAR-p62 experiments, COS-7 cells were transfected with CKAR-p62 and mCherry-PKC constructs, and the analysis used CFP/FRET as the FRET ratio so that a phosphorylation event is shown as an increase in FRET ratio.
Fluorescence Lifetime Imaging
Fluorescence lifetime imaging microscopy (FLIM) was performed as described previously (40). Briefly, live COS-7 cells were imaged with a SliceScope two-photon microscope (Scientifica) using a 60× water immersion objective (LUMPLFLN 60XW, NA = 1.0, Olympus) and a Chameleon Ultra II IR laser (Coherent) tuned at 930 nm for the excitation of GFP. Fluorescence lifetime images were acquired with a hybrid photomultiplier tube detector (HPM-100-40, Becker and Hickl) and a time-correlated single photon counting module (SPC-150, Becker and Hickl). FLIM images were analyzed in SPCImage (41) with a single exponential model. Curve offset and shift were not fixed; this allowed for better goodness of fit values, particularly in images that had a broad range of pixel intensities. For further analysis, each FLIM image was exported as a matrix of lifetimes, photon counts, and goodness of fit values (χ-squared). A custom MATLAB program was used to output a mean lifetime for each selected region of interest (1–4 regions of interest per cell). To calculate FRET efficiency, the following formula was used: EFRET = 1 − TDA/TD (TDA = lifetime of the donor in the presence of the acceptor, TD = average lifetime of the donor alone) (42).
Peptide Array
Peptide spot arrays were generated by the INTAVIS MultiPep RS peptide synthesizer (INTAVIS Bioanalytical Instruments AG, Koeln, Germany) using standard Fmoc (9-fluorenylmethoxycarbonyl) protection-based solid phase peptide synthesis to produce short, overlapping 18-mer peptides directly conjugated onto amino-PEG modified cellulose membranes (ACS01, Intavis AG). Each peptide spot sequence corresponds to a segment of the PB1 domain of p62, scanning residues 1–105 by shifting in increments of three amino acids.
Each pair of array strips was blocked with 5% BSA in TBS-T (0.0005% Tween 20) for 1 h at room temperature. Each strip was incubated with 2 μg/ml (27 nm) of PKCλ purified from insect cells plus either 5 μm Cy5-conjugated ZIP or 5 μm Cy5 as a control for 1 h at room temperature. The arrays were washed three times with TBS-T for 10 min and once with TBS and then incubated with anti-PKCλ antibodies for 30 min at room temperature. Standard Western blotting method was used to visualize PKCλ binding to the array. A FluorChem Q imaging system (ProteinSimple) was used for visualization and quantification of Cy5-conjugated ZIP (Cy5-ZIP) binding and PKCλ binding to the peptide arrays.
Results
The Basic Pseudosubstrate of aPKC Is Predicted to Bind to an Acidic Patch within the PB1 Domain of p62
Given the inability of ZIP, a myristoylated peptide comprising the PKCζ pseudosubstrate sequence (SIYRRGARRWRKL; this sequence is conserved in PKCι/λ), to directly inhibit the catalytic activity of the kinase domain of PKCζ in cells, despite its profound cellular effects, we reasoned that the pseudosubstrate segment of aPKCs may bind a unique surface to mediate a biological function. To identify potential targets, we interrogated the STRING database for binding partners for PKCζ because the pseudosubstrate sequence is from this protein. Because the biological effects of ZIP are not observed with the general kinase inhibitor staurosporine, we focused on non-kinase interaction partners. Thus, we ruled out interaction partners such as PDK1, Akt1, and RAF1. Of the remaining partners in the interaction network, we focused on p62, a PB1 domain-containing scaffold that binds the PB1 domain of aPKCs. We noted that the pseudosubstrate immediately follows the PB1 domain (Fig. 1A). We next docked the pseudosubstrate segment on the PB1 domain of p62. Because there is no homologous structure template for the ZIP containing region, we used ab initio structure prediction to model the peptide and then used the ZDock (36) and HEX (37) programs to predict the putative binding pose between the pseudosubstrate of PKCζ and the human p62 PB1 dimer; the structure was refined using RosettaDock (38). This resulted in the identification of a negatively charged surface on the PB1 domain of p62, distinct from the surface involved in the canonical binding to the aPKC PB1 domain, which is complimentary to the positively charged surface of ZIP (Fig. 1B). Notably, acidic segments from two distinct loops within the p62 PB1 domain, one containing Glu32 and the other containing Glu81, came together to form this previously unidentified potential binding surface.
FIGURE 1.

The basic pseudosubstrate of aPKC interacts with an acidic surface on the PB1 domain of the scaffold p62. A, schematic representation showing the domain structure of atypical PKC members (PKCλ/ι and PKCζ) and p62, each of which contains a PB1 domain (lavender and tan, respectively). aPKCs also have a pseudosubstrate segment (green rectangle; sequence indicated below) immediately followed by a C1 domain (orange) and a kinase domain (cyan). p62 also has a ZZ domain (pink) and ubiquitin-associated (UBA) domain (yellow). B, structural modeling showing predicted binding pose of PKCζ pseudosubstrate to a PB1 domain dimer of p62 (rat; Protein Data Bank ID 2KTR). A fragment of PKCζ sequence (residues 79–146) containing the pseudosubstrate segment (green helix) flanked by a partial C1 domain and a partial PB1 (gray) was docked onto the structure of a PB1 dimer of p62 (blue and purple). The positively charged Arg and Lys residues (shown in dot representation) within the pseudosubstrate are predicted to form an electrostatic interaction interface with the negatively charged Glu and Asp residues of the p62 PB1 (shown in red stick and dot representation); these residues include Glu81 and Glu32 on two separate segments of the p62 PB1 domain. C, peptide overlay of the p62 PB1 domain with Cy5-ZIP or Cy5 and PKCλ. Overlapping 18-mer peptides covering the PB1 domain of p62 (residues 1–105) and staggered by three amino acids were overlaid with 5 μm Cy5 or Cy5-ZIP, and binding was detected by a FluorChem Q imaging system. The sequence of residues 1–54 is shown above and that for residues 43–99 below the array. Glu residues present in the spots to which ZIP bound are indicated in red; Glu32 and Glu81 are indicated in bold. Lys residues known to bind the PB1 domain of p62 are colored in blue. Spots are numbered on the left. D, peptide overlay of the p62 PB1 domain with PKCλ (27 nm) and Cy5 or PKCλ and Cy5-ZIP (5 μm). PKCλ binding to the array was detected with an anti-PKCλ antibody. Spots in clusters 1 and 2 (blue boxes) were quantified in E. The graph presents the mean ± S.E. of three separate strips. The data were analyzed by non-paired Student's t test: *, p < 0.05.
ZIP Binds Acidic Segments on the PB1 Domain of p62 and Competes with the Binding of PKCλ
To validate the predicted interaction of ZIP with the complimentary acidic surface on p62, we overlaid Cy5-conjugated ZIP on a peptide array containing overlapping 18-residue peptides covering the PB1 domain (residues 1–105) of p62 (Fig. 1C). ZIP bound two regions of the PB1 domain of p62, both enriched in acidic residues. Binding to the first cluster (spots 2–12) was observed when Glu19 was incorporated in the peptides (spot 2) and was no longer observed when Glu36 was removed from the peptides (spot 13). This cluster contained Glu32, identified in the docked model as a key determinant for binding ZIP. Binding to the second cluster (spots 18–22) was observed when Asp69 was present (spot 18) and no longer observed when the segment QAH was dropped (spot 23). No significant binding of Cy5 alone was observed (left strip). Thus, the segment 19EIRRFSFCCSPEPEAEAE36 was identified as a binding region for ZIP; the underlined residue corresponds to Glu32 (Fig. 1B) on one of the two acidic loops comprising the predicted ZIP-binding surface.
We next examined the binding of full-length PKCλ to the p62 PB1 domain peptide array (Fig. 1D). Purified PKCλ bound the peptides that ZIP bound, in addition to spot 1 and sequences between theses two clusters (corresponding to residues 34–66, spots 12–17). Spot 1 contains Lys7, a key determinant in mediating the canonical binding of the PB1 domain of aPKCs to that of p62 (43); there was also particularly strong interaction with all peptides that contained two other key determinants, Arg21 and Arg22 (spots 3–8). Thus, in the primary sequence, these peptides contained determinants from the acidic cluster that bind the pseudosubstrate segment (Fig. 1B) as well as the basic cluster that binds an acidic surface on the PB1 domain of PKCλ (15). In the presence of ZIP, binding of PKCλ to the same regions where ZIP bound was reduced (Fig. 1D, boxed areas labeled cluster 1 and cluster 2). Quantification of the signal from three independent overlays revealed that ZIP decreased the binding of PKCλ to cluster 1 (spots 8–11) by ∼32% and to cluster 2 (spots 18–22) by ∼73%, with no effect on an area that did not bind ZIP (normalization patch, spots 15–17) (Fig. 1E). The greater effect of ZIP on patch 2 is likely because the pseudosubstrate of PKCλ is the primary determinant driving binding to these peptides, whereas the PB1 domain of PKCλ also contributes to binding of the peptides in cluster 1. These data confirm that ZIP binds an acidic segment on the PB1 domain of p62 and that this interaction competes with binding of full-length PKCλ.
Atypical PKCλ Interacts with Scaffold Protein p62 via PB1-PB1 Interface and Prevents p62 Aggregation
To explore the role of the pseudosubstrate of PKCζ in binding p62, we generated YFP-tagged constructs of full-length PKCλ, a construct in which the PB1 domain was deleted (YFP-PKCλ ΔPB1), one in which the pseudosubstrate was deleted (YFP-PKCλ ΔPS), and a construct of PKCι containing its PB1 domain through the pseudosubstrate segment (PKCι PB1-PS, residues 16–131) (Fig. 2A). First, we examined whether ZIP inhibits the known PB1-PB1 interaction of p62 and aPKC. The effect of treating COS-7 cells with ZIP on the interaction between HA-tagged p62 PB1 (residues 3–102) and the PKCι PB1-PS construct was examined by co-immunoprecipitation. When using an HA antibody, the YFP-tagged PKCι construct (PKCι PB1-PS) was effectively co-immunoprecipitated with the HA-tagged p62 PB1 domain (Fig. 2B, lane 2), and this interaction was not affected by treatment of cells with ZIP (Fig. 2B, lane 3). As a positive control, this interaction was dissociated by ANF, a molecule that forms a thio-gold adduct with Cys69 of the PB1 domain of PKCι to disrupt its interaction with PB1 domain partners (44, 45) (Fig. 2B, lane 4). ZIP treatment did not enhance the ability of ANF to disrupt the interaction of the two PB1 domains (Fig. 2B, lane 5). Thus, ZIP does not dissociate the PKCι PB1-PS construct from the p62 PB1 domain, but the PB1 domain disruptor, ANF, does.
FIGURE 2.

Atypical PKC physically interacts with scaffold p62. A, schematic of YFP-tagged PKCλ constructs used in this study: PKCλ with PB1 domain deleted (PKCλ ΔPB1), PKCλ with pseudosubstrate deleted (PKCλ ΔPS), and N-terminal portion of PKCι containing PB1 domain through pseudosubstrate (PKCι PB1-PS). Indicated are the phosphorylation sites (P) at the activation loop and turn motif sites; occupancy of these sites serves as a measure for proper folding of the enzyme (see Refs. 5 and 6). B, COS-7 cells co-expressing YFP-tagged PKCι PB1-PS and HA-tagged p62 PB1 were treated with 5 μm ZIP for 20 min and/or 10 μm ANF for 1 h before lysing. Immunoprecipitation (IP) was performed with a HA monoclonal antibody or IgG control. Immunoprecipitates (entire sample) and 2.5% of total lysates were analyzed by Western blots (IB) probing for a GFP antibody to detect PKCι and a HA antibody to detect the PB1 domain of p62. The PKCι PB1-PS detected in the immunoprecipitates was normalized to the HA signal in the immunopellet and quantified in the graph below. Data represent the mean ± S.E. of 6 independent experiments and were analyzed by one-way analysis of variance with Holm-Šídák multiple comparison test: n.s., no significance; *, p < 0.05; **, p < 0.01. C, fluorescence micrographs of COS-7 cells showing the localization of CFP-tagged p62 expressed alone (panel i) or with PKCλ (panel ii), PKCλ ΔPB1 (panel iii) or PKCλ ΔPS (panel iv). Images were adjusted for an optimal dynamic range using ImageJ. D, Western blot analysis of YFP-tagged constructs showing immunoreactivity with antibodies to the activation loop (Thr410) and turn motif sites (Thr560) as well as total GFP to detect the YFP-tagged PKCλ. p-410 PKCλ, phospho-PKCλ (Thr410); p-560 PKCλ, phospho-PKCλ (Thr560). E, Western blot of COS-7 cells expressing the constructs in panel C and probed with antibodies to PKCλ, p62, and β-actin.
We noted that p62 formed aggregates when expressed in COS-7 cells but that co-expression with PKCλ prevented this aggregation and resulted in a more even cytosolic distribution of p62, as observed using fluorescence microscopy (Fig. 2C, compare panels i and ii). The ability to prevent aggregation of p62 was abolished upon deletion of the PB1 domain (YFP-PKCλ ΔPB1) to prevent the binding of PKC to p62, indicating that the binding of PKCλ to p62 prevented p62 aggregation (Fig. 2C, panel iii). We took advantage of this effect to address whether deletion of the pseudosubstrate segment affected the binding of PKCλ to p62. Fig. 2C (panel iv) shows that a construct lacking the pseudosubstrate (YFP-PKCλ ΔPS) effectively prevented p62 aggregation. The inability of PKCλ ΔPB1 to de-aggregate p62 was not a result of misfolding of the construct, as both deletion constructs were processed by phosphorylation, evident by immunoreactivity with phospho-antibodies to the activation loop phosphorylation site, Thr410, and the turn motif phosphorylation site, Thr560 (Fig. 2D), nor was it a result of greater overexpression of CFP-p62 in the conditions promoting aggregation because similar levels of CFP-p62 were expressed under non-aggregating (co-expression with YFP-PKCλ; Fig. 2E, lane 2) and aggregating (co-expression with YFP-PKCλ ΔPB1; Fig. 2E, lane 3) conditions. Note that co-expression with any of the PKCλ constructs resulted in a modest increase in the expression of both endogenous and CFP-tagged p62, suggesting that the presence of PKCλ may stabilize p62 (Fig. 2E). These data are consistent with the canonical PB1 domain interaction driving the binding of PKCλ to p62, independent of an intact pseudosubstrate, an event that prevents the aggregation of p62.
ZIP Displaces the Pseudosubstrate Segment from p62
To address whether ZIP disrupts the predicted interaction of the pseudosubstrate segment of scaffolded PKCλ with the acidic surface on p62, we asked whether FRET between CFP-p62 and YFP-PKCλ changed upon treatment of cells with ZIP. Treatment of cells with 5 μm ZIP resulted in a statistically significant decrease in FRET between CFP-p62 and YFP-PKCλ (Fig. 3A, black circles, and graph). The scrambled ZIP peptide (RLYRKRIWRSAGR) caused a similar drop in the FRET ratio as ZIP (Fig. 3A, gray circles, and graph), suggesting that net charge rather than exact sequence is important to cause the FRET drop. Supporting this possibility, molecular modeling revealed that the scrambled peptide is also predicted to form an amphipathic helix with a basic face (data not shown). In contrast, ZIP was ineffective at disrupting a CFP-p62-YFP-p62 interaction (brown trace, Fig. 3A). These results are consistent with ZIP competing for the binding of the pseudosubstrate segment of PKCλ on p62, but not p62-p62 interactions.
FIGURE 3.

ZIP disruption of p62/aPKC complex requires an intact pseudosubstrate segment. A, the ability of ZIP to disrupt the p62/PKCλ complex was assessed by monitoring real-time FRET between fluorescent pair CFP and YFP using live cell imaging. The FRET/CFP ratio of COS-7 cells co-expressing CFP-p62 and YFP-PKCλ (black circles, n = 26) or YFP-p62 as control (brown circles, n = 13) was monitored before and after treatment with 5 μm ZIP or scrambled ZIP (Scr. ZIP) (gray circles, n = 24). Data were plotted as a p62-aPKC interaction and represent the normalized FRET/CFP ratio mean ± S.E. of the indicated number of cells from at least 3 independent experiments. The graph shows the ratio values immediately prior to ZIP treatment (basal) and 10 min past treatment (+ ZIP) analyzed by paired Student's t test: n.s., no significance; *, p < 0.05; **, p < 0.01. B, FRET/CFP ratio changes were monitored as in panel A for COS-7 cells co-expressing YFP-PKCλ and either CFP-tagged wild-type p62 (black circles, n = 26) or a construct in which Glu32 and Glu81 were mutated to Gln (p62E32/81Q; open circles, n = 24); cells were treated with 5 μm ZIP. C, FRET/CFP ratios were monitored for COS-7 cells expressing CFP-p62 and YFP-PKCλ ΔPS (red circles, n = 31), PKCλ ΔPB1 (blue circles, n = 12), or full-length YFP-PKCλ (black circles, n = 26). D, FRET/CFP ratios were measured for COS-7 cells expressing CFP-p62 and YFP-PKCλ (black circles, n = 15), YFP-PKCλ ΔPS (red circles, n = 16), or CFP-PKCλ ΔPB1 (blue circles, n = 15) before and after treatment with 10 μm ANF. E, fluorescence micrographs of COS-7 cells showing the localization of CFP-tagged p62 co-expressed with PKCλ (panels i and iv), PKCλ ΔPB1 (panels ii and v), or PKCλ ΔPS (panels iii and vi) before or after 1 h ANF treatment.
Next, we determined whether the predicted acidic surface on the p62 PB1 domain is responsible for the ZIP-sensitive binding of PKCλ. Based on the predicted docking pose in Fig. 1B, we selected two glutamate residues, Glu32 and Glu81, predicted to form ion pairs with basic residues in the pseudosubstrate sequence to mutate to Gln. Fig. 3B reveals that ZIP was ineffective in altering FRET between the p62E32/81Q construct and PKCλ (Fig. 3B). These results validate the acidic cluster illustrated in Fig. 1A as the relevant binding surface for the ZIP-sensitive interaction of PKCλ with p62.
Pseudosubstrate of aPKC Is Necessary for ZIP to Modulate the aPKC-p62 Interaction
To test the hypothesis that ZIP competes with the binding of the PKCλ pseudosubstrate segment to p62, as suggested by in vitro peptide array data, we examined the effect of ZIP on the interaction of p62 with a PKCλ construct lacking the pseudosubstrate region (PKCλ ΔPS). This construct still binds p62, as indicated by its ability to prevent p62 aggregation (Figs. 2C, panel iv, and 3E, panel iii). FRET between this construct and p62 was insensitive to ZIP (Fig. 3C, red circles), revealing that the pseudosubstrate segment mediates the ZIP-sensitive interaction with p62. A construct lacking the PB1 domain (PKCλ ΔPB1) retained ZIP sensitivity, revealing that even in the absence of the canonical association of PKCλ to p62 via the PB1 domain, there was sufficient weak binding via the pseudosubstrate that could be displaced by ZIP. As a control, the association of the PKCλ ΔPS construct was sensitive to ANF, the PB1-PB1 disruptor (red circles in Fig. 3D). Its release from the scaffold was also revealed by the appearance of aggregates upon treatment with ANF (Fig. 3E, compare panels iii and vi). In contrast, ANF had no effect on either the FRET between p62 and the PKCλ ΔPB1 (blue circles in Fig. 3D) or the aggregation of p62 (Fig. 3E, compare panels ii and v). Interestingly, ANF produced a biphasic effect on FRET between wild-type PKCλ and p62; the compound caused an initial increase in FRET followed by a decrease (Fig. 3D, black circles). Long-term treatment with ANF also resulted in the appearance of p62 aggregates (Fig. 3E, compare panels i and iv), consistent with release of PKCλ from the scaffold. One possibility is that displacing the PB1 domain allows a transient adjustment of PKCλ on the scaffold in which the pseudosubstrate-p62 interaction is enhanced, followed by the release of PKCλ from the scaffold and, as indicated in Fig. 3E, the aggregation of p62.
ZIP Releases the Pseudosubstrate of PKCλ from p62, Allowing It to Autoinhibit PKC
We reasoned that if the acidic surface on p62 tethers the pseudosubstrate of the PB1-bound aPKCs, then the scaffold-bound enzyme should be catalytically active. To test this, we fused our genetically encoded PKC activity reporter (CKAR) (33, 34) to the N terminus of p62, as illustrated in Fig. 4A. To ensure that CKAR-p62 measured only aPKC activity, we pretreated COS-7 cells with 1 μm Gö6983 to inhibit conventional and novel PKC isozymes (46). The addition of 5 μm ZIP caused a reduction in the FRET ratio, reflecting basal activity of PKCλ on p62 scaffold that was inhibited by ZIP (Fig. 4B, black circles). This drop in FRET was lower than the baseline drift observed using a control non-phosphorylatable reporter in which an Ala occupies the phospho-acceptor site (Fig. 4B, T/A, blue triangles). To verify that ZIP was not inhibiting activity by directly binding the substrate-binding site of the aPKC, we tested the effect of ZIP on constructs in which the pseudosubstrate was deleted (Fig. 4B, red squares) or the Ala at the phospho-acceptor site was mutated to Glu (PKCι A129E; Fig. 4B, green triangles), a mutation that is commonly used to generate constitutively active aPKCs (47). Neither of these constructs was inhibited by ZIP, consistent with our previous report that ZIP is unable to inhibit the catalytic activity of the kinase domain of aPKCs (1) and establishing that ZIP inhibition requires an intact pseudosubstrate. Note that ZIP also had no effect on the interaction of the PKCι A129E construct with p62 (Fig. 4C), recapitulating the effect of deleting the pseudosubstrate (Fig. 4C). These results underscore the requirement for an intact pseudosubstrate for ZIP both to exert its effect on the interaction of aPKCs with p62 and to inhibit the activity of the aPKC on the scaffold. These results are consistent with ZIP releasing the pseudosubstrate from p62, prompting the intramolecular autoinhibition that maintains PKCs in an inactive state.
FIGURE 4.

ZIP releases the pseudosubstrate of PKCλ from p62 to allow intramolecular autoinhibition of the kinase. A, schematic showing fusion of the PKC activity reporter CKAR to the N terminus of p62. Phosphorylation causes an intramolecular clamp of the phosphorylated segment (pink) with a phospho-peptide-binding FHA2 module (orange) that results in a decrease in FRET between the flanking CFP (blue) and YFP (yellow) (see Ref. 33 for more details). B, COS-7 cells co-expressing CKAR-p62 and mCherry-tagged wild-type PKCλ (black circles, n = 44), PKCλ ΔPS (red squares, n = 19), or PKCι A129E (green triangles, n = 41) were pretreated with 1 μm Gö6983, an inhibitor of conventional and novel PKC isozymes, and then monitored for CFP/FRET ratio changes following 5 μm ZIP treatment. The control CKAR-p62 T/A (blue triangles, n = 23), where its phospho-acceptor Thr was mutated to Ala, was also examined under the same experimental conditions. Data were plotted as PKC activity and represent the normalized FRET/CFP ratio mean ± S.E. of the indicated number of cells from at least 3 independent experiments. C, the FRET/CFP ratio of COS-7 cells co-expressing CFP-p62 and YFP-PKCι (black circles, n = 31) or YFP-PKCι A129E (green diamonds, n = 28) was monitored before and after 5 μm ZIP treatment. Data were plotted as a p62-aPKC interaction and represent the normalized FRET/CFP ratio mean ± S.E. of the indicated number of cells from at least 3 independent experiments. D, COS-7 cells co-expressing CKAR-p62 and mCherry-tagged wild-type PKCλ (black circles, n = 28), PKCλ ΔPS (red squares, n = 30), or PKCι A129E (green triangles, n = 18) were monitored for CFP/FRET ratio changes before and after treatment with 5 μm PZ09, an atypical PKC-selective inhibitor (48). E, COS-7 cells co-expressing CKAR-p62 and either mCherry-tagged wild-type PKCλ (black circles, n = 33) or PKCλ ΔPS (red squares, n = 38) were monitored for CFP/FRET ratio changes when treated with 50 nm calyculin A, a Ser/Thr protein phosphatase inhibitor (56). Data were plotted as PKC activity and represent the normalized FRET/CFP ratio mean ± S.E. of the indicated number of cells from at least 3 independent experiments. F, quantification of basal and phosphatase-suppressed activities of PKCλ and PKCλ ΔPS was calculated as a fraction of FRET ratio change. The data were extrapolated from the plateau portion of the curves in panels D and E, 20 min after the addition of PZ09 or calyculin A.
We next assessed the level of basal activity PKCλ on the p62 scaffold by comparing the activity of wild-type enzyme with that of enzyme with unrestrained activity resulting from deletion or mutation of the autoinhibitory pseudosubstrate segment. Cells expressing wild-type PKCλ, PKCλ ΔPS, or PKCι A129E were treated with the aPKC-specific inhibitor PZ09 (32, 48). This compound caused a modest drop in the activity of the wild-type enzyme, reflecting basal activity, and a larger drop in the activity of the PKCλ ΔPS, reflecting the maximal unrestrained activity of PKCλ (Fig. 4D, black circles for wild type and red squares for ΔPS). The A129E construct displayed the same sensitivity to PZ09 (Fig. 4D, green triangles) as the PKCλ ΔPS construct, validating that this mutation effectively expels the pseudosubstrate to result in maximal activity of aPKCs. To measure the full amplitude of the PKCλ signal at the scaffold, cells were treated with the phosphatase inhibitor, calyculin A (Fig. 4E), to cause maximal phosphorylation of the reporter (see Ref. 49). Phosphatase inhibition caused only a minimal increase in the FRET signal in cells expressing the PKCλ ΔPS construct, consistent with nearly maximal phosphorylation of the reporter. In contrast, suppression of phosphatase activity resulted in a large increase in FRET for cells expressing wild-type PKCλ. Quantification of the data from multiple experiments (n > 18) revealed 17% maximal phosphorylation of the reporter in cells expressing wild-type enzyme as compared with 75% in cells expressing the PKCλ ΔPS construct. Thus, the wild-type enzyme displayed ∼25% of maximal, unrestrained activity (Fig. 4F). These data support a model in which p62-scaffolded PKCλ has significant basal activity because of tethering of the pseudosubstrate segment to the acidic patch on p62. ZIP competes with this interaction to re-engage the pseudosubstrate and suppress signaling at the scaffold.
ZIP Peptide Also Targets the Interacting Surface between p62 and AMPA Receptor Subunits, GluR1 and GluR2
We next addressed whether the ZIP-binding surface on p62 could be an interaction surface for other signaling molecules. Because ZIP peptide has been previously shown to block LTP and reduce learning in animal models (21, 26–29), coupled with the reports that p62 is required for LTP and memory formation and associates with the AMPA receptor subunits GluR1 and GluR2 (50), we asked whether the interaction of GluR1 and GluR2 with p62 was sensitive to ZIP. FLIM was used to examine the effect of ZIP on the interaction between EGFP-p62 and mCherry-tagged GluR1, GluR2, PKCι (positive control), or p62 (negative control). The addition of ZIP to COS-7 cells co-expressing p62 and GluR1 receptor subunit caused a significant drop in FRET efficiency (Fig. 5A, white bars, and Fig. 5B, compare panels i and v), similar to that observed for the ZIP disruption of the p62-PKCι interaction (Fig. 5A, red bars, and Fig. 5B, compare panels iii and vii). ZIP also decreased the FRET efficiency between p62 and GluR2 (Fig. 5A, blue bars, and Fig. 5B, compare panels ii and vi), although to a lesser extent than the effect on GluR1. In contrast, ZIP had no effect on the FRET efficiency detected between EGFP-p62 and mCherry-p62 (Fig. 5A, brown bars, and Fig 5B, compare panels iv and viii), as observed using the FRET interaction assay in Fig. 3A. These data are consistent with ZIP displacing GluR1 and GluR2 from p62.
FIGURE 5.
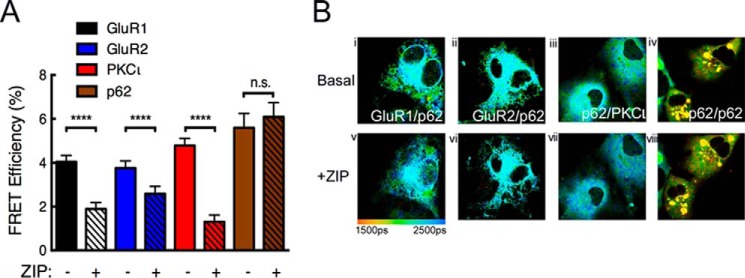
ZIP targets the association of GluR1 or GluR2 with the p62 scaffold. A, FRET efficiency was measured from COS-7 cells co-expressing mCherry-p62 and GFP-tagged GluR1 (white bars, n = 114), GluR2 (blue bars, n = 167), PKCι (red bars, n = 94), or p62 (brown bars, n = 53), both before and after 5 μm ZIP treatment. Solid bars represent before ZIP treatment, and bars with black slashes indicate FRET efficiency of post-ZIP treatment. The data were analyzed by paired Student's t test: n.s., no significance; ****, p < 0.0001. B, representative fluorescence lifetime images of COS-7 cells expressing the indicated constructs before and after ZIP treatment. Pseudocolor scale indicates GFP lifetime at each pixel.
Discussion
Using bioinformatics, peptide arrays, and cellular FRET assays, we have identified an acidic surface on the PB1 domain of the scaffold p62, distal to the known interaction surfaces of the domain, that tethers the pseudosubstrate of aPKCs to maintain them in an active conformation. This surface comprising acidic residues on two loops in the PB1 domain of p62 was uncovered in our efforts to identify the cellular target of the biologically active peptide ZIP. Mechanistic studies support a model in which ZIP binds to this surface, displacing the pseudosubstrate of p62-bound aPKCs and resulting in the re-engagement in the substrate-binding cavity for intramolecular autoinhibition. Furthermore, we show that ZIP also displaces AMPA receptor subunits from p62, consistent with this acidic patch being a key interaction surface for other proteins poised on the scaffold. Thus, this study uncovers a cellular target, and molecular mechanism, for the cellular effects of ZIP, and identifies a novel mechanism to maintain a kinase in an active conformation on a scaffold.
Fig. 6 presents a model consistent with our data for the regulation of aPKC function by binding to p62. The dotted line between the PB1 domains of p62 and aPKCs represents the canonical interaction that has been previously characterized. Specifically, basic residues in the PB1 domain of p62 bind a complimentary acidic patch on the PB1 domain of aPKCs (15, 43). Recent elucidation of a crystal structure of the PB1 domain of p62 complexed to that of PKCζ (51) validates biochemical analysis identifying Lys7, Arg21, and Arg22 as key residues on p62 mediating the binding to the PB1 domain of aPKCs (52). Here we identify a second interaction surface: acidic residues in two adjacent loops of the p62 PB1 domain bind the basic pseudosubstrate segment (green rectangle) of aPKCs to lock the enzyme in an active conformation. This acidic segment is a distinct surface that is not involved in either homodimerization of the p62 PB1 domains or heterodimerization with the PB1 domain of other binding partners. ZIP binds the acidic surface of p62 to displace the pseudosubstrate, freeing the segment to bind the substrate-binding cavity of aPKCs, resulting in autoinhibition (lower panel). The pseudosubstrate segment of aPKCs immediately follows their PB1 domain. Whether the pseudosubstrate and the PB1 domains simultaneously engage, or whether the interactions are in dynamic equilibrium with both binding surfaces, awaits structural determination. Our finding that the net activity of the scaffolded aPKC corresponds to ∼25% of its maximal unrestrained activity suggests that not all pseudosubstrates are tethered open at any one time, favoring the latter possibility. Tethering of the pseudosubstrate of aPKCs by their scaffold partners may be a general mechanism for regulating aPKC activity. Prehoda and co-workers (53) have shown that the PB1 domain of another aPKC scaffold, Par6, is able to activate purified PKCζ in in vitro kinase assays. Because our study examined overexpressed proteins, it is unclear what fraction of aPKCs signal at the p62 scaffold physiologically. Nonetheless, our data provide insight into the conformational mechanisms that allow pseudosubstrate release and activation of aPKCs.
FIGURE 6.
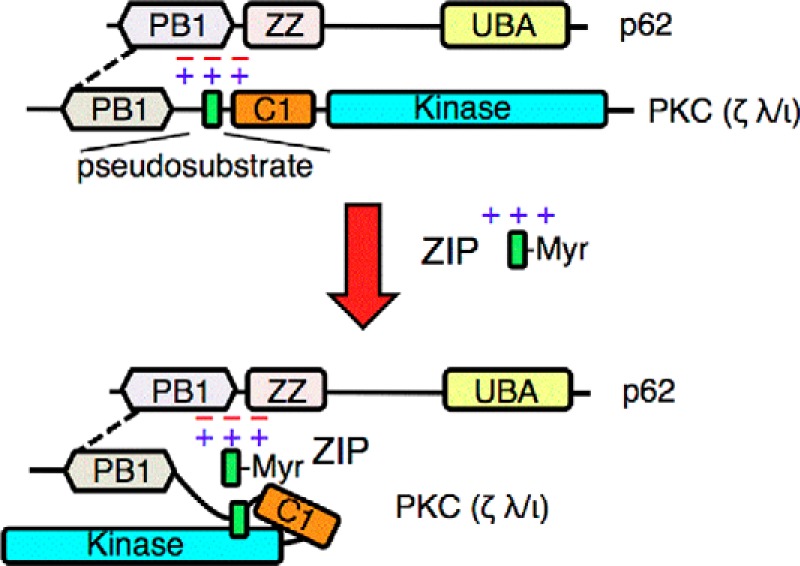
The acidic surface of p62 tethers the pseudosubstrate segment of aPKCs to maintain its active conformation. The schematic shows the canonical interaction of the PB1 domain of aPKCs with that of p62 (dotted line) and the previously undescribed interaction between the basic pseudosubstrate and acidic surface on p62. This interaction tethers the pseudosubstrate out of the substrate-binding cavity of aPKCs, allowing constitutive activity on the scaffold. The positively charged peptide ZIP competes with the pseudosubstrate for binding to the acidic surface of p62, displacing the pseudosubstrate and allowing it to re-engage in the substrate-binding site of aPKC to affect intramolecular autoinhibition of the kinase. UBA, ubiquitin-associated domain; Myr, myristoylated peptide.
The mechanism of activation of conventional and novel PKCs is well understood: binding of the lipid second messenger diacylglycerol promotes a conformational change that expels the pseudosubstrate, resulting in activation (see Ref. 5). Atypical PKCs do not bind diacylglycerol, and the mechanism by which the pseudosubstrate is released to allow activity is poorly understood. aPKCs differ from other family members by the presence of a PB1 domain and a predicted Type III PDZ ligand, suggesting that positioning on protein scaffolds drives downstream signaling. Indeed, mechanisms to allow coordination next to protein substrates is likely critical to their function given their exceptionally low catalytic rate. However, how the pseudosubstrate is released in cells is poorly understood. Our finding that the p62-scaffolded aPKC is tethered in an open conformation by an electrostatic interaction of the basic pseudosubstrate with an acidic surface on p62, distinct from the basic surface on p62 that binds a complimentary surface on the PB1 domain of aPKCs, provides a unique mechanism for how aPKCs signal at protein scaffolds.
An important ramification of this study is the identification of a cellular target for ZIP. This molecule has been used extensively as a pharmacological tool, and its effects are attributed to its ability to inhibit the catalytic activity of atypical members of the PKC family, especially PKMζ, the short neuron-specific form of PKCζ. Although the peptide effectively inhibits all the PKCs in vitro, it is ineffective at inhibiting their catalytic activity in cells (1), likely because the affinity of the peptide for the substrate-binding cavity is too low for effective binding in cells. Also, in this study, we confirm that ZIP does not inhibit aPKCs in cells by competing for substrate binding in the kinase domain: constitutively active constructs lacking the pseudosubstrate segment are insensitive to ZIP. However, ZIP has profound cellular effects, most notably, blocking LTP and learning and memory (21, 26–29). Thus, uncovering the bona fide target for ZIP has potential clinical relevance. Our discovery that p62 is a cellular target of ZIP may shed light on several studies in the learning and memory field. First, knock-out mice of p62 have significantly reduced LTP (50, 54), phenocopying the effect of ZIP. This is in contrast to knock-out mice of aPKCs, which continue to display ZIP-sensitive LTP (30, 31), an effect that is also observed with scrambled ZIP (30, 31). Second, AMPA receptor subunits, which play key roles in plasticity (55), bind p62 (50). Most strikingly, the small Arg-containing dodecapeptide loop between transmembrane spans 2 and 3 of GluR contributes to this interaction. It is worth noting that the juxtaposed sequence immediately after the fourth transmembrane span is also basic, suggesting that a basic juxtamembrane patch could be the relevant interaction site with the acidic patch on p62. Lastly, our FRET data reveal that ZIP reduces the interaction of GluR1, and to a lesser extent GluR2, with p62. The ability of ZIP to inhibit aPKCs on the p62 scaffold and to displace AMPA receptor subunits, which are regulated by aPKCs (50), provides a potential molecular mechanism for the effects of ZIP on LTP.
Author Contributions
L. L. T. and A. C. N. conceived the experiments and wrote the manuscript. L. L. T. performed and analyzed most of the experiments. K. D. designed, performed, and analyzed the experiments shown in Figure 5 with advice of R. M. Under the guidance of P. E. B., Lei X., and Li X. performed the computational studies to identify the acidic surface on p62. J. C. D. R. and C. C. K. synthesized the peptide arrays. G. M. A and C. H. synthesized and provided the atypical PKC inhibitor PZ09. All authors reviewed and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Alan Fields for the generous gifts of Auranofin and PKCι plasmid, Dr. Jorge Moscat for the p62 plasmid, and members of the Newton laboratory for helpful comments.
This work was supported by National Institutes of Health Grant P01 DK54441 (to A. C. N.). The authors declare that they have no conflicts of interest with the contents of this article.
I. S. Tobias, M. Kaulich, P. K. Kim, N. Simon, E. Jacinto, S. Dowdy, C. C. King, and A. C. Newton, submitted for publication.
- aPKC
- atypical protein kinase C
- ZIP
- zeta inhibitory peptide
- CKAR
- C kinase activity reporter
- FLIM
- fluorescence lifetime imaging microscopy
- GluR
- glutamate receptor
- LTP
- long-term potentiation
- ANF
- Auranofin
- PS
- pseudosubstrate.
References
- 1. Wu-Zhang A. X., Schramm C. L., Nabavi S., Malinow R., Newton A. C. (2012) Cellular pharmacology of protein kinase Mζ (PKMζ) contrasts with its in vitro profile: implications for PKMζ as a mediator of memory. J. Biol. Chem. 287, 12879–12885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Langeberg L. K., Scott J. D. (2015) Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol. 16, 232–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rosse C., Linch M., Kermorgant S., Cameron A. J., Boeckeler K., Parker P. J. (2010) PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 11, 103–112 [DOI] [PubMed] [Google Scholar]
- 4. Antal C. E., Newton A. C. (2013) Spatiotemporal dynamics of phosphorylation in lipid second messenger signaling. Mol. Cell. Proteomics 12, 3498–3508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Newton A. C. (2010) Protein kinase C: poised to signal. Am. J. Physiol. Endocrinol. Metab. 298, E395–E402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parker P. J., Murray-Rust J. (2004) PKC at a glance. J. Cell Sci. 117, 131–132 [DOI] [PubMed] [Google Scholar]
- 7. Lopez-Garcia L. A., Schulze J. O., Fröhner W., Zhang H., Süss E., Weber N., Navratil J., Amon S., Hindie V., Zeuzem S., Jørgensen T. J., Alzari P. M., Neimanis S., Engel M., Biondi R. M. (2011) Allosteric regulation of protein kinase PKCζ by the N-terminal C1 domain and small compounds to the PIF-pocket. Chem. Biol. 18, 1463–1473 [DOI] [PubMed] [Google Scholar]
- 8. Antal C. E., Callender J. A., Kornov A. P., Taylor S. S., Newton A. C. (2015) Intramolecular C2 domain-mediated autoinhibition of protein kinase CbII. Cell Rep., 10.1016/j.celrep.2015.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Orr J. W., Keranen L. M., Newton A. C. (1992) Reversible exposure of the pseudosubstrate domain of protein kinase C by phosphatidylserine and diacylglycerol. J. Biol. Chem. 267, 15263–15266 [PubMed] [Google Scholar]
- 10. Nalefski E. A., Newton A. C. (2001) Membrane binding kinetics of protein kinase C βII mediated by the C2 domain. Biochemistry 40, 13216–13229 [DOI] [PubMed] [Google Scholar]
- 11. Evans J. H., Murray D., Leslie C. C., Falke J. J. (2006) Specific translocation of protein kinase Cα to the plasma membrane requires both Ca2+ and PIP2 recognition by its C2 domain. Mol. Biol. Cell 17, 56–6616236797 [Google Scholar]
- 12. Sánchez-Bautista S., Marín-Vicente C., Gómez-Fernández J. C., Corbalán-García S. (2006) The C2 domain of PKCα is a Ca2+-dependent PtdIns(4,5)P2 sensing domain: a new insight into an old pathway. J. Mol. Biol. 362, 901–914 [DOI] [PubMed] [Google Scholar]
- 13. Kazanietz M. G., Bustelo X. R., Barbacid M., Kolch W., Mischak H., Wong G., Pettit G. R., Bruns J. D., Blumberg P. M. (1994) Zinc finger domains and phorbol ester pharmacophore: analysis of binding to mutated form of protein kinase C z and the vav and c-raf proto-oncogene products. J. Biol. Chem. 269, 11590–11594 [PubMed] [Google Scholar]
- 14. Pu Y., Peach M. L., Garfield S. H., Wincovitch S., Marquez V. E., Blumberg P. M. (2006) Effects on ligand interaction and membrane translocation of the positively charged arginine residues situated along the C1 domain binding cleft in the atypical protein kinase C isoforms. J. Biol. Chem. 281, 33773–33788 [DOI] [PubMed] [Google Scholar]
- 15. Sumimoto H., Kamakura S., Ito T. (2007) Structure and function of the PB1 domain, a protein interaction module conserved in animals, fungi, amoebas, and plants. Sci. STKE 2007, re6. [DOI] [PubMed] [Google Scholar]
- 16. Seibenhener M. L., Geetha T., Wooten M. W. (2007) Sequestosome 1/p62: more than just a scaffold. FEBS Lett. 581, 175–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cariou B., Bereziat V., Moncoq K., Kasus-Jacobi A., Perdereau D., Le Marcis V., Burnol A. F. (2004) Regulation and functional roles of Grb14. Front. Biosci. 9, 1626–1636 [DOI] [PubMed] [Google Scholar]
- 18. Kim J., Kim I., Yang J. S., Shin Y. E., Hwang J., Park S., Choi Y. S., Kim S. (2012) Rewiring of PDZ domain-ligand interaction network contributed to eukaryotic evolution. PLoS Genet. 8, e1002510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ron D., Kazanietz M. G. (1999) New insights into the regulation of protein kinase C and novel phorbol ester receptors. FASEB J. 13, 1658–1676 [PubMed] [Google Scholar]
- 20. Nakanishi H., Brewer K. A., Exton J. H. (1993) Activation of the ζ isozyme of protein kinase C by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 268, 13–16 [PubMed] [Google Scholar]
- 21. Ling D. S., Benardo L. S., Serrano P. A., Blace N., Kelly M. T., Crary J. F., Sacktor T. C. (2002) Protein kinase Mζ is necessary and sufficient for LTP maintenance. Nat. Neurosci. 5, 295–296 [DOI] [PubMed] [Google Scholar]
- 22. Scott J. D., Glaccum M. B., Fischer E. H., Krebs E. G. (1986) Primary-structure requirements for inhibition by the heat-stable inhibitor of the cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. U.S.A. 83, 1613–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu R., Dalgalan D., Mandell E. K., Parker S. S., Ghosh S., Wilson J. M. (2015) PKCι interacts with Rab14 and modulates epithelial barrier function through regulation of claudin-2 levels. Mol. Biol. Cell 26, 1523–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bandyopadhyay G., Sajan M. P., Kanoh Y., Standaert M. L., Quon M. J., Lea-Currie R., Sen A., Farese R. V. (2002) PKC-ζ mediates insulin effects on glucose transport in cultured preadipocyte-derived human adipocytes. J. Clin. Endocrinol. Metab. 87, 716–723 [DOI] [PubMed] [Google Scholar]
- 25. Imamura T., Huang J., Usui I., Satoh H., Bever J., Olefsky J. M. (2003) Insulin-induced GLUT4 translocation involves protein kinase C-lambda-mediated functional coupling between Rab4 and the motor protein kinesin. Mol. Cell. Biol. 23, 4892–4900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Migues P. V., Hardt O., Wu D. C., Gamache K., Sacktor T. C., Wang Y. T., Nader K. (2010) PKMζ maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat. Neurosci. 13, 630–634 [DOI] [PubMed] [Google Scholar]
- 27. Pastalkova E., Serrano P., Pinkhasova D., Wallace E., Fenton A. A., Sacktor T. C. (2006) Storage of spatial information by the maintenance mechanism of LTP. Science 313, 1141–1144 [DOI] [PubMed] [Google Scholar]
- 28. Serrano P., Friedman E. L., Kenney J., Taubenfeld S. M., Zimmerman J. M., Hanna J., Alberini C., Kelley A. E., Maren S., Rudy J. W., Yin J. C., Sacktor T. C., Fenton A. A. (2008) PKMζ maintains spatial, instrumental, and classically conditioned long-term memories. PLoS Biol. 6, 2698–2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shema R., Sacktor T. C., Dudai Y. (2007) Rapid erasure of long-term memory associations in the cortex by an inhibitor of PKMζ. Science 317, 951–953 [DOI] [PubMed] [Google Scholar]
- 30. Lee A. M., Kanter B. R., Wang D., Lim J. P., Zou M. E., Qiu C., McMahon T., Dadgar J., Fischbach-Weiss S. C., Messing R. O. (2013) Prkcz null mice show normal learning and memory. Nature 493, 416–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Volk L. J., Bachman J. L., Johnson R., Yu Y., Huganir R. L. (2013) PKM-ζ is not required for hippocampal synaptic plasticity, learning and memory. Nature 493, 420–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Trujillo J. I., Kiefer J. R., Huang W., Thorarensen A., Xing L., Caspers N. L., Day J. E., Mathis K. J., Kretzmer K. K., Reitz B. A., Weinberg R. A., Stegeman R. A., Wrightstone A., Christine L., Compton R., Li X. (2009) 2-(6-Phenyl-1H-indazol-3-yl)-1H-benzo[d]imidazoles: design and synthesis of a potent and isoform selective PKC-ζ inhibitor. Bioorg. Med. Chem. Lett. 19, 908–911 [DOI] [PubMed] [Google Scholar]
- 33. Violin J. D., Zhang J., Tsien R. Y., Newton A. C. (2003) A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J. Cell Biol. 161, 899–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gallegos L. L., Kunkel M. T., Newton A. C. (2006) Targeting protein kinase C activity reporter to discrete intracellular regions reveals spatiotemporal differences in agonist-dependent signaling. J. Biol. Chem. 281, 30947–30956 [DOI] [PubMed] [Google Scholar]
- 35. Xu D., Zhang Y. (2012) Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 80, 1715–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen R., Li L., Weng Z. (2003) ZDOCK: an initial-stage protein-docking algorithm. Proteins 52, 80–87 [DOI] [PubMed] [Google Scholar]
- 37. Macindoe G., Mavridis L., Venkatraman V., Devignes M. D., Ritchie D. W. (2010) HexServer: an FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 38, W445–W449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gray J. J., Moughon S., Wang C., Schueler-Furman O., Kuhlman B., Rohl C. A., Baker D. (2003) Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 331, 281–299 [DOI] [PubMed] [Google Scholar]
- 39. Kunkel M. T., Ni Q., Tsien R. Y., Zhang J., Newton A. C. (2005) Spatio-temporal dynamics of protein kinase B/Akt signaling revealed by a genetically encoded fluorescent reporter. J. Biol. Chem. 280, 5581–5587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Viger M. L., Sheng W., Doré K., Alhasan A. H., Carling C. J., Lux J., de Gracia Lux C., Grossman M., Malinow R., Almutairi A. (2014) Near-infrared-induced heating of confined water in polymeric particles for efficient payload release. ACS Nano 8, 4815–4826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Becker W., Bergmann A., Hink M. A., König K., Benndorf K., Biskup C. (2004) Fluorescence lifetime imaging by time-correlated single-photon counting. Microsc. Res. Tech. 63, 58–66 [DOI] [PubMed] [Google Scholar]
- 42. Lakowicz J. R. (2006) Principles of Fluorescence Spectroscopy, 3rd Ed., Springer, New York [Google Scholar]
- 43. Lamark T., Perander M., Outzen H., Kristiansen K., Øvervatn A., Michaelsen E., Bjørkøy G., Johansen T. (2003) Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J. Biol. Chem. 278, 34568–34581 [DOI] [PubMed] [Google Scholar]
- 44. Erdogan E., Lamark T., Stallings-Mann M., Lee J., Pellecchia M., Thompson E. A., Johansen T., Fields A. P. (2006) Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Cι. J. Biol. Chem. 281, 28450–28459 [DOI] [PubMed] [Google Scholar]
- 45. Parker P. J., Justilien V., Riou P., Linch M., Fields A. P. (2014) Atypical protein kinase Cι as a human oncogene and therapeutic target. Biochem. Pharmacol. 88, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu-Zhang A. X., Newton A. C. (2013) Protein kinase C pharmacology: refining the toolbox. Biochem. J. 452, 195–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goode N. T., Parker P. J. (1994) A phorbol ester-responsive PKC-ζ generated by fusion with the regulatory domain of PKC-δ. FEBS Lett. 340, 145–150 [DOI] [PubMed] [Google Scholar]
- 48. Kusne Y., Carrera-Silva E. A., Perry A. S., Rushing E. J., Mandell E. K., Dietrich J. D., Errasti A. E., Gibbs D., Berens M. E., Loftus J. C., Hulme C., Yang W., Lu Z., Aldape K., Sanai N., Rothlin C. V., Ghosh S. (2014) Targeting aPKC disables oncogenic signaling by both the EGFR and the proinflammatory cytokine TNFα in glioblastoma. Sci. Signal. 7, ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gallegos L. L., Newton A. C. (2011) Genetically encoded fluorescent reporters to visualize protein kinase C activation in live cells. Methods Mol. Biol. 756, 295–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jiang J., Parameshwaran K., Seibenhener M. L., Kang M. G., Suppiramaniam V., Huganir R. L., Diaz-Meco M. T., Wooten M. W. (2009) AMPA receptor trafficking and synaptic plasticity require SQSTM1/p62. Hippocampus 19, 392–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ren J., Wang J., Wang Z., Wu J. (2014) Structural and biochemical insights into the homotypic PB1-PB1 complex between PKCζ and p62. Sci. China Life Sci. 57, 69–80 [DOI] [PubMed] [Google Scholar]
- 52. Hirano Y., Yoshinaga S., Ogura K., Yokochi M., Noda Y., Sumimoto H., Inagaki F. (2004) Solution structure of atypical protein kinase C PB1 domain and its mode of interaction with ZIP/p62 and MEK5. J. Biol. Chem. 279, 31883–31890 [DOI] [PubMed] [Google Scholar]
- 53. Graybill C., Wee B., Atwood S. X., Prehoda K. E. (2012) Partitioning-defective protein 6 (Par-6) activates atypical protein kinase C (aPKC) by pseudosubstrate displacement. J. Biol. Chem. 287, 21003–21011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ramesh Babu J., Lamar Seibenhener M., Peng J., Strom A. L., Kemppainen R., Cox N., Zhu H., Wooten M. C., Diaz-Meco M. T., Moscat J., Wooten M. W. (2008) Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J. Neurochem. 106, 107–120 [DOI] [PubMed] [Google Scholar]
- 55. Malinow R., Malenka R. C. (2002) AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126 [DOI] [PubMed] [Google Scholar]
- 56. Ishihara H., Martin B. L., Brautigan D. L., Karaki H., Ozaki H., Kato Y., Fusetani N., Watabe S., Hashimoto K., Uemura D., Hartshorne D. J. (1989) Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem. Biophys. Res. Commun. 159, 871–877 [DOI] [PubMed] [Google Scholar]