

Background: Flavin-based electron bifurcation is a vital process in microbial energy metabolism.
Results: The NfnAB complex structure determines the positions of the prosthetic groups and the substrates.
Conclusion: The environment of the central FAD and its distance to the next redox centers of the two electron routes control electron bifurcation.
Significance: The first complete structure of a flavin-based electron bifurcating enzyme provides insights into this ancient catalytic process.
Keywords: crystal structure, energy metabolism, enzyme catalysis, flavin, iron-sulfur protein, NADH-dependent reduced ferredoxin-NADP oxidoreductase, electron bifurcation
Abstract
NADH-dependent reduced ferredoxin:NADP oxidoreductase (NfnAB) is found in the cytoplasm of various anaerobic bacteria and archaea. The enzyme reversibly catalyzes the endergonic reduction of ferredoxin with NADPH driven by the exergonic transhydrogenation from NADPH onto NAD+. Coupling is most probably accomplished via the mechanism of flavin-based electron bifurcation. To understand this process on a structural basis, we heterologously produced the NfnAB complex of Thermotoga maritima in Escherichia coli, provided kinetic evidence for its bifurcating behavior, and determined its x-ray structure in the absence and presence of NADH. The structure of NfnAB reveals an electron transfer route including the FAD (a-FAD), the [2Fe-2S] cluster of NfnA and the FAD (b-FAD), and the two [4Fe-4S] clusters of NfnB. Ferredoxin is presumably docked onto NfnB close to the [4Fe-4S] cluster distal to b-FAD. NAD(H) binds to a-FAD and NADP(H) consequently to b-FAD, which is positioned in the center of the NfnAB complex and the site of electron bifurcation. Arg187 is hydrogen-bonded to N5 and O4 of the bifurcating b-FAD and might play a key role in adjusting a low redox potential of the FADH•/FAD pair required for ferredoxin reduction. A mechanism of FAD-coupled electron bifurcation by NfnAB is proposed.
Introduction
In 2008, a novel mechanism of energy coupling was discovered, namely flavin-based electron bifurcation that changed our views of the energy metabolism of many anaerobic microorganisms, both bacteria and archaea (1–3). The first example was the coupling of the endergonic reduction of ferredoxin (Fdox) with NADH to the exergonic reduction of crotonyl-CoA with NADH via the cytoplasmic butyryl-CoA dehydrogenase/electron transfer flavoprotein (Bcd/EtfAB)6 complex from Clostridium kluyveri and other butyric acid forming Gram-positive bacteria (4). The Bcd/EtfAB complex contains per subunit one FAD and no other cofactors. The crystal structure of the EtfAB subcomplex was recently solved, and a mechanism of flavin-based electron bifurcation was proposed (5). The proposal is based on the property of free flavins (F) to have three redox potentials: Eo′ (F/FH−) = −219 mV; Eo′ (FH•/FH−) = −124 mV; and Eo′ (FH•/F−) = −314 mV (6). Upon downhill one-electron transfer from the fully reduced bifurcating FAD bound to EtfB to the oxidized FAD bound to EtfA, a flavin radical FADH• at EtfB is generated that has a redox potential sufficiently negative to drive the reduction of Fdox (Eo′ = −400 mV).
Since 2008, many other electron-bifurcating enzyme complexes have been discovered in the cytoplasm of anaerobic bacteria and archaea (7). Among them are caffeyl-CoA reductase (8) and lactate dehydrogenase (9) in Acetobacterium woodii, [FeFe]-hydrogenases, both NAD- and NADP-specific ones, in Thermotoga maritima (10), A. woodii (11), Moorella thermoacetica (12), and Clostridium autoethanogenum (13), formate dehydrogenase in Clostridium acidurici (14), heterodisulfide reductase in methanogenic archaea (2, 15–18), and electron-bifurcating transhydrogenase in C. kluyveri (19) and M. thermoacetica (20). The electron-bifurcating enzyme complexes known to date have in common that at least one of their subunits contains an FAD or FMN and that they are all ferredoxin-dependent, shown as follows,
 |
with the redox potential (E) of the low potential 2e donor lying between that of ferredoxin (Fd) and that of the high potential 2e acceptor.
Dependent on the redox potential difference between the low potential 2e donor (H2, formate, NAD(P)H) and high potential 2e acceptor (crotonyl-CoA, caffeyl-CoA, pyruvate, CoM-S-S-CoB, NAD+), Reaction 1 is more or less reversible. In case of reversibility, the back reaction is electron-confurcating rather than electron-bifurcating (7).
As mentioned above, the Bcd/EtfAB complex contains only FAD as prosthetic group, and therefore only FAD can be the site of electron bifurcation. All other electron-bifurcating complexes analyzed to date also contain, in addition to FAD or FMN, iron-sulfur clusters that, however, can accept and donate electrons only one at a time and are therefore not likely sites of electron bifurcation. In the case of electron-bifurcating hydrogenases and formate dehydrogenases, the site of H2 activation ([FeFe] center) and that of formate activation (molybdopterin center) function as reversible 2e/1e switches and could therefore principally also be sites of electron bifurcation (21).
The electron-bifurcating enzyme complexes mentioned above are all composed of at least three different subunits with one exception: the electron-bifurcating transhydrogenase from C. kluyveri and M. thermoacetica is only a heterodimer of subunit NfnA (∼32 kDa) and subunit NfnB (∼50 kDa). The complex contains two FAD, one [2Fe-2S] cluster, and two [4Fe-4S] clusters. NfnA shows sequence similarities to plant ferredoxin:NADP+ reductase (Fnr) and bacterial dihydroorotate dehydrogenase (Dodh) from bacteria and NfnB to the β-subunit of bacterial NADPH-dependent glutamate synthase (Gls) (22) and eukaryotic dihydropyrimidine dehydrogenase (Dpdh) (23). The transhydrogenase complex catalyzes the reversible reduction of ferredoxin and NAD+ with 2 NADPH (Reaction 2). In vivo, its function is rather to catalyze the back reaction, the NADH-dependent ferredoxin:NADP reductase reaction (19, 20). In both directions, the bifurcation process is based on the low NADH/NAD+ ratio (1/30) and the high NADPH/NADP+ ratio (50/1) in the cell (24).
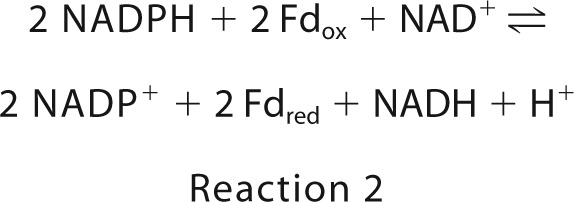 |
NfnAB appears to be an ideal model system to explore the mechanism of flavin-based electron bifurcation because it has the simplest structure. Therefore, we overproduced the NfnAB complex from T. maritima and characterized it biochemically, kinetically, and structurally to acquire insights in its mode of action. NfnAB is the first flavin-based electron-bifurcating enzyme whose complete x-ray structure is characterized.
Experimental Procedures
Ferredoxin and ferredoxin-dependent [FeFe]-hydrogenase used in the experiments were purified from Clostridium pasteurianum (25, 26) and pyruvate:ferredoxin oxidoreductase from M. thermoacetica (27).
Heterologous Expression of nfnAB Genes and Purification of NfnAB
The nfnAB genes from T. maritima genomic DNA were amplified with the forward primer 5′-GACGACGACAAGATGGGGGGGACGGCTTTG-3′ and the reverse primer 5′-GAGGAGAAGCCCGGTCACTTTTGCCACGGATTCCATTC-3′ (the inserted sequences specific for ligation-independent cloning are underlined). After purification with MinElute PCR purification kit, the blunt PCR product was treated with T4 DNA polymerase in the presence of dATP to generate specific vector-compatible overhangs. Then it was annealed into the linear pET-51b(+)Ek/LIC vector (Merck) and thereby 5′-tagged with a Strep cassette. The vector was transformed into Escherichia coli C41 (DE3), which already harbored pCodonPlus and pRKISC vectors successfully used for the production of iron-sulfur cluster proteins (28, 29).
Before inoculation, the medium was supplemented with carbenicillin (50 mg liter−1), chloramphenicol (25 mg liter−1), and tetracycline (10 mg liter−1) to maintain the plasmids and with cysteine (0.12 g liter−1), ferrous sulfate (0.1 g liter−1), ferric citrate (0.1 g liter−1), and ferric ammonium citrate (0.1 g liter−1) for the enhancement of iron-sulfur cluster synthesis. The recombinant E. coli C41 (DE3) cells were aerobically grown in 2 liters of Terrific broth (TB medium), and after nfnAB expression induced by isopropyl β-d-thiogalactopyranoside, the solution was continuously stirred at a speed of 250 rpm for 20 h at 37 °C under aerobic conditions and further for 2 h under anaerobic conditions. After incubation for further 20 h at 4 °C, the recombinant cells were harvested by centrifugation and washed with anaerobic 100 mm Tris-HCl, pH 7.5. The obtained cell pellet was stored at −80 °C under N2.
All steps of purification were performed at room temperature under strictly anaerobic conditions in a Coy type B vinyl anaerobic chamber (Coy, Grass Lake, MI) that was filled with a mixture of N2 and H2 (95%:5%) and contained a palladium catalyst for continuous O2 reduction with H2. All buffers were boiled to remove O2 and then evacuated and filled with N2 under slight overpressure. The E. coli cell pellet (10 g of wet cells) was resuspended in 100 mm Tris-HCl, pH 7.5, containing 2 mm DTT and 10 μm FAD. The cells were disrupted by sonication, and the generated cell debris was removed by ultracentrifugation. The supernatant was then heated for 30 min at 80 °C (growth temperature optimum of T. maritima). After removing the denatured protein by centrifugation at 10,000 × g at 25 °C for 10 min, the supernatant was applied to a 5-ml Strep-Tactin Superflow column (IBA, Goettingen, Germany), which was equilibrated with 20 ml of buffer A (100 mm Tris-HCl, 150 mm NaCl, 2 mm DTT, 10 μm FAD, pH 7.5). Then the column was washed with 35 ml of buffer A. The recombinant protein was eluted with 15 ml of buffer A supplemented with 2.5 mm desthiobiotin. The fractions containing NfnAB activity were pooled and concentrated by ultrafiltration with 50-kDa cut-off Amicon filters. Subsequently, the enzyme was subjected to iron-sulfur cluster reconstitution in 100 mm Tris-HCl, pH 7.5, containing 8 mm DTT, 10 μm FAD, 2 mm cysteine, and 1.5 mm FeSO4 at room temperature for 1 h under strictly anaerobic conditions. After centrifugation at 52,000 × g at 4 °C for 30 min, the supernatant with the reconstituted enzyme was concentrated via 50-kDa cut-off Amicon filters to a volume of ∼1 ml and then loaded on a HiPrep 16/60 Sephacryl S-200 high resolution column equilibrated with 100 mm Tris-HCl, pH 7.5, containing 500 mm NaCl, 2 mm DTT, and 10 μm FAD. NfnAB activity was found in two peaks (Fig. 1A). Peak A eluted first was smaller and not always detectable. The fractions of peak B that eluted last were pooled, concentrated via 50-kDa cut-off Amicon filters, and washed with 10 mm MOPS-KOH, pH 7.0, 2 mm DTT, and 10 μm FAD. The purified enzyme was then concentrated to ∼25 mg/ml and stored at −80 °C under N2.
FIGURE 1.
Purification of the recombinant NfnAB from T. maritima. A, SDS-PAGE. Lane M, low molecular weight SDS marker (GE Healthcare); lane 1, cell extract of recombinant E. coli cells; lane 2, supernatant after heat treatment of 80 °C for 30 min; lane 3, elution fraction of Strep-Tactin column; lane 4, NfnAB after iron-sulfur cluster reconstitution in vitro; lane 5, concentrate of elution peak A from HiPrep Sephacryl S-200; lane 6, concentrate of elution peak B from HiPrep Sephacryl S-200. After purification with the Strep-Tactin column, the preparation still contained some contaminating proteins, which turned out to be NfnB proteolysis products as shown by tandem MALDI-TOF-TOF MS analysis. B, UV-visible absorption spectra of NfnAB (1.2 mg/ml) in 100 mm Tris-HCl, pH 7.5, before and after iron-sulfur reconstitution in vitro (black and red lines). 6.5 and 9.0 mol iron per mol NfnAB was determined, respectively.
Cofactor Content Determination
The flavin was identified by thin-layer chromatography on an RP-18 F254 aluminum sheet (Merck) using the supernatant of the denaturated protein. The thin-layer chromatography plate was developed with an aqueous solution containing 85% (v/v) 5 mm ammonium formate and 15% (v/v) methanol. The presence of flavin was determined under UV light. FAD was quantified by UV-visible spectroscopically quantified with ϵ450 nm = 11.3 mm−1 cm−1 (30). The iron content of the enzyme was measured colorimetrically with 3-(2-pyridyl)-5,6-bis (5-sulfo-2-furyl)-1,2,4-triazinedisodium trihydrate (Ferene) using Mohr's salt as standard (31, 32).
Activity Assays
The assays were performed at 45 °C in 1.5-ml anaerobic cuvettes closed with a rubber stopper and filled with 0.8-ml reaction mixtures and 0.7 ml of 100% N2 or 100% H2 at 1.2 × 105 Pa as gas phase. The reaction mixtures contained 100 mm MOPS-KOH, pH 7.0, 10 mm 2-mercaptoethanol, and 12 μm FAD as basal ingredients. The reactions were followed photometrically at the wavelength indicated. Measured activities (see Table 1) remained essentially constant for several days at 4 °C under strictly anaerobic conditions and for more than 2 weeks when frozen at −20 or −80 °C. (For estimated redox potentials under physiological conditions of the electron donors and acceptors, see “Results and Discussion”).
TABLE 1.
Reactions catalyzed by NfnAB from T. maritima
| Reaction | Apparent Vmax at 45 °C | Apparent Km |
|---|---|---|
| units/mg | mm | |
| NADP+ reduction with | ||
| Fdred50%a + NADH | 4.8 | 0.2 (NADP+)e |
| 0.02 (NADH)f | ||
| Fdred50%a | 0.05 | |
| NADH | 0.04 | |
| NADH + Fdoxb | 0.04 | |
| Fdox reduction with | ||
| NADPHc + NAD+ | 3.9 | 0.002 (NADPH)g |
| 0.24 (NAD+)h | ||
| NADPHc | 0.01 | |
| NAD+ reduction with | ||
| NADPHc + Fdoxb | 2.2 | 0.001 (NADPH)i |
| 1.0 (NAD+)j | ||
| Fdred100%d | 0.4 | |
| NADPHc + Fdred50%a | 1.8 | |
| NADPHc + Fdred100%d | 0.4 | |
| NADPHc | 0.34 | |
a Fdred50%-regenerating system (Fd, hydrogenase, and 100% H2 keeping Fd about 50% reduced).
b Fdox-regenerating system (Fd, hydrogenase, and 100% N2 keeping Fd to almost 100% oxidized).
c NADPH-regenerating system (NADP+, glucose-6-phosphate dehydrogenase, and glucose-6-phosphate keeping NADP to almost 100% reduced).
d Fdred100%-regenerating system (Fd, pyruvate, CoA, phosphotransacetylase, phosphate, thiamin pyrophosphate, pyruvate:ferredoxin oxidoreductase keeping Fd about 100% reduced).
e 0.5 mm NADH, 25 μm Fdred50%.
f 2 mm NADP+, 25 μm Fdred50%.
g 10 mm NAD+, 31 μm Fdox.
h 0.5 mm NADP+, 31 μm Fdox.
i 10 mm NAD+, 10 μm Fdox.
j 10 mm NAD+, 10 μm Fdox.
For the determination of the NADH-dependent NADP+ reduction with reduced ferredoxin, the basal reaction mixture was supplemented with 2 mm NADP+, 0.5 mm NADH, and a Fdred50% regenerating system composed of Fdox (25 μm), ferredoxin-dependent [FeFe]-hydrogenase (1 unit), and 100% H2 as a gas phase. The reaction was started by adding NfnAB, and NADP+ reduction was monitored at 380 nm (ϵ = 1.2 mm−1 cm−1). The NAD+-dependent Fdox reduction with NADPH was measured by supplementing the basal reaction mixture with 10 mm NAD+ and 30 μm Fdox and an NADPH-regenerating system composed of NADP+ (0.5 mm), glucose-6-phosphate (40 mm), and glucose-6-phosphate dehydrogenase from Baker's yeast (Sigma) (2 units). The gas phase was 100% N2. The reaction was started by adding of NfnAB, and Fdox reduction was monitored at 430 nm (ϵΔox-red ≈ 13.1 mm−1 cm−1). To record the ferredoxin-dependent NAD+ reduction with NADPH, the NADPH-regenerating system (see above), 10 mm NAD+, and the Fdox-regenerating system composed of ferredoxin (10 μm) and ferredoxin-dependent [FeFe]-hydrogenase (2 units) was added to the basal reaction mixture. The gas phase was 100% N2. The reaction was started by adding of NfnAB, and NAD+ reduction was monitored at 380 nm (ϵ = 1.2 mm−1 cm−1). In some experiments, a Fdred100%-regenerating system was employed that was composed of Fdox (10 μm), pyruvate (10 mm), CoA (1 mm), pyruvate:ferredoxin oxidoreductase from M. thermoacetica (6 units), thyamin pyrophosphate (1 mm), phosphate (100 mm), and phosphotransacetylase (from Sigma) (5 units).
Crystallization and Structure Determination
NfnAB was crystallized with the sitting drop method at room temperature inside an anaerobic chamber equipped with an OryxNano crystallization robot (Douglas Instruments Ltd.). Protein solution contained 17.5 mg/ml NfnAB, 10 mm MOPS-KOH, pH 7.0, 2 mm DTT, and 10 μm FAD. For screening experiments, the JBScreen Pentaerythritol, JBScreen Classic 1–10 and SaltRx kits were applied. The best crystallization conditions were listed in Table 2. X-ray diffraction data were collected at Beamline PXII at the Swiss Light Source (Villigen, Switzerland) using XDS (33) for data processing. Multiple wavelength anomalous dispersion data sets were measured at the iron absorption edge at a wavelength of 1.73 Å. The iron positions were identified with SHELXD (34), and the phases were calculated with SHARP (35) and improved by solvent flattening with SOLOMON (36). Coot was used for electron density analysis and model building (37). Several secondary structure elements were clearly visible in the experimental electron density, and the related subunits of Dodh from Lactococcus lactis (38) and Gls from Azospirillum brasilense (22) were fitted into the electron density of NfnA and NfnB, respectively. Automatic model building in Phenix (39) correctly positioned ∼50% of the residues of NfnA and NfnB into the electron density when incorporating the superimposed Dodh and Gls as reference models. The Rfree factor was 37%. Because refinement in space group P43212 did not converge, the data were reprocessed in P43, and partial twinning was introduced into refinement with Phenix. Partial twinning was also detectable with Phenix.xtriage (40). The temperature factors of several segments were higher than 100 Å2, and accurate fitting of the amino acids was not always possible, although the polypeptide chain could be traced except for segment C170-C182. Parameters describing the structure quality are listed in Table 2. The structure of the NfnAB-NADH complex was determined in the same crystal form after soaking the crystals with 5 mm NADH for 40 min. In addition to the highly occupied NADH in NfnA, one NfnB subunit of the asymmetric unit also contains a weakly occupied NADH. Its adenosine part is, however, highly disordered (B ∼ 100 Å2). Figs. 2–5 were generated with PYMOL (Schrödinger, LLC).
TABLE 2.
Crystallographic data for NfnAB from T. maritima
| NfnAB-phosphate | NfnAB-NADH | |
|---|---|---|
| Crystallization conditions | 0.2 m NH4H2PO4 | 0.225 m NH4H2PO45% (v/v) ethanol, 20% (v/v) glycerol |
| Soaking conditions | Soaked for 40 min in 5 mm NADH | |
| Cryo conditions | 30% (v/v) 1,2-propanediol 0.2 m NH4H2PO4 2 mm DTT, 10 μm FAD | 10% (v/v) 1,2-propanediol, 5% (v/v) ethanol, 20% (v/v) glycerol, 0.225 m NH4H2PO4, 2 mm DTT, 10 μm FAD |
| Data collection | ||
| Space group | P43 | P43 |
| Wavelength (Å) | 1.0 | 0.979 |
| Resolution range (Å) | 50.0–2.3 (2.4–2.3) | 50.0–2.4 (2.5–2.4) |
| Unit cell a, b, c (Å) | 81.5, 81.5, 311.8 | 81.4, 307.5 |
| Redundancy | 2.9 (2.2) | 5.2 (5.0) |
| Completeness (%) | 96.5 (86.2) | 99.1 (97.1) |
| Rsym (%) | 7.8 (64.0) | 11.3 (89.6) |
| I/σ(I) | 8.4 (1.4) | 14.2 (2.3) |
| Refinement statistics | ||
| Molecular asymmetric unit | 2 NfnAB | 2 NfnAB |
| No. of residues, cofactors, waters, phosphates, NADH | 1475, 10, 159, 2, / | 1454, 10, 131, 1, 1 |
| Rworking, Rfree (%) | 18.7, 21.9 | 20.0, 24.7 |
| Baverage (Å2) polypeptide, cofactor water, phosphate, NADH | 52.4, 34.8, 26.3, 59.6, / | 51.3, 42.7 30.6, 51.7, 45.1 |
| Segments with B factors of ∼100 Å2 | A138–A162, B130–132, B361–B374, C1–C6, C98–C117, C139–C150, C161–C163, C190–C207, C252–C257, D135–D137, D362–D371 | A138–A140, A160–A166, B126–B135, B360–B372, C1–C6, C14–C17, C99–C104, C112–C116, C163–C167, C268–C272, D236–D237, D361–D369 |
| Disordered segments | C170–C182 | C174–C179, C201–C205 |
| Root mean square deviation | ||
| Bond lengths (Å) | 0.008 | 0.005 |
| Bond angles (°) | 1.1 | 1.1 |
| Ramachandran plot | ||
| Favored (%) | 92.2 | 93.8 |
| Outliers (%) | 1.4 | 0.8 |
FIGURE 2.
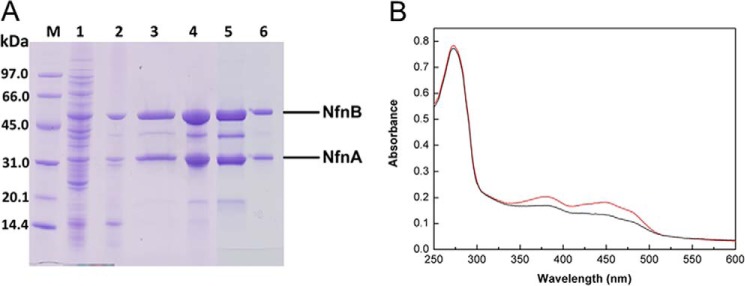
X-ray structure of the NfnAB complex. A, overall structure: NfnA is subdivided into the FAD and NAD domains and an iron-sulfur cluster binding extension (light blue, deep sky blue, and dark blue) and NfnB into the FAD, NADP, and iron-sulfur binding domains (green, dark green, and pale green). The iron-sulfur clusters and FAD are drawn as stick models. Yellow, carbon; red, oxygen; blue, nitrogen; gray, phosphorus; yellow, sulfur; brown, iron. B, the electron transfer chain. NfnAB is shown in a transparent molecular surface representation. A continuous electron transfer route (orange dotted line) is visible from a-FAD, via the [2Fe-2S] cluster, b-FAD, and the proximal and the distal [4Fe-4S] clusters. Ferredoxin binds at the bottom of the iron-sulfur domain of NfnB.
FIGURE 3.

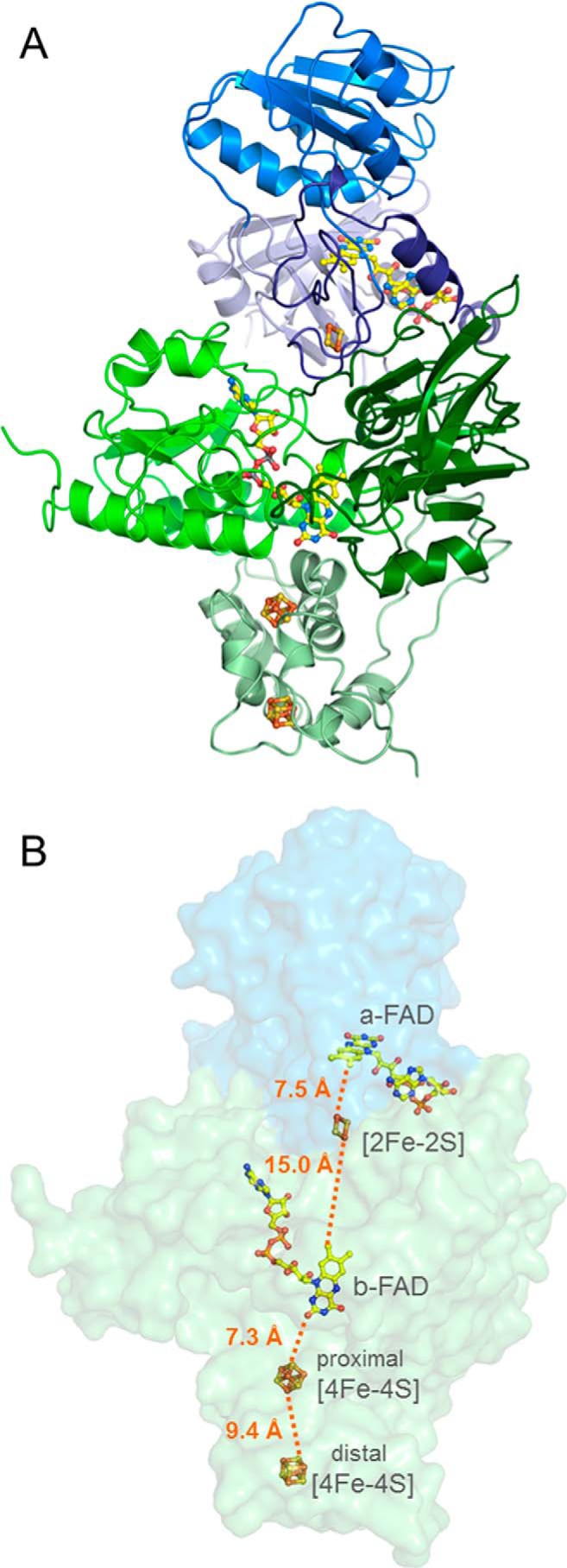
Binding of FAD and the iron-sulfur clusters in the NfnAB complex. A, a-FAD (yellow) is attached to the β-barrel domain of NfnA (deep sky blue) in a bent conformation, the shortest distance between its N6A and C9 being 5.0 Å (green dots). Hydrogen bonds are indicated with orange dashed lines. B, b-FAD is bound in an elongated conformation at the C-terminal end of the β-sheet of the FAD domain. Arg187 directly interacts with N5 and O4 of the isoalloxazine ring that stabilize, in particular, the oxidized state. C, the [2Fe-2S] cluster is bound to NfnA. D and E, the proximal and distal [4Fe-4S] clusters are located in NfnB. The [2Fe-2S] and proximal [4Fe-4S] clusters are coordinated by unusual aspartate and glutamate ligands, respectively.
FIGURE 4.
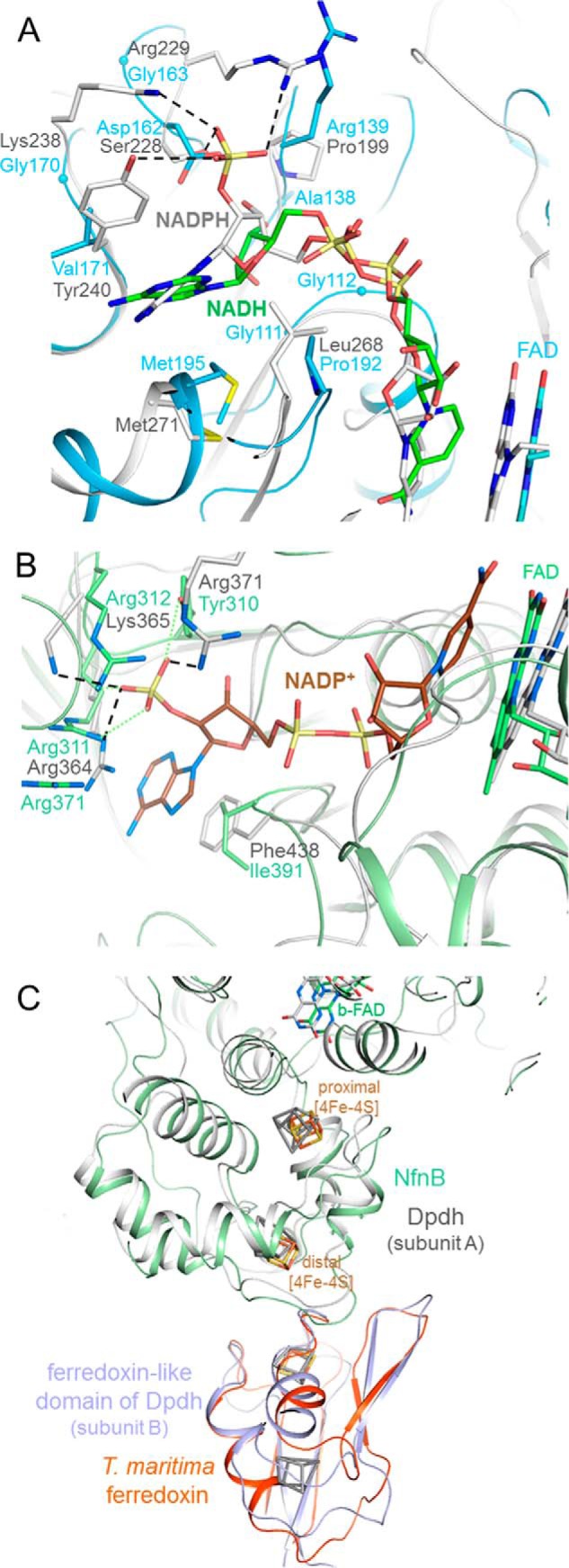
Substrate binding sites of the NfnAB complex. A, NADH (carbon in green) is bound to NfnA (deep sky blue) at the C-terminal side of the central β-sheet of the NAD domain. The different adenosine binding sites in NfnAB and Fnr are highlighted by superimposing the NfnAB-NADH and the Fnr-NADPH complex structures. A hypothetical 2-phosphate group of NADPH in NfnAB would sterically and electrostatically interfere with Asp162 and is not surrounded by positively charged residues as in Fnr. B, modeled NADP+ bound to NfnB. The superimposed NADP binding domains of the Dpdh-NADP+ complex (gray) and NfnAB (green) reveal a highly conserved site for binding NADP(H) (carbons in brown) and not NAD(H). C, binding of ferredoxin. Ferredoxin (orange-red) is modeled close to the distal [4Fe-4S] cluster of NfnB based on the structure of Dpdh, which contains both a NfnB-like and a ferredoxin-like domain (gray and blue). Its iron-sulfur clusters are drawn in gray.
FIGURE 5.

Mechanism of FAD-based electron bifurcation proposed for the NfnAB complex. The reaction cycle proceeds in two runs (top and bottom rows). In the bifurcating direction, b-FAD is reduced with NADPH, and b-FADH− subsequently donates its first electron to a-FAD and its second electron to Fdox. Therefore, a capability has to exist to block or conduct the downhill electron transfer (ET) from b-FADH• toward a-FADH•. The rearrangement of NfnA relative to NfnB would be a plausible model. The oxidation state of the iron-sulfur clusters in NfnAB is not indicated because of the instant change in electron delivery. In comparison with the Bcd/EtfAB complex, EtfB corresponds to NfnB carrying the bifurcating FAD, and Bcd corresponds to NfnA carrying the FAD for hydride transfer to the high potential 2e acceptor (crotonyl-CoA, NAD+). The varying distances between the bifurcating FAD and FAD of EtfA or the [2Fe-2S] cluster of NfnA, respectively, control the electron transfer route.
Results and Discussion
Attempts to obtain a crystal structure of heterologously produced NfnAB from C. kluyveri and from M. thermoacetica, in which this enzyme was first discovered, have failed. Crystallization of the two enzymes without a tag and tagged with His6 or Strep cassettes were tried. We have therefore screened heterologously produced NfnAB complexes from various organisms, both bacteria (Thermoanaerobacter tengcongensis and T. maritima) and archaea (Pyrococcus furiosus) and were finally successful with NfnAB from the hyperthermophile T. maritima.
Molecular Mass and Cofactor Content of the NfnAB Complex from T. maritima
Expression of the genes from T. maritima in E. coli with a Strep tag at its N terminus led to a soluble and intact NfnAB complex after purification (Fig. 1). Purification of NfnAB under strictly anaerobic conditions from 10 g (wet mass) of recombinant E. coli cells resulted in ∼8 mg of enzyme with a specific activity of ∼4 units/mg at 45 °C (Fig. 1A and Table 1). Via gel filtration chromatography on HiPrep Sephacryl S-200, NfnAB was separated in two peaks. The smaller peak 1 and the larger peak 2 elute at apparent molecular masses of ∼170 and 80–90 kDa, respectively, corresponding to a Nfn(AB)2 heterotetramer and NfnAB heterodimer. The specific activity of the enzyme from peak B was 35% higher than that from peak A, contained much less NfnB proteolysis products, and was therefore used for all further studies.
The flavin content was calculated by UV-visible absorbance spectroscopy to be approximately two FAD per NfnAB heterodimer, which fits well to two flavin binding sites detectable in the protein sequence. From the 10 irons predicted in NfnAB, 6–8 were detectable before and ∼9 were detectable after in vitro iron-sulfur cluster reconstitution, which also led to an increase in absorbance of the enzyme between 350 and 500 nm (Fig. 1B) and a slight increase in the specific activity.
Catalytic Properties of T. maritima NfnAB
The enzymatic activity of NfnAB of T. maritima was routinely measured at 45 °C in 100 mm MOPS-KOH at the pH optimum of 7.0 by following the NAD+-dependent reduction of ferredoxin with NADPH (regenerating system). Under these conditions, the specific activity of the most active fractions was near 4 units/mg, which is rather low (Table 1). Measurements at 80 °C, the optimal growth temperature of T. maritima, resulted in a 10 times higher specific activity of NAD+-dependent ferredoxin reduction, which is in the order of the specific activities measured for NfnAB complexes from other organisms (19, 20). NfnAB was found to catalyze the NADH-dependent reduction of NADP+ with reduced ferredoxin (Fdred), the NAD+-dependent reduction of Fdox with NADPH, and the Fdox-dependent reduction of NAD+ with NADPH (Table 1). These findings indicate a complete reversibility of the reaction. The apparent Km value of NADPH is lower than that of NADH, and that of NADP+ is lower than that of NAD+. Notably, the NADP+-dependent reduction of NAD+ with Fdred is not feasible.
Normally, Fdred was continuously regenerated via reduction with 100% H2 catalyzed by ferredoxin-dependent [FeFe]-hydrogenase from C. pasteurianum. At pH 7, the redox potential of the C. pasteurianum ferredoxin is near −400 mV, and that of the H+/H2 couple is −414 mV. Therefore, in the experiments, the ferredoxin was only reduced to ∼50%, the rest remained oxidized. This is indicated in Table 1 by Fdred50%. In some experiments, reduced ferredoxin was regenerated in a system composed of ferredoxin, pyruvate, CoA, pyruvate:ferredoxin oxidoreductase from M. thermoacetica, phosphotransacetylase, and phosphate (100 mm). The redox potential of the acetyl-CoA + CO2/pyruvate couple is near −500 mV. Therefore, ferredoxin was reduced to almost 100% indicated in Table 1 by Fdred100%. In addition, NAD+ or NADP+ cannot be reduced by Fdred without a high potential electron donor, although this is thermodynamically feasible and performed by plant Fnr.
The X-ray Structure
The crystal structure of the NfnAB complex was determined by the multiple anomalous dispersion method using the irons of the iron-sulfur clusters as anomalous scatterers and refined to R/Rfree factors of 18.7/21.9% at 2.3 Å resolution (Table 2). In the crystalline state, the enzyme is present as a NfnAB heterodimer (Fig. 2), which is most likely also the oligomeric state in solution despite the ambiguous gel filtration profile. In agreement with the biochemically determined FAD and iron contents of the T. maritima (see above) and also of the C. kluyveri and M. thermoacetica enzymes (19, 20), NfnA contains one FAD (termed a-FAD) and one [2Fe-2S] cluster and NfnB one FAD (b-FAD) and two [4Fe-4S] clusters (Fig. 3).
NfnA architecturally belongs to the Fnr family (41), the most related structurally known member being the L. lactis Dodh with a root mean square deviation of 2.0 Å by a sequence identity of 24% (38). Accordingly, NfnA is composed of an FAD-binding six-stranded antiparallel β-barrel domain (1–97), an NAD(P) binding α/β domain (98–216), and a C-terminal extension (217–277) subdivided into a segment carrying the [2Fe-2S] cluster and a long C-terminal α-helix (Fig. 2A). The planar isoalloxazine ring of a-FAD is sandwiched between the FAD and NAD(P) domains in the canonical manner; the si-face is attached to strand 48:54 of the β-barrel, and the re-face points to the NAD(P) binding site (Fig. 3A). In high agreement with other family members, N5 of FAD is hydrogen-bonded to Thr53-OG and -NH, O2 is hydrogen-bonded to Lys69-NH, and N1 has no contact to the polypeptide. Therefore, the redox potential of FAD from Fnr and Dodh of −342 mV (E′o(FAD/FADH•) = −350 mV; E′o(FADH•/FADH−) = −335 mV; pH 7, 10 °C) (42) and −304 mV (E′o(FAD/FADH•) = −312 mV; E′o(FADH•/FADH−) = −297 mV) (43) might be approximate guidelines for NfnAB. For both enzymes, a neutral blue semiquinone was detectable. Fe1 and Fe2 of the [2Fe-2S] cluster are ligated to Cys228 and Cys240, as well as to Cys225 and Asp220, respectively (Fig. 3C). The aspartate might implicate a lower redox potential than Eo′ = −212 mV measured for the [2Fe-2S] cluster of Dodh, which contains a cysteine at the equivalent position (43).
NfnB is a member of a disulfide oxidoreductase superfamily, and the structurally most related representatives are domains I–III of subunit A of pig Dpdh (23) and the β-subunit of A. brasilense Gls (22) with root mean square deviations of 2.1 and 2.3 Å and sequence identities of 32 and 35%, respectively. Accordingly, NfnB is composed of an helical domain (positions 1–130) containing two [4Fe-4S] clusters and two Rossmann-type nucleotide binding domains for FAD(131–237, 393–468) and NAD(P)(238–392) (Fig. 2A) each basically built up of a five-stranded α/β structures. The binding mode of b-FAD is essentially conserved in NfnAB, Dpdh, and Gls. The planar isoalloxazine ring, placed in a pocket between all three domains, is flanked from the si-face by the side chains of Val89, Val178, Ile183, and Val441 (Fig. 3B). The NAD(P) binding site lies at the re-face. Interestingly, the guanidinium group of Arg187 is positioned roughly parallel to the isoalloxazine ring and points toward the pyrazine and pyrimidine rings. Its guanidino group fixed by a bidentate salt bridge to Asp293 interacts with O4 and N5 of FAD by its NH2 and NH1 groups, respectively. N10 and O2 are hydrogen-bonded to Val441-NH, the N-terminal residue of helix 441:460 (Fig. 3B). The redox potential of the equivalent FAD of Gls was determined to be −300 mV (44). A semiquinone was not detectable.
The proximal [4Fe-4S] cluster is embedded into a rather hydrophilic pocket, and the irons are ligated to three cysteines (Cys51, Cys90, and Cys96) and Glu117 (Fig. 3D); the glutamate is conserved in Gls and replaced by a glutamine in Dpdh. A glutamate carboxylate as iron ligand is also reported for a few other [4Fe-4S] clusters (45) and should lower the redox potential compared with a cysteine thiolate ligand. The distal [4Fe-4S] cluster is flanked by predominantly hydrophobic residues and ligated to four cysteines (Fig. 3E). Although located close to the surface of the iron-sulfur cluster domain, it is completely shielded from bulk solvent. The redox potentials of both [4Fe-4S] clusters investigated in Gls and Dpdh are very low (≤−400 mV) (44, 46), and one of them could not be determined in Dpdh.
The Binding Site of the Substrates NADH, NADP+, and Ferredoxin
Both NfnA and NfnB contain a nicotinamide coenzyme binding site, but the respective location of NAD(H) and NADP(H) has to be identified. An x-ray structure of an NfnAB-NADH complex at 2.4 Å resolution clearly determined NfnA as binding site for NADH (Fig. 4A and Table 2). The nicotinamide ring of NADH is sandwiched between the isoalloxazine and Gly114, Gly191, and Pro192. Because the distances of 3.8 Å between their C4 and N5 atoms are relatively long, and the angle of ∼45° between the nicotinamide and isoalloxazine ring significantly deviates from planar, the hydride transfer geometry appears not to be optimally adjusted. However, the corresponding rings in the Fnr-NADPH complex are also tilted 30° against each other.
The structural basis for the different NAD(H) and NADP(H) specificity of NfnA and Fnr (47), respectively, can be extracted from the NfnAB-NADH and Fnr-NADPH complex structure (Fig. 4A). The adenosine ribose group in NfnA is oriented in a manner that its hydroxy groups point toward strands 106:111 and 131:137, and their subsequent loops thereby interact with the main chain of Gly111, Gly112, Gly138, and Arg139. In comparison, the ribose-2′-phosphate of NADP in Fnr is rotated ∼90°, and the phosphate group is hydrogen-bonded to Ser228, Arg229, Lys238, and Tyr240. In NfnA, These residues are replaced by Asp162, Gly163, Val170, and Val171, which are unsuitable for phosphate group binding. The different ribose or ribose-phosphate arrangement in NfnA and Fnr also implies a rotation of ∼90° of their adenine rings relative to each other, indicating a redesign of the entire adenosine binding site in the absence of the extra phosphate group (Fig. 4A). Because of the described similarity between NfnAB and Dodh, the NADH binding mode of NfnAB also provides a reasonable model for Dodh that was not structurally characterized in complex with NADH yet.
The unambiguous identification of the NADH binding site in NfnA defines NfnB as the binding site for NADP+. This indirect identification is corroborated by the finding that the related Dpdh is also NADP dependent, and a superposition of NfnB and the Dpdh-NADP+ complex (48) indicates only a few collisions but several favorable and conserved side chain interactions between NADP+ and NfnB. Notably, the 2′ phosphate group of ribose of Dpdh is hydrogen-bonded with Arg364 and Lys365, which are equivalently exchanged by Arg311 and Arg312 in NfnB. A third salt bridge to the 2′-phosphate group is formed by Arg371 in Dpdh and by Arg371 in NfnB (Fig. 4B).
The binding site of the third substrate ferredoxin has not been experimentally explored. Structure comparison studies immediately exclude the known ferredoxin binding sites of Fnr because its position is occupied in NfnA (and Dodh) by the iron-sulfur cluster carrying extension. A more conclusive model for ferredoxin binding to NfnAB could be derived from the Dpdh structure, which contains the mentioned NfnB-like domain in subunit A but also a ferredoxin-like domain in subunit B. A superposition of NfnB, and the structurally known ferredoxin of T. maritima carrying a single [4Fe-4S] cluster (49) onto Dpdh results in a model of the NfnAB-ferredoxin complex (Fig. 4C). The root mean square deviation between the Cα atoms of T. maritima and Dpdh ferredoxin is 1.6 Å using 92% of the residues for calculation. One main chain collision occurs between loop 11:14 in T. maritima ferredoxin and loop 40:46 in NfnB, which have to evade each other during complex formation. Moreover, this model for the NfnB-ferredoxin complex is in a high agreement with that based on protein-protein docking calculations (50). Accordingly, ferredoxin is attached to the outside margin of the iron-sulfur cluster domain of NfnB, and the edge to edge distance between its distal [4Fe-4S] cluster and the [4Fe-4S] cluster of ferredoxin is ∼8 Å (Fig. 4C), which is sufficiently close to allow efficient electron transfer (51). The postulated ferredoxin binding site is also plausible because all redox centers are thus integrated into the electron transfer chain (see below).
Mechanism of FAD-based Electron Bifurcation
Kinetic characterization provided evidence that the NfnAB complex of T. maritima is an electron-bifurcating enzyme and reversibly catalyzes the reduction of NAD+ and Fdox with 2 NADPH (Reaction 2 and Table 1). The obtained structural data revealed valuable information about how this enzymatic process works on an atomic basis (1). NAD(H) specifically binds to a-FAD and NADP(H) to b-FAD. Ferredoxin is most likely docked to the iron-sulfur cluster domain of NfnB adjacent to the distal [4Fe-4S] cluster (Fig. 4) (2). A one-electron transfer chain extends from a-FAD via the [2Fe-2S] cluster (8.5 Å) to b-FAD (15.0 Å) and from there via the proximal and distal [4Fe-4S] clusters (7.3 + 9.4 Å) to the [4Fe-4S] cluster of ferredoxin (∼8 Å) (Fig. 2B). The edge to edge distances (in parentheses) are sufficiently short to ensure a fast electron transfer process (51) except for the borderline value between the [2Fe-2S] cluster and b-FAD (3). b-FAD is assigned as the bifurcating FAD because of its central position in the electron transfer chain (Fig. 2) and because of its binding site for the high potential 2e donor NADPH (Fig. 4B) whose redox potential (−370 mV) under physiological conditions is between that of NAD (−280 mV) and ferredoxin (<−400 mV). Kinetic measurement also demonstrated that NADH cannot replace NADPH as high potential 2e donor (Table 1) and NADP+ not NAD+ as high potential 2e acceptor, indicating specific binding sites for them in agreement with the structural data (Fig. 4) (4). Because of related isoalloxazine binding modes, the determined redox potentials of Dodh and Gls were applied as approximate guidelines in the following mechanistic proposal.
In the bifurcating direction (Fig. 5), NADPH binds to NfnB and transfers a hydride to the bifurcating b-FAD serving as a 2 to 1e switch. According to energetic considerations, the first electron flows toward the site of the exergonic reaction and subsequently the second electron to the site of the endergonic reaction (21), implicating a high FADH−/FADH• and a low FADH•/FAD redox potential. The corresponding redox potentials of Gls (44) structurally related to NfnB qualitatively follow with E′o(FADH•/FADH−) ≥ −240 mV and E′o(FAD/FADH•) ≤ −360 mV this specification. Thus, b-FADH− donates one electron via the [2Fe-2S] cluster to a-FAD. Its redox potential should be approximately −240 mV, which approximately corresponds to that of the homologous Dodh (E′o(FAD/FADH•) = −312 mV). Subsequently, the generated b-FADH• delivers the second electron via the two low potential [4Fe-4S] clusters of NfnB to the [4Fe-4S] cluster (−453 mV; 80 °C) of Fdox (52). Therefore, the redox potential of the b-FAD/b-FADH• pair have to be lower than −400 mV, which is in the range of the estimated value of Gls. In a second round, the described process is repeated (Fig. 5). Finally, NAD+ is reduced to NADH from a-FADH−, and a second Fdox is reduced to Fdred.
Kinetic data clearly indicate that NADPH can only reduce NAD+ in the presence of Fdox, despite the more unfavorable thermodynamics. Therefore, the downhill flow of the second electron from electron-rich b-FADH• toward a-FADH• has to be prevented in order to direct the electron to the uphill electron transfer branch for producing the energy-rich Fdred. How this process is governed is, so far, unresolved. According to the architecture of the NfnAB structure, our current model assumes a conformational rearrangement of NfnA relative to NfnB that prolongs or shortens the distance between the [2Fe-2S] cluster and b-FAD, thereby blocking or conducting electron flow (Figs. 5 and 6). The distance of 15.0 Å between them in the NfnAB structure is on the borderline for an efficient electron transfer. We speculate that NADPH binding shortens the distance and thus enables electron conduction to a-FAD, whereas the subsequent formation of b-FADH• associated with the loss of a negative charge and perhaps with the release of NADP+ implicates the corresponding return movement. An analysis of the NfnA-NfnB interface reveals rather few intersubunit contacts, which would allow a concerted rigid body movement perhaps induced by NADPH binding and NADP+ release (Fig. 6). In parallel, the thermodynamically unfavorable electron flow to Fdox is promoted by the short distance found between b-FAD and the proximal [4Fe-4S] cluster.
FIGURE 6.
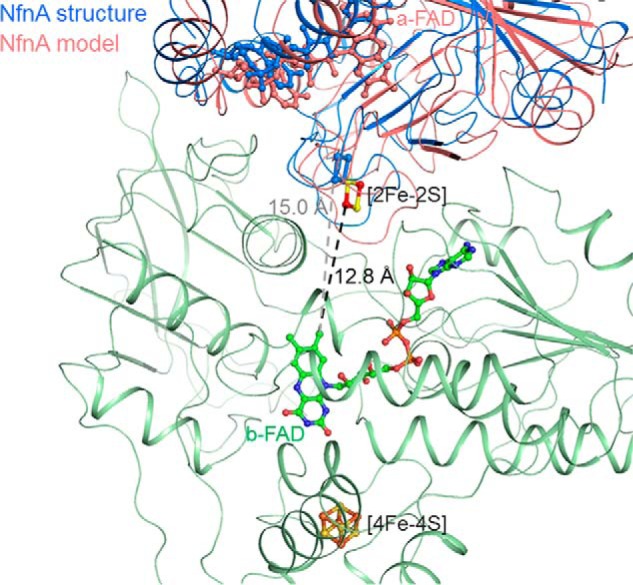
Blocking (blue) and conducting (salmon) states of the exergonic electron transfer branch. Assuming the experimentally determined NfnAB structure in the blocking state, a conducting state might be adjusted by a conformational change of NfnA relative to NfnB (green), which reduces the distance between the bifurcating b-FAD of NfnB and the [2Fe-2S] cluster of NfnA. Modeling was performed in a manner to minimize the interference between the fixed NfnB and the moving NfnA.
Bcd and EtfAB as separated proteins and the NfnAB complex are the only flavin-based electron bifurcating proteins structurally studied yet. In both systems, the reactions are embedded into a modularly composed protein apparatus with redox-center carrying domains/subunits that are adjustable relative to each other. Both enzymes contain bifurcating FADs that accept 2e from the high potential 2e donor NAD(P)H and are the starting point for two 1e transfer pathways into different directions to a high potential 2e acceptor and the low potential Fdox. Additional 1e redox centers, i.e. iron-sulfur clusters or FAD, are spaced between the active sites of the three redox reactions if their distances are too long. It appears that the electron transfer routes between the bifurcating FAD and the [4Fe-4S] cluster of ferredoxin are optimized perhaps to enable a moderate uphill electron transfer and/or to compete successfully with the electron flow to the exergonic NAD+ reduction. The binding pocket for bifurcating isoalloxazine rings has to be designed to adjust a low redox potential of the FADH•/FAD pair. In EtfAB and the NfnAB complex, the oxidized state might be stabilized by an arginine placed in front of the N5 and O4 atoms that preferably interacts with a deprotonated oxidized FAD (Fig. 3B). For both Bcd/EtfAB and NfnAB, a subunit/domain rearrangement is postulated to switch on or off the electron flow between the bifurcating FAD and the first redox center of the exergonic electron transfer branch (Fig. 6). This scenario is related to that established for the quinone-based electron bifurcation process where the second electron transfer between the semiquinone and the high potential [2Fe-2S] center is interrupted by a conformational change of the Riske protein.
Author Contributions
J. K. D. determined and refined the x-ray structures. H. H. overproduced, purified, partly crystallized, and kinetically characterized NfnAB supported by S. W. U. D. crystallized NfnAB and mounted crystals. R. K. T. initiated the project. R. K. T. and U. E. analyzed the data and wrote the paper. All authors approved the final version of the manuscript.
Acknowledgments
We thank Hartmut Michel for continuous support, the staff of the Swiss Light Source for help in data collection, and Michael Adams for providing us with the genomic DNA of T. maritima.
This work was supported by the Max-Planck Society. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 4YLF and 4YRY) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Bcd/EtfAB
- butyryl-CoA dehydrogenase/electron transfer flavoprotein
- NfnAB
- NADH-dependent reduced ferredoxin:NADP oxidoreductase
- Fnr
- ferredoxin:NADP reductase
- Dodh
- dihydroorotate dehydrogenase B
- Gls
- β-subunit of bacterial NADPH-dependent glutamate synthase
- Dpdh
- eukaryotic dihydropyrimidine dehydrogenase.
References
- 1. Herrmann G., Jayamani E., Mai G., Buckel W. (2008) Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J. Bacteriol. 190, 784–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thauer R. K., Kaster A. K., Seedorf H., Buckel W., Hedderich R. (2008) Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591 [DOI] [PubMed] [Google Scholar]
- 3. Seedorf H., Fricke W. F., Veith B., Brüggemann H., Liesegang H., Strittmatter A., Miethke M., Buckel W., Hinderberger J., Li F., Hagemeier C., Thauer R. K., Gottschalk G. (2008) The genome of Clostridium kluyveri, a strict anaerobe with unique metabolic features. Proc. Natl. Acad. Sci. U.S.A. 105, 2128–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li F., Hinderberger J., Seedorf H., Zhang J., Buckel W., Thauer R. K. (2008) Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J. Bacteriol. 190, 843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chowdhury N. P., Mowafy A. M., Demmer J. K., Upadhyay V., Koelzer S., Jayamani E., Kahnt J., Hornung M., Demmer U., Ermler U., Buckel W. (2014) Studies on the mechanism of electron bifurcation catalyzed by electron transferring flavoprotein (Etf) and butyryl-CoA dehydrogenase (Bcd) of Acidaminococcus fermentans. J. Biol. Chem. 289, 5145–5157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anderson R. F. (1983) Energetics of the one-electron reduction steps of riboflavin, FMN and FAD to their fully reduced forms. Biochim. Biophys. Acta 722, 158–162 [DOI] [PubMed] [Google Scholar]
- 7. Buckel W., Thauer R. K. (2013) Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochim. Biophys. Acta 1827, 94–113 [DOI] [PubMed] [Google Scholar]
- 8. Bertsch J., Parthasarathy A., Buckel W., Müller V. (2013) An electron-bifurcating caffeyl-CoA reductase. J. Biol. Chem. 288, 11304–11311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weghoff M. C., Bertsch J., Müller V. (2015) A novel mode of lactate metabolism in strictly anaerobic bacteria. Environ. Microbiol. 17, 670–677 [DOI] [PubMed] [Google Scholar]
- 10. Schut G. J., Adams M. W. (2009) The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production. J. Bacteriol. 191, 4451–4457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schuchmann K., Müller V. (2012) A bacterial electron-bifurcating hydrogenase. J. Biol. Chem. 287, 31165–31171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang S., Huang H., Kahnt J., Thauer R. K. (2013) A reversible electron-bifurcating ferredoxin- and NAD-dependent [FeFe]-hydrogenase (HydABC) in Moorella thermoacetica. J. Bacteriol. 195, 1267–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang S., Huang H., Kahnt J., Mueller A. P., Köpke M., Thauer R. K. (2013) NADP-specific electron-bifurcating [FeFe]-hydrogenase in a functional complex with formate dehydrogenase in Clostridium autoethanogenum grown on CO. J. Bacteriol. 195, 4373–4386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang S., Huang H., Kahnt J., Thauer R. K. (2013) Clostridium acidurici electron-bifurcating formate dehydrogenase. Appl. Environ. Microbiol. 79, 6176–6179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaster A. K., Goenrich M., Seedorf H., Liesegang H., Wollherr A., Gottschalk G., Thauer R. K. (2011) More than 200 genes required for methane formation from H2 and CO2 and energy conservation are present in Methanothermobacter marburgensis and Methanothermobacter thermautotrophicus. Archaea 2011, 973848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Costa K. C., Lie T. J., Xia Q., Leigh J. A. (2013) VhuD facilitates electron flow from H2 or formate to heterodisulfide reductase in Methanococcus maripaludis. J. Bacteriol. 195, 5160–5165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Costa K. C., Wong P. M., Wang T., Lie T. J., Dodsworth J. A., Swanson I., Burn J. A., Hackett M., Leigh J. A. (2010) Protein complexing in a methanogen suggests electron bifurcation and electron delivery from formate to heterodisulfide reductase. Proc. Natl. Acad. Sci. U.S.A. 107, 11050–11055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lie T. J., Costa K. C., Lupa B., Korpole S., Whitman W. B., Leigh J. A. (2012) Essential anaplerotic role for the energy-converting hydrogenase Eha in hydrogenotrophic methanogenesis. Proc. Natl. Acad. Sci. U.S.A. 109, 15473–15478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang S., Huang H., Moll J., Thauer R. K. (2010) NADP+ reduction with reduced ferredoxin and NADP+ reduction with NADH are coupled via an electron-bifurcating enzyme complex in Clostridium kluyveri. J. Bacteriol. 192, 5115–5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang H., Wang S., Moll J., Thauer R. K. (2012) Electron bifurcation involved in the energy metabolism of the acetogenic bacterium Moorella thermoacetica growing on glucose or H2 plus CO2. J. Bacteriol. 194, 3689–3699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nitschke W., Russell M. J. (2012) Redox bifurcations: mechanisms and importance to life now, and at its origin: a widespread means of energy conversion in biology unfolds. Bioessays 34, 106–109 [DOI] [PubMed] [Google Scholar]
- 22. Cottevieille M., Larquet E., Jonic S., Petoukhov M. V., Caprini G., Paravisi S., Svergun D. I., Vanoni M. A., Boisset N. (2008) The subnanometer resolution structure of the glutamate synthase 1.2-MDa hexamer by cryoelectron microscopy and its oligomerization behavior in solution: functional implications. J. Biol. Chem. 283, 8237–8249 [DOI] [PubMed] [Google Scholar]
- 23. Dobritzsch D., Schneider G., Schnackerz K. D., Lindqvist Y. (2001) Crystal structure of dihydropyrimidine dehydrogenase, a major determinant of the pharmacokinetics of the anti-cancer drug 5-fluorouracil. EMBO J. 20, 650–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bennett B. D., Kimball E. H., Gao M., Osterhout R., Van Dien S. J., Rabinowitz J. D. (2009) Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 5, 593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schönheit P., Wäscher C., Thauer R. K. (1978) A rapid procedure for the purification of ferredoxin from Clostridia using polyethyleneimine. FEBS Lett. 89, 219–222 [DOI] [PubMed] [Google Scholar]
- 26. Chen J. S., Mortenson L. E. (1974) Purification and properties of hydrogenase from Clostridium pasteurianum W5. Biochim. Biophys. Acta 371, 283–298 [DOI] [PubMed] [Google Scholar]
- 27. Wahl R. C., Orme-Johnson W. H. (1987) Clostridial pyruvate oxidoreductase and the pyruvate-oxidizing enzyme specific to nitrogen fixation in Klebsiella pneumoniae are similar enzymes. J. Biol. Chem. 262, 10489–10496 [PubMed] [Google Scholar]
- 28. Hamann N., Mander G. J., Shokes J. E., Scott R. A., Bennati M., Hedderich R. (2007) A cysteine-rich CCG domain contains a novel [4Fe-4S] cluster binding motif as deduced from studies with subunit B of heterodisulfide reductase from Methanothermobacter marburgensis. Biochemistry 46, 12875–12885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakamura M., Saeki K., Takahashi Y. (1999) Hyperproduction of recombinant ferredoxins in Escherichia coli by coexpression of the ORF1-ORF2-iscS-iscU-iscA-hscB-hscA-fdx-ORF3 gene cluster. J. Biochem. 126, 10–18 [DOI] [PubMed] [Google Scholar]
- 30. Aliverti A., Curti B., Vanoni M. A. (1999) Identifying and quantitating FAD and FMN in simple and in iron-sulfur-containing flavoproteins. Methods Mol. Biol. 131, 9–23 [DOI] [PubMed] [Google Scholar]
- 31. Dickert S., Pierik A. J., Linder D., Buckel W. (2000) The involvement of coenzyme A esters in the dehydration of (R)-phenyllactate to (E)-cinnamate by Clostridium sporogenes. Eur. J. Biochem. 267, 3874–3884 [DOI] [PubMed] [Google Scholar]
- 32. Pierik A. J., Wolbert R. B., Mutsaers P. H., Hagen W. R., Veeger C. (1992) Purification and biochemical characterization of a putative [6fe-6s] prismane-cluster-containing protein from Desulfovibrio-Vulgaris (Hildenborough). Eur. J. Biochem. 206, 697–704 [DOI] [PubMed] [Google Scholar]
- 33. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schneider T. R., Sheldrick G. M. (2002) Substructure solution with SHELXD. Acta Crystallogr. D 58, 1772–1779 [DOI] [PubMed] [Google Scholar]
- 35. De La Fortelle E., Bricogne G. (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol. 276, 472–494 [DOI] [PubMed] [Google Scholar]
- 36. Abrahams J. P., Leslie A. G. (1996) Methods used in the structure determination of bovine mitochondrial F-1 ATPase. Acta Crystallogr. D 52, 30–42 [DOI] [PubMed] [Google Scholar]
- 37. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 38. Rowland P., Nørager S., Jensen K. F., Larsen S. (2000) Structure of dihydroorotate dehydrogenase B: electron transfer between two flavin groups bridged by an iron-sulphur cluster. Structure 8, 1227–1238 [DOI] [PubMed] [Google Scholar]
- 39. Afonine P. V., Grosse-Kunstleve R. W., Chen V. B., Headd J. J., Moriarty N. W., Richardson J. S., Richardson D. C., Urzhumtsev A., Zwart P. H., Adams P. D. (2010) phenix.model_vs_data: a high-level tool for the calculation of crystallographic model and data statistics. J. Appl. Crystallogr. 43, 669–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lebedev A. A., Vagin A. A., Murshudov G. N. (2006) Intensity statistics in twinned crystals with examples from the PDB. Acta Crystallogr. D 62, 83–95 [DOI] [PubMed] [Google Scholar]
- 41. Karplus P. A., Faber H. R. (2004) Structural aspects of plant ferredoxin:NADP+ oxidoreductases. Photosynth. Res. 81, 303–315 [DOI] [PubMed] [Google Scholar]
- 42. Corrado M. E., Aliverti A., Zanetti G., Mayhew S. G. (1996) Analysis of the oxidation-reduction potentials of recombinant ferredoxin-NADP+ reductase from spinach chloroplasts. Eur. J. Biochem. 239, 662–667 [DOI] [PubMed] [Google Scholar]
- 43. Mohsen A. W., Rigby S. E., Jensen K. F., Munro A. W., Scrutton N. S. (2004) Thermodynamic basis of electron transfer in dihydroorotate dehydrogenase B from Lactococcus lactis: analysis by potentiometry, EPR spectroscopy, and ENDOR spectroscopy. Biochemistry 43, 6498–6510 [DOI] [PubMed] [Google Scholar]
- 44. Ravasio S., Curti B., Vanoni M. A. (2001) Determination of the midpoint potential of the FAD and FMN flavin cofactors and of the 3Fe-4S cluster of glutamate synthase. Biochemistry 40, 5533–5541 [DOI] [PubMed] [Google Scholar]
- 45. Lee M., Gräwert T., Quitterer F., Rohdich F., Eppinger J., Eisenreich W., Bacher A., Groll M. (2010) Biosynthesis of isoprenoids: crystal structure of the [4Fe-4S] cluster protein IspG. J. Mol. Biol. 404, 600–610 [DOI] [PubMed] [Google Scholar]
- 46. Hagen W. R., Vanoni M. A., Rosenbaum K., Schnackerz K. D. (2000) On the iron-sulfur clusters in the complex redox enzyme dihydropyrimidine dehydrogenase. Eur. J. Biochem. 267, 3640–3646 [DOI] [PubMed] [Google Scholar]
- 47. Deng Z., Aliverti A., Zanetti G., Arakaki A. K., Ottado J., Orellano E. G., Calcaterra N. B., Ceccarelli E. A., Carrillo N., Karplus P. A. (1999) A productive NADP+ binding mode of ferredoxin-NADP+ reductase revealed by protein engineering and crystallographic studies. Nat. Struct. Biol. 6, 847–853 [DOI] [PubMed] [Google Scholar]
- 48. Dobritzsch D., Ricagno S., Schneider G., Schnackerz K. D., Lindqvist Y. (2002) Crystal structure of the productive ternary complex of dihydropyrimidine dehydrogenase with NADPH and 5-iodouracil. Implications for mechanism of inhibition and electron transfer. J. Biol. Chem. 277, 13155–13166 [DOI] [PubMed] [Google Scholar]
- 49. Macedo-Ribeiro S., Darimont B., Sterner R., Huber R. (1996) Small structural changes account for the high thermostability of 1[4Fe-4S] ferredoxin from the hyperthermophilic bacterium Thermotoga maritima. Structure 4, 1291–1301 [DOI] [PubMed] [Google Scholar]
- 50. Kozakov D., Beglov D., Bohnuud T., Mottarella S. E., Xia B., Hall D. R., Vajda S. (2013) How good is automated protein docking? Proteins 81, 2159–2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Page C. C., Moser C. C., Chen X., Dutton P. L. (1999) Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature 402, 47–52 [DOI] [PubMed] [Google Scholar]
- 52. Smith E. T., Blamey J. M., Zhou Z. H., Adams M. W. (1995) A variable-temperature direct electrochemical study of metalloproteins from hyperthermophilic microorganisms involved in hydrogen production from pyruvate. Biochemistry 34, 7161–7169 [DOI] [PubMed] [Google Scholar]