

Background: CXCR4 promotes melanoma metastasis.
Results: FOXD3 and PAX3 drive CXCR4 expression. Blocking these factors inhibits cell migration, but this is rescued by ectopic CXCR4 expression.
Conclusion: PAX3- and FOXD3-mediated melanoma cell migration is dependent on promoting the expression of CXCR4.
Significance: This study identifies a new pathway and targets for therapeutic strategies to treat invasive melanoma.
Keywords: DNA-protein interaction, gene expression, gene regulation, melanoma, transcription factor
Abstract
Metastatic melanoma is an aggressive and deadly disease. The chemokine receptor CXCR4 is active in melanoma metastasis, although the mechanism for the promotion and maintenance of CXCR4 expression in these cells is mostly unknown. Here, we find melanoma cells express two CXCR4 isoforms, the common version and a variant that is normally restricted to cells during development or to mature blood cells. CXCR4 expression is driven through a highly conserved intronic enhancer element by the transcription factors PAX3 and FOXD3. Inhibition of these transcription factors slows melanoma cell growth, migration, and motility, as well as reduces CXCR4 expression. Overexpression of these transcription factors drives the production of increased CXCR4 levels. Loss of PAX3 and FOXD3 transcription factor activity results in a reduction in cell motility, migration, and chemotaxis, all of which are rescued by CXCR4 overexpression. Here, we discover a molecular pathway wherein PAX3 and FOXD3 promote CXCR4 gene expression in melanoma.
Introduction
Melanoma is an aggressive cancer where metastasis occurs early in disease progression. There is currently no cure for metastatic melanoma, and the 5-year survival rates are less than 12% (1). It is therefore essential that the molecular pathways governing melanoma metastasis be identified to drive new therapeutic discoveries. CXC chemokine receptor 4 (CXCR4)3 has been linked to metastasis in a number of different tumor types and is detected in the invasive edge of melanoma primary lesions, regional lymph nodes, and distal metastases (2, 3). In addition, CXCR4 actively promotes melanoma cell motility, invasion, and metastasis (4–7). Inhibition of CXCR4 through small molecule inhibitors AMD3100 and AMD11070 (3, 8–10) or peptides T22 or a variant of the CXCR4 ligand CXCL12 (11, 12) attenuate melanoma progression and metastasis but do not alter primary tumor growth. Whereas CXCR4 is expressed in melanoma and drives disease progression, very little is known about what initiates, promotes, and maintains CXCR4 expression in melanoma. Although there is some evidence that supports regulation of CXCR4 expression in melanoma through the SMAD/TGFβ pathway (13), any direct regulation of the CXCR4 promoter in this tumor type is previously unknown.
PAX3, a transcription factor that actively drives melanoma progression, is aberrantly expressed in both melanoma cell lines and in primary tumors. PAX3 inhibition leads to a decrease in melanoma cell proliferation and survival (14–17). PAX3 has also been implicated in melanoma migration, invasion, and metastasis (18). The mechanism by which PAX3 acts as an oncogene is poorly understood, although it most likely parallels its role during development by regulating growth and differentiation. PAX3 likely exerts its oncogenic potential through the regulation of downstream effector genes. A limited number of PAX3 direct targets have been discovered in melanoma including the MET tyrosine kinase receptor as well as transcription factors TBX2 and BRN2 (15, 18, 19). The full scope of PAX3 activity in melanoma cannot be explained by the regulation of these factors alone, therefore, PAX3 must also regulate other downstream effectors.
PAX3 is generally a weak transcription factor on its own, and often recruits other proteins to act in activator or repressor complexes (20). A small number of PAX3 transcriptional cofactors have been identified, but only SOX10 and ETS1 have been verified within melanoma cells (15, 21, 22). PAX3 binds directly to both SOX10 and ETS1, resulting in a synergistic activation of downstream targets (21, 23). Due to the propensity of PAX3 to recruit cofactors, it is logical to assume that PAX3 has several cofactors in melanoma. One potential cofactor for PAX3 is the transcription factor FOXD3. FOXD3 is expressed in melanoma, regulated by B-RAF, and overexpressed as a possible adaptive response during BRAF-inhibitor drug resistance (24–26). Although the functionality of FOXD3 in melanoma is not clear, it acts as a transcription factor and binds DNA as a monomer or with other proteins during development (27). There is some evidence to support that FOXD3 is upstream of CXCR4 and that FOXD3 and PAX3 interact. FOXD3-related proteins FOXC1 and FOXC2 up-regulate CXCR4 through an element in the 5′ proximal promoter, and an increase in FOXC2 within endothelial progenitor cells is correlated with a rise in CXCR4 expression (28, 29). Another FOXD3-related protein, FOXO1, is involved in a translocation mutation in alveolar rhabdomyosarcomas, resulting in a chimeric protein that fuses the DNA-binding domains of PAX3 with the transactivation domain of FOXO1 (30, 31). This translocation product promotes CXCR4 expression through an undefined binding site (32, 33). There is also evidence of a functional interaction between PAX3 and FOXD3 during development, as well as support that the proteins may physically interact (34, 35). Although there is evidence of an interaction between PAX3 and FOXD3 and these proteins are linked to similar developmental functionalities such as cellular specification, migration, and survival, a shared pathway in melanoma is not characterized.
In this report, we determine that PAX3 and FOXD3 interact and activate the CXCR4 promoter through a highly conserved island of sequence homology located within intron 1. Although both FOXD3 and PAX3 modestly activate the CXCR4 promoter alone, together these transcription factors synergistically activate this promoter. This molecular pathway is active in melanoma cells with PAX3 and FOXD3 overexpression resulting in an increase of CXCR4 protein levels. Conversely, inhibition FOXD3 and PAX3 transcription factor activity through dominant-negative constructs resulted in a reduction of CXCR4 protein levels. The loss of FOXD3 and PAX3 activity negatively affected cell motility, migration, and chemotaxis, with reintroduction of CXCR4 rescuing these phenotypes. Here, we discover a molecular pathway where the transcription factors PAX3 and FOXD3 drive CXCR4 expression, thereby promoting melanoma cell motility, migration, and chemotaxis.
Experimental Procedures
Cell Culture and Transfection
Human melanoma lines (A375, mel-537, mel-624, mel-888, SKMEL-5, SKMEL-23, and SKMEL-28), 293T, and 3T3 cells (ATCC and University of Chicago Comprehensive Cancer Center Core Facilities) were cultured in DMEM, 10% FBS (Sigma). Morphology and melanoma marker testing verified melanoma cell identity. All cells were negative for the presence of mycoplasma. Cells were transfected with Effectene (Qiagen) or Lipofectamine 2000 (Life Technologies) reagent following the manufacturer's protocols.
Quantitative Real-time Polymerase Chain Reaction
The expression of CXCR4-A, CXCR4-B, and GAPDH transcripts was measured utilizing SYBR Green mixture and the CFX Connect Real-time System (Bio-Rad). Each reaction was performed in triplicate with values normalized to GAPDH mRNA levels. Primer sequences are provided in Table 1.
TABLE 1.
Primer and probe sequences
| Primer name | Application | Sequence |
|---|---|---|
| pGexFOXD3 forward | Cloning | ATG GAT CCA TGA CCC TCT CCG GC |
| pGexFOXD3 reverse | Cloning | CTG AAT TCA TAT GAC CCT CTC CGG C |
| FOXDN forward | Cloning | CAG GAT CCA TGC GGC GCC GGA AAC GCT TCA A |
| FOXDN reverse | Cloning | CAG AAT TCC TAT TGC GCC GGC CAT TTG GCT T |
| FOX enhancer forward | Cloning | TCG AGT GTT TGC CCG CTA TTG TTA TTT TCC CCC GTT TGT TGT TTT CCC C |
| FOX enhancer reverse | Cloning | CAT GGG GGA AAA CAA CAA ACG GGG GAA AAT AAC AAT AGC GGG CAA ACA C |
| hGAPDH forward | RT-PCR | ACA TCA TCC CTG CCT CTA CT |
| hGAPDH reverse | RT-PCR | CTC TCT TCC TCT TGT GCT CTT G |
| CXCR4 canonical forward | RT-PCR | CCG AGG GCC TGA GTG CTC CAG |
| CXCR4 noncanical forward | RT-PCR | GCA GAG GAG TTA GCC AAG ATG |
| CXCR4 reverse | RT-PCR | ATC CAT TGC CCA CAA TGC CAG |
| CXCR4 round1 forward | ChIP | TTT GTT GGC TGC GGC AGC AGG |
| CXCR4 round1 reverse | ChIP | TTT TGG AGT ACG GGT ACC TCC |
| CXCR4 nested forward | ChIP | TAG CAA AGT GAC GCC GAG GG |
| CXCR4 nested reverse | ChIP | AAT GTC CTG GCC GCT TCT GC |
| β-Tubulin round1 forward | ChIP | AAA GGC CAC TAC ACA GAG GG |
| β-Tubulin round1 reverse | ChIP | TAC CAA CTG ATG GAC GGA GAG G |
| β-Tubulin nested forward | ChIP | TTG ATT CTG TCC TGG ATG TGG |
| β-Tubulin nested reverse | ChIP | TCA GAC ACT TTG GGT GAA GGC |
| CXCR4 WT forward | EMSA | CTT GAG TCC CGC CGC GCG CGG CGG CTT GCA CGC TGT TTG CAA A |
| CXCR4 WT reverse | EMSA | CTT ACG TTT GCA AAC AGC GTG CAA GCC GCC GCG CGC GGC GGG ACT |
| CXCR4 ΔP1 forward | EMSA | CTT GAT TAA TTA CGC GCG CGG CGG CTT GCA CGC TGT TTG CAA A |
| CXCR4 ΔP1 reverse | EMSA | CTT ACG TTT GCA AAC AGC GTG CAA GCC GCC GCG CGC GTA ATT AAT |
| CXCR4 ΔP2 forward | EMSA | CTT GAG TCC CGC CGC GCG CGG CGG CTT TAA TTA TGT TTG CAA A |
| CXCR4 ΔP2 reverse | EMSA | CTT ACG TTT GCA AAC ATA ATT AAA GCC GCC GCG CGC GGC GGG ACT |
| CXCR4 ΔF forward | EMSA | CTT GAG TCC CGC CGC GCG CGG CGG CTT GCA CGC GGC CGG CAA A |
| CXCR4 ΔF reverse | EMSA | CTT ACG TTT GCC GGC CGC GTG CAA GCC GCC GCG CGC GGC GGG ACT |
| CXCR4 ΔP1ΔP2 forward | EMSA | CTT GAA CTC AGC CGC GCG CGG CGG CTT GGG CGA TGT TTG CAA A |
| CXCR4 ΔP1ΔP2 reverse | EMSA | ACG TTT GCA AAC ATC GCC CAA GCC GCC GCG CGC GGC TGA GTT |
Western Blots
Cells were lysed in RIPA buffer and 50 μg of total protein was separated on 7.5% BisTris gels, transferred to nitrocellulose membranes (Bio-Rad), then probed with 1:200 Pax3 (University of Iowa Hybridoma Bank), 1:500 FOXD3 (BioLegend), 1:1,000 CXCR4 (AbCam), 1:10,000 GAPDH (Cell Signaling), or 1:1,000,000 vinculin antibody (Sigma).
Phylogenetic Footprinting/in Silico Promoter Analysis
For promoter analysis, human and mouse candidate gene loci were blasted against each other (NCBI/Blast), or multiple species were compared with human (dcode) or blat search, Ensemble) to identify genetic islands of homology. For sequence analysis, the match (reward score) to mismatch (penalty score) ratio was set at 1 to −1 to locate islands with 75% conservation between species, with high stringency for wordsize (of 11) for small conserved segments, and a low stringency for gaps between conserved nucleotides. After identification of potential enhancer islands, these sequences were analyzed for potential PAX3 homeodomain TAAT(N2–3)ATTA (36) or paired (G)T(T/C)(C/A)(C/T)(G/C)(G/C) (20, 37–40) cis-response elements, and FOXD3 long high-affinity sites (A/T)(A/T)T(A/G)TTTN(T/C)N(T/C) or short core FOX DNA recognition sites T(A/G)TT(T/G)(A/G)(T/C) (27, 41).
Plasmid Construction
The human CXCR4 reporter constructs shown schematically in Fig. 3 contain either 572 or 393 base pairs (bp) (pGL2-CXCR4pm572 or pGL2-CXCR4pm393, respectively) of proximal promoter and 92 bp of exon 1 including the start codon (42) cloned in-frame with the luciferase gene. These constructs produced a protein containing the first 10 codons of CXCR4-B and the entire luciferase enzyme. The pGL2-CXCR4pm393L construct contains 1781 bp of intron 1 and the first three codons of exon 2 cloned in-frame with the luciferase reporter. In the pGL2-CXCR4 pmΔISH construct, a 267-bp segment flanked by BstXI and AgeI sites was removed from intron 1, which included a 52-bp island of sequence homology (ISH) shown in Fig. 3E. Specific point mutations in the P1, P2, and/or F sites were created by site-directed mutagenesis.
FIGURE 3.

The CXCR4 locus contains a conserved element within intron 1 that is PAX3 and FOXD3 responsive. A, 393 bp of 5′ sequence proximal to the CXCR4-B transcriptional start site, exon 1, and partial intron 1 is shown. Within the promoter region, the previously defined TATA box and FOXC1/C2 site are underlined and labeled (28, 42). Exon 1 is highlighted with gray, and the translational start site ATG is boxed. The region deleted in the CXCR4-ΔISH constructs is contained within the dotted area. A highly conserved 52-bp region is highlighted with black. B, schematics of CXCR4 luciferase reporter constructs. The pGL2-CXCR4pm572 and pGL2-CXCR4pm393 vectors contain 572 or 393 bp of 5′ promoter sequence, respectively, as well as a partial 5′ UTR fused to the luciferase gene. The pGL2-CXCR4pm393L construct contains 393 bp of promoter, exon 1, intron 1, and a partial exon 2 cloned in-frame with a luciferase reporter cassette. The dotted and block box areas shown in A are highlighted. C, intron 1 of the CXCR4 gene contains a PAX3 response element. Reporter constructs shown in B were transfected into 293T cells without (white bars, control) or with (black bars) a PAX3 expression construct. Luciferase levels shown as fold-units over controls. D, intron 1 of the CXCR4 gene contains a 52-bp enhancer element possessing PAX and FOX sites that is highly conserved between humans, gorillas, rats, and mice (ISH). E, comparison between pGL2-CXCR4pm393L and pGL2-CXCR4pm393L-ΔISH constructs. The constructs are identical except that the region containing the ISH removed in the latter. F, reporter constructs shown in E were transfected into 293T cells with PAX3 (white), FOXD3 (gray), or both PAX3 and FOXD3 (black) expression constructs. Luciferase levels are shown as fold-units over controls (levels without PAX3 or FOXD3 expression).
For EMSAs the pGex2T-PAX3 construct was a gift from J. Epstein (University of Pennsylvania). The FOXD3 coding sequence was cloned in-frame into pGex2T (GE Healthcare) using primers pGexFOXD3 forward and reverse (Table 1), subjected to restriction digest with BamHI and EcoRI, followed by ligation.
The PAX3 expression, PAX3 dominant-negative, and MET promoter constructs were created as described (21, 23). The pcS2-mFoxD3 expression construct, containing 75 bp of 5′ UTR, 600 bp of 3′ UTR, and a complete cDNA for mouse FOXD3, was kindly provided by P. Labosky, Vanderbilt University (43). The FOXD3 dominant-negative construct (FOXD3-DN) was created by cloning the C-terminal end of FOXD3 (amino acids 232–369) into pcDNA3 using primers FOX-DN forward and reverse (Table 1). The FOXD3 reporter construct (FOX-rep) was created including three FOX binding motifs in primers FOX enhancer forward and reverse (Table 1) and cloned into pGL2 containing the minimal Ret promoter (23).
Luciferase Assays
Cells were transfected with pGL2-CXCR4 reporter constructs, pCMV-β-galactosidase (Clontech) as an internal control, with or without PAX3 and/or FOXD3 expression constructs. The total amount of DNA transfected was kept constant by the addition of empty pBluescript vector (Stratagene). Cells were transfected, incubated for 48 h, then luciferase and β-galactosidase levels were measured (Promega). Luciferase activity was normalized to the internal control β-galactosidase. For all luciferase experiments shown, fold-induction was calculated by the measurement of the arbitrary light units produced by luciferase reporter protein divided by the measurements obtained from the reporter vector alone. Each bar represents n = 9, with mean ± S.E.
Chromatin Immunoprecipitation (ChIP) Assays
SKMEL-28 and A375 cells were fixed in 1% formaldehyde, quenched in 0.125 m glycine, then processed according to the manufacturer's protocol (EMD Millipore). For immunoprecipitation of PAX3 or FOXD3 DNA complexes, 1 μl of PAX3 or FOXD3 antibody (Cell Signaling) was added per experimental reaction. Normal IgG (Sigma) was used as a negative control against nonspecific DNA precipitation by an antibody. Nested PCR was performed with primers to the CXCR4 enhancer (Table 1). All ChIP samples were tested for false positive PCR amplification using primers that amplify a sequence from within the fourth coding exon of the β-tubulin gene to control against genomic DNA contamination. The nested primer set for this control is provided in Table 1.
Electrophoretic Mobility Shift Assay (EMSA)
GST-tagged proteins were expressed in BL21 cells and purified with glutathione-Sepharose 4B beads according to the manufacturer's protocol (GE Healthcare). GST-tagged proteins or melanoma protein lysates were mixed in reaction buffer (10 mm Tris-HCl, pH 8.0, 150 mm KCl, 0.5 mm EDTA, 0.1% Triton X-100, 12.5% glycerol, 0.2 mm DTT, and 100 μg/ml of poly(dI-dC)) for 30 min at room temperature, then incubated 15 min after the addition of ×100 cold competitor probe. Probe sequences are listed in Table 1. Cold (unlabeled) and hot (radiolabeled) probe sets contain identical sequences. Probes were prepared by annealing primers (Fig. 4B), labeled by end filling with DNA polymerase I Large (Klenow) fragment (New England Biolabs), purified on Illustra ProbeQuant G-50 Micro columns, then added to the mixture for 15 min. Gels were run as stated above on 5% native gels.
FIGURE 4.

The CXCR4 gene intronic enhancer region has specific PAX and FOX binding sites. A, mutations in putative PAX and FOX sites within the CXCR4 intronic enhancer alter the ability of PAX3 and FOXD3 to drive expression. pGL2-CXCR4pm393L or constructs with mutant P1, P2, and/or F sites were transfected into 293T cells with PAX3 (white), FOXD3 (gray), or both PAX3 and FOXD3 (black) expression constructs. Luciferase levels shown as fold-units over controls (levels without PAX3 or FOXD3 expression). B, sequence of the probe used in C with the PAX and FOX sites boxed. C, EMSA analysis for PAX3 and FOXD3 binding in vitro. Labeled probe encompassing the CXCR4 enhancer region as shown in B was incubated without any protein (lane 2), or with GST alone (lane 3) as negative controls. Lane 1 is without protein or probe (empty lane, negative control). The labeled probe was incubated with GST-PAX3 (lanes 4–8) or GST-FOXD3 (lane 9-11), or both (lane 12), and in the presence of ×100 cold probes with or without PAX or FOX sites mutated as indicated (lanes 5–8, 10, and 11), which may inhibit protein binding to the labeled probe.
Immunoprecipitation
For in vivo co-immunoprecipitations, cells were sonicated in 2× GS buffer (40 mm HEPES, 100 mm KCl, 40% glycerol, 2 mm 2-mercaptoethanol) supplemented with protease inhibitor mixture (Sigma) and 1 mm PMSF. Cell lysates were mixed with either FOXD3 or normal human IgG (Sigma) antibodies for 2 h at 4 ºC, then with Protein G/Protein A-agarose beads (Calbiochem) for an additional 2 h. Precipitates were spun down, washed 3 times in lysis buffer, resolved on 10% SDS-PAGE gels, and evaluated by standard Western blot analysis for PAX3 expression.
Scratch Cell Migration Assay
A375 and mel-624 cells were transfected with pcDNA3 (as a negative control), CXCR4, PAX3-DN, FOXD3-DN, PAX3-DN, and FOXD3-DN, or all three expression constructs and were allowed to grow for 48 h at which time the cell monolayer was scratched with a 100-μl pipette tip. Photographs were taken at 0, 6, and 24 h then the distance migrated was measured and graphed as percent of wound area closed (wound width at time 6 or 24 h divided by the wound width at time 0, multiplied by 100). Six wound areas were quantified per experiment, and each experiment was repeated in triplicate.
Transwell Chemotactic Assay
Transwell chambers with 8-μm pores were prepared by coating the bottom of the top chamber with 15 μg/ml of fibronectin (Fisher) overnight at 4 °C. A375 and mel-624 cells were transfected with expression constructs pcDNA3 (negative control), CXCR4, PAX3-DN, FOXD3-DN, PAX3-DN, and FOXD3-DN, or all three constructs and trypsinized 24 h post-transfection. The A375 and mel-624 cells were seeded into the top of the chamber in DMEM without FBS at 120,000 or 75,000 cells, respectively. DMEM supplemented with 10% FBS and 100 ng/ml of SDF-1α (Sigma) was added to the bottom chamber as a chemotactic agent. Cells were allowed to migrate for 48 h and then cells in the top chamber were removed and the remaining cells were fixed in 4% paraformaldehyde, 0.1% gluteraldehyde and photographed. Data were graphed as number of cells per microscopic field. Six independent areas per transwell filter were photographed and cell numbers were recorded. Each experiment was performed in triplicate.
Statistical Analysis
Significance of the differences between variable conditions was determined by GraphPad Prism statistical software (version 5.0 GraphPad Software) utilizing the Student's t test and Chi-square analysis with a confidence interval of 95%. All values stated as significant have p values of less than or equal to 0.05 unless indicated.
Results
Melanoma Cell Lines Express Two CXCR4 Transcript Isoforms
Comparative genomics between the human and mouse genomes reveal conservation of several regions of the CXCR4 gene (Fig. 1A). Significant conservation (>70% identical) 5′ proximal to the exon 1 untranslated region (UTR) is less than 100 bp. Even with this region being relatively small, other studies find that this proximal promoter (−191 to +88 from the 5′ UTR start) is biologically active (44, 45). The genomic region 5′ to the promoter region contains simple repeats and low sequence conservation. The intronic region of the CXCR4 gene has several regions of high homology (>70%) between the human and mouse.
FIGURE 1.

The CXCR4 locus is highly conserved between human and mouse genomes and encodes two CXCR4 isoforms, CXCR-A and CXCR4-B. A, comparison of human and mouse CXCR4 gene loci (analyzed using NCBI DCODE comparative genomics resources). The y axis indicates levels of homology, with the x axis level at ≤50% homology. Exons are shaded, with coding regions indicated in black and untranslated regions in gray. Schematic of two transcripts generated, CXCR4-B and CXCR4-A, are shown above the alignment graph. Length of genomic region is indicated in the scale bar below the graph in kilobases (kb), with the transcriptional start of exon 1/CXCR4-B transcript assigned as zero. B, schematics of genomic segments in construct utilized in studies. The diagrams are in alignment with their genomic locations in A. C, schematic of CXCR4-B and CXCR4-A transcripts, with the amino acid sequences indicated. The protein sequences are identical except for the most N-terminal residues. D and E, both CXCR4 transcripts are expressed in melanoma cells. All cell lines in the panel expressed the canonical CXCR4-B transcript (D) as detected by quantitative PCR analysis, whereas 3/7 expressed the non-canonical CXCR4-A transcript (E).
Two human alternate spliced transcripts have been identified (46) that differ from one another at the N-terminal end of the resulting protein (Fig. 1C). The more common isoform (CXCR4-B NM_003467, also known as CXCR4 variant 2 or canonical CXCR4) is coded from exons 1 and 2 of the CXCR4 gene and is expressed in a wide variety of tissues (46). For this transcript, exon 1 is 92 bp and includes the 5′ UTR plus the first five codons (Figs. 1C and 3A). The other transcript encodes a longer protein (CXCR4-A NM_001008540, also known as CXCR4-Lo or CXCR4 variant 1) and is encoded in its entirety from exon 2 and has a distinct start codon and 5′ UTR. CXCR4-A expression is restricted in normal tissues to peripheral blood lymphocytes and spleen cells. Although the impact of these differences has not been comprehensively studied, there is some evidence that the receptors have differential responses to ligands (46, 47). Differential expression of CXCR4 isoforms in melanoma and cancer in general is nearly unexplored. In a panel of melanoma cell lines, the CXCR4-B/canonical transcript is expressed in all cell lines tested (Fig. 1D). In addition, the variant transcript is expressed to some degree in 3/7 melanoma cell lines (Fig. 1E). How these two isoforms are differentially regulated, and any functional difference between CXCR4 isotype proteins in melanoma, is unknown.
Melanoma Cell Lines Express PAX3, FOXD3, and CXCR4
Expression of PAX3, FOXD3, and CXCR4 was determined in seven melanoma cell lines. These proteins are expressed in all lines tested to variant degrees (Fig. 2). CXCR4 in the melanoma lysates is ∼47 kDa, and a slower migrating band at 52 kDa may also be present. Although the predicted molecular mass of CXCR4 is ∼40 kDa, multiple sizes have been documented due to post-transcriptional modification including phosphorylation and glycosylation (48). Cell line 3T3 is shown as negative controls for PAX3 and FOXD3.
FIGURE 2.

PAX3, FOXD3, and CXCR4 are expressed in melanoma cells. Western analysis for PAX3, FOXD3, CXCR4, and GAPDH was performed in a panel of cell lines (lanes 1–7). Negative controls for PAX3 and FOXD3 were included (lane 8).
PAX3 Activates the CXCR4 Gene
Published data support, albeit indirectly, that PAX3 is directly upstream of CXCR4. During embryonic muscle development, CXCR4 and PAX3 have similar but not identical expression patterns (49). All CXCR4 positive cells expressed PAX3, however, not all PAX3 positive cells expressed CXCR4. In rhabdomyosarcoma, the translocation product PAX3-FOXO1 (PAX3-FHKD) also promotes CXCR4 expression, although it was not determined if PAX3 alone was able to activate this gene (32, 33). To determine whether PAX3 activates the CXCR4 5′ proximal promoter and/or intronic elements, plasmids containing regions from the CXCR4 locus driving luciferase expression were transfected into 293T cells in the presence or absence of a PAX3 expression vector. Three constructs were initially tested: pGL2-CXCR4pm572, CXCR4pm393, and pGL2-CXCR4pm393L, containing either 572 or 393 bp of the 5′ proximal promoter sequence with or without the full intronic sequence. The sequence of the CXCR4-B promoter is shown in Fig. 3A, and the constructs created are shown schematically in Fig. 3B. PAX3 activated the reporter constructs significantly only with the construct containing intron 1 (Fig. 3C, CXCR4pm393L). This induction, whereas significant, was modest (3.4 ± 0.23-fold light units over levels without PAX3). This weak induction is unsurprising because PAX3 is generally not a strong transcription factor alone, and often recruits other cofactors to effectively activate promoter sequences (20). Here, we find that PAX3 activates genomic elements within intron 1 of CXCR4.
A Highly Conserved Enhancer Region within Intron 1 Contains PAX3 and FOXD3 Consensus Motifs
Because PAX3 often works with co-factors, potential binding partner candidates were tested. The ability of two known PAX3 co-factors, SOX10 and ETS1, failed to synergistically activate pGL2-CXCR4pm393L with PAX3 (data not shown). In silico promoter analysis and phylogenetic footprinting was performed to identify any islands of sequence homology conserved between species in which the PAX3 binding site as well as potential recognition motifs of PAX3 co-activators may be located. An island of sequence homology between human and mouse CXCR4 loci is situated within intron 1, 3′ proximal to exon 1 (black highlight, Fig. 3A). This 52-bp sequence, which is highly conserved between Homo sapiens, Gorilla gorilla, Mus musculus, and Rattus noregicus (Fig. 3D), contains two putative PAX paired binding sites (P1 and P2) and a short FOX core recognition sequence (FOX). The pGL2-CXCR4pm393L vector, as well as pGL2-CXCR4pm393L-ΔISH, a construct with a section of intron 1 deleted (spliced region shown in Fig. 3A, with a dotted line, schematic of plasmids shown in Fig. 3E), were transfected into 293T cells in the presence or absence of PAX3 expression construct (Fig. 3F). PAX3 was able to drive the complete CXCR4 promoter luciferase reporter construct but this activity was abolished in the ΔISH construct where the island of sequence homology was removed.
Due to the presence of a FOXD3 binding site within the CXCR4 intronic enhancer (Fig. 3D) and expression of FOXD3 in all seven melanoma lines (Fig. 2), luciferase assays were performed to verify if FOXD3 drives expression of luciferase from the CXCR4 reporter constructs. The pGL2-CXCR4pm393L and pGL2-CXCR4pm393L-ΔISH reporter constructs were transfected into 293T cells with PAX3, FOXD3, or both expression constructs. The addition of FOXD3 protein lead to a 4.0 ± 0.23-fold induction of light units over reporter vector alone, and luciferase levels increased significantly after the addition of PAX3 to 16.6 ± 2.0-fold (Fig. 3F) but was abolished if the reporter construct lacked the intronic enhancer. These data support that FOXD3 activates the CXCR4 enhancer element and this induction is significantly enhanced in the presence of PAX3.
Mutation of Specific Binding Sites within the CXCR4 Intronic Enhancer Leads to a Loss of Activation by PAX3 and FOXD3
Specific mutations were created in the identified P1, P2, and/or F sites (Fig. 3D) within the pGL2-CXCR4pm393L reporter construct and transfected into 293T cells with or without PAX3 and/or FOXD3 expression constructs. In 293T cells, the P1 site was dispensable for activation by PAX3 alone or synergistically with FOXD3 (Fig. 4A, set 2, p = 0.194 and 0.206, respectively). Conversely, mutation of the P2 site resulted in a loss of the ability of PAX3 to drive luciferase expression alone or in combination with FOXD3 (Fig. 4A, sets 3, 5, 7, and 8, p < 0.05 and 0.005, respectively). Furthermore, mutation of the F site lead to a loss of FOXD3-dependent luciferase transcription either alone or in combination with PAX3 (Fig. 4A, sets 4 and 6–8, p < 0.05 and 0.0005, respectively). These findings support that, in 293T cells, the P2 and F sites within the CXCR4 intronic enhancer are necessary for PAX3- and FOXD3-dependent gene expression and synergistic activation.
PAX3 and FOXD3 Directly Bind to the CXCR4 Enhancer Element
Electromobility shift assays (EMSA) were performed to determine whether PAX3 and FOXD3 directly bind to the CXCR4 intronic enhancer element. GST-tagged PAX3 and FOXD3 were able to bind to the probe specific to the enhancer element (Fig. 4B) and cold probe inhibited these interactions (Fig. 4C, lanes 4 and 9, lanes 5 and 10, respectively). Probe alone and GST alone were run as negative controls and did not produce bands (Fig. 4C, lanes 2 and 3). Specific mutations in each of the putative binding sites further identified which sites are utilized in vitro. Cold probe with mutations in the P1 site was still able to compete (Fig. 4C, lane 6), whereas cold probes with mutations in the P2 site lost their ability to compete (Fig. 4C, lanes 7 and 8). These results demonstrate that the P2 site is utilized in vitro over the P1 site. When the F site was mutated (Fig. 4C, lane 11), there was an incomplete competition, suggesting that this site is in use as well. The addition of both PAX3 and FOXD3 proteins resulted in a supershift (Fig. 4C, lane 12). These data show that FOXD3 and PAX3 directly interact with the conserved enhancer element of CXCR4.
The CXCR4 Intronic Enhancer Is Active in Melanoma Cells
To determine whether the CXCR4 intronic enhancer drives expression in melanoma cells, CXCR4 reporter constructs were transfected into A375, SKMEL-28, and mel-624 cells (Fig. 5A). CXCR4pm393L produced significant levels of luciferase reporter activity in all melanoma cells tested (Fig. 5A, black bars, 11.3 ± 1.6 (A375), 9.5 ± 2.0 (SKMEL-28), and 6.72 ± 1.1 (mel-624)-fold induction over pGL2 vector p < 0.001). The levels of reporter expression were attenuated when the intronic enhancer was removed (Fig. 5A, gray bars, 5.1 ± 0.8 (A375), 4.4 ± 1.4 (SKMEL-28), and 3.59 ± 0.5 (mel-624)-fold induction over pGL2 vector alone, p < 0.02). These experiments support that the CXCR4pm393L reporter construct is able to drive gene expression in melanoma cells, and this function was reduced in the absence of the intronic enhancer element.
FIGURE 5.

The CXCR4 enhancer is active in melanoma cells through the PAX and FOX sites. A, the CXCR4 intronic enhancer is active in melanoma cells. Empty vector (pGL2) or CXCR4 reporter constructs shown in Fig. 3E were transfected into A375, SKMEL-28, and mel-624 melanoma cells. Luciferase levels are shown as fold-units over controls (levels without PAX3 or FOXD3 expression). B, mutation of PAX and FOX sites attenuates the activity of the CXCR4 enhancer in melanoma cells. A375 and mel-624 cells were transfected with pGL2-CXCR4pm393L constructs with or without mutations in P1, P2, and/or F sites. Luciferase levels are shown as percent of control luciferase levels (pGL2-CXCR4pm393L). C, EMSA analysis using a probe with the CXCR4 enhancer element with melanoma cell lysates produces slow migrating bands. Labeled probe encompassing the CXCR4 enhancer region as shown in Fig. 4B was incubated with SKMEL-28, A375, and mel-624 cell lysates without (lane 2) or with (lanes 3–7) ×100 cold probes with or without PAX or FOX sites mutated as indicated, which may inhibit protein binding to the labeled probe. Lane 1 contains samples with labeled probe without added cell lysate (negative control). Major shift band is indicated with arrows. D and E, PAX3 and FOXD3 are located at the CXCR4 locus in A375 (D) and SKMEL-28 (E) cells. ChIP analysis was performed by precipitating proteins with antibodies specific for PAX3 (lane 1), FOXD3 (lane 2), or with nonspecific normal mouse IgG (lane 3) as a negative control. Additional controls were DNA input (positive control, lane 4) and no template DNA/water blank (negative control, lane 5). Precipitated DNA fragments were utilized as PCR templates with primers specific for the CXCR4 intronic region (top gels) or exon 4 of the β-tubulin gene (bottom gels, negative controls). F, PAX3 and FOXD3 directly interact in melanoma cells. Lysates from mel-624 (top bands) and SKMEL-28 (bottom bands) cells are immunoprecipitated (IP) with a FOXD3-specific antibody (lane 1), no antibody/beads only (lane 2), or with a nonspecific human IgG (lane 3), and probed for PAX3 expression. WB, Western blot.
To determine which specific sites are responsible for the loss of activity of the intronic enhancer in melanoma cells, reporter constructs with specific mutations in P1, P2, and/or F sites were transfected into A375 and mel-624 melanoma cells (Fig. 5B). For both cell lines, mutation in either P1 or P2 site was dispensable (Fig. 5B, sets 2 and 3, all p > 0.07). Mutation of both P1 and P2 lead to a significant loss in luciferase activity when compared with a reporter with a wild-type sequence (Fig. 5B, sets 1 and 5, p = 0.001 (A375) and 0.0015 (mel-624)). Loss of the F site did not lead to a significant reduction in luciferase activity when compared with controls alone (set 4) or in combination with the P2 site (set 7). Mutation of the F site in combination with the P1 site or both the P1 and P2 site mutated resulted in a significant loss of luciferase activity (Fig. 5B, set 6, p = 0.0045 (A375) and p = 0.0016 (mel-624), and set 8, p = 0.018 (A375) and p = 0.0021 (mel-624)). In these experiments, mutation of the P1 site in combination with a loss of either the P2 or the F site leads to a significant loss of the CXCR4 intronic activity of the enhancer.
Endogenous Proteins Do Not Bind Completely to Probes Containing Mutations of Specific Binding Sites within the CXCR4 Intronic Enhancer
Radioactive probes containing the wild-type CXCR4 intronic enhancer sequence (Fig. 4B) generate bands in an EMSA analysis that are lost when ×100-fold excess cold probe is added in melanoma cell lysates (Fig. 5C, lanes 2 and 3). When the P1, P2, or F sites were mutated, the ability to completely inhibit binding was lost (lanes 4–7). The probe with a mutated P2 site was only partially able to compete in A375 cells (lane 5). In addition, probes containing an F site mutation only partially competed against the wild-type probe in all cell lines tested (lane 7). These findings demonstrate that proteins within melanoma cells bind to DNA sequences from the CXCR4 intronic enhancer that require wild-type P1, P2, and F sites.
PAX3 and FOXD3 Reside on the Endogenous CXCR4 Locus in Melanoma Cells
PAX3 and FOXD3 are also located within the CXCR4 locus in melanoma cells, as determined by chromatin immunoprecipitation (ChIP) assays. A PCR amplicon was generated utilizing primers specific for the CXCR4 gene region near the intronic enhancer when proteins were precipitated with antibodies specific for either PAX3 or FOXD3 (Fig. 5, D and E, lanes 1 and 2). No product was generated when the immunoprecipitation was performed with normal mouse IgG, when the primers were sequence specific for the coding region of the β-tubulin gene that does not have PAX or FOX enhancer sites, or when no ChIP sample template was added (PCR water blank, Fig. 5, D and E, lane 5), supporting that the amplified bands are specifically generated from the ChIP analysis. As a positive control of the input sample, DNA was isolated from the cell lysate before immunoprecipitation and utilized as a template for the PCR (Fig. 5, D and E, lane 4). These experiments support that both PAX3 and FOXD3 are bound to the CXCR4 gene locus in melanoma cells.
PAX3 and FOXD3 Directly Interact in Melanoma Cells
PAX3 and FOXD3 are both expressed in melanoma cells, and activate the CXCR4 reporter constructs more robustly together than alone, form a supershift complex in EMSA experiments, and are both located on the endogenous CXCR4 gene (Figs. 2, 3, 4C, and 5, D and E). Immunoprecipitation assays were performed to determine whether these proteins interact within melanoma cells. Immunoprecipitation of protein lysates from mel-624 or SKMEL-28 melanoma cells utilizing an anti-FOXD3 antibody yielded a band when probed for PAX3 by Western analysis (Fig. 5F, lane 1), but not in the absence of a FOXD3-specific antibody (Fig. 5F, lane 2) or with nonspecific antibody (Fig. 5F, lane 3). We find that PAX3 and FOXD3 directly interact in melanoma cells.
Dominant-negative PAX3 and FOXD3 Proteins Inhibit the Ability of the Wild-type Proteins to Promote Transcription
For cell biology experiments, a technical roadblock occurred when introducing both expression constructs and siRNA molecules, due to difficulties arising in differences in kinetics of inhibition of expression and ideal methodologies of transfection. As an alternative approach, dominant-negative constructs were employed and tested. To validate that the dominant-negative proteins functioned as inhibitors, proteins were expressed with or without the wild-type proteins with reporter constructs containing known PAX or FOX binding sites (Fig. 6, A–F). The C terminus of PAX3, which contains a transactivation domain, was deleted to create a PAX3 dominant-negative protein (PAX3-DN) (Fig. 6A). A reporter construct containing a segment of the MET gene with a validated PAX3 binding site was used as a control (Fig. 6B). As previously reported (15), PAX3 was able to promote luciferase expression, and this was attenuated with the addition of PAX3-DN (Fig. 6C). A previously described FOXD3 dominant-negative protein (FOXD3-DN) comprised of only the C-terminal end of FOXD3 (Fig. 6D) (50), was transfected with a synthetic reporter construct (FOX reporter construct) (Fig. 6E). The FOX reporter construct contains three FOXD3 binding motifs cloned upstream of a DNA segment from the RET gene locus that functions as a minimum promoter but does not have significant basal activity on its own (23, 27). The FOXD3-DN completely blocked the ability of wild-type FOXD3 to drive luciferase expression from the FOX reporter construct (Fig. 6F). In parallel to these validation studies, both dominant-negative proteins completely inhibited the ability of PAX3 and FOXD3 to activate expression utilizing the CXCR4 enhancer (Fig. 6G). The dominant-negative constructs were transfected into cells to measure consequences on cell numbers. Using siRNA molecules, inhibition of PAX3 results in a significant reduction of cell numbers, whereas FOXD3 loss leads to an increase in cell growth in both A375 and mel-624 cells (Fig. 6, H and I), following previous observations (24, 51). Introduction of PAX3-DN or FOXD3-DN lead to a significant change in cell numbers (p < 0.05) following the same pattern as the siRNA-treated cells, albeit to a lesser degree. In these experiments, we find that PAX3-DN and FOXD3-DN inhibit the ability of the wild-type proteins to act as transcription factors, and result in similar cellular effects when compared with siRNA-mediated transcript-specific interference.
FIGURE 6.

Dominant-negative PAX3 and FOXD3 proteins inhibit wild-type protein transcriptional function and melanoma cell growth. A, a schematic comparison of PAX3 wild-type and dominant-negative (PAX3-DN) proteins. B, schematic diagram of a PAX3-responsive reporter construct, containing sequence comprised of the UTR and 5′ proximal promoter segments of the MET gene. This reporter is PAX3 responsive, as previously reported (15). C, PAX3-DN protein inhibits the ability of wild-type PAX3 to activate a reporter construct containing the MET promoter. PAX3 promoted luciferase activity 4.47 ± 0.43-fold over reporter levels alone, whereas the addition of PAX3-DN reduced these levels (all p < 0.0005). D, schematic comparison of FOXD3 wild-type and dominant-negative (FOXD3-DN) proteins. E, schematic of a synthetic FOX reporter construct. The gene cassette contains three FOX binding motifs (as shown, boxed) cloned 5′ to a minimum promoter element that is upstream of a luciferase open reading frame. F, FOXD3-DN blocks the ability of FOXD3 to drive luciferase expression. FOXD3 drove luciferase activity 4.48 ± 0.18-fold over reporter levels alone, whereas the addition of FOXD3-DN inhibited this activation (all p < 0.0005). G, dominant-negative PAX3 and FOXD3 block the ability of the wild-type proteins to activate expression through the CXCR4 intronic enhancer. PAX3-DN and FOXD3-DN alone did not drive luciferase activity from the CXCR4 reporter vector (sets 3 and 5, respectively, p > 0.2). The dominant-negative proteins significantly inhibited the synergistic activation of the CXCR4 reporter by PAX3 and FOXD3 (PAX3-DN, set 7; FOXD3-DN, set 8; p < 0.0005 compared with set 6). H and I, dominant-negative protein expression or siRNA targeting of PAX3 or FOXD3 results in similar consequences in melanoma cell growth. A375 (H) and mel-624 (I) cells were transfected with Scramble siRNA (control, first lanes), or siRNA targeted against PAX3 or FOXD3 (second and third lanes). Cells were also transfected with empty vector (fourth lanes) or with PAX3-DN or FOXD3-DN expression constructs (fifth and sixth lanes). Growth changes seen for PAX3 or FOXD3 inhibition are significant when compared with controls (*, p < 0.05; ***, p < 0.0005).
PAX3 and FOXD3 Proteins Are Sufficient at Driving CXCR4 Expression in Melanoma Cells
FOXD3 and PAX3 were overexpressed in melanoma cell lines to determine whether CXCR4 protein levels would be altered. Melanoma cell lines A375 and SKMEL-28 were transfected with empty vector pcDNA3, or with expression constructs for PAX3 and/or FOXD3. Protein lysates from these cells were analyzed for the expression of CXCR4 and vinculin (Fig. 7, A–D). In A375 cells, CXCR4 levels increased significantly with the addition of either PAX3 or FOXD3, and to even higher levels (383.0 ± 72.6-fold over cDNA3 controls) when both PAX3 and FOXD3 were added (Fig. 7, A and B). In SKMEL-28 cells, addition of PAX3 and FOXD3 alone did not promote a significant increase in CXCR4 expression. However, together PAX3 and FOXD3 increased CXCR4 expression 8.3 ± 1.4-fold over control levels (Fig. 7, C and D). CXCR4 protein was increased at multiple molecular masses, including 47, 52, and 75 kDa. Here, we find that the addition of exogenous PAX3 and/or FOXD3 protein was sufficient to increase the level of CXCR4 receptor in melanoma cells.
FIGURE 7.

PAX3 and FOXD3 are sufficient to overexpress CXCR4 protein in melanoma cells, and PAX3 with or without FOXD3 is necessary for full CXCR4 expression. A–D, the addition of exogenous PAX3 and FOXD3 in A375 (A and B) or SKMEL-28 (C and D) cells leads to an increased level of CXCR4 protein. In A375 cells, endogenous CXCR4 is expressed in multifold lower levels in comparison to transfected cells and is not detectable at the exposure presented in A. A representative Western analysis is shown (A and C), and densitometry of four independent experiments are displayed in graph form (B and D). PAX3, FOXD3, or both proteins together were able to overexpress significant levels of CXCR4 protein in A375 cells (B, p < 0.002 all groups), whereas only the expression of both PAX3 and FOXD3 overexpressed CXCR4 at significant levels in SKMEL-28 (D, p < 0.0005 PAX3 + FOXD3 group). E–G, the inhibition of PAX3, or PAX3 and FOXD3, leads to a reduction of CXCR4 protein. Dominant-negative proteins shown in Fig. 6 are transfected into A375 (E) or mel-624 (F) cells. Exogenous PAX3-DN protein is detected by the HA tag, whereas endogenous PAX3 levels are revealed using an antibody that recognizes the C terminus that is lacking in the PAX3-DN. Endogenous FOXD3 levels are expressed at significantly lower levels in comparison to FOXD3-DN protein and is not detectable at the exposure shown in E and F. A representative Western analysis is shown (E and F), and densitometry of four independent experiments are displayed in graph form (G). Significant decreases in CXCR4 was seen when PAX3-DN alone (44.7 ± 16.5% of control levels, p = 0.006) or with PAX3-DN and FOXD3-DN together (28.4 ± 9.2%, p = 0.009) were added to A375 cells (top graph), or both dominant-negative proteins in mel-624 cells (47.8 ± 15.4%, p = 0.002, bottom graph). Decrease in CXCR4 intensity was predominantly seen in the slower migrating 52-kDa band.
PAX3 and FOXD3 Are Necessary to Maintain Full CXCR4 Levels in Melanoma Cells
To determine whether inhibition of PAX3 and/or FOXD3 function alters CXCR4 levels, vectors encoding PAX3 and/or FOXD3 dominant-negative proteins were transfected into melanoma cells (Fig. 7, E–G). Levels of CXCR4 were reduced when FOXD3-DN was present in A375 cells, but not significantly, whereas PAX3-DN lead to significant decreases (44.7 ± 16.5% of control levels, p = 0.006). Presence of both PAX3-DN and FOXD3-DN lead to significant decreases in CXCR4 levels in both A375 (28.4 ± 9.2%, p = 0.009) and mel-624 (47.8 ± 15.4%, p = 0.002) melanoma cells. Reduction of CXCR4 was not found with either dominant-negative protein alone in mel-624 cells. Decrease in CXCR4 intensity was predominantly seen in the 52-kDa band. These dominant-negative findings trend with experiments where siRNAs are employed (data not shown). These data support that FOXD3 alone is not necessary for CXCR4 expression in the melanoma cells analyzed, but that PAX3 is necessary, either alone or together with FOXD3.
Inhibition of PAX3 and FOXD3 Leads to a Reduction of Motility, Migration, and Chemotaxis
To determine whether inhibition of PAX3 and FOXD3 leads to an inhibition of cellular motility, migration, and chemotaxis, cells were transfected with empty vector (pcDNA3), or constructs expressing dominant-negative PAX3, dominant-negative FOXD3, or both (Fig. 8). Cells were transfected at high density and an open region was introduced by scratching the cell layer, creating a “wound” area. Cells were photographed after the creation of the wound area, as well as 6 (A375 cells) or 24 h (mel-624 cells) post-wound generation. Percent wound area closed was calculated to determine motility rates compared with control groups. Although control groups demonstrated significant wound closing (24.70 ± 9.37%, A375, and 66.7 ± 3.54%, mel-624), there were significant decreases in motility for both cell lines with either dominant-negative protein or both (p ≤ 0.05, Fig. 8A). To test if the presence of dominant-negative proteins would affect cellular migration and chemotaxis, cells were seeded in transwell chambers, with transfected cells and serum-free media in the top chamber and media with FBS and the CXCR4 ligand SDF1 in the lower chamber (Fig. 8C). Presence of both dominant-negative proteins significantly affected migration and chemotaxis for both cell lines (Fig. 8D, pcDNA3 group versus PAX3-DN + FOXD3-DN group, p ≤ 0.05). At longer time points, detrimental affects on cellular growth and survival were observed, consistent with previous observations for both PAX3 and FOXD3 (14, 24). These data support that inhibition of PAX3 and/or FOXD3 in melanoma cells leads to a significant reduction in cellular motility, migration, and chemotaxis.
FIGURE 8.
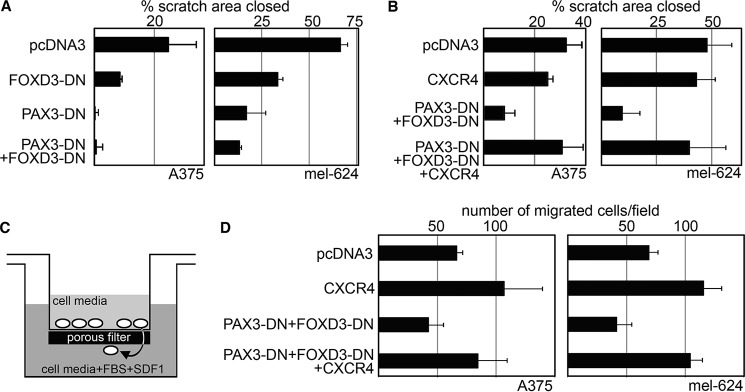
Inhibition of PAX3 and FOXD3 function leads to a significant decrease in motility, migration, and chemotaxis in melanoma cells, but this inhibition is reversed with the addition of exogenous CXCR4. A and B, dominant-negative PAX3 and FOXD3 proteins reduce cellular motility (A), whereas added CXCR4 restores this loss (B). Cellular scratch assays of cells were transfected with either pcDNA3 (control group), or with FOXD3-DN, PAX3-DN, and/or CXCR4 expression constructs. Graphs are shown as percent scratch area closed, calculated as measurements of the wound area at 6 (A375) or 24 h (mel-624) post-creation of the scratch, divided by the measurements at the start of the experiment and multiplied by 100. Dominant-negative proteins demonstrated decreased motility compared with control groups (graphs in A, A375 p ≤ 0.05, mel-624 p ≤ 0.005). The addition of CXCR4 restored cellular motility (CXCR4 group versus PAX3-DN + FOXD3-DN + CXCR4 groups, p > 0.17 both cell types). C and D, dominant-negative PAX3 and FOXD3 proteins decrease cellular migration and chemotaxis, whereas added CXCR4 restores this loss. Melanoma cells were tested in transwell assays with FBS and SDF1 added to the lower chamber as a chemoattractant (shown schematically in C) that were transfected with either pcDNA3 (control groups), or with vectors expressing CXCR4, PAX3-DN and FOXD3-DN, or both (D). For each group, six fields of cells for each experiment was counted and averaged. The addition of CXCR4 increased migration significantly (66.72 ± 5.45 cells/field (pcDNA3) to 107.17 ± 32.19 (CXCR4) for A375, and 69.33 ± 7.54 (pcDNA3) to 115.67 ± 15.09 for mel-624, p ≤ 0.05 both cell lines). The presence of both dominant-negative proteins significantly inhibited migration (42.72 ± 12.45 for A375, 41.94 ± 12.70 for mel-624, p ≤ 0.05 both cell lines). Adding CXCR4 to cells transfected with both dominant-negative proteins restored cellular migration (84.50 ± 24.90 for A375, 104.28 ± 10.22 for mel-624, p ≥ 0.19 when comparing CXCR4 groups to PAX3-DN + FOXD3-DN + CXCR4 groups, both cell lines).
Reintroduction of CXCR4 Expression Rescues Decrease in Motility, Migration, and Chemotaxis Due to Inhibition of PAX3 and FOXD3 in Melanoma Cells
We find that inhibition of PAX3 and FOXD3 results in both a loss of cellular movement and CXCR4 expression. To determine how much of a role CXCR4 expression is playing in PAX3- and FOXD3-dependent cell migration, motility, and chemotaxis, CXCR4 expression was reintroduced in cells expressing PAX3 and FOXD3 dominant-negative proteins. In scratch assays, no significant increase in cell motility was noted for either cell line when exogenous CXCR4 protein expression was present (Fig. 8B, p > 0.10). Although the presence of both dominant-negative proteins significantly inhibited cell motility, the addition of CXCR4 restored motility to control levels. Furthermore, whereas the addition of CXCR4 alone increased cell migration and chemotaxis in transwell assays (Fig. 8D, p ≤ 0.05 both cell lines), CXCR4 expression maintained this migration ability when both dominant-negative proteins were present (p > 0.19 both cell lines). We find that, in scratch and transwell assays, the addition of exogenous CXCR4 expression rescues the effects of PAX3 and FOXD3 inhibition on cellular motility, migration, and chemotaxis. In summary, we define a pathway involving PAX3 and FOXD3 promoting CXCR4 expression in melanoma through a highly conserved intronic enhancer, thereby promoting melanoma cell motility and migration.
Discussion
Melanoma Cells Express Two CXCR4 Isoforms, One Commonly Expressed in Cells (CXCR4-B), Whereas the Other Is Normally Restricted to Cells during Development or Mature Blood Cells (CXCR4-A)
In humans, two alternate spliced transcripts of CXCR4 have been identified (1). The two isoforms differ from one another by 9 amino acids in the functionally important N-terminal extracellular region of the receptor (Fig. 1 and Ref. 11). The common isoform, called CXCR4-B, is expressed in all of the melanoma cell lines. The other variant, CXCR4-A, is expressed in a subset of the cell lines screened (3/7 lines tested). The expression of the alternate transcript is not expected, because it has been reported to have a restricted expression pattern in mature tissues to peripheral blood lymphocytes and spleen cells (11). Although the impact of these differences between isoforms have not been extensively studied, it appears that this alteration in the N-terminal region results in CXCR4-A being less efficient in its response to CXCL12/SDF1 as measured by calcium ion flux and chemotactic responses (11). We did not detect any differential expression between CXCR4 isoforms in our studies (data not shown). The role of CXCR4-A in cancer will be a focus of future studies, due to the potential clinical relevance. Although CXCR4 is presently a target in current cancer therapy trials, the expression of a more cell-type restricted isoform may provide a unique drug target. Because CXCR4-A features a unique extracellular N-terminal region, it is plausible that a small molecule inhibitor could be designed to specifically inhibit CXCR4-A while sparing the function of the more widely expressed canonical CXCR4 protein. Furthermore, CXCR4-A may serve as a unique diagnostic marker in melanoma if it is associated with cells that are more prone to metastasis. Thus, CXCR4-A may represent a more focused therapeutic target and should be further studied in the context of melanoma and other cancers.
PAX3 and FOXD3 Are Expressed in Melanoma, Directly Interact, and Promote the Expression of CXCR4 through a Highly Conserved Genomic Element
Here, we identified a highly conserved enhancer located within intron 1 of the CXCR4 gene, containing both PAX and FOX binding sites (Figs. 3 and 4). These sites are active in melanoma cells (Fig. 5). In our experiments, loss of this enhancer element, or the P1 and P2 sites specifically, abolished the ability of PAX3 to activate the CXCR4 regulatory element (Figs. 3F, 4A, and 5). The deletion of the intronic enhancer region or the F site within the CXCR4 locus reduced but did not eliminate the ability of FOXD3 to activate a CXCR4 reporter construct (Figs. 3F, 4A, and 5). This may be due to a secondary FOXD3 binding site within the genomic segment contained in the construct, perhaps the previously identified FOXC binding site just 5′ to the first exon (see Ref. 28 and marked in Fig. 3A). However, loss of the intronic enhancer abrogated synergistic activation of FOXD3 and PAX3 together. Our data support that FOXD3 and PAX3 synergistically activate the CXCR4 gene through the intronic enhancer in melanoma cells.
In this report, we find that PAX3 and FOXD3 directly interact and form a super-shifted complex in a gel shift containing the CXCR4 enhancer element (Figs. 4C and 5C). This is a parallel finding to what occurs during development in the neural crest, which are the precursors to future melanocyte cells. In the neural crest, FOXD3 acts as a regulator of PAX3 transcriptional function and represses a pathway that drives cells to differentiate into a melanocytic fate (35). It is unclear if this mechanism is recycled in melanoma cells. If so, FOXD3 may be an inhibitor to melanocyte differentiation and promotes a more stem-cell like fate. In development, FOXD3 binds to PAX3 and acts as a repressor to PAX3-mediated MITF activation (35). This is a different mechanism than what we discovered here, where the two factors bind and act together as transcriptional activators of CXCR4. Potential future studies will focus on other potential downstream genes that are both PAX3 and FOXD3 activated in addition to CXCR4, and if FOXD3 retains its ability to act as a repressor of PAX3 activity for other genes.
CXCR7, an Alternate SDF1 Receptor in Melanoma
In this report, we focus on the CXCR4 receptor, an established driver of melanoma metastasis (4–7). Recently, expression of CXCR7 was found through candidate gene RT-PCR and RNAseq analysis in human melanoma cell lines (52, 53), as well as medaka aquarium fish melanomas (54) and normal melanocytes (55). Furthermore, CXCR7 is necessary for melanocyte and medaka melanoma SDF1-dependent migration (54, 55). These works conclude that CXCR4 is not necessary in their models; however, expression of CXCR4-A is overlooked. In this report, we discovered that both the canonical CXCR4-B and the variant CXCR4-A are expressed in melanoma cells (Fig. 1). Medaka has two separate genes for CXCR4, cxcr4a and cxcr4b, and although both are expressed in medaka melanomas at similar levels, only cxcr4b was blocked in this study (54). In normal melanocytes, CXCR4 was inhibited with neutralizing antibody Abcam ab2074 (55). This antibody targets the most N-terminal epitopes of CXCR4-B, and many of these amino acids are not in the CXCR4-A protein. In our experiments, we do not detect CXCR4-A with this antibody, but do with another antibody (Thermo Scientific, PA3-305) that targets residues 338–359 of CXCR4 (data not shown). Although there is good evidence that demonstrates CXCR7 role in migration, CXCR4-A cannot be ruled out.
In our studies, the role of CXCR7 is unknown. Fig. 8 is focused on the very specific question if FOXD3- and PAX3-dependent migration was due to the loss of CXCR4. In this figure, we were able to rescue the reduction in cell migration with the addition of CXCR4. It may be that the residual migration seen with loss of CXCR4 (Fig. 8D) is CXCR7 dependent, and inhibition of this second receptor will fully block cell migration. Alternatively, PAX3 and/or FOXD3 may also regulate CXCR7 and levels of this second receptor were altered. Furthermore, there is some suggestion of cross-talk between receptors in other cell types, and CXCR7 may be affected by changes in CXCR4 levels. Only limited studies now exist that examine CXCR7 in melanoma (52, 54) and this receptor may complicate clinical approaches to CXCR4-specific therapeutic targeting.
FOXD3 and PAX3 Involvement in Melanoma Metastasis through the Promotion of CXCR4 Expression
Although overexpression of FOXD3 may inhibit migration and invasion, FOXD3 also promotes metastasis through the activation of ERBB3 (26, 56). PAX3 drives melanoma migration and invasiveness, and this is due at least in part by the activation of the BRN2 gene (18).
CXCR4 has been clearly implicated in melanoma invasion and metastasis (4–7). Here, both FOXD3 and PAX3 are linked to melanoma progression and metastasis by driving CXCR4 expression. In melanoma, CXCR4 is primarily correlated with melanoma lung metastasis (6). At present, it is not known what role PAX3 and FOXD3 plays in tissue-specific metastases. Here, we find that inhibition of PAX3 and FOXD3 reduce cellular motility and migration, at least in the in cellulo models tested. Our findings support that this reduction of cell movement through PAX3 and FOXD3 inhibition is due to a decrease in CXCR4 expression. Other studies have made similar findings, where inhibition of CXCR4 in both transwell and scratch assays lead to an impaired motility and migration (10). If these transcription factors purely drove metastasis through CXCR4, it would be expected that PAX3 and FOXD3 driven melanoma would preferentially direct metastasis to the lung. However, if these transcription factors promoted metastasis to other distal sites commonly seen as metastatic destinations for melanoma, such as skin, liver, lymph nodes, and brain, some other CXCR4-independent pathway would be responsible. If this second possibility is true, this supports that targeting PAX3 and FOXD3 expression in melanoma would inhibit melanoma metastasis. Although our data support nearly full rescue of the PAX3/FOXD3-dependent loss by reintroducing exogenous CXCR4 in the model systems tested, it is not clear if this will translate to in vivo studies. Future studies will aim to determine whether PAX3 and FOXD3 actively promote metastasis, if this is solely due to the expression of CXCR4, or if there are other downstream targets mediating cellular invasion and metastatic spread.
Author Contributions
J. D. K. and D. L. conceived and coordinated the study and wrote the paper, and performed the cell biology, protein biology, luciferase assays, EMSAs, ChIPs, and gene analysis experiments. J. W. L. designed, performed, and analyzed the protein interaction studies. E. C. L. and S. K. designed, performed, and analyzed the alternate splice transcripts experiments. E. C. L. also performed and analyzed the quantitative PCR experiments. A. E. L. designed and created the CXCR4 promoter luciferase constructs. R. S. and A. E. A. provided technical and scientific assistance for studies focused on CXCR4 and FOXD3, respectively. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Patricia Labosky for providing for FOXD3 expression construct. The monoclonal PAX3 antibody was developed by C. P. Ordahl and obtained from the Developmental Studies Hybridoma Bank, created by the NICHD, National Institutes of Health, and maintained at The University of Iowa, Department of Biology, Iowa City, IA.
This work was supported, in whole or in part, by National Institutes of Health Grants T32 GM007183 (to J. W. L.), T35 DK062719 (TO S. K.), R01 CA160495 (to A. E. A.), and R01 CA130202, R01-AR062547, and R01CA184001 (to D. L.), American Cancer Society Research Scholar Award RSG-CSM-121505 (to D. L.), Friends of Dermatology-University of Chicago, the Wendy Will Case Foundation, and the University of Chicago Comprehensive Cancer Center. The authors declare that they have no conflicts of interest with the contents of this article.
- CXCR4
- CXC chemokine receptor 4
- DN
- dominant-negative
- F1
- FOX binding site 1
- F2
- FOX binding site 2
- FOXD3
- Forkhead box D3
- ISH
- island of sequence homology
- PAX3
- Paired box transcription factor 3
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Thompson J. A. (2012) Ten years of progress in melanoma. J. Natl. Compr. Canc. Netw. 10, 932–935 [DOI] [PubMed] [Google Scholar]
- 2. Longo-Imedio M. I., Longo N., Treviño I., Lázaro P., Sánchez-Mateos P. (2005) Clinical significance of CXCR3 and CXCR4 expression in primary melanoma. Int. J. Cancer 117, 861–865 [DOI] [PubMed] [Google Scholar]
- 3. Scala S., Giuliano P., Ascierto P. A., Ieranò C., Franco R., Napolitano M., Ottaiano A., Lombardi M. L., Luongo M., Simeone E., Castiglia D., Mauro F., De Michele I., Calemma R., Botti G., Caracò C., Nicoletti G., Satriano R. A., Castello G. (2006) Human melanoma metastases express functional CXCR4. Clin. Cancer Res. 12, 2427–2433 [DOI] [PubMed] [Google Scholar]
- 4. Bartolomé R. A., Gálvez B. G., Longo N., Baleux F., Van Muijen G. N., Sánchez-Mateos P., Arroyo A. G., Teixidó J. (2004) Stromal cell-derived factor-1α promotes melanoma cell invasion across basement membranes involving stimulation of membrane-type 1 matrix metalloproteinase and Rho GTPase activities. Cancer Res. 64, 2534–2543 [DOI] [PubMed] [Google Scholar]
- 5. Cardones A. R., Murakami T., Hwang S. T. (2003) CXCR4 enhances adhesion of B16 tumor cells to endothelial cells in vitro and in vivo via β1 integrin. Cancer Res. 63, 6751–6757 [PubMed] [Google Scholar]
- 6. Murakami T., Maki W., Cardones A. R., Fang H., Tun Kyi A., Nestle F. O., Hwang S. T. (2002) Expression of CXC chemokine receptor-4 enhances the pulmonary metastatic potential of murine B16 melanoma cells. Cancer Res. 62, 7328–7334 [PubMed] [Google Scholar]
- 7. Robledo M. M., Bartolome R. A., Longo N., Rodríguez-Frade J. M., Mellado M., Longo I., van Muijen G. N., Sánchez-Mateos P., Teixidó J. (2001) Expression of functional chemokine receptors CXCR3 and CXCR4 on human melanoma cells. J. Biol. Chem. 276, 45098–45105 [DOI] [PubMed] [Google Scholar]
- 8. Vianello F., Papeta N., Chen T., Kraft P., White N., Hart W. K., Kircher M. F., Swart E., Rhee S., Palù G., Irimia D., Toner M., Weissleder R., Poznansky M. C. (2006) Murine B16 melanomas expressing high levels of the chemokine stromal-derived factor-1/CXCL12 induce tumor-specific T cell chemorepulsion and escape from immune control. J. Immunol. 176, 2902–2914 [DOI] [PubMed] [Google Scholar]
- 9. Kim M., Koh Y. J., Kim K. E., Koh B. I., Nam D. H., Alitalo K., Kim I., Koh G. Y. (2010) CXCR4 signaling regulates metastasis of chemoresistant melanoma cells by a lymphatic metastatic niche. Cancer Res. 70, 10411–10421 [DOI] [PubMed] [Google Scholar]
- 10. O'Boyle G., Swidenbank I., Marshall H., Barker C. E., Armstrong J., White S. A., Fricker S. P., Plummer R., Wright M., Lovat P. E. (2013) Inhibition of CXCR4-CXCL12 chemotaxis in melanoma by AMD11070. Br. J. Cancer 108, 1634–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee C. H., Kakinuma T., Wang J., Zhang H., Palmer D. C., Restifo N. P., Hwang S. T. (2006) Sensitization of B16 tumor cells with a CXCR4 antagonist increases the efficacy of immunotherapy for established lung metastases. Mol. Cancer Ther. 5, 2592–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takekoshi T., Ziarek J. J., Volkman B. F., Hwang S. T. (2012) A locked, dimeric CXCL12 variant effectively inhibits pulmonary metastasis of CXCR4-expressing melanoma cells due to enhanced serum stability. Mol. Cancer Ther. 11, 2516–2525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Javelaud D., Mohammad K. S., McKenna C. R., Fournier P., Luciani F., Niewolna M., André J., Delmas V., Larue L., Guise T. A., Mauviel A. (2007) Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res. 67, 2317–2324 [DOI] [PubMed] [Google Scholar]
- 14. Kubic J. D., Mascarenhas J. B., Iizuka T., Wolfgeher D., Lang D. (2012) GSK-3 promotes cell survival, growth, and PAX3 levels in human melanoma cells. Mol. Cancer Res. 10, 1065–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mascarenhas J. B., Littlejohn E. L., Wolsky R. J., Young K. P., Nelson M., Salgia R., Lang D. (2010) PAX3 and SOX10 activate MET receptor expression in melanoma. Pigment Cell Melanoma Res. 23, 225–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Plummer R. S., Shea C. R., Nelson M., Powell S. K., Freeman D. M., Dan C. P., Lang D. (2008) PAX3 expression in primary melanomas and nevi. Mod. Pathol. 21, 525–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scholl F. A., Kamarashev J., Murmann O. V., Geertsen R., Dummer R., Schäfer B. W. (2001) PAX3 is expressed in human melanomas and contributes to tumor cell survival. Cancer Res. 61, 823–826 [PubMed] [Google Scholar]
- 18. Bonvin E., Falletta P., Shaw H., Delmas V., Goding C. R. (2012) A phosphatidylinositol 3-kinase-Pax3 axis regulates Brn-2 expression in melanoma. Mol. Cell. Biol. 32, 4674–4683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu F., Cao J., Lv J., Dong L., Pier E., Xu G. X., Wang R. A., Xu Z., Goding C., Cui R. (2013) TBX2 expression is regulated by PAX3 in the melanocyte lineage. Pigment Cell Melanoma Res. 26, 67–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chalepakis G., Jones F. S., Edelman G. M., Gruss P. (1994) Pax-3 contains domains for transcription activation and transcription inhibition. Proc. Natl. Acad. Sci. U.S.A. 91, 12745–12749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kubic J. D., Little E. C., Lui J. W., Iizuka T., Lang D. (2014) PAX3 and ETS1 synergistically activate MET expression in melanoma cells. Oncogene 10.1038/onc.2014.420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kubic J. D., Young K. P., Plummer R. S., Ludvik A. E., Lang D. (2008) Pigmentation PAX-ways: the role of Pax3 in melanogenesis, melanocyte stem cell maintenance, and disease. Pigment Cell Melanoma Res. 21, 627–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lang D., Epstein J. A. (2003) Sox10 and Pax3 physically interact to mediate activation of a conserved c-RET enhancer. Hum. Mol. Genet. 12, 937–945 [DOI] [PubMed] [Google Scholar]
- 24. Abel E. V., Aplin A. E. (2010) FOXD3 is a mutant B-RAF-regulated inhibitor of G1-S progression in melanoma cells. Cancer Res. 70, 2891–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Basile K. J., Abel E. V., Aplin A. E. (2012) Adaptive upregulation of FOXD3 and resistance to PLX4032/4720-induced cell death in mutant B-RAF melanoma cells. Oncogene 31, 2471–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Katiyar P., Aplin A. E. (2011) FOXD3 regulates migration properties and Rnd3 expression in melanoma cells. Mol. Cancer Res. 9, 545–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sutton J., Costa R., Klug M., Field L., Xu D., Largaespada D. A., Fletcher C. F., Jenkins N. A., Copeland N. G., Klemsz M., Hromas R. (1996) Genesis, a winged helix transcriptional repressor with expression restricted to embryonic stem cells. J. Biol. Chem. 271, 23126–23133 [DOI] [PubMed] [Google Scholar]
- 28. Hayashi H., Kume T. (2008) Forkhead transcription factors regulate expression of the chemokine receptor CXCR4 in endothelial cells and CXCL12-induced cell migration. Biochem. Biophys. Res. Commun. 367, 584–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li D., Yan D., Liu W., Li M., Yu J., Li Y., Qu Z., Ruan Q. (2011) Foxc2 overexpression enhances benefit of endothelial progenitor cells for inhibiting neointimal formation by promoting CXCR4-dependent homing. J. Vasc. Surg. 53, 1668–1678 [DOI] [PubMed] [Google Scholar]
- 30. Fredericks W. J., Galili N., Mukhopadhyay S., Rovera G., Bennicelli J., Barr F. G., Rauscher F. J. 3rd. (1995) The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol. Cell. Biol. 15, 1522–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Galili N., Davis R. J., Fredericks W. J., Mukhopadhyay S., Rauscher F. J. 3rd, Emanuel B. S., Rovera G., Barr F. G. (1993) Fusion of a forkhead domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 5, 230–235 [DOI] [PubMed] [Google Scholar]
- 32. Tarnowski M., Grymula K., Reca R., Jankowski K., Maksym R., Tarnowska J., Przybylski G., Barr F. G., Kucia M., Ratajczak M. Z. (2010) Regulation of expression of stromal-derived factor-1 receptors: CXCR4 and CXCR7 in human rhabdomyosarcomas. Mol. Cancer Res. 8, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tomescu O., Xia S. J., Strezlecki D., Bennicelli J. L., Ginsberg J., Pawel B., Barr F. G. (2004) Inducible short-term and stable long-term cell culture systems reveal that the PAX3-FKHR fusion oncoprotein regulates CXCR4, PAX3, and PAX7 expression. Lab. Invest. 84, 1060–1070 [DOI] [PubMed] [Google Scholar]
- 34. Nelms B. L., Pfaltzgraff E. R., Labosky P. A. (2011) Functional interaction between Foxd3 and Pax3 in cardiac neural crest development. Genesis 49, 10–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thomas A. J., Erickson C. A. (2009) FOXD3 regulates the lineage switch between neural crest-derived glial cells and pigment cells by repressing MITF through a non-canonical mechanism. Development 136, 1849–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wilson D., Sheng G., Lecuit T., Dostatni N., Desplan C. (1993) Cooperative dimerization of paired class homeo domains on DNA. Genes Dev. 7, 2120–2134 [DOI] [PubMed] [Google Scholar]
- 37. Chalepakis G., Goulding M., Read A., Strachan T., Gruss P. (1994) Molecular basis of splotch and Waardenburg Pax-3 mutations. Proc. Natl. Acad. Sci. U.S.A. 91, 3685–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chalepakis G., Gruss P. (1995) Identification of DNA recognition sequences for the Pax3 paired domain. Gene 162, 267–270 [DOI] [PubMed] [Google Scholar]
- 39. Epstein J. A., Shapiro D. N., Cheng J., Lam P. Y., Maas R. L. (1996) Pax3 modulates expression of the c-Met receptor during limb muscle development. Proc. Natl. Acad. Sci. U.S.A. 93, 4213–4218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jun S., Desplan C. (1996) Cooperative interactions between paired domain and homeodomain. Development 122, 2639–2650 [DOI] [PubMed] [Google Scholar]
- 41. Overdier D. G., Porcella A., Costa R. H. (1994) The DNA-binding specificity of the hepatocyte nuclear factor 3/forkhead domain is influenced by amino-acid residues adjacent to the recognition helix. Mol. Cell. Biol. 14, 2755–2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Caruz A., Samsom M., Alonso J. M., Alcami J., Baleux F., Virelizier J. L., Parmentier M., Arenzana-Seisdedos F. (1998) Genomic organization and promoter characterization of human CXCR4 gene. FEBS Lett. 426, 271–278 [DOI] [PubMed] [Google Scholar]
- 43. Labosky P. A., Kaestner K. H. (1998) The winged helix transcription factor Hfh2 is expressed in neural crest and spinal cord during mouse development. Mech. Dev. 76, 185–190 [DOI] [PubMed] [Google Scholar]
- 44. Zhu Z. B., Makhija S. K., Lu B., Wang M., Kaliberova L., Liu B., Rivera A. A., Nettelbeck D. M., Mahasreshti P. J., Leath C. A. 3rd, Yamamoto M., Yamaoto M., Alvarez R. D., Curiel D. T. (2004) Transcriptional targeting of adenoviral vector through the CXCR4 tumor-specific promoter. Gene Ther. 11, 645–648 [DOI] [PubMed] [Google Scholar]
- 45. Wegner S. A., Ehrenberg P. K., Chang G., Dayhoff D. E., Sleeker A. L., Michael N. L. (1998) Genomic organization and functional characterization of the chemokine receptor CXCR4, a major entry co-receptor for human immunodeficiency virus type 1. J. Biol. Chem. 273, 4754–4760 [DOI] [PubMed] [Google Scholar]
- 46. Gupta S. K., Pillarisetti K. (1999) Cutting edge: CXCR4-Lo: molecular cloning and functional expression of a novel human CXCR4 splice variant. J. Immunol. 163, 2368–2372 [PubMed] [Google Scholar]
- 47. Duquenne C., Psomas C., Gimenez S., Guigues A., Carles M. J., Barbuat C., Lavigne J. P., Sotto A., Reynes J., Guglielmi P., Mettling C., François V., Corbeau P. (2014) The two human CXCR4 isoforms display different HIV receptor activities: consequences for the emergence of X4 strains. J. Immunol. 193, 4188–4194 [DOI] [PubMed] [Google Scholar]
- 48. Sloane A. J., Raso V., Dimitrov D. S., Xiao X., Deo S., Muljadi N., Restuccia D., Turville S., Kearney C., Broder C. C., Zoellner H., Cunningham A. L., Bendall L., Lynch G. W. (2005) Marked structural and functional heterogeneity in CXCR4: separation of HIV-1 and SDF-1α responses. Immunol. Cell Biol. 83, 129–143 [DOI] [PubMed] [Google Scholar]
- 49. Vasyutina E., Stebler J., Brand-Saberi B., Schulz S., Raz E., Birchmeier C. (2005) CXCR4 and Gab1 cooperate to control the development of migrating muscle progenitor cells. Genes Dev. 19, 2187–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sasai N., Mizuseki K., Sasai Y. (2001) Requirement of FoxD3-class signaling for neural crest determination in Xenopus. Development 128, 2525–2536 [DOI] [PubMed] [Google Scholar]
- 51. He S., Li C. G., Slobbe L., Glover A., Marshall E., Baguley B. C., Eccles M. R. (2011) PAX3 knockdown in metastatic melanoma cell lines does not reduce MITF expression. Melanoma Res. 21, 24–34 [DOI] [PubMed] [Google Scholar]
- 52. Izraely S., Klein A., Sagi-Assif O., Meshel T., Tsarfaty G., Hoon D. S., Witz I. P. (2010) Chemokine-chemokine receptor axes in melanoma brain metastasis. Immunol. Lett. 130, 107–114 [DOI] [PubMed] [Google Scholar]
- 53. Widmer D. S., Cheng P. F., Eichhoff O. M., Belloni B. C., Zipser M. C., Schlegel N. C., Javelaud D., Mauviel A., Dummer R., Hoek K. S. (2012) Systematic classification of melanoma cells by phenotype-specific gene expression mapping. Pigment Cell Melanoma Res. 25, 343–353 [DOI] [PubMed] [Google Scholar]
- 54. Liedtke D., Erhard I., Abe K., Furutani-Seiki M., Kondoh H., Schartl M. (2014) Xmrk-induced melanoma progression is affected by Sdf1 signals through Cxcr7. Pigment Cell Melanoma Res. 27, 221–233 [DOI] [PubMed] [Google Scholar]
- 55. Lee E., Han J., Kim K., Choi H., Cho E. G., Lee T. R. (2013) CXCR7 mediates SDF1-induced melanocyte migration. Pigment Cell Melanoma Res. 26, 58–66 [DOI] [PubMed] [Google Scholar]
- 56. Ueno Y., Sakurai H., Tsunoda S., Choo M. K., Matsuo M., Koizumi K., Saiki I. (2008) Heregulin-induced activation of ErbB3 by EGFR tyrosine kinase activity promotes tumor growth and metastasis in melanoma cells. Int. J. Cancer 123, 340–347 [DOI] [PubMed] [Google Scholar]