

Background: Expansion of the polyQ tract in ataxin 7 is the main pathology of SCA7.
Results: The aggregates formed by polyQ-expanded ataxin 7 sequester USP22 through specific interactions.
Conclusion: Sequestration of USP22 impairs its deubiquitinating function in the SAGA complex.
Significance: This provides evidence for the hijacking model in which specific sequestration leads to cytotoxicity and neurodegeneration.
Keywords: aggregation, deubiquitylation (deubiquitination), polyglutamine, protein-protein interaction, zinc finger, DUB module, SAGA complex, USP22, ataxin 7, sequestration
Abstract
Human ataxin 7 (Atx7) is a component of the deubiquitination module (DUBm) in the Spt-Ada-Gcn5-acetyltransferase (SAGA) complex for transcriptional regulation, and expansion of its polyglutamine (polyQ) tract leads to spinocerebellar ataxia type 7. However, how polyQ expansion of Atx7 affects DUBm function remains elusive. We investigated the effects of polyQ-expanded Atx7 on ubiquitin-specific protease (USP22), an interacting partner of Atx7 functioning in deubiquitination of histone H2B. The results showed that the inclusions or aggregates formed by polyQ-expanded Atx7 specifically sequester USP22 through their interactions mediated by the N-terminal zinc finger domain of Atx7. The mutation of the zinc finger domain in Atx7 that disrupts its interaction with USP22 dramatically abolishes sequestration of USP22. Moreover, polyQ expansion of Atx7 decreases the deubiquitinating activity of USP22 and, consequently, increases the level of monoubiquitinated H2B. Therefore, we propose that polyQ-expanded Atx7 forms insoluble aggregates that sequester USP22 into a catalytically inactive state, and then the impaired DUBm loses the function to deubiquitinate monoubiquitinated histone H2B or H2A. This may result in dysfunction of the SAGA complex and transcriptional dysregulation in spinocerebellar ataxia type 7 disease.
Introduction
There are nine inherited neurodegenerative diseases that have been identified to date to be caused by polyglutamine (polyQ)2 expansion of the related proteins (1–3). Spinocerebellar ataxia type 7 (SCA7) is caused by expansion of the polyQ tract in ataxin 7 (Atx7) (4, 5). It is the only polyQ disease that leads to visual failure and eventual blindness, except for neurodegeneration (6). Many studies have revealed that there are nuclear inclusions in neurons and photoreceptor cells of SCA7 patients (7–9). These inclusions contain not only polyQ-expanded Atx7 but also other proteins, such as Cbl-associated proteins (10, 11); CREB-binding protein, HSP70 chaperones, and 19S subunit of proteasome (12, 13); and GCN5 (14). Dysfunction of these proteins might be related to the pathogenesis of SCA7.
Human Atx7 is a subunit of the SAGA (Spt-Ada-Gcn5-acetyltransferase) complex, a highly conserved coactivator involved in RNA polymerase II transcription (15). SAGA contains two enzymes, GCN5 and ubiquitin-specific protease 22 (USP22) (16, 17). GCN5 endows the histone acetyltransferase activity of SAGA (18), whereas USP22, together with Atx7, Atx7L3, and ENY2, assembles into the deubiquitination module (DUBm), which is responsible for deubiquitinating monoubiquitinated histone H2A (H2Aub) and H2B (H2Bub) (19–22). Both H2Aub and H2Bub participate in many cellular processes, including transcription initiation and elongation, gene silencing, and DNA repair (23, 24).
Transcriptional dysregulation may be central to the pathogenesis of SCA7, especially affecting a subset of genes involved in neuronal function, such as the genes specifically expressed in rod photoreceptors (25–27). However, it is controversial whether polyQ expansion of Atx7 affects the histone acetyltransferase activity of SAGA (27–29). Moreover, it is reported that polyQ expansion may alter Atx7 binding and the level of H2Bub at the RELN promoter and, therefore, reduce reelin expression (14). Recently, Mohan et al. (30) reported that loss of Atx7 in Drosophila reduces the level of H2Bub and causes neural and retinal degeneration similar to the phenotype when polyQ-expanded Atx7 is overexpressed in Drosophila (31). Although many efforts have been made to reveal the pathogenesis of SCA7, how polyQ-expanded Atx7 causes transcription alterations still needs to be elucidated.
Our previous studies have focused on the neurodegenerative disease-related proteins that sequester their interacting partners into insoluble aggregates or inclusions (11, 32–34). Recently, we revealed that aggregation of polyQ-expanded ataxin 3 (Atx3) specifically sequesters ubiquitin chains and P97/VCP into inclusions and impairs the normal function of P97/VCP (35). We investigated the effects of polyQ expansion in Atx7 on USP22 and found that polyQ-expanded Atx7 specifically sequesters USP22 into aggregates or inclusions and reduces the deubiquitination level of histone H2Bub. Therefore, we propose that aggregation of polyQ-expanded Atx7 specifically sequesters USP22 and deteriorates its deubiquitinating function in the SAGA complex.
Experimental Procedures
Plasmids, Antibodies, and Reagents
For prokaryotic expression, the coding sequence for USP22-N (1–170) was PCR-amplified and cloned into a pGEX-4T3 vector. The cDNA for Atx7-ZnF (residues 75–172) was cloned into pET-MG (36). For eukaryotic expression, full-length USP22, USP22-N (1–170), and USP22-USPD (172–525) were cloned into the FLAG-pcDNA3.0 vector, and USP5 was cloned into the FLAG-pCMV-Tag2B vector. Atx710Q-N (1–172) and its ZnF mutants were PCR-amplified and cloned into the FLAG-pcDNA3.0 vector or the HA-pcDNA3.0 vector. To obtain Atx7EPQ-N and its ZnF mutants, we digested the DNA of full-length Atx7100Q (stored by our laboratory) with restriction enzymes and retrieved the small fragment that contains the polyQ tract. We PCR-amplified the posterior fragment of Atx7 and then ligated the whole cDNA into the FLAG-pcDNA3.0 vector. To obtain the mutants of Atx7EPQ, we applied a similar method as for Atx7EPQ-N but cloned into pEGFP-N1. The polyQ tracts are variable during cloning of the plasmids, but the lengths are around 100Q. All the constructs were verified by DNA sequencing. For Western blotting, antibodies against FLAG and USP22 were from Sigma, anti-HA antibody was from Santa Cruz Biotechnology, anti-H2Bub was from Cell Signaling Technology, anti-H2B was from Bioworld, and anti-GAPDH was from Zen BioScience. All secondary antibodies were purchased from Jackson ImmunoResearch Laboratories. PVDF membranes were from PerkinElmer Life Sciences, and proteins were visualized using an ECL detection kit (Thermo Scientific). Ubiquitin vinyl sulfone was purchased from Boston Biochem. The FLAG peptide was obtained from ChinaPeptides Co.
Cell Culture and Transfection
Human HEK 293T cells or HeLa cells were cultured in DMEM (HyClone) supplemented with 10% fetal bovine serum (Gibco) and penicillin-streptomycin at 37 °C under a humidified atmosphere containing 5% CO2. All transfections of HEK 293T cells were performed by using PolyJetTM reagent (SignaGen). To test H2Bub, HeLa cells were transfected with the respective plasmids by using Lipofectamine 2000 (Invitrogen) and harvested about 48 h (for full-length Atx7) or 72 h (for Atx7-N) after transfection.
Immunoprecipitation and Immunofluorescence Microscopy
For immunoprecipitation, HEK 293T cells were harvested and lysed in radioimmune precipitation assay (RIPA) buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, and protease inhibitor mixture) on ice for 30 min, and then the lysates were incubated with FLAG antibody-conjugated agarose (Abmart) for 2 h at 4 °C. The beads were washed with RIPA buffer four times and subjected to Western blot analysis.
For immunofluorescence microscopy, HEK 293T cells grown on cover slides were transfected with PolyJetTM. For cotransfection, about 48 h later, the cells were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 1 min, and blocked with 5% BSA for 1 h. Then the cells were incubated with an antibody against FLAG (1:100, Sigma) for 1 h at room temperature. After washing with PBS, the cells were labeled with a TRITC-conjugated anti-mouse antibody (1:100, Jackson ImmunoResearch Laboratories) for 1 h, and the nuclei were stained with Hoechst (Sigma). The cells were visualized on a Leica Microsystems TCS SP8 confocal microscope. To detect endogenous USP22, immunocytochemistry experiments were performed about 96 h after transfection.
Supernatant/Pellet Fractionation for Cell Lysates
About 72 h after transfection, HEK 293T cells were lysed in 100 μl of radioimmune precipitation assay buffer on ice for 30 min and centrifuged at 16,200 × g for 15 min at 4 °C. Then the supernatant was added to 100 μl of 2× loading buffer (2% SDS), and the pellet was washed sufficiently with RIPA buffer three times at 4 °C and added to 40 μl of 4× loading buffer (4% SDS). Finally the samples were subjected to Western blotting. For time course experiments, the cultured cells were harvested 24, 36, 48, 60, or 72 h after transfection.
Ubiquitin Vinyl Sulfone (Ub-VS) Assay
HEK 293T cells were harvested and then subjected to immunoprecipitation for 2 h. The beads were incubated with FLAG peptide to competitively elute the immunoprecipitated material for 1 h at 4 °C in Ub-VS reaction buffer (100 mm Tris-HCl, 100 mm KCl, 3 mm DTT, and 5% glycerol (pH 8.0)). The immunoprecipitated material was concentrated with 3-kDa centrifugal filter devices (Millipore), incubated with 5 μm Ub-VS in Ub-VS reaction buffer for 30 min at 37 °C, and then used for Western blotting.
Results
PolyQ-expanded Atx7 Specifically Sequesters USP22 into Inclusions in Cells
To define whether polyQ expansion of Atx7 affects the function of USP22 in the SAGA complex, we first investigated by confocal microscopy whether polyQ-expanded Atx7 (namely Atx7EPQ) could sequester USP22 into inclusions. When overexpressed in HEK 293T cells, both normal Atx7 (Atx710Q) and USP22 diffused in the nucleus (Fig. 1A, first row). However, Atx7EPQ (100Q) formed nuclear inclusion bodies and sequestered coexpressed USP22 into the inclusions (Fig. 1A, second row). In contrast, the Atx7EPQ inclusions were not able to sequester another deubiquitinating enzyme, USP5 (37) (Fig. 1A, third row). Moreover, USP22 could not be sequestered to the inclusions formed by another polyQ-expanded protein, Atx371Q (38) (Fig. 1A, fourth row). This demonstrates that polyQ-expanded Atx7 specifically sequesters USP22 into inclusions. We further examined whether the inclusions of Atx7EPQ sequestered endogenous USP22. Similarly, the inclusions formed by Atx7EPQ could sequester endogenous USP22 (Fig. 1B). This implies that polyQ-expanded Atx7 could also sequester USP22 in SCA7 patients. Because the N-terminal region of Atx7 mediates its interaction with other subunits of the DUBm (20) (Fig. 2A), we tested whether the N terminus of Atx7EPQ (Atx7EPQ-N, residues 1–172) could sequester USP22. As the full-length Atx7EPQ, Atx7EPQ-N could also sequester USP22 into inclusions (data not shown), suggesting that sequestration of USP22 by the inclusions of polyQ-expanded Atx7 may be attributed to the interaction between the N terminus of Atx7 and USP22.
FIGURE 1.
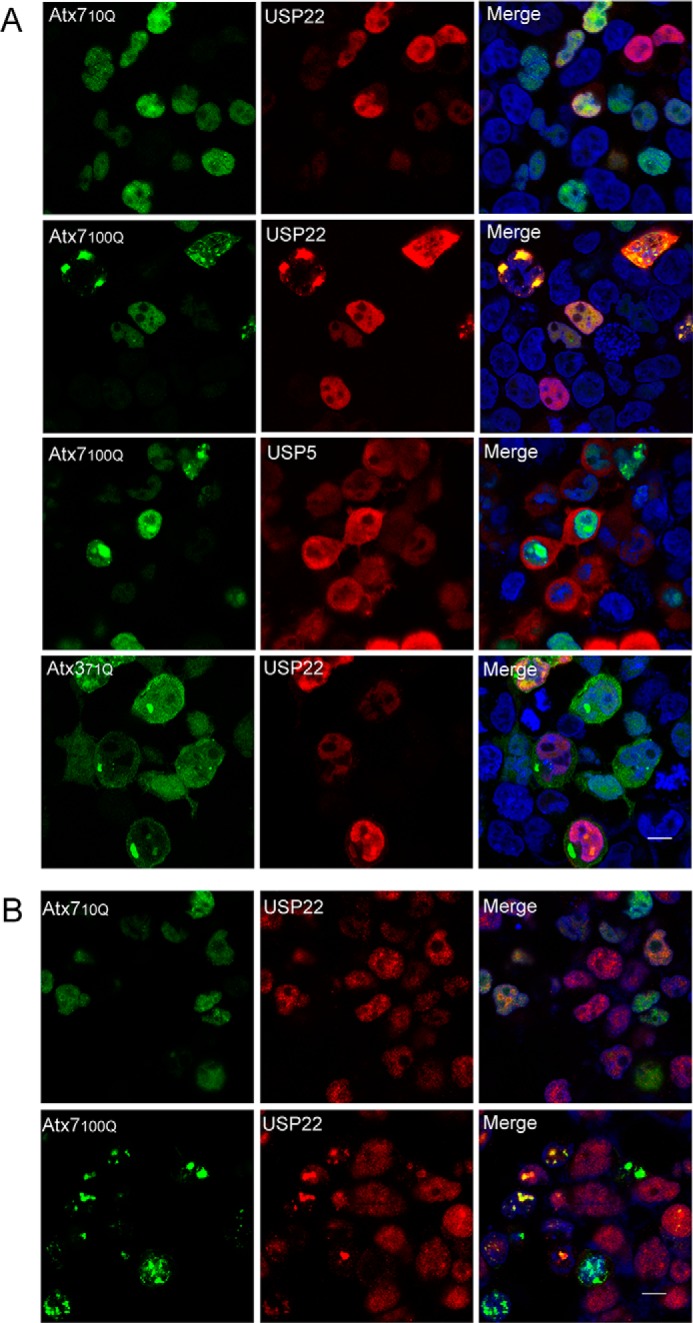
PolyQ-expanded Atx7 sequesters USP22 into inclusions in cells as visualized by confocal microscopy. A, sequestration of overexpressed USP22. Atx710Q-GFP or Atx7EPQ-GFP (100Q) was cotransfected with FLAG-USP22 into HEK 293T cells, and the cells were visualized by confocal fluorescence microscopy. Atx371Q-GFP and FLAG-USP5 were set as controls. USP22 and USP5 were stained with anti-FLAG antibody (red), and nuclei were stained with Hoechst (blue). Scale bar =10 μm. B, sequestration of endogenous USP22. HEK 293T cells transfected with Atx710Q-GFP or Atx7EPQ-GFP (100Q) were visualized by confocal fluorescence microscopy. USP22 was stained with anti-USP22 antibody (red), and nuclei were stained with Hoechst (blue). Scale bar = 10 μm.
FIGURE 2.

Characterization of the interaction between Atx7 and USP22. A, the domain architecture of Atx7 and USP22. Qn, polyQ tract; PRR, proline-rich region; ZnF, zinc finger domain; SCA7, SCA7 domain; USPD, ubiquitin-specific protease domain. B, co-IP experiment for the interaction between the N-terminal fragment of Atx7 and endogenous USP22. HEK 293T cells were transfected with FLAG-Atx710Q-N and FLAG-Atx7EPQ-N (115Q), respectively, and the cell lysates were subjected to coimmunoprecipitation with anti-FLAG antibody. USP22 was detected with an anti-USP22 antibody. Vec, vector. C, co-IP experiment for the interacting fragment in USP22. HA-Atx710Q-N was cotransfected with FLAG-tagged USP22 or its fragments into HEK 293T cells, and the cell lysates were subjected to coimmunoprecipitation with anti-FLAG antibody. USP22-N, residues 1–170; USP22-C, residues 172–525. D, GST pulldown experiment showing that Atx7-ZnF interacts directly with the N-terminal ZnF domain of USP22. Top panel, SDS-PAGE graph with Coomassie Blue (CBB) staining. Bottom panel, Western blotting (WB) to detect GB1-His6-tagged Atx7-ZnF with anti-His antibody (10% input). Atx7-ZnF, residues 75–172 of Atx7; USP22-N, residues 1–170 of USP22 containing a ZnF domain.
The Interaction between Atx7 and USP22 Depends on the N-terminal ZnF Domain of Atx7
To better understand the mechanism underlying the sequestration of USP22 by polyQ-expanded Atx7, we explored the specific interaction between Atx7 and USP22. Atx7 contains a polyQ tract (Qn), a proline-rich region, a ZnF domain, and a SCA7 domain (39), whereas USP22 is comprised of an N-terminal ZnF domain and a USP domain (40) (Fig. 2A). A previous study has indicated that the N-terminal ZnF domain of Atx7 is critical for assembly of the DUB module (20), so we used Atx7-N (residues 1–172) to investigate the interaction with USP22. Atx710Q-N or Atx7EPQ-N was transfected into HEK 293T cells, and the cell lysates were subjected to coimmunoprecipitation (co-IP). The data showed that endogenous USP22 could be coimmunoprecipitated by both the normal and polyQ-expanded proteins (Fig. 2B). Furthermore, we identified the specific region of USP22 associating with Atx7-N. Although not strictly quantitative, the co-IP experiment showed that the N terminus of USP22 (USP22-N, residues 1–170) could immunoprecipitate a considerable amount of Atx710Q-N whereas the C terminus of USP22 (USP22-C, residues 172–525) could not (Fig. 2C). This interaction between Atx7-N and USP22-N was also confirmed by a GST pulldown experiment (Fig. 2D). As shown, the ZnF domain of Atx7 interacts directly with the N-terminal domain of USP22, which is mainly comprised of a ZnF domain (Fig. 2A).
A Mutation in the ZnF Domain of Atx7 Disrupts Its Interaction with and Sequestration of USP22
Our previous study indicated that specific interaction is important for the sequestration of its interacting partners by polyQ proteins (33, 35). We proposed that the sequestration of USP22 by Atx7EPQ is also closely associated with their interaction. To test this hypothesis, we prepared the Atx7 mutants to disrupt the interaction with USP22 and re-examined the sequestration. The three ZnF mutants of Atx7 were C2* (C150S/C153S), H2* (H165N/H170N), and C2H2* (C150S/C153S + H165N/H170N) (Fig. 3A). Co-IP experiment showed that these mutations largely disrupted the interaction between Atx710Q-N and endogenous USP22 (Fig. 3B). Therefore, we performed confocal microscopy to detect the sequestration of USP22 by Atx7EPQ or its ZnF mutants (C2*, H2*, and C2H2*). Compared with wild-type Atx7EPQ, the three mutants lost the ability to sequester endogenous USP22 into inclusions (Fig. 3C), suggesting that polyQ-expanded Atx7 sequesters USP22 into inclusions depending on the specific interaction between them.
FIGURE 3.
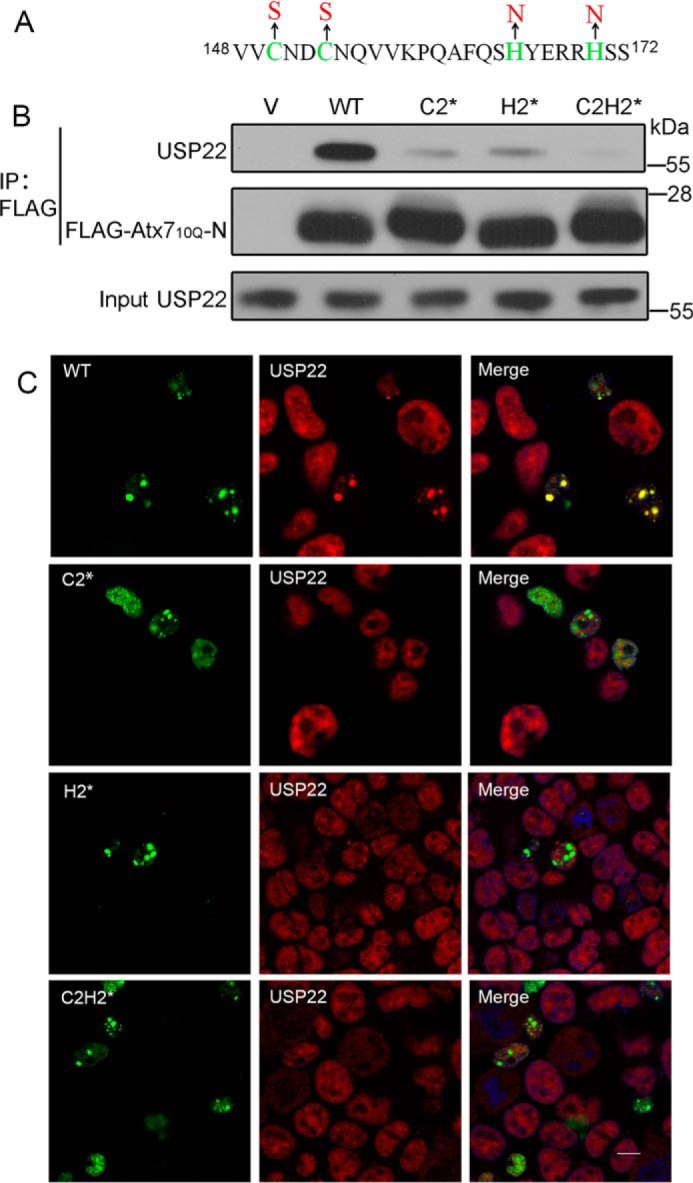
Characterizing the role of the ZnF domain in Atx7 in interaction with and sequestration of USP22 by mutagenesis. A, sequence for the ZnF domain of Atx7 showing the mutation sites. The three ZnF mutants are as follows: C2*, C150S/C153S; H2*, H165N/H170N; and C2H2*, C150S/C153S + H165N/H170N. B, co-IP experiment for the interaction of Atx710Q-N or its mutants with endogenous USP22. HEK 293T cells were transfected with FLAG-Atx710Q-N or its mutants, and the cell lysates were subjected to immunoprecipitation with anti-FLAG antibody. V, vector. C, sequestration of endogenous USP22 by Atx7EPQ or its ZnF mutants as visualized by confocal microscopy. Atx7EPQ-GFP or its ZnF mutants were transfected into HEK 293T cells. The polyQ lengths of Atx7EPQ-GFP used were as follows: WT, 100Q; C2*, 109Q; H2*, 90Q; C2H2*, 99Q. USP22 was stained with anti-USP22 antibody (red), and nuclei were stained with Hoechst (blue). Scale bar = 10 μm.
PolyQ-expanded Atx7 Sequesters Endogenous USP22 into Insoluble Aggregates
To further examine whether Atx7EPQ can redistribute endogenous USP22, we performed supernatant/pellet fractionation of the cell lysates. The data showed that overexpression of Atx7EPQ-N could increase the amount of USP22 in the pellet fraction because of the presence of Atx7EPQ-N in the pellet (Fig. 4A) but Atx710Q-N could not. This hijacking effect was also confirmed by the supernatant/pellet fractionation experiments in a dose-dependent manner (Fig. 4, B and C). Moreover, we also performed time course experiments on the sequestration of USP22 into the pellet fraction by Atx7EPQ-N in cells. With the aggregation of Atx7EPQ-N progressing, the amounts of USP22 sequestered into insoluble aggregates increased gradually (Fig. 4, D and E).
FIGURE 4.

PolyQ-expanded Atx7 sequesters endogenous USP22 into aggregates. A, sequestration of endogenous USP22 into insoluble aggregates by Atx7EPQ-N. FLAG-tagged Atx710Q-N or Atx7EPQ-N (115Q) was transfected into HEK 293T cells, and the cell lysates were subjected to fractionation and Western blot analysis with an anti-FLAG or anti-USP22 antibody. Sup, supernatant; Pel, pellet; Vec, vector. B and C, dose-dependent experiments for the sequestration of USP22 into aggregates by Atx710Q-N (B) or Atx7EPQ-N (C). FLAG-tagged Atx710Q-N or Atx7EPQ-N (115Q) was transfected into HEK 293T cells in a dose-dependent manner. D, time course experiments for the sequestration of USP22 into aggregates by Atx7EPQ-N. FLAG-tagged Atx7EPQ-N (115Q) was transfected into HEK 293T cells, and the cell lysates were subjected to fractionation in a time course manner. E, quantification of endogenous USP22 and Atx7EPQ-N in pellets (data from D). Data are mean ± S.E. (n = 3). F, comparison of the sequestration of USP22 by Atx7EPQ-N and its ZnF mutants. FLAG-tagged Atx7EPQ-N and its mutants were transfected into HEK 293T cells, respectively. The polyQ lengths of Atx7EPQ-N used were as follows: WT, 115Q; C2*, 115Q; H2*, 115Q; C2H2*, 106Q.
Next, we investigated whether Atx7EPQ-N sequestered USP22 into aggregates depending on the ZnF domain. The result showed that, in comparison with the wild type, the amount of USP22 sequestered by the ZnF mutants decreased considerably (Fig. 4F). Taken together, these results demonstrate that polyQ-expanded Atx7 sequesters USP22 into insoluble aggregates depending on the interaction through the ZnF domain of Atx7.
PolyQ Expansion in Atx7 Reduces the Catalytic Activity of USP22
To detect whether polyQ-expanded Atx7 affects the catalytic activity of USP22, we immunoprecipitated endogenous USP22 with FLAG-tagged Atx7-N with different polyQ lengths and examined its catalytic activity using Ub-VS. Ub-VS is a C-terminally modified vinyl sulfone derivative of ubiquitin that can irreversibly modify an active deubiquitinating enzyme with a covalent bond (20, 41, 42). Compared with Atx710Q-N, which led to the formation of a considerable amount of USP22Ub-VS derivative, Atx7EPQ-N resulted in less product (Fig. 5, A and B), indicating that polyQ-expanded Atx7 reduces the catalytic activity of USP22.
FIGURE 5.

Detection of the catalytic activities of USP22 immunoprecipitated by Atx7-N variants. A, Ub-VS modification of catalytically active USP22 immunoprecipitated by Atx710Q-N and Atx7EPQ-N (100Q). HEK 293T cells were transfected with FLAG-tagged Atx710Q-N or Atx7100Q-N, and then the cell lysates were subjected to immunoprecipitation. The immunoprecipitated species were incubated with or without Ub-VS (5 μm), and the reaction mixtures were detected by Western blotting. B, quantification of the ratio of USP22Ub-VS against total USP22 (data from A). Data are mean ± S.E. (n = 3). **, p < 0.01.
PolyQ-expanded Atx7 Impairs the Deubiquitination of Histone H2Bub
Our studies revealed that polyQ-expanded Atx7 could affect USP22 not only by sequestration but also by impairing its catalytic activity. We wondered whether polyQ expansion of Atx7 affects the normal function of USP22. It has been demonstrated that incorporation of USP22 into the DUBm endows the SAGA complex with the main deubiquitinating function for histone H2B (19, 20, 22, 43). Therefore, we investigated the effect of polyQ expansion in Atx7 on the H2Bub level. Compared with the mock vector, overexpression of Atx710Q-N had no influence on the H2Bub level, whereas overexpression of Atx7EPQ-N significantly increased the level of H2Bub (Fig. 6, A and B). However, overexpression of the ZnF mutants of Atx7EPQ-N caused a significant decrease of the level of H2Bub compared with the wild type (Fig. 6, C and D). To further verify the above results, we also applied full-length Atx7 and repeated the experiment. The data showed that overexpression of full-length Atx7EPQ significantly increased the level of H2Bub, whereas its C2H2* mutant restored H2Bub to the mock level (Fig. 6, E and F). Together, these findings corroborate that polyQ expansion in Atx7 impairs the deubiquitinating function of USP22 by specific interaction mediated by the N-terminal ZnF domain of Atx7.
FIGURE 6.

Effects of various Atx7-N forms on cellular H2Bub levels. A, assay of the H2Bub level affected by Atx710Q-N or Atx7EPQ-N. FLAG-tagged Atx710Q-N or Atx7EPQ-N (115Q) was transfected into HeLa cells, and the cell lysates were subjected to Western blotting with an anti-H2Bub or anti-H2B antibody. Vec, vector. B, quantification of the H2Bub levels affected by Atx7-N forms. Data are mean ± S.E. (n = 3). *, p < 0.05; **, p < 0.01; N.S., no significance. C, as in A, by Atx7EPQ-N and its ZnF mutants. The polyQ lengths of Atx7EPQ-N used were as follows: WT, 115Q; C2*, 115Q; H2*, 115Q; C2H2*, 106Q. D, quantification of the H2Bub levels affected by Atx7100Q-N and its ZnF mutants. Data are mean ± S.E. (n = 3). **, p < 0.01. E, assay of the H2Bub level affected by full-length Atx7. Various HA-Atx7-GFP forms were transfected into HeLa cells, and the cell lysates were subjected to Western blotting with an anti-H2Bub or anti-H2B antibody. C2H2*, 99Q. F, quantification of the H2Bub levels affected by full-length Atx7. Data are mean ± S.E. (n = 3). *, p < 0.05.
Discussion
The development of neurodegenerative diseases is a progressive process. Protein aggregation and inclusion formation are considered to be a hallmark of these diseases. Accumulating evidence has revealed that protein aggregates or inclusions lead to cellular dysfunction through the sequestration of essential proteins (33, 35, 44–47). We investigated the effect of polyQ-expanded Atx7 on its interacting partner, USP22, and found that the aggregates or inclusions sequester USP22, which leads to dysfunction of USP22 and, consequently, an altered level of monoubiquitinated histone H2B. Moreover, the sequestration is an ongoing process over time. With the aggregation of polyQ-expanded Atx7, the amounts of USP22 in the insoluble aggregates increase gradually.
A number of studies demonstrate that the inclusions formed by polyQ-expanded proteins such as Atx7 contain various other cellular essential proteins (12, 13, 48). We discovered, for the first time, that the inclusions or aggregates of polyQ-expanded Atx7 specifically sequester USP22, as in the case of polyQ-expanded Atx3 aggregates, which specifically sequester P97/VCP and ubiquitin conjugates (35). It has also been reported that GCN5 is present in the nuclear inclusions of polyQ-expanded Atx7 in a SCA7 astrocyte model (14). Given these findings, we speculate that the inclusions or aggregates of polyQ-expanded Atx7 also sequester other subunits of the SAGA complex, especially the components of the DUBm, which may ultimately ruin the function of the SAGA complex.
Specific Interaction Is Vital for the Sequestration of USP22 by polyQ-expanded Atx7
The N-terminal region of Atx7 (residues 75–172), together with USP22, Atx7L3, and ENY2, can assemble into a minimal recombinant DUBm (20). Our studies indicate that sequestration of USP22 by polyQ-expanded Atx7 relies on the specific interaction between their harboring domains. We confirmed that the N-terminal ZnF domain of Atx7, whether normal or polyQ-expanded, can interact with USP22 and that the N-terminal region of USP22, including its harbored ZnF domain, contributes to the interaction and sequestration of USP22 into the inclusions formed by polyQ-expanded Atx7. Mutation of the ZnF domain in Atx7 abolishes the interaction with and sequestration of USP22 into inclusions or insoluble aggregates. All of these data demonstrate that specific interaction is critical for the sequestration of USP22 by polyQ-expanded Atx7. This is also consistent with previous studies of the sequestration of interacting partners by polyQ (33, 35, 49–51) and other amyloidogenic proteins (34, 45, 52).
We focused on the sequestration that is mediated by domain-domain interactions, whereas the polyQ tract is just the source for protein aggregation. On the other hand, some studies have revealed that the sequestration is mediated by coaggregation of polyQ tracts (53–55). Cooperative polyQ-polyQ interactions also prompt aggregation of the polyQ proteins. In this case, a polyQ protein can sequester another polyQ-containing protein, and they coaggregate into inclusions or insolubilities (56, 57). This nonspecific sequestration may sometimes also deteriorate the functions of other polyQ proteins.
Aggregation of polyQ-expanded Atx7 Impairs the Function of USP22
The best known function of USP22 is deubiquitinating monoubiquitinated histone H2B (19, 20, 22). We revealed that polyQ-expanded Atx7 can increase the level of H2Bub, depending on the specific interaction through its ZnF domain, suggesting that polyQ expansion of Atx7 interferes with the normal function of USP22 and, therefore, the DUB module. Recently, studies have reported that polyQ expansion of Atx7 increases the global level of H2Bub without changing the bulk levels of H3 Lys-9/14 acetylation (14, 58). Our result corroborates the finding that expansion of the polyQ tract in Atx7 leads to formation of aggregates and that the aggregates specifically sequester USP22 and other components of DUBm into a dysfunctional state. This process leads to loss of DUBm function in deubiquitinating histone H2Bub (Fig. 7). Because H2Bub is important for both transcription initiation and elongation (23, 24), dysfunction of the DUBm in the SAGA complex caused by polyQ-expanded Atx7 may be a major source for transcriptional dysregulation in SCA7 disease.
FIGURE 7.
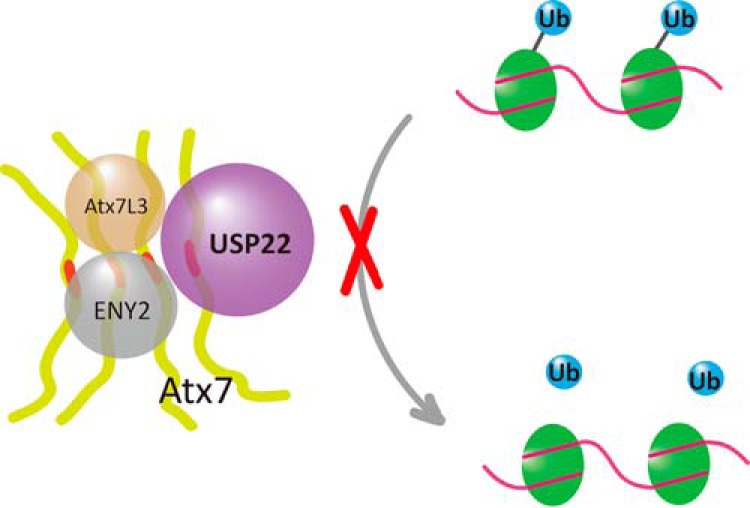
Schematic of the aggregation of polyQ-expanded Atx7 impairing the deubiquitination of H2Bub. PolyQ-expanded Atx7 forms aggregates that sequester USP22 into a catalytically inactive state in the DUB module. The impaired DUB module loses the function to deubiquitinate monoubiquitinated histone H2B or H2A. The polyQ tract of Atx7 is highlighted in red. Atx7, USP22, Atx7L3, and ENY2 are the components of the DUBm in the SAGA complex.
The Generality and Individuality of polyQ Diseases
Our previous work has demonstrated that polyQ-expanded Atx3 specifically sequesters P97/VCP and influences its function in down-regulating neddylation and proposed a hijacking model for the cytotoxicity and neurodegeneration caused by polyQ-expanded proteins (35). Here we report that polyQ-expanded Atx7 sequesters USP22 into aggregates or inclusions through a specific interaction that impairs the function of the DUBm in deubiquitinating H2Bub. Generally, for a polyQ-disease protein, its expanded polyQ-tract region leads to the formation of aggregates or inclusions, and its flanking domains or motifs are responsible for specific interactions with other proteins. During aggregation, its interacting partners are sequestered into insoluble aggregates and become dysfunctional, whereas the soluble fraction of the active partners are decreased consequently, which is probably the major source of cytotoxicity and neurodegeneration. On the other hand, it is also mentioned that some polyQ disease proteins are able to sequester the components involved in cell quality control through nonspecific sequestration (44, 47, 59, 60). However, different polyQ proteins have different domains or motifs that mediate specific interactions with respective partners. A specific flanking domain or motif determines its interacting proteins to be sequestered and dysfunctional, which, in our opinion, leads to the individualities of polyQ diseases.
Author Contributions
H. Y. H. and H. Y. designed the research and wrote the manuscript. H. Y. and S. L. performed and analyzed the experiments. H. Y. and W. T. H. purified the proteins. L. L. J. provided technical assistance. H. Y. and J. Z. conceived and drew Fig. 7. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Drs. X. C. Gao, Y. J. Jiang and A. X. Song for valuable discussions, and Drs. M. X. Che and Y. G. Gao for technical help.
This work was supported by National Basic Research Program of China Grant 2012CB911003 and National Natural Science Foundation of China Grants 31270773, 31200570, and 31470758. The authors declare that they have no conflicts of interest with the contents of this article.
- polyQ
- polyglutamine
- SCA7
- spinocerebellar ataxia type 7
- CREB
- cAMP response element-binding protein
- SAGA
- Spt-Ada-Gcn5-acetyltransferase
- DUBm
- deubiquitination module
- ZnF
- zinc finger
- VCP
- vasolin-containing protein
- TRITC
- tetramethylrhodamine isothiocyanate
- RIPA
- radioimmune precipitation assay
- Ub-VS
- ubiquitin vinyl sulfone
- USP
- ubiquitin-specific protease
- IP
- immunoprecipitation.
References
- 1. Zoghbi H. Y., Orr H. T. (2000) Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 23, 217–247 [DOI] [PubMed] [Google Scholar]
- 2. Orr H. T., Zoghbi H. Y. (2007) Trinucleotide repeat disorders. Annu. Rev. Neurosci. 30, 575–621 [DOI] [PubMed] [Google Scholar]
- 3. Blum E. S., Schwendeman A. R., Shaham S. (2013) PolyQ disease: misfiring of a developmental cell death program? Trends Cell Biol. 23, 168–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. David G., Abbas N., Stevanin G., Dürr A., Yvert G., Cancel G., Weber C., Imbert G., Saudou F., Antoniou E., Drabkin H., Gemmill R., Giunti P., Benomar A., Wood N., Ruberg M., Agid Y., Mandel J. L., Brice A. (1997) Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 17, 65–70 [DOI] [PubMed] [Google Scholar]
- 5. David G., Dürr A., Stevanin G., Cancel G., Abbas N., Benomar A., Belal S., Lebre A. S., Abada-Bendib M., Grid D., Holmberg M., Yahyaoui M., Hentati F., Chkili T., Agid Y., Brice A. (1998) Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7). Hum. Mol. Genet. 7, 165–170 [DOI] [PubMed] [Google Scholar]
- 6. Enevoldson T. P., Sanders M. D., Harding A. E. (1994) Autosomal dominant cerebellar ataxia with pigmentary macular dystrophy. A clinical and genetic study of eight families. Brain 117, 445–460 [DOI] [PubMed] [Google Scholar]
- 7. Einum D. D., Townsend J. J., Ptácek L. J., Fu Y. H. (2001) Ataxin-7 expression analysis in controls and spinocerebellar ataxia type 7 patients. Neurogenetics 3, 83–90 [DOI] [PubMed] [Google Scholar]
- 8. Ansorge O., Giunti P., Michalik A., Van Broeckhoven C., Harding B., Wood N., Scaravilli F. (2004) Ataxin-7 aggregation and ubiquitination in infantile SCA7 with 180 CAG repeats. Ann. Neurol. 56, 448–452 [DOI] [PubMed] [Google Scholar]
- 9. Holmberg M., Duyckaerts C., Dürr A., Cancel G., Gourfinkel-An I., Damier P., Faucheux B., Trottier Y., Hirsch E. C., Agid Y., Brice A. (1998) Spinocerebellar ataxia type 7 (SCA7): a neurodegenerative disorder with neuronal intranuclear inclusions. Hum. Mol. Genet. 7, 913–918 [DOI] [PubMed] [Google Scholar]
- 10. Lebre A. S., Jamot L., Takahashi J., Spassky N., Leprince C., Ravisé N., Zander C., Fujigasaki H., Kussel-Andermann P., Duyckaerts C., Camonis J. H., Brice A. (2001) Ataxin-7 interacts with a Cbl-associated protein that it recruits into neuronal intranuclear inclusions. Hum. Mol. Genet. 10, 1201–1213 [DOI] [PubMed] [Google Scholar]
- 11. Jiang Y. J., Zhou C. J., Zhou Z. R., Wu M., Hu H. Y. (2013) Structural basis for recognition of the third SH3 domain of full-length R85 (R85FL)/ponsin by ataxin-7. FEBS Lett. 587, 2905–2911 [DOI] [PubMed] [Google Scholar]
- 12. Takahashi J., Fujigasaki H., Zander C., El Hachimi K. H., Stevanin G., Dürr A., Lebre A. S., Yvert G., Trottier Y., de Thé H., Hauw J. J., Duyckaerts C., Brice A. (2002) Two populations of neuronal intranuclear inclusions in SCA7 differ in size and promyelocytic leukaemia protein content. Brain 125, 1534–1543 [DOI] [PubMed] [Google Scholar]
- 13. Janer A., Werner A., Takahashi-Fujigasaki J., Daret A., Fujigasaki H., Takada K., Duyckaerts C., Brice A., Dejean A., Sittler A. (2010) SUMOylation attenuates the aggregation propensity and cellular toxicity of the polyglutamine expanded ataxin-7. Hum. Mol. Genet. 19, 181–195 [DOI] [PubMed] [Google Scholar]
- 14. McCullough S. D., Xu X., Dent S. Y., Bekiranov S., Roeder R. G., Grant P. A. (2012) Reelin is a target of polyglutamine expanded ataxin-7 in human spinocerebellar ataxia type 7 (SCA7) astrocytes. Proc. Natl. Acad. Sci. U.S.A. 109, 21319–21324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Helmlinger D., Hardy S., Sasorith S., Klein F., Robert F., Weber C., Miguet L., Potier N., Van-Dorsselaer A., Wurtz J. M., Mandel J. L., Tora L., Devys D. (2004) Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum. Mol. Genet. 13, 1257–1265 [DOI] [PubMed] [Google Scholar]
- 16. Baker S. P., Grant P. A. (2007) The SAGA continues: expanding the cellular role of a transcriptional co-activator complex. Oncogene 26, 5329–5340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rodríguez-Navarro S. (2009) Insights into SAGA function during gene expression. EMBO Rep. 10, 843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nagy Z., Tora L. (2007) Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene 26, 5341–5357 [DOI] [PubMed] [Google Scholar]
- 19. Zhao Y., Lang G., Ito S., Bonnet J., Metzger E., Sawatsubashi S., Suzuki E., Le Guezennec X., Stunnenberg H. G., Krasnov A., Georgieva S. G., Schüle R., Takeyama K., Kato S., Tora L., Devys D. (2008) A TFTC/STAGA module mediates histone H2A and H2B deubiquitination, coactivates nuclear receptors, and counteracts heterochromatin silencing. Mol. Cell 29, 92–101 [DOI] [PubMed] [Google Scholar]
- 20. Lang G., Bonnet J., Umlauf D., Karmodiya K., Koffler J., Stierle M., Devys D., Tora L. (2011) The tightly controlled deubiquitination activity of the human SAGA complex differentially modifies distinct gene regulatory elements. Mol. Cell. Biol. 31, 3734–3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang X. Y., Pfeiffer H. K., Thorne A. W., McMahon S. B. (2008) USP22, an hSAGA subunit and potential cancer stem cell marker, reverses the polycomb-catalyzed ubiquitylation of histone H2A. Cell Cycle 7, 1522–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang X. Y., Varthi M., Sykes S. M., Phillips C., Warzecha C., Zhu W., Wyce A., Thorne A. W., Berger S. L., McMahon S. B. (2008) The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol. Cell 29, 102–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Johnsen S. A. (2012) The enigmatic role of H2Bub1 in cancer. FEBS Lett. 586, 1592–1601 [DOI] [PubMed] [Google Scholar]
- 24. Weake V. M., Workman J. L. (2008) Histone ubiquitination: triggering gene activity. Mol. Cell 29, 653–663 [DOI] [PubMed] [Google Scholar]
- 25. Helmlinger D., Tora L., Devys D. (2006) Transcriptional alterations and chromatin remodeling in polyglutamine diseases. Trends Genet. 22, 562–570 [DOI] [PubMed] [Google Scholar]
- 26. Chou A. H., Chen C. Y., Chen S. Y., Chen W. J., Chen Y. L., Weng Y. S., Wang H. L. (2010) Polyglutamine-expanded ataxin-7 causes cerebellar dysfunction by inducing transcriptional dysregulation. Neurochem. Int. 56, 329–339 [DOI] [PubMed] [Google Scholar]
- 27. Helmlinger D., Hardy S., Abou-Sleymane G., Eberlin A., Bowman A. B., Gansmüller A., Picaud S., Zoghbi H. Y., Trottier Y., Tora L., Devys D. (2006) Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLos Biol. 4, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Palhan V. B., Chen S., Peng G. H., Tjernberg A., Gamper A. M., Fan Y., Chait B. T., La Spada A. R., Roeder R. G. (2005) Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc. Natl. Acad. Sci. U.S.A. 102, 8472–8477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McMahon S. J., Pray-Grant M. G., Schieltz D., Yates J. R. 3rd, Grant P. A. (2005) Polyglutamine-expanded spinocerebellar ataxia-7 protein disrupts normal SAGA and SLIK histone acetyltransferase activity. Proc. Natl. Acad. Sci. U.S.A. 102, 8478–8482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mohan R. D., Dialynas G., Weake V. M., Liu J., Martin-Brown S., Florens L., Washburn M. P., Workman J. L., Abmayr S. M. (2014) Loss of Drosophila Ataxin-7, a SAGA subunit, reduces H2B ubiquitination and leads to neural and retinal degeneration. Genes Dev. 28, 259–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Latouche M., Lasbleiz C., Martin E., Monnier V., Debeir T., Mouatt-Prigent A., Muriel M. P., Morel L., Ruberg M., Brice A., Stevanin G., Tricoire H. (2007) A conditional pan-neuronal Drosophila model of spinocerebellar ataxia 7 with a reversible adult phenotype suitable for identifying modifier genes. J. Neurosci. 27, 2483–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gao Y. G., Yang H., Zhao J., Jiang Y. J., Hu H. Y. (2014) Autoinhibitory structure of the WW domain of HYPB/SETD2 regulates its interaction with the proline-rich region of huntingtin. Structure 22, 378–386 [DOI] [PubMed] [Google Scholar]
- 33. Jiang Y. J., Che M. X., Yuan J. Q., Xie Y. Y., Yan X. Z., Hu H. Y. (2011) Interaction with polyglutamine-expanded huntingtin alters cellular distribution and RNA processing of huntingtin yeast two-hybrid protein A (HYPA). J. Biol. Chem. 286, 25236–25245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xie Y. Y., Zhou C. J., Zhou Z. R., Hong J., Che M. X., Fu Q. S., Song A. X., Lin D. H., Hu H. Y. (2010) Interaction with synphilin-1 promotes inclusion formation of α-synuclein: mechanistic insights and pathological implication. FASEB J. 24, 196–205 [DOI] [PubMed] [Google Scholar]
- 35. Yang H., Li J. J., Liu S., Zhao J., Jiang Y. J., Song A. X., Hu H. Y. (2014) Aggregation of polyglutamine-expanded ataxin-3 sequesters its specific interacting partners into inclusions: implication in a loss-of-function pathology. Sci. Rep. 4, 6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bao W. J., Gao Y. G., Chang Y. G., Zhang T. Y., Lin X. J., Yan X. Z., Hu H. Y. (2006) Highly efficient expression and purification system of small-size protein domains in Escherichia coli for biochemical characterization. Protein Expr. Purif. 47, 599–606 [DOI] [PubMed] [Google Scholar]
- 37. Reyes-Turcu F. E., Horton J. R., Mullally J. E., Heroux A., Cheng X., Wilkinson K. D. (2006) The ubiquitin binding domain ZnF UBP recognizes the C-terminal diglycine motif of unanchored ubiquitin. Cell 124, 1197–1208 [DOI] [PubMed] [Google Scholar]
- 38. Chai Y., Wu L., Griffin J. D., Paulson H. L. (2001) The role of protein composition in specifying nuclear inclusion formation in polyglutamine disease. J. Biol. Chem. 276, 44889–44897 [DOI] [PubMed] [Google Scholar]
- 39. Bonnet J., Wang Y. H., Spedale G., Atkinson R. A., Romier C., Hamiche A., Pijnappel W. W., Timmers H. T., Tora L., Devys D., Kieffer B. (2010) The structural plasticity of SCA7 domains defines their differential nucleosome-binding properties. EMBO Rep. 11, 612–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lin Z., Yang H., Kong Q., Li J., Lee S. M., Gao B., Dong H., Wei J., Song J., Zhang D. D., Fang D. (2012) USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol. Cell 46, 484–494 [DOI] [PubMed] [Google Scholar]
- 41. Borodovsky A., Kessler B. M., Casagrande R., Overkleeft H. S., Wilkinson K. D., Ploegh H. L. (2001) A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J. 20, 5187–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Verma R., Aravind L., Oania R., McDonald W. H., Yates J. R. 3rd, Koonin E. V., Deshaies R. J. (2002) Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 298, 611–615 [DOI] [PubMed] [Google Scholar]
- 43. Henry K. W., Wyce A., Lo W. S., Duggan L. J., Emre N. C., Kao C. F., Pillus L., Shilatifard A., Osley M. A., Berger S. L. (2003) Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev. 17, 2648–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Park S. H., Kukushkin Y., Gupta R., Chen T., Konagai A., Hipp M. S., Hayer-Hartl M., Hartl F. U. (2013) PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 154, 134–145 [DOI] [PubMed] [Google Scholar]
- 45. Olzscha H., Schermann S. M., Woerner A. C., Pinkert S., Hecht M. H., Tartaglia G. G., Vendruscolo M., Hayer-Hartl M., Hartl F. U., Vabulas R. M. (2011) Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144, 67–78 [DOI] [PubMed] [Google Scholar]
- 46. Fujita K., Nakamura Y., Oka T., Ito H., Tamura T., Tagawa K., Sasabe T., Katsuta A., Motoki K., Shiwaku H., Sone M., Yoshida C., Katsuno M., Eishi Y., Murata M., Taylor J. P., Wanker E. E., Kono K., Tashiro S., Sobue G., La Spada A. R., Okazawa H. (2013) A functional deficiency of TERA/VCP/p97 contributes to impaired DNA repair in multiple polyglutamine diseases. Nat. Commun. 4, 1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yu A., Shibata Y., Shah B., Calamini B., Lo D. C., Morimoto R. I. (2014) Protein aggregation can inhibit clathrin-mediated endocytosis by chaperone competition. Proc. Natl. Acad. Sci. U.S.A. 111, E1481–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McCampbell A., Taylor J. P., Taye A. A., Robitschek J., Li M., Walcott J., Merry D., Chai Y., Paulson H., Sobue G., Fischbeck K. H. (2000) CREB-binding protein sequestration by expanded polyglutamine. Hum. Mol. Genet. 9, 2197–2202 [DOI] [PubMed] [Google Scholar]
- 49. Damrath E., Heck M. V., Gispert S., Azizov M., Nowock J., Seifried C., Rüb U., Walter M., Auburger G. (2012) ATXN2-CAG42 sequesters PABPC1 into insolubility and induces FBXW8 in cerebellum of old ataxic knock-in mice. PLoS Genet. 8, e1002920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Friedman M. J., Shah A. G., Fang Z. H., Ward E. G., Warren S. T., Li S., Li X. J. (2007) Polyglutamine domain modulates the TBP-TFIIB interaction: implications for its normal function and neurodegeneration. Nat. Neurosci. 10, 1519–1528 [DOI] [PubMed] [Google Scholar]
- 51. Choi Y. J., Kim S. I., Lee J. W., Kwon Y. S., Lee H. J., Kim S. S., Chun W. (2012) Suppression of aggregate formation of mutant huntingtin potentiates CREB-binding protein sequestration and apoptotic cell death. Mol. Cell Neurosci 49, 127–137 [DOI] [PubMed] [Google Scholar]
- 52. Rantanen K., Pursiheimo J. P., Högel H., Miikkulainen P., Sundström J., Jaakkola P. M. (2013) p62/SQSTM1 regulates cellular oxygen sensing by attenuating PHD3 activity through aggregate sequestration and enhanced degradation. J. Cell Sci. 126, 1144–1154 [DOI] [PubMed] [Google Scholar]
- 53. Kayatekin C., Matlack K. E., Hesse W. R., Guan Y., Chakrabortee S., Russ J., Wanker E. E., Shah J. V., Lindquist S. (2014) Prion-like proteins sequester and suppress the toxicity of huntingtin exon 1. Proc. Natl. Acad. Sci. U.S.A. 111, 12085–12090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao X., Park Y. N., Todor H., Moomau C., Masison D., Eisenberg E., Greene L. E. (2012) Sequestration of Sup35 by aggregates of huntingtin fragments causes toxicity of [PSI+] yeast. J. Biol. Chem. 287, 23346–23355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hsu T. C., Wang C. K., Yang C. Y., Lee L. C., Hsieh-Li H. M., Ro L. S., Chen C. M., Lee-Chen G. J., Su M. T. (2014) Deactivation of TBP contributes to SCA17 pathogenesis. Hum. Mol. Genet. 23, 6878–6893 [DOI] [PubMed] [Google Scholar]
- 56. Holmes W. M., Klaips C. L., Serio T. R. (2014) Defining the limits: protein aggregation and toxicity in vivo. Crit. Rev. Biochem. Mol. Biol. 49, 294–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Perez M. K., Paulson H. L., Pendse S. J., Saionz S. J., Bonini N. M., Pittman R. N. (1998) Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J. Cell Biol. 143, 1457–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lan X., Koutelou E., Schibler A. C., Chen Y. C., Grant P. A., Dent S. Y. (2015) Poly(Q) Expansions in ATXN7 affect solubility but not activity of the SAGA deubiquitinating module. Mol. Cell. Biol. 35, 1777–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chai Y., Koppenhafer S. L., Shoesmith S. J., Perez M. K., Paulson H. L. (1999) Evidence for proteasome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum. Mol. Genet. 8, 673–682 [DOI] [PubMed] [Google Scholar]
- 60. Schmidt T., Lindenberg K. S., Krebs A., Schöls L., Laccone F., Herms J., Rechsteiner M., Riess O., Landwehrmeyer G. B. (2002) Protein surveillance machinery in brains with spinocerebellar ataxia type 3: redistribution and differential recruitment of 26S proteasome subunits and chaperones to neuronal intranuclear inclusions. Ann. Neurol. 51, 302–310 [DOI] [PubMed] [Google Scholar]