

Background: NMDA receptor hyperactivity results in mitochondrial dysfunction in neurons promoting neurodegenerative disorders.
Results: Short polyarginine peptides target mitochondria to promote neuronal survival.
Conclusion: Short polyarginine peptides reduce mitochondrial respiration, membrane hyperpolarization, and generation of reactive oxygen species.
Significance: Treatment with polyarginine has the potential to minimize neuronal damage resulting from stroke or traumatic brain injury and may be therapeutic to ameliorate multiple sclerosis and Parkinson disease.
Keywords: apoptosis; mitochondrial membrane potential; N-methyl-d-aspartate receptor (NMDA receptor, NMDAR); necrosis (necrotic death); neuroprotection; retina; retinal ganglion cells
Abstract
It is widely accepted that overactivation of NMDA receptors, resulting in calcium overload and consequent mitochondrial dysfunction in retinal ganglion neurons, plays a significant role in promoting neurodegenerative disorders such as glaucoma. Calcium has been shown to initiate a transient hyperpolarization of the mitochondrial membrane potential triggering a burst of reactive oxygen species leading to apoptosis. Strategies that enhance cell survival signaling pathways aimed at preventing this adverse hyperpolarization of the mitochondrial membrane potential may provide a novel therapeutic intervention in retinal disease. In the retina, brain-derived neurotrophic factor has been shown to be neuroprotective, and our group previously reported a PSD-95/PDZ-binding cyclic peptide (CN2097) that augments brain-derived neurotrophic factor-induced pro-survival signaling. Here, we examined the neuroprotective properties of CN2097 using an established retinal in vivo NMDA toxicity model. CN2097 completely attenuated NMDA-induced caspase 3-dependent and -independent cell death and PARP-1 activation pathways, blocked necrosis, and fully prevented the loss of long term ganglion cell viability. Although neuroprotection was partially dependent upon CN2097 binding to the PDZ domain of PSD-95, our results show that the polyarginine-rich transport moiety C-R(7), linked to the PDZ-PSD-95-binding cyclic peptide, was sufficient to mediate short and long term protection via a mitochondrial targeting mechanism. C-R(7) localized to mitochondria and was found to reduce mitochondrial respiration, mitochondrial membrane hyperpolarization, and the generation of reactive oxygen species, promoting survival of retinal neurons.
Introduction
Mitochondrial impairment is believed to play a prominent role in the pathogenesis of retinal diseases such as glaucoma (1, 2), diabetic retinopathy (3), and retinal ischemia (4). Implicated in these disorders, the N-methyl-d-aspartate subtype of glutamate receptor (NMDAR),2 which contributes to ON and OFF light-evoked responses in retinal ganglion cells (RGC) (5), has also been linked to mitochondrial dysfunction and RGC death when hyperstimulated (6–8). Both RGCs and a large population of NMDAR-expressing amacrine cells (9, 10) are highly susceptible to cell death in the presence of elevated levels of glutamate (11, 12), whereby excessive calcium perturbs the mitochondrial membrane potential (ΔΨm) promoting both programmed and nonprogrammed cell death pathways (7, 13). In particular, the C terminus of the NMDAR GluN2B subunit contains a motif with high affinity for the PSD-95/Discs-large/ZO-1 homology (PDZ) scaffold protein PSD-95 (14) that is believed to couple PSD-95 to neuronal nitric oxide synthase (NOS), resulting in a pro-death decrease in CREB activity (15). However, long term blockade of NMDAR activity has proven to be detrimental to neuronal survival, as synaptic GluN2A NMDARs stimulate pro-survival AKT and CREB phosphorylation (16, 17).
NMDARs have been shown to colocalize with PSD-95 at RGC synapses (18). GluN2A-containing NMDARs are localized post-synaptically on OFF RGC and perisynaptically on ON RGC dendrites, whereas GluN2B-containing NMDARs are primarily found perisynaptically on ON RGC (18, 19). Uncoupling NMDARs from PSD-95 using a peptide composed of the C terminal nine amino acids of the GluN2B subunit fused to the cell-permeable HIV-1 Tat peptide (Tat-NR2B9c) has been reported to provide neuroprotection (20). In an effort to develop high affinity PSD-95/PDZ modulators, we synthesized a selective cyclic peptidomimetic (CN2097), based on a previously reported peptide (21), designed to target the PDZ domains of PSD-95 (22, 23). We found that CN2097 increased the association of PSD-95 with the neurotrophin receptor TrkB to augment brain-derived neurotrophic factor (BDNF)-induced signaling, leading to increased AKT and CREB activation, and facilitation of long term potentiation (LTP) (24). As the signaling pathways by which NMDARs induce LTP overlap with those involved in cell survival (25), we postulated that CN2097 would promote neuroprotection. Here, using an in vivo retinal toxicity model (26), we show that CN2097 blocks all aspects of NMDA-induced cell death to fully rescue RGC viability. In addition, the polyarginine-rich transport moiety C-R(7) alone, independent of the PDZ-PSD-95-binding cyclic peptide of CN2097, was identified to attenuate mitochondrial dysfunction resulting in protection against NMDA-induced death.
Materials and Methods
Reagents
Rabbit anti-poly-ADP-ribosyl(ation) (PAR) polyclonal antibody was from Pharmingen. 4-Nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl-phosphate was from Roche Diagnostics, and anti-β-actin mouse monoclonal antibody (AC-15) was from Sigma. Fmoc-l-amino acids and unnatural d-amino acids, such as Fmoc-d-Arg (Pbf)-OH and d-Cys-OH, preloaded amino acids on Wang resin, Rink amide AM resin, biotin NovaTagTM resin, 5-carboxytetramethylrhodamine, and 5-carboxyfluorescein (FAM) were purchased from Novabiochem (EMD Chemicals). The coupling reagents, 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate and benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate were purchased from Chem-Impex International Inc. Fmoc-N-amido-dPEG2 acid was purchased from Quanta BioDesign, Ltd. Piperidine, N-methylmorpholine, and N,N-diisopropylethylamine were purchased from Sigma. The other chemicals, such as cleavage mixture reagents trifluoroacetic acid (TFA), anisole, thioanisole, ethanedithiol, triisopropylsilane, and 2,2′-dithiodipyridine, anhydrous solvents (i.e. dichloromethane), and N,N-dimethylformamide were purchased from Fisher.
Synthesis of Peptides Used for This Study
Peptides were synthesized using N-(9-fluorenyl)methoxycarbonyl (Fmoc)-solid phase peptide synthesis protocols employing microwave protocols (27). Some were manually prepared using PS3 automated peptide synthesizer (Ranin Instrument Co., Inc.) at room temperature. The linear peptides in Table 1 were synthesized on Rink amide AM resin with the relevant Fmoc-protected amino acids, coupling, activating, and deprotecting reagents using 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate, N-methylmorpholine (0.4 m), and piperidine in N,N-dimethylformamide (20% v/v), respectively. The linear peptides (R(N)7, hs-CR(7), K(7), Tat, and Tat-NR2B9c) were synthesized on Rink amide AM resin followed by cleavage using reagent R (TFA/thioanisole/anisole/ethanedithiol (90:5:2:3, v/v/v/v). The peptides were purified by reverse phase HPLC, lyophilized, and exchanged with HCl. Disulfide linkage between the peptides was conducted in the solution phase using cysteine-containing linear peptides. The activated cysteine-containing peptides were either coupled with cysteine or other cysteine-containing peptides in solution phase in an equimolar amount at room temperature in (3–48 h) using water as a solvent. For example, the individual peptides containing cysteine were synthesized on a solid support. The activation of cysteine was performed during cleavage using 2,2′-dithiodipyridine (5 eq) in cleavage mixture (TFA/triisopropylsilane/H2O, 95:2.5:2.5, v/v/v). The peptides were purified and lyophilized. A number of peptides were synthesized using this strategy, such as C-s-s-C-R(7), C-s-s-C-R(5), C-s-s-C-R(3), C-s-s-C-K(7), DC-s-s-DCDR(7), C-s-s-C-R(7)-biotin, C-s-s-C-R(7)-FAM, C-s-s-C-Tat, and C-R(7). The macrocyclic peptide in CN2097, CN5135, and CN1105 was synthesized, as reported previously (22, 24), followed by cleavage and purification to afford the cyclic peptides containing a cysteine residue at N terminus that was further coupled onto on-bead CR(7) containing the activated cysteine. The fatty acyl derivatives of macrocyclic peptides CN2180 and CN3200 were synthesized with coupling of myristic anhydride at the N terminus. The coupling of carboxyfluorescein in CN3205 was achieved at the polyethylene linker attached to the side chain of the lysine residue at the N terminus followed by coupling of the myristic acid chain at the N terminus. Similarly, the rhodamine derivative CN6005 was synthesized by coupling of 6-carboxy-N,N,N′,N′-tetramethylrhodamine at the polyethylene glycol linker at the lysine side chain residue followed by coupling of multiple arginines at the N terminus.
TABLE 1.
Nomenclature for linear peptides
| R(7) | RRRRRRR-amide |
| C-R(7) | CRRRRRRR-amide |
| C-s-s-C-R(7) | C-s-s-CRRRRRRR-amide |
| C-s-s-C-R(5) | C-s-s-CRRRRR-amide |
| C-s-s-C-R(3) | C-s-s-CRRR-amide |
| DC-s-s-DCDR(7) | DC-s-s-DCDRDRDRDRDRDRDR-amide |
| C-s-s-C-R(7)-biotin | C-s-s-CRRRRRRR- (C2-linker)-biotin |
| C-s-s-C-R(7)-FAM | C-s-s-CRRRRRRRK- (apen-FAM) |
| Tat | YGRKKRRQRR-amide |
| C-s-s-C-Tat | C-s-s-CYGRKKRRQRR-amide |
| Tat-NR2B9c | YGRKKRRQRRKLSSIESDV-amide |
| Lin-CN2097 | Amide-RRRRRRRC-s-s-CKNYKKTEV-amide |
| Lin-CN5135 | Amide-RRRRRRRC-s-s-CKNYKKAEA-amide |
All of the peptides were purified by reverse phase HPLC, lyophilized, and exchanged with HCl. Peptide purity was in the range of 90–95% as determined using high resolution time of flight AXIMA performance MALDI TOF-TOF mass spectrometer (Shimadzu).
Animals
Male Sprague-Dawley rats of 225–250 g were obtained from Charles River. All animal procedures and care were performed following IACUC-approved protocols in compliance with United States Department of Agriculture guidelines and with those prescribed by the Association for Research in Vision and Ophthalmology.
Intravitreal Injections
Rats were anesthetized with isoflurane after which the eyes were given a 3-μl intravitreal pre-injection containing either the designated peptide/vehicle or vehicle alone (0.01 m phosphate buffer, pH 7.2), as described previously (26). Unless otherwise stated, 2 h following the pre-injection, the reanesthetized animals were given a second intravitreal injection containing 20 nmol of NMDA dissolved in vehicle, with or without the inclusion of the designated peptide. Survival times and retinal processing following NMDA insult are detailed below.
Determining Optimal Intravitreal Dosing of Lead Neuroprotective Compounds
Optimal dosing of the CN2097, CN2180, and negative control CN3200 for the in vivo retinal toxicity model were determined by quantitatively assessing the attenuation of NMDA-induced PAR formation by performing Western blot analyses, as described previously (26). All blots were digitally scanned for densitometric analysis using ImageJ software. Quantification of PAR immunoreactivity for each individually treated retina was normalized to β-actin signal.
Ethidium Bromide Staining
Freshly obtained posterior eyecups from treated animals were stained with ethidium bromide (EtBr) as described previously (26, 29). Equal 1.5-mm-wide swatch across each retina, through the optic nerve head, was digitally photographed in montage format using a Zeiss Apotome microscope equipped with a rhodamine fluorescence filter pack, using ×20 objective. Labeling distributions were determined by manually counting EtBr-stained cells in the GCL and normalized to the area sampled. Previous studies have shown that multiple populations of ganglion and displaced amacrine cells are susceptible to NMDA-induced loss of plasma membrane selectivity (LPMS) and that their distributions (converted to number of EtBr cells/mm2) surrounding the optic nerve head between 0.5 and 3 mm eccentricities are relatively constant (29).
Fluorogold Retrograde Labeling of Retinal Ganglion Cells
For long term protection studies, retinal ganglion cell viability was assessed using the retrograde dye fluorogold (Fluorochrome LLC, Denver, CO). Fourteen days following intravitreal injections of the designated peptide and NMDA, rats were anesthetized with xylazine/ketamine, and the incision site was prepped for surgery following IACUC-approved guidelines. The superior surface of the cranium was exposed, whereby two portals, corresponding to the positioning of the right and left superior colliculi, were obtained using a high speed drill. The dura mater was cut away, and a 2-mm diameter core of overlying cortex was aspirated off to expose the superior surface of each colliculi. Twenty five microliters of a 4% aqueous solution of fluorogold was absorbed into an equal sized portion of sterile powered gel foam with half being applied onto the exposed surface of each superior colliculi. The access portals were sealed with bone wax followed by the scalp being sutured closed. All animals were closely monitored for both food and water intake, pain/distress, weight loss, and inactivity according to IACUC guidelines, and those few that did present complications were removed from the study. Four days following this surgery, rats were euthanized, and the retinas were quickly dissected, fixed, and flat mounted onto glass slides. All retinas were digitally photographed in their entirety using montage formatting with a Zeiss Apotome, microscope equipped with a DAPI filter pack, and ×10 objective. Each reconstructed retinal montage was regionally subdivided into four regions defined by spacing of 1-mm concentric rings, centered about the optic nerve head and used to bin fluorogold-labeled ganglion cell distributions.
In Vivo Caspase Activation
Previous studies have shown that caspase-3 activation in the retina peaks 2 h following an intravitreal injection of 20 nmol of NMDA (29). Utilizing the membrane-permeable peptide carboxyfluorescein-peptide valylalanyl aspartic acid-fluoromethyl ketone (FAM-VAD-FMK), which specifically and irreversibly binds in situ to activated caspases 1, 3, 4 and 7, we quantitatively evaluated selected peptides for their respective ability to block NMDA-induced caspase activation using our established in vivo retinal toxicity model. Rat eyes were given a single intravitreal injection containing one of the following treatments, 3 nmol of the designated peptide in the presence of 20 nmol of NMDA, 20 nmol of NMDA alone, or vehicle alone. One hour following the intravitreal injection, each rat received a 100-μl intravenous tail vein injection containing ∼8 μg of FAM-FLIVO (ImmunoChemistry Technologies, Bloomington, MN), freshly prepared under sterile conditions. Two hours following intravitreal injections, the eyes were quickly dissected and fixed as above. Retinas were then flat mounted, ganglion cell side up, and photographed in montage format incorporating a 1-mm-thick band that passed though the optic nerve head on a temporal-nasal path using ×20 objective and a FITC filter pack. The distribution of FLAVO-positive cells was manually counted from each image, converted to number of cells/mm2, and averaged for each treated retina.
Multielectrode Array (MEA) Recording
Male Sprague-Dawley rats about 2 months of age were used. Prior to each experiment, the animal was dark-adapted overnight in a ventilated lightproof box. Under dim red light, the animal was euthanized using CO2 inhalation followed by pneumothorax. Eyes were removed and hemisected. Following vitrectomy using forceps, each eyecup was put in Ames' medium at room temperature gassed with 95% O2, 5% CO2, cut in two halves, and allowed to dark-adapt for at least 1 h. Under infrared illumination using night vision devices (NiteMate NAV-3, Litton Industries, Watertown, CT) attached to the eyepieces of a dissecting microscope, a piece of retina was isolated from an eye cup using a paint brush and flattened on an MEA with the RGC side down. The MEA contained 60 30-μm diameter electrodes at a center-to-center spacing of 200 μm (Multi Channel Systems, Germany). The retina was continuously superfused at 3 ml/min with Ames' medium gassed with 95% O2 5% CO2 and maintained at 33 °C with a temperature controller, and it was kept in darkness except during light stimulation. All photoresponse data were collected after the retina had been superfused for at least 100 min. The stimuli were 1-s full-field 480 nm light generated by a monochromator (Optical Building Blocks, Birmingham, NJ). The timing of stimulus presentation was controlled by an electromechanical shutter built into this monochromator. Light intensity was adjusted by a continuously variable neutral density filter (Newport Corp., Franklin, MA). The intensity-adjusted light was delivered via a fiber optic cable to the retina from below the MEA superfusion chamber. The unattenuated light intensity (i.e. −0 log I) was 4.1 × 1015 photons cm−2 s−1 at the retina. Drugs were applied by adding stock solutions to the superfusion reservoir. For each drug condition, the retina was superfused with the drug for 25–30 min prior to light testing.
MEA recordings were amplified, filtered with cutoffs at 200 and 3 kHz, and digitized at 10 kHz using MC Rack software (Multi Channel Systems). The raw recordings from all 60 channels were saved onto a computer for off-line analysis. To quantify photoresponse amplitudes, traditional cluster analysis of spiking activity could not be used because in many channels the effect of NMDA application was a dramatic reduction in spike amplitude, not just a change in spike rate (see Fig. 9a). Instead, we used MathWorks MATLAB software (Natick, MA) to calculate the variance of the recording during the 1-s light stimulus and of the recording during the second before light-stimulus onset. The difference between these variances was used as the photoresponse amplitude (Fig. 9a). For each data point in the plots, the light-induced variance differences for all 60 channels were averaged, and the error bars represent S.E.
FIGURE 9.
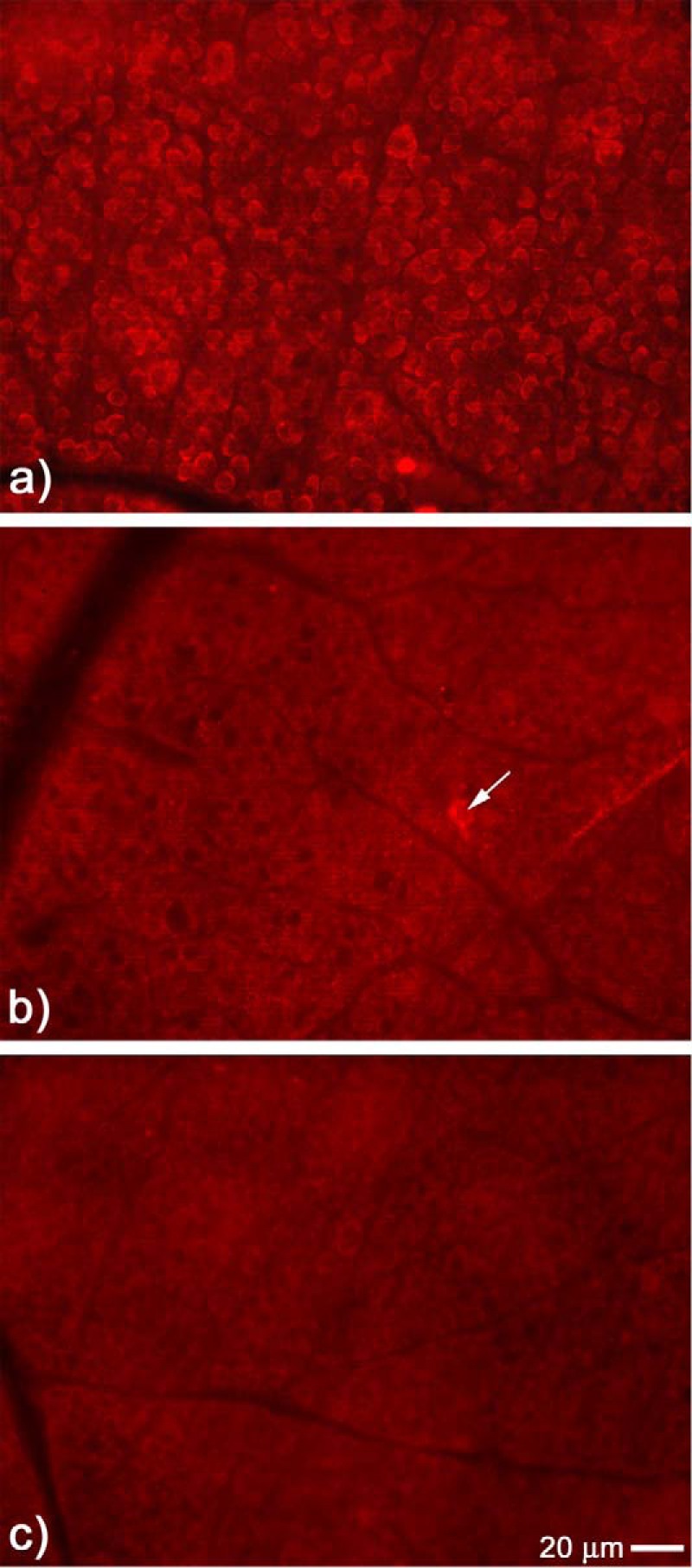
C-s-s-C-R(7) decreases NMDA-induced mitochondrial distress in the retina. Mitosox detection of mitochondrial distress was monitored 2 h following intravitreal injection with NMDA. a, flat mount view of the GCL exposed to NMDA alone displaying intense cytoplasmic mitosox fluorescent staining surrounding the majority of cells located in the GCL. b, in the presence of C-s-s-C-R(7), NMDA-induced mitosox staining was significantly attenuated (arrow shows one cell positive for staining). c, sham injection with PBS does not activate mitosox staining in the GCL.
Colocalization of C-R(7)-Biotin and MitoTracker in HEK293 Cells
HEK293 cells were grown to 50% confluence in DMEM (Gibco), supplemented with 10% fetal bovine serum. Cells were incubated with 1 nm C-R(7)-biotin for 2.5 h in fresh media at 37 °C. MitoTracker Red was added to the media to a final concentration of 500 nm, for an additional 30 min. Following aspiration, the cells were trypsinized, pelleted, and washed with PBS. Slides were prepared by cytospin (Cyto-Tek, Rankin) at 600 rpm. Cells were fixed in 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, dehydrated with graded alcohol, and cured overnight at room temperature. Mounted slides were preincubated with ChemiBLOCKER (Millipore) solution for 1 h, exposed to the streptavidin-Alexa 488 antibody (1:250 dilution) for 2 h, and stained with DAPI. Images were digitally photographed using a Leica TCS SP8 laser scanning confocal microscope (Leica Microsystems Inc, Buffalo Grove IL) with ×100 water objective.
Mitochondria Respiration Measurements
HEK293 cells were seeded 24 h before the measurements, trypsinized, washed, and resuspended in DMEM (without phenol red, FBS, and antibiotics) on ice. DMEM was removed after centrifugation, and the pellet was resuspended in measuring buffer (120 mm KCl, 3 mm HEPES, 5 mm KPi, 3 mm MgSO4, 1 mm EGTA, pH 7.2) to a protein concentration of 1 mg/ml. Respiration was measured at 35 °C in a closed oxygen electrode chamber (Oxygraph System, Hansatech) with a final volume of 500 μl following addition of 10 mm succinate as substrate, 3 mm ADP to induce state 3 respiration, and 6 μg of digitonin to solubilize the cell membrane. Increasing amounts of peptide were added from 0 to 500 nm.
Mitochondrial Membrane Potential Measurements
The mitochondrial membrane potential of intact cells was measured as described (28). Briefly, HEK cells were washed with PBS and trypsinized. The concentration of cells was adjusted to 0.2 mg/ml protein using DMEM without phenol red (21063, Gibco-Invitrogen) and without fetal bovine serum and antibiotics. Twenty nanomolar tetramethylrhodamine methyl ester (TMRM, T-668, Invitrogen) was added to the cells. As a control, the mitochondrial membrane potential was dissipated using 1 μm carbonyl cyanide 4-(trifluoromethoxy)-phenylhydrazone. The samples were incubated at 35 °C for 30 min in the dark under slow rotation. Yellow fluorescence (excitation 532-nm laser; emission, 585 nm, bandpass, 42 nm) was measured using a BD FACS Array (BD Biosciences), and data were analyzed with WinMDI version 2.9 software.
Reactive Oxygen Species Measurements
ROS were analyzed as described with minor modifications (28). In brief, HEK293 cells were seeded on a 12-well plate (Costar 3513; 1.1 × 105 cells per well) and cultured for 18 h, washed once with PBS, and incubated with the ROS-sensitive probe 2′,7′-dichlorodihydrofluorescein diacetate (D-399, Molecular Probes) at a final concentration of 8 μm and 240 nm CN2097, R(7), or R(7)-C or vehicle for 1 h at 37 °C in the dark in serum-free media. Cells were analyzed on an Ascent Fluoroskan plate reader (485 nm excitation; 527 nm emission) as described (28). Experiments were performed in quadruplicate, and data were standardized to the protein concentration as determined with the DC protein assay kit (Bio-Rad) after trypsinization of the cells.
Statistics
Unless otherwise indicated, statistical significance between treatment conditions were assessed using one-way analysis of variance (ANOVA) with post hoc comparison testing analysis using Tukey-Kramer multiple comparison test (GraphPad software).
Results
Polyarginine-linked TMR-CN2097 and the Myristoylated-linked Analog FITC-CN2180 Accumulate in Retinal Neurons
The retina has been found to be an excellent model for studying the mechanisms of glutamate-induced toxicity, with manyof the resulting studies contributing to our current understanding of the cell death signaling process in neurons (7). Utilizing an established in vivo retinal neurotoxicity rat model (26, 29) and following intravitreal injection, we compared the uptake properties of two different cell-penetrating compounds, a myristoylated linked (termed CN3205, Fig. 1) and a polyarginine-linked PSD-95 PDZ-binding cyclic peptide tagged with FITC (termed CN6005), to determine whether they could cross the inner limiting membrane of the retina and accumulate within retinal neurons. Results show that both CN3205 and CN6005 are not restricted by the inner limiting membrane (Fig. 2, a–e, respectively), with detectable uptake into retinal neurons noted as early as 2 h following injection (Fig. 2a). Retinas sampled at 4 and 6 h (Fig. 2, b and c, respectively) show CN3205 accumulation in GCL neurons increased in both number and staining intensity. Radial sectioning of the 6-h exposed retinas (Fig. 2d) shows that CN3205 accumulation is restricted to the inner half of the retina and was localized to cells in the GCL and in the innermost row of the inner nuclear layer. Substituting the myristoylated tail with a different transport moiety, consisting of a covalently linked peptide containing seven arginine residues (CN6005), resulted in a similar neuronal uptake pattern in the flat mount view of the GCL (Fig. 2e), signifying that attachment of polyarginine or a myristoylated tail to the PSD-95/PDZ domain-binding cyclic moiety is equally effective in permeating the vitreal/retinal barrier and accumulating within retinal neurons. These findings are in agreement with previous studies showing that CN2097 readily crosses the pia/glial barrier to accumulate within select populations of neurons in the spinal cord (22) and brain (24).
FIGURE 1.

Chemical structures of PSD-95/PDZ-binding peptidomimetic, control, and fluorescent-tagged compounds.
FIGURE 2.

Uptake of PDZ-PSD-95 binding peptides following intravitreal injection in the retina. Intravitreal injection of the fluorescent-tagged PSD-95/PDZ-binding peptide mimics CN3205 and CN6005 were monitored for uptake into retinal neurons located in the GCL. a, retinal flat mounts showing specific localization of the myristoyl-linked peptide CN3205 in the cytoplasm of neurons (white arrows) 2 h following intravitreal injection. An increased number and staining intensity of CN3205-labeled cells were noted between 4 h (b) and 6 h (c). d, radial sectioning of the retina shows numerous CN3205-labeled cells in the GCL, including larger ganglion cells (G) and in cells located in the innermost row of the inner nuclear layer (INL) indicated by white arrows. e, similarly, the fluorescent-tagged polyarginine-linked PSD-95-binding peptide mimic CN6005 displayed a comparable uptake pattern when viewed in retinal flat mounts at 6 h, demonstrating that both uptake carriers are equally effective in transporting the peptide mimic across the vitreal/retina barrier and into classes of neurons shown to be highly susceptible to NMDA-induced excitotoxity. Os, outer segments; ONL, outer nuclear layer; OPL, outer plexiform layer; IPL, inner plexiform layer. Bars, 10 μm.
PSD-95-binding Cyclic Peptide CN2097 Blocks NMDA-induced Poly(ADP-ribosyl)ation
The activity of poly(ADP-ribose) polymerase-1 (PARP-1), a nuclear enzyme facilitating DNA repair, is dramatically elevated following NMDA- and NO-induced neurotoxicity, significantly increasing poly(ADP-ribosyl)ation (PAR) of nuclear proteins in the distressed neurons (26, 29). In the retina, NMDA-induced PARP-1 activation is a divergent cell death pathway, whereby its blockade offers only partial neuroprotection against NMDA-induced cell death (26). Nevertheless, quantifying the decline in the levels of NMDA-induced PAR formation has proven to be a useful measure for monitoring the effectiveness of neuroprotective compounds targeting upstream cell death signaling initiator pathways (26). The PSD-95/PDZ-binding cyclic compound CN2097 (Fig. 1) was found to attenuate NMDA-induced PAR of high molecular weight proteins in the retina (Fig. 3a), with an optimal dose between 1.5 and 3 nmol to fully attenuate a 20-nmol NMDA insult (Fig. 3d). In contrast, the myristoylated version of this same compound CN2180 (Fig. 1) was ineffective in attenuating PAR activity using either 0.6 nmol (Fig. 3b) or 3 nmol (data not shown), and at 6 nmol it was shown to only marginally lower PAR (Fig. 3b). Increasing the amount of CN2180 to 60 nmol decreased NMDA-induced PAR formation by 45% (Fig. 3, b and e), further demonstrating that CN2180 was considerably less effective in blocking PAR formation compared with CN2097. These results suggest that the cyclic PSD-95 PDZ-binding motif, common to both CN2097 and CN2180, only partially contributes to protection against the NMDA receptor-induced PAR activation, raising the possibility that the noncyclic component of the chemical structure of CN2097 contributes to neuroprotection. To confirm that the partial attenuation of PAR activation by CN2180 is dependent upon the PSD-95/PDZ-binding cyclic peptide KTEV-β-alanine, we substituted the T and V residues with alanine (KAEA-β-alanine, CN3200 (Fig. 1), designed to render the myristoylated cyclic peptide ineffective in binding to PSD-95 (30, 31). Results show that CN3200 up to 6 nmol had no effect in attenuating NMDA-induced PAR activation (Fig. 3, c and f), showing that the PSD-95/PDZ-binding cyclic moiety itself plays a role in attenuating NMDA-induced PAR activation. Overall, these results show CN2097 is ∼40–100 times more effective than CN2180 in blocking NMDA-induced PAR activation, and as described below, CN2097 was further examined to monitor its effects on both short and long term neuronal cell distress and death.
FIGURE 3.

Dose response of PDZ-peptidomimetic ligands CN2180 and CN2097 in blocking NMDA-induced PAR in the retina. Intravitreal injections of myristoyl-linked PDZ-binding CN2180, myristoyl-linked non-PDZ binding control CN3200, and polyarginine-linked PDZ binding CN2097 compounds delivered into the left eye of each rat, at the indicated dose (nanomoles), 2 h prior to a second injection of compound in the presence of 20 nmol of NMDA, and the right eye received vehicle followed 2 h later with NMDA alone. PAR was detected using Western blot 6 h after the NMDA insult. a, polyarginine-linked PSD-95/PDZ-binding ligand CN2097 significantly attenuated NMDA-induced PAR formation at 1.5, 3, and 6 nmol, as compared with the companion eye exposed to NMDA alone (indicated by -). b, CN2180 (a myristoyl-linked PSD-95/PDZ-binding ligand) did not significantly block NMDA-induced PAR formation compared with the matched NMDA-treated retina using 0.6 or 6 nmol but did reduce PAR levels by ∼40% using 60 nmol. c, control CN3200 (myristoyl-linked non-PSD-95/PDZ domain-binding peptide) was ineffective in blocking NMDA-induced PAR formation at 1.5, 3.0, and 6.0 nmol. CN2097 (d), CN2180 (e), and CN3200 (f) show quantification of PAR levels normalized to the corresponding β-actin using a minimum of four retinas per dose tested. Error bars, S.E. * indicates a significant decline in PAR formation, as compared with NMDA treatment alone, n = 4 retinas/treatment condition. *, p < 0.01.
CN2097 Blocks NMDA-induced LPMS
A hallmark of NMDA-induced neuronal distress (nonprogrammed cell death pathway, e.g. necrosis) is the inability of NMDA-susceptible neurons to exclude the influx of ethidium bromide (EtBr). As reported previously (26), retinas exposed to 20 nmol of NMDA cause RGCs and displaced amacrine cells to exhibit LPMS against EtBr influx, which peaks 4 h post-insult (Fig. 4a). Treatment with CN2097 significantly blocked NMDA-induced LPMS in GCL cells (Fig. 4, b and h), as compared with NMDA-treated retinas (p < 0.0001, ANOVA, F(15/65) = 142.00; Tukey-Kramer multiple comparison test; Fig. 4, a and h), and CN2097-treated retinas were indistinguishable from untreated (Fig. 4, g and h) or PBS-treated (Fig. 4h) retinas (p > 0.05). For comparison, we also tested the Tat-based NR2B fusion peptide Tat-NR2B9c, previously shown to protect against excitotoxicity by uncoupling the GluN2B subunit from PSD-95-associated downstream cell death-signaling molecules, such as neuronal NOS (20) and p38 MAPK (32). Although Tat-NR2B9c attenuated NMDA-induced LPMS by roughly 50%, p < 0.001 (Fig. 4, c and h), it was significantly elevated compared with the untreated retina (p < 0.001), indicating that at this dose full protection was not achieved.
FIGURE 4.

Assessing the neuroprotective properties of the PSD-95/PDZ-binding peptidomimetic compounds against NMDA-induced LPMS in the retina. Representative flat mount views of the GCL were taken from an area 1–2 mm from the optic nerve head 4 h following NMDA insult. a, retinas exposed to NMDA alone (n = 5) displayed a densely labeled distribution of EtBr-positive cells. b, retinas exposed to the PSD/PDZ domain-binding peptide CN2097 (n = 4) fully attenuated NMDA-induced LPMS. c, PSD-95-interacting peptide Tat-NR2B9c (n = 4) was also shown to reduce but not fully attenuate NMDA-induced LPMS. The non-PDZ-binding cyclic controls containing the polyarginine uptake carrier CN5125 (n = 4) (d) and CN5135 (n = 3) (e) both fully attenuated NMDA-induced LPMS. f, CN2180 (n = 3), a myristoyl-linked PSD-95-binding cyclic peptide, did not block LPMS. g, no cell staining was detected in the untreated retina (n = 5). h, histogram displaying the average labeling distributions from the sample areas above. Error bars S.E. *, treatment condition is significantly different from NMDA alone, p < 0.0001.
PSD-95 PDZ Domain Binding Ligand of CN2097 Is Not Required for Mitigating NMDA-induced LPMS
As shown above, CN2097 pretreatment completely blocked NMDA-induced LPMS (Fig. 4, b and h), and the PSD-95 PDZ-binding peptide Tat-NR2B9c (Fig. 4c) provided significant attenuation of NMDA-induced LPMS, suggesting that ligand binding to PSD-95 is responsible for blocking this NMDA receptor-activated event. However, at variance with this conclusion is the observation above that CN2180, which has the identical PDZ-targeting cyclic ligand as CN2097 but with an uptake carrier linkage substituted with a myristoyl moiety in place of polyarginine (see Fig. 1), only partially protected against NMDAR-induced PAR activation (Fig. 3, b and e), raising the possibility that the noncyclic component of the chemical structure of CN2097 may also provide neuroprotective properties. To resolve this inconsistency, the PSD-95 PDZ-binding ligand of CN2097 (KTEV-β-alanine) was modified to prevent binding, so that the Thr and Val amino acids key to PDZ binding (23, 30) were replaced with either glycine residues (KGEG-β-alanine, termed CN5125) or with alanine residues (KAEA-β-alanine, termed CN5135), with the remaining composition of CN2097 unaltered, including the disulfide-linked polyarginine tail (Fig. 1); and they were tested for their ability to mitigate NMDA-induced LPMS. Supporting the proposal that the noncyclic component of CN2097 exhibits neuroprotective properties, both of the non-PDZ-binding cyclic analogs CN5125 (Fig. 4, d and h) and CN5135 (Fig. 4, e and h) significantly attenuated NMDA-induced LPMS in the retina and produced a distribution pattern of EtBr-labeled cells that was statistically the same as for CN2097-treated and -untreated retinas (p > 0.05, Fig. 4h). Furthermore, the myristoylated PDZ-binding cyclic peptide CN2180 did not block NMDA-induced LPMS in the retina (Fig. 4, f and h), being statistically indistinguishable from NMDA-treated retinas (p > 0.05, Fig. 4, a and h). From these results, we conclude that the ability of CN2097 to block NMDA-induced LPMS is not primarily mediated by the interaction of the cyclic ligand with PSD-95.
Polyarginine Blocks NMDA-induced LPMS
Having discounted the PDZ domain-binding moiety of CN2097 as being responsible for attenuating NMDA-induced LPMS, we tested whether the polyarginine uptake carrier peptide, common to all the protective compounds (e.g. CN2097, CN5135, and CN5125: Fig. 1), plays a role in mediating this blockade. Replicating the treatment conditions above, we found that R(7) (Table 1 and Fig. 5a) and R(7) peptides retaining the N-terminal cysteine used to link R(7) via a disulfide bridge to PSD-95-binding ligands, e.g. C-s-s-C-R(7) (Table 1 and Fig. 5b) and C-R(7) (Table 1 and Fig. 5c), all attenuated NMDA-induced LPMS (p < 0.0001) to a significance indistinguishable from CN2097, CN5135, CN5125, and untreated retina (p > 0.05, Fig. 5h). These data demonstrate that the polyarginine-rich peptide is responsible for providing short term protection against NMDA-induced LPMS.
FIGURE 5.

Assessment of polyarginine derivatives for attenuation of NMDA-induced LPMS. a, R(7) (n = 5). b, C-s-s-C-R(7) (n = 4), and c, C-R(7) (n = 4) fully attenuated NMDA-LPMS. d, DC-s-s-DCDR(7) (n = 4), containing substituted d-isomers of arginine and cysteine, was found equally effective in blocking NMDA-induced LPMS as the l-isoform (a–c). e, C-s-s-C-R(5) (n = 4), a derivative containing five arginine residues, was effective in attenuating NMDA-LPMS. f, C-s-s-C-R(3) (n = 4), a derivative containing three arginine residues, had no visible effect in preventing NMDA-LPMS. g, Tat (n = 4), an arginine-rich Tat peptide, produced full attenuation of NMDA-LPMS. h, histogram of all data shows average distribution of EtBr cells in NMDA-treated retinas (dashed line) and untreated retina (dotted line). Error bars S.E., * treatment condition is significantly different from NMDA alone, *, p < 0.0001.
We next examined whether the C-s-s-C-R(7) ability to block NMDA-induced LPMS was stereospecific by substituting d-isomers of cysteine and arginine. We tested the effects of DC-s-s-DC-DR(7) (Table 1) and found that it was equally effective in blocking NMDA-induced LPMS (p > 0.05, n = 5; Fig. 5, d and h) as the l-form C-s-s-C-R(7), indicating that the protective response produced by polyarginine is not stereospecific. With R(7), C-R(7), and C-s-s-C-R(7) each providing full blockade against NMDA-induced LPMS, we sought to determine the minimal number of sequential arginine amino acids required to provide full protection against NMDA-induced LPMS. Results show that C-s-s-C-R(5) significantly attenuated NMDA-induced LPMS (p < 0.001: Fig. 5, e and h); however, full protection was not obtained, as the level of EtBr staining was significantly higher than retinas treated with C-s-s-C-R(7) or untreated retinas (p < 0.01, and p < 0.001 respectively, Fig. 5j). In contrast, C-s-s-C-R(3) (Fig. 5, f and h) did not attenuate NMDA-induced LPMS (p > 0.05). We also found that a Tat peptide (Table 1, YGRKK7R6R5Q4R3R2R1), lacking the GluN2B-PDZ-binding motif present on Tat-NR2B9c, was equally effective in blocking NMDA-induced LPMS as R(7), C-s-s-C-R(7), C-R(7) (p > 0.05, Fig. 5, g and h), and Tat-NR2B9c (Fig. 4h). This suggests that Tat-NR2B9c blockade of NMDA-induced LPMS does not require the PSD-95/PDZ domain-binding interaction.
Polyarginine Blocks NMDA-induced Caspase Activation
We have previously shown that intravitreally injected NMDA induces peak caspase-3 activation in the rat retina 2 h following insult (29). We next examined the effects of CN2097, CN2180, R(7), and C-R(7) in blocking NMDA-induced caspase activation using the in vivo administered fluorescent substrate FAM-VAD-FMK, a carboxyfluorescein derivative of valylalanyl-aspartic acid fluoromethyl ketone, which provides a green fluorescent signal in the presence of activated caspases. Retinas treated with NMDA alone showed positive caspase activation within numerous GCL cells (Fig. 6a). Results show that CN2097 fully blocked NMDA-induced caspase activation in the retina (Fig. 6b), with a staining pattern indistinguishable from the untreated retina (ANOVA, Tukey-Kramer Multiple Comparison Test, F(5/31) = 269.75, p > 0.05; Fig. 6, f and g). Similarly, treatment with R(7) or C-R(7) fully blocked NMDA-induced caspase activation (Fig. 6, c and d, respectively), showing no significant differences in the labeling distributions between the untreated retina, C-R(7) and R(7) (Fig. 6g, ANOVA, p > 0.05). In contrast, the myristoylated cyclic analog CN2180 did not significantly block NMDA-induced caspase activation (Fig. 6e) and displayed a cellular distribution identical to retinas treated with NMDA alone (Fig. 6a). These data are consistent with our findings that blockade of NMDA-induced cell death signaling is polyarginine-dependent.
FIGURE 6.

Assessment of PSD-95/PDZ-binding peptidomimetic compound inhibition of NMDA-induced caspase activation in the rat retina. Caspase activation was monitored by increased fluorescence in retinal flat mounts 6 h post-NMDA treatment. a, caspase activity was noted within numerous cells located in the GCL in the NMDA-treated retina (n = 6) but was absent in retinas treated with CN2097/NMDA (n = 6) (b), R(7)/NMDA (n = 5) (c), and C-R(7)/NMDA (n = 5) (d). CN2180/NMDA (n = 5)-treated retinas failed to block NMDA-induced caspase activation (e) compared with untreated retina (n = 5) (f). g, quantification of caspase-positive cells for each treatment condition, sampled 1–2 mm from the optic nerve head. Error bars S.E. * indicates a significant difference from NMDA-treated retina, p < 0.0001.
Long Term Protection by Polyarginine Requires a Terminal Cysteine Residue
Although R(7) and its derivatives C-R(7) and C-s-s-C-R(7) provide short term blockade of NMDA-induced LPMS and caspase activation, it is possible that the PSD-95 PDZ-domain binding motif of CN2097 is required for long term protection against NMDA-induced loss of functional retinal ganglion cells. To access RGCs that maintain functional axon connections to the superior colliculus (33), we performed retrograde labeling of these cells using fluorogold, as described previously (26, 29).
In agreement with previous work (29), a 20-nmol NMDA insult results in a significant loss in the ability of RGCs to transport the dye to their somas (Fig. 7a). Retinas treated with CN2097 provided full protection against NMDA-induced RGC loss (Fig. 7b), whereas the myristoylated cyclic analog CN2180 had no effect (Fig. 7c). Regional quantification from montages of retinal flat mounts illustrated in Fig. 7p showed that retinas exposed to CN2097/NMDA treatment displayed regional cell distributions that were not significantly different from untreated retinas (Fig. 7, d and q) (ANOVA, Tukey-Kramer multiple comparisons test, F(13/77) = 37.249, p > 0.05; Fig. 7, q and r), whereas treatment with CN2180 did not show a statistically significant difference from retinas exposed to NMDA alone (Fig. 7q). Similar to CN2180, and consistent with other studies using a strong NMDA insult (32), Tat-NR2B9c was ineffective in attenuating NMDA-induced loss of retinal ganglion viability (Fig. 7h), and the distributions of labeled RGCs in Tat-NR2B9c-treated retinas were not significantly different from NMDA-treated retinas (p > 0.05, Fig. 7, a and r).
FIGURE 7.


Assessing long term viability of retinal ganglion cells 14–18 days post-NMDA insult by monitoring retrograde uptake of fluorogold into retinal ganglion cells. Representative images of fluorogold uptake into viable RGCs sampled 1–2 mm from the optic nerve head are shown for each treatment condition. a, NMDA treatment alone (n = 5) shows a dramatic reduction in the distribution of labeled RGCs. b, CN2097 (n = 5) provided full protection against the NMDA-induced loss of viable RGCs. c, CN2180 (n = 3), the myristoylated PSD-95/PDZ domain-binding ligand, shows a reduction in labeled RGCs comparable with NMDA treatment alone. d, untreated retina (n = 6) showing the distribution of all viable RGCs. Retinas treated with non-PDZ-binding control peptides were linked via a cysteine/cysteine disulfide bridge to C-R(7). CN5135 (n = 5) (e), CR1105 (n = 3) (f), and lin-CN2097 (n = 5) (g) all attenuated NMDA-induced loss of the RGC uptake of fluorogold. Tat-NR2B9c (n = 4) (h) and R(7) (n = 4) (i) were ineffective in attenuating the effects of NMDA. However, adding a terminal cysteine to R(7) to generate C-R(7) (n = 4) (j) or stabilizing it via disulfide bridge forming C-s-s-C-R(7) (n = 5) (k) were all fully protective. Shortening the number of consecutive arginines to five, C-s-s-C-R(5) (l) (n = 3) was still protective; however, reducing to three arginines (m) C-s-s-C-R(3)/NMDA (n = 3) had no effect in blocking NMDA. Substituting d-amino acids showed that DC-s-s-DC-DR(7) (n = 4); n was equally effective in blocking NMDA effects. o, Tat-C-s-s-C (n = 3) also fully blocked the effects of NMDA. Bar, 20 μm. p, schematic of a typical photo-montage from a fluorogold-labeled retinal flat mount illustrates the regions sampled for obtaining labeled cell distributions. Each montage was binned into 1-mm concentric ring increments centered about the optic nerve head (ONH) (area I, 0–1 mm; area II, 1–2 mm; area III, 2–3 mm; area IV, 3–4 mm). q, distributions of fluorogold uptake into RGCs were assessed across the full extent of each retina following treatments with NMDA alone, CN2097/NMDA, and CN2180/NMDA and compared with untreated retina. Bars represent the average cell distribution per mm2 ± S.E. Average areas sampled for each region were as follows: area I, 2.48 ± 0.69 mm2; area II, 7.07 ± 1.51 mm2; area III, 10.44 ± 2.20 mm2; area IV, 16.26 ± 4.37 mm2; n = 90 retinas sampled. r, for ease of comparison of all the treatment conditions, area II (1–2 mm from the optic nerve head) was chosen to evaluate neuroprotection (quantification of the number of retinas/treatment condition in graphs q and r are listed following each treatment condition described in legends a–o above). Error bars S.E., * indicates a significant difference from NMDA-treated retina, p < 0.001.
As only the polyarginine-containing compound CN2097 provided long term protection against NMDA-induced loss of RGC viability, we next examined the protective ability of polyarginine moiety-containing peptides, which had their PDZ domain binding altered, either by amino acid substitutions of the cyclic backbone (CN5135 and CR1105, Fig. 1) or by linearizing the cyclic component of CN2097 (designated lin-CN2097 in Table 1). In this next series of studies, we chose to quantify RGC distributions between 1 and 2 mm from the center of the optic nerve head, where peak distributions in untreated retina are located (Fig. 7r). Results show that CN5135 (Fig. 7e), CR1105 (Fig. 7f), and lin-CN2097 (Fig. 7g) provided full protection that was not significantly different from retinas treated with CN2097 or untreated retinas, p > 0.05 (Fig. 7r). These data, together with the ability of these peptides to fully block NMDA-induced LPMS, support a role for the polyarginine linker in mediating RGC viability in the presence of NMDA.
Having ruled out the requirement for a PSD-95/PDZ domain-targeting motif for short and long term neuroprotection against NMDA-induced cell distress and death in the retina, we next compared the effects of R(7), C-R(7), and C-s-s-C-R(7) on preserving long term RGC viability. Although R(7) provided short term protection against NMDA-induced LPMS and caspase activation (Figs. 5a and 6f), it was ineffective in blocking NMDA-induced loss of RGC viability (Fig. 7i). Quantification of fluorogold labeling showed that these retinas were not significantly different from retinas treated with NMDA alone (Fig. 7, a and r, p > 0.05). In contrast, both C-R(7) (Fig. 7j) and C-s-s-C-R(7) (Fig. 7k) fully attenuated NMDA-induced inhibition of fluorogold uptake and were indistinguishable from untreated retinas (Fig. 7r). Additionally, with both d- and l-isoforms of C-s-s-C-R(7) shown to provide short term protection (Fig. 5h), we found that DC-s-s-DCDR(7) was also effective in blocking the effects produced by an NMDA insult (Fig. 7, o and r, p < 0.05), indicating that neuroprotection is not stereospecific. Similar to short term neuroprotection, decreasing the number of consecutive arginine amino acids compromised long term protection against NMDA-induced loss of viable ganglion cells. Thus, C-s-s-C-R(5), although still able to significantly attenuate NMDA-loss of ganglion cell viability (Fig. 7, l and r, p < 0.05), was less effective than C-s-s-C-R(7) (Fig. 7k), and as predicted from its inability to block NMDA-induced LPMS (Fig. 5, h and j), C-s-s-C-R(3) provided no protection (Fig. 7, m and r).
To further examine the role of a terminal cysteine, we attached to a Tat-peptide a C-terminal cysteine linked to a free cysteine via a disulfide bridge (C-s-s-C-Tat, Table 1). Results show that C-s-s-C-Tat peptide was equally as effective as C-R(7) and C-s-s-C-R(7) in blocking the loss of RGC viability by NMDA (p > 0.05, Fig. 7, o and r). As the Tat peptide consists of a mix of arginine and lysine amino acids (Table 1), these data suggest that provided the peptides are cell-penetrating, which optimally necessitates the guanidinium headgroup of arginine-rich peptides (34), it is the positive charge of the peptide that is the driving force for providing blockade from an NMDA-excitotoxic challenge. Thus, it may be the polycationic character of the peptide, rather than a consecutive string of five or more arginines, together with a terminal cysteine that is necessary to attenuate long term NMDA-induced neuronal distress.
Polyarginine Does Not Block NMDAR Activation in the Retina
With full protection being provided by C-R(7) derivatives against NMDA-induced LPMS and on maintaining long term viability of RGCs, we sought to determine whether this protection was the result of polyarginine acting as a direct NMDAR antagonist. To test this possibility, rat retinas were prepared for multielectrode array (MEA) recording of RGCs, and their ability to respond to light-driven synaptic input was assessed. Responses to an intensity series of light stimuli were first recorded while the retinas were superfused with either normal Ames' medium or Ames' medium supplemented with R(7), C-s-s-C-R(7), or the NMDAR antagonist MK-801. Robust light responses were observed in all MEA channels under all four conditions (see Fig. 8, B, panel i, C, panel i, D, panel i, and E, panel i for examples and the open circles in Fig. 8, B, panel iii, C, panel iii, D, panel iii, and E, panel iii for population data), indicating that R(7), C-s-s-C-R(7), or MK-801 alone do not block rod/cone signaling through RGCs. NMDA (500 μm) was then added to the bathing solutions, and the same intensity series of lights presented again. Results show that all light-evoked responses were virtually eliminated when NMDA was applied alone (Fig. 8B, panels ii and iii, black triangles) or when it was applied in the presence of R(7) or C-s-s-C-R(7) (Fig. 8, C, panel ii, and D, panel ii, C, panel iii, and D, panel iii, black triangles). In contrast, light responses persisted when NMDA was applied on top of MK-801 (Fig. 8, E, panels ii and iii). Collectively, these data confirm that R(7) and C-s-s-C-R(7) do not block NMDAR function in the intact retina.
FIGURE 8.

Effects of select peptides in altering NMDA-induced loss of light responses in the retina. A, method for quantifying ganglion cell photoresponses. Example recordings from an MEA channel, obtained while the retina was superfused with Ames' medium containing 2 μm R(7) (top recording trace) and after the addition of 500 μm NMDA (bottom recording trace). The stimulus was a 1-s light step with an intensity of 4.1 × 1012 photons cm−2 s−1. Photoresponse amplitude was quantified as the difference in variance between the 1 s during light treatment and the 1 s immediately before light onset (B–E). Panels i, example recordings obtained when retinas were superfused with normal Ames' medium (B, panel i), Ames containing 2 μm R(7) (C, panel i), Ames containing 2 μm C-s-s-C-R(7) (D, panel i), and Ames containing 2 μm MK-801 (E, panel i). Stimulus intensity was 4.1 × 1011 photons cm−2 s−1 in all cases. Panels ii, recordings from the same MEA channels after 500 μm NMDA had been added. Panels iii, photoresponse amplitudes averaged from all MEA channels, before (open circles) and during (black triangles) NMDA treatment. For each MEA channel, the amplitude of the response to the highest intensity stimulus in the pre-NMDA condition (open circles) was normalized to 1.
C-s-s-C-R(7) Rescues NMDA-induced Mitochondrial Oxidative Stress in the Retina
It is well documented that a strong NMDA insult causes mitochondrial failure and, depending upon the degree of stimulation, will initiate cell death within the susceptible neurons by way of programmed and/or nonprogrammed cell death pathways (32, 35, 36). Using the mitochondrion-specific probe MitoSOX, which becomes fluorescently activated by the generation of superoxide resulting from mitochondrial oxidative stress, we examined retinas exposed to 20 nmol of NMDA alone, C-s-s-C-R(7)/NMDA, or PBS-sham 2 h following intravitreal injection. Results from retinal flat mounts showed that NMDA alone elicited an intense MitoSOX staining surrounding the majority of cells in the GCL (Fig. 9a), which resembled the EtBr-staining pattern observed in NMDA-treated retinas (Fig. 4a). In contrast, retinas exposed to C-s-s-C-R(7)/NMDA (Fig. 9b) showed diffuse labeling in small puncta surrounding the majority of neurons in the GCL, similar to the staining pattern displayed by PBS-sham-treated retinas (Fig. 9c). These data demonstrate that C-s-s-C-R(7) provides protection against excitotoxin-induced mitochondrial oxidative stress in NMDA-receptive neurons.
Localization of C-s-s-C-R(7) to Mitochondria
As all cyclic derivatives containing the C-R(7) uptake peptide demonstrated full protection against an NMDA insult in our in vivo retinal toxicity model, we next addressed whether the neuronal localization of fluorescent-tagged C-s-s-C-R(7) was unique from the uptake pattern produced by the two PSD-95/PDZ domain-binding peptides CN3205 and CN6005 (e.g. CN2180 and CN2097) shown in Fig. 2. Results show that C-s-sC-R(7)-FITC localizes to small membrane-bound structures ranging in size from 0.2–0.8 μm in diameter 6 h following intravitreal injection (Fig. 10a). These labeled structures were located in the perinuclear cytoplasm of GCL cells (see arrows in Fig. 10a), as well as within RGC axon bundles (designated ab). The relative size and distribution within the RGCs suggested that C-s-s-C-R(7) may be targeted to the mitochondria in these neurons. To verify mitochondrial targeting by C-R(7), immortalized kidney epithelial cells (HEK293 cells) were cultured and treated with C-R(7)-biotin (Table 1) in the presence of the mitochondrion-specific marker MitoTracker followed by post-fixation visualization of the biotin probe with streptavidin-Alexa 488. Results show that C-R(7)-staining in HEK293 cells localized to numerous membrane-bound structures within the cytoplasm of HEK293 cells (Fig. 10b). MitoTracker staining of the same cells (Fig. 10c) was shown to colocalize with the majority of C-R(7)-stained structures (Fig. 10e) demonstrating that C-R(7) is targeted to the mitochondria.
FIGURE 10.

Localization of C-s-s-C-R(7) in the retina and colocalization in mitochondria of HEK293 cells. a, uptake of C-s-s-C-R(7)-FITC was visualized in retinal flat mounts and shown to preferentially accumulate in the perinuclear cytoplasm of cells located in the GCL (arrows), as well as in small vesicle-contained structures that are consistent with the positioning on mitochondria within retinal ganglion cell axon bundles (indicated ab in the figure). b–e, HEK293 cells were incubated with biotin-labeled C-s-s-C-R(7) followed by incubation with the mitochondrion-specific dye Mitotracker, with post-fixation visualization of biotin with streptavidin-Alexa 488, and cell nuclei with DAPI. Confocal imaging shows localization of biotin-labeled C-s-s-C-R(7) (green) (b), Mitotracker staining (red) (c), and DAPI staining (blue) (d). Merged image shows that the majority of biotin C-s-s-C-R(7) uptake in HEK293 cells colocalize with Mitotracker (e).
Polyarginine Derivatives Attenuate Mitochondrial Hyperpolarization
Mitochondrial dysfunction has been implicated in acute and chronic degenerative disease (37). It is believed that activation of the proton pumps in the electron transport chain, followed by ΔΨm hyperpolarization, leads to excessive reactive oxygen species (ROS) production signaling cell death (37). With R(7), C-R(7), and CN2097 displaying strong neuroprotective properties, and the C-R(7)-biotin peptide demonstrating affinity to mitochondria, we next tested these three compounds directly for key readouts of mitochondrial function. We first analyzed mitochondrial respiration and observed a concentration-dependent decrease in respiration in the presence of each of the tested compounds. Results show that C-R(7), R(7), and CN2097 all significantly reduced mitochondrial respiration as compared with untreated control samples in a concentration-dependent manner (Fig. 11a, ANOVA, p < 0.0001, F(5/29) = 64.51).
FIGURE 11.

C-R(7) containing peptides all reduce mitochondrial respiration, mitochondrial membrane potential, and generation of radical oxygen species in a dose-dependent matter. a, HEK293 cells were incubated with peptides (CN2097, C-R(7), R(7), n = 4 for each) at the indicated concentrations for 30 min at 37 °C. Mitochondrial respiration was analyzed in digitonin-permeabilized cells in measuring buffer (see “Materials and Methods”) in the presence of succinate and ADP using a computer-controlled closed micro-oxygen electrode chamber. Respiration rates were normalized to untreated controls and are presented as % change relative to control (n = 4). b, HEK293 cells were incubated with peptides CN2097, C-R(7), and R(7) (n = 4 for each) for 30 min at 37 °C, at the concentrations indicated. TMRM fluorescence was analyzed by flow cytometry. Results show that all three peptides resulted in a significant reduction in TMRM fluorescence, indicating a reduction of the mitochondrial membrane potential. c, detection of ROS in cultured HEK293 cells by the ROS-sensitive probe 2′,7′-dichlorodihydrofluorescein diacetate was assessed in the presence of 240 nm of either CN2097, C-R(7), or R(7) and compared with matched seeded and untreated cells. Results showed that all three peptides significantly reduced detectable ROS generation (n = 4 for each condition). *, significance from control p < 0.0001; p, error bars S.D.
Based on this observation, we predicted that all three peptides would also reduce ΔΨm. This was assessed using the voltage-dependent mitochondrial probe TMRM, which accumulates in the mitochondria as a function of the magnitude of the ΔΨm. Results showed ΔΨm was significantly reduced as compared with controls in a concentration-dependent manner (Fig. 11b) by CN2097 (ANOVA, p < 0.001, F(4/19) = 169.59), C-R(7) (ANOVA, p < 0.001, F = 28.602), and R(7) (ANOVA, p < 0.0001, F(4/19) = 37.286). These data demonstrate that the neuroprotection provided by the R(7) and C-R(7) peptides, including CN2097, is the result of their affinity for targeting the mitochondria and their resulting reduction in the resting ΔΨm.
Given the direct correlation between the ΔΨm and ROS (38) and the demonstration that R(7) and all of the C-R(7) derivatives tested provided full protection against NMDA-induced short term neurotoxicity, we predicted that all three compounds would also reduce mitochondrial generation of ROS. To test the effect of the compounds on ROS production, we analyzed CN2097, R(7), and C-R(7) in conjunction with the ROS probe CM-H2DCFA. We observed, even under baseline conditions (i.e. without exogenous stress), a significant decrease in fluorescent signal indicating that the polyarginine derivatives are responsible for reducing ROS production (Fig. 11c, ANOVA p < 0.0001, F(3/23) = 23.768).
Discussion
A number of studies show that synaptic and extrasynaptic NMDARs impose counteracting effects on cell death with synaptic NMDA receptors stimulating pro-survival AKT and CREB phosphorylation signaling (13, 16, 17). Synaptic activity has been shown to promote the release of BDNF (39) to activate synaptic TrkB signaling (40, 41) required for the induction and maintenance of LTP and the pro-survival PI3K/Akt signaling pathway (42–46). We have developed a new cyclic peptide-based compound, CN2097, that selectively binds the synaptic scaffold protein PSD-95 to potentiate BDNF signaling and promote LTP (24). Here, using an in vivo retinal toxicity model (26), we investigated the neuroprotective effects of CN2097, which we predicted would augment the neuroprotective effects of endogenous BDNF (47). We found that CN2097 significantly attenuated the activation of NMDA-induced cell death pathways linked to caspase 3-dependent, independent, and PARP-1-activated cell death pathways in the retina. Furthermore, CN2097 provided full protection against NMDA-induced LPMS and fully blocked the loss of long term RGC viability. However, when the polyarginine cationic cell-penetrating peptide moiety of CN2097 was substituted with a myristoyl group, this analog, CN2180, only attenuated NMDA receptor-induced PAR activation by 45% (Fig. 3, b and e), was insufficient to prevent LPMS and caspase activation, but more importantly failed to prevent loss of RGC viability and long term survival. Nevertheless, the neuroprotective efficacy of the cyclic PSD-95 PDZ-binding moiety of CN2180 is comparable with that of the PSD-95-binding Tat-NR2B9c peptide (Fig. 7r) (32), which is also not protective at higher NMDA concentrations (32). Similar to the modulatory effect of CN2097 on TrkB signaling, Tat-NR2B9c also acts to selectively enhance CaMKIV and CREB activation (48). These results suggest that although the PSD-95-dependent pro-survival pathway is sufficient to maintain and protect neurons from a low level NMDA insult, this pathway cannot overcome the strong NMDA insult administered in our model. As discussed below, further studies revealed that the polyarginine-rich (Arg-rich) transport moiety, C-R(7), independently provided complete protection against NMDA-induced death.
Increasing evidence demonstrates that Arg-rich peptides are neuroprotective in excitotoxic models (49, 50). In in vitro studies, cell-penetrating peptides were recently reported to protect dissociated cortical neurons against excitotoxic doses of NMDA (50, 51). Furthermore, Meloni et al. (49) reported that only long Arg-rich peptides, containing nine or more arginines, demonstrated neuroprotective properties. Extending these studies, we show using an in vivo retinal neuroprotection model in which the retina is subjected to a strong NMDA insult (20 nmol of NMDA into the vitreal chamber corresponding to ∼300 μm) that the Arg-rich peptides Tat, Tat-NR2B9c, R(7), C-R(7), both d- and l-isoforms of C-s-s-C-R(7) and the cyclic peptides that were linked via a disulfide bridge to C-R(7), including CN2097, all blocked “short term” NMDA-induced LPMS. Consistent with an earlier report that the Tat-NR2B9c peptide is not protective at higher NMDA concentrations (32), Tat-NR2B9c, Tat, and R(7) did not prevent NMDA-induced “longer term” loss of RGC viability with this strong NMDA insult (Fig. 7r). However, in a novel finding we show that adding a terminal cysteine to these short polyarginine-rich peptides (e.g. C-s-s-C-R(7), Tat-C-s-s-C) provided long term RGC protection at levels comparable with those produced by CN2097, in which R(7) is linked to the PSD95/PDZ-binding cyclic peptide via a cysteine-cysteine disulfide bridge (Fig. 7r).
The neuroprotective mechanism of polyarginine peptides has been hypothesized to involve the suppression of calcium influx resulting from peptide-induced internalization of the GluN2B NMDA receptor and transporters (49). Testing this hypothesis, we recorded NMDAR activity using multielectrode-array recording of RGCs and found that polyarginine compounds do not interfere with NMDA blockade of light-driven synaptic responses, confirming that the neuroprotection provided by R(7), C-R(7), and CN2097 is not the result of NMDA-receptor antagonism in the intact retina. Supporting this conclusion, it has been reported that Tat does not induce internalization of the NMDA NR2B subunit and the sodium calcium exchanger (NCX) proteins, whereas the neuroprotective peptide Tat-CBD3, a 15-amino acid peptide from CRMP2 fused to Tat, attenuated NMDAR activity and protected neurons against glutamate-induced Ca2+ dysregulation (52).
As mitochondria are key players in signaling neuronal death (35, 53, 54), and with biotin-C-R(7) peptide localizing to mitochondria (Fig. 10, b–d) and structures within the retina resembling mitochondria (Fig. 10a), we assessed whether this specific mitochondrial localization could influence mitochondrial function. Novel to this study, we showed that R(7), C-R(7), and CN2097 all significantly lowered the ΔΨm, an outcome consistent with the observed decreases in mitochondrial respiration and generation of ROS in resting HEK293 cells (Fig. 11). These data strongly suggest that the short term neuroprotection provided by the R(7) and C-R(7) peptides, including CN2097, are the result of their affinity for mitochondria and their ability to reduce the resting ΔΨm.
Stimulation of extrasynaptic NMDA receptors leads to Ca2+ cycling across the mitochondrial membranes, collapse of the proton gradient, and loss of ΔΨm at a later stage, leading to cell death by apoptosis or necrosis (16, 53). An initial trigger for mitochondrial apoptosis, which precedes the release of cytochrome c, is a transient hyperpolarization of the ΔΨm after cells are exposed to stress (55). We and others have proposed that stress signals lead to calcium release, followed by changes of the phosphorylation state of the proton pumps (i.e. complexes I, II, and IV) in the mitochondrial respiratory chain, which generate ΔΨm (37, 56). Under such conditions the proton pumps are hyperactive, leading to a hyperpolarization of the mitochondrial membrane potential. This in turn leads to a burst in ROS production. Importantly, the connection between the ΔΨm and ROS is exponential when ΔΨm values exceed 140 mV (38). We therefore propose that under conditions of stress the following sequence of events takes place: cellular stress↑ → mitochondrial respiration↑ → ΔΨm ↑ → ROS↑ → cell death↑. Thus, under stress conditions polyarginine compounds may act to reduce the activity of the proton pumps resulting in a normalization (i.e. a beneficial reduction) of ΔΨm during stress and a decrease in ROS production favoring cell survival.
Although the mitochondrial target of C-R(7) has yet to be identified, we speculate that the guanidinium groups in the poly-l-arginine peptide could act in a similar fashion to guanidinium-containing compounds known for their important biological roles on mitochondria. During ischemia-reperfusion injury or stroke, oxidative damage from elevated production of ROS results in a sudden increase in the mitochondrial permeability transition pore (mPTP) to solutes (53), and guanidinium-like inhibitors of ATP hydrolysis have been reported to protect against myocardial ischemia (57). The biguanide metformin, widely prescribed for type II diabetes, is reported to inhibit calcium-induced mitochondrial permeability transition and cell death (58). The positive charge of metformin was proposed to account for its accumulation within the matrix driven by ΔΨm (59). Our results indicate that the local positive charge of C-R(7) plays a role in attenuating NMDA-induced neuronal distress, as decreasing the number of consecutive arginine bases significantly reduces protection. These results parallel previous reports in isolated mitochondria, showing that polycations, including tetrapeptides containing four basic amino acids of arginine, lysine, or ornithine attenuated the loss of ΔΨm induced by calcium, whereas three residues provided partial inhibition (60).
It has been reported that the mitochondrial permeability transition pore can be regulated by the oxidation of sulfhydryl groups and membrane surface potential (61) and following ΔΨm depolarization, would likely favor the formation of a disulfide bridge with C-R(7) within the pore, as supported by studies showing that dithiol, through dithiol-disulfide interconversions, control the pore conformation of the mPTP (62). The anchoring of the positively charged C-R(7) peptide within the channel could then interfere with the otherwise massive calcium entry into the mitochondrial matrix. Another possible target for C-R(7) to interfere with NMDAR-induced calcium influx into the mitochondria would be the mitochondrial calcium uniporter (Mcu). Recent studies established the role of Mcu in coupling NMDA receptor stimulation to neuronal excitotoxicity (35), where calcium entry through Mcu mediates mPTP opening. Thus C-R(7) could act similarly to the hexavalent cation ruthenium red and the derivative Ru360, which have been reported to selectively block the Mcu in isolated mitochondria and to suppress the eventual glutamate-induced loss of ΔΨm (63).
Relative to maintaining RGC function, RGCs have long thin axons (0.2–0.7 μm in diameter) with the unmyelinated region positioned on the inner surface of the retina. They are metabolically demanding and susceptible to mitochondrial dysfunction and neuropathy under conditions of ischemia, glutamate excitotoxicity, and glaucoma (33). Recent studies have identified several novel cell-penetrating peptides that limit mitochondrial damage. The Szeto-Schiller peptides consist of alternating aromatic and basic residues that concentrate in the mitochondria to reduce ROS (64). Further evidence of mitochondrial interactions of cell-penetrating peptides with mitochondria is described by Guo et al. (65), where Tat-conjugated peptide inhibitors may potentially reduce the excessive mitochondrial fusion associated with neurological disorders, including Parkinson disease.
Glutamate-induced oxidative stress and retinal ischemia promote mitochondrial dysfunction (66, 67), and novel strategies eliciting prolonged mitochondrial stabilization, such as regimented C-R(7) treatment, may protect RGC function against glaucomatous damage (7). On a broader scale, therapeutic treatment with C-R(7) may have the potential to minimize mitochondrially induced damage resulting from stroke or traumatic brain injury (55, 68) and possibly serve to ameliorate slow progressive neurodegenerative disorders such as multiple sclerosis and Parkinson disease (69).
Author Contributions
J. M., M.H., K. Y. W., and D. J. G. conceived and coordinated this study and wrote/edited the manuscript. D. J. G. designed and analyzed the experiments displayed in Figs. 2–7 and 9. K. Y. W. in coordination with D. J. G. and X. Z. designed the retina multielectrode array experiments and its analysis shown in Fig. 8. C. N. R., R. T., K. P., and M. R. S. designed and synthesized all of the peptides used in this study. M. R. S. and K. P. also contributed to the editing of the manuscript. M. H. designed and coordinated the mitochondrial studies shown in Fig. 11, and J. L. and I. L. were responsible for their execution and analysis. C. S. was responsible for designing and implementing the colocalization of CR(7) in mitochondria of the HEK293 cells shown in Fig. 10. E. D. B. was responsible for the Western blot data displayed in Fig. 2 and devoted countless hours assisting D. J. G. with experimentation and quantifying the majority of data presented in Figs. 3 and 7.
This work was supported, in whole or in part, by National Institutes of Health Grants R21 NS061176 (to D. J. G., M. R. S., and J. M.) from NINDS, Grants R01 EY014430 (to D. J. G.) and GM63021 (to M. R. S.) from NEI, NEI Core Grants P30EY0468 (Wayne State University) and P20-EY00700 (University of Michigan), and NHLBI Training Grant T32HL120822 (to C. S.). J. M. owns shares in Angelus Therapeutics. No materials or support were received from this company. United States patent application entitled “Neuroprotective Composition and Method of Use” (United States Patent Application WO2013158739) has been filed by Brown University, with D. J. G. and J. M. listed as inventors.
- NMDAR
- NMDA receptor
- BDNF
- brain-derived neurotrophic factor
- RGC
- retinal ganglion cell
- CREB
- cAMP-response element-binding protein
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- PAR
- poly(ADP-ribosyl)ation
- FAM
- 5-carboxyfluorescein
- TMRM
- tetramethylrhodamine methyl ester
- LPMS
- loss of plasma membrane selectivity
- Mcu
- mitochondrial calcium uniporter
- ROS
- reactive oxygen species
- MEA
- multielectrode array
- ANOVA
- analysis of variance
- LTP
- long term potentiation
- GCL
- ganglion cell layer
- mPTP
- mitochondrial permeability transition pore.
References
- 1. Tezel G. (2006) Oxidative stress in glaucomatous neurodegeneration: mechanisms and consequences. Prog. Retin. Eye Res. 25, 490–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mittag T. W., Danias J., Pohorenec G., Yuan H. M., Burakgazi E., Chalmers-Redman R., Podos S. M., Tatton W. G. (2000) Retinal damage after 3 to 4 months of elevated intraocular pressure in a rat glaucoma model. Invest. Ophthalmol. Vis. Sci. 41, 3451–3459 [PubMed] [Google Scholar]
- 3. Hernández C., Simó R. (2012) Neuroprotection in diabetic retinopathy. Curr. Diab. Rep. 12, 329–337 [DOI] [PubMed] [Google Scholar]
- 4. Lee D., Kim K. Y., Shim M. S., Kim S. Y., Ellisman M. H., Weinreb R. N., Ju W. K. (2014) Coenzyme Q10 ameliorates oxidative stress and prevents mitochondrial alteration in ischemic retinal injury. Apoptosis 19, 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stafford B. K., Park S. J., Wong K. Y., Demb J. B. (2014) Developmental changes in NMDA receptor subunit composition at ON and OFF bipolar cell synapses onto direction-selective retinal ganglion cells. J. Neurosci. 34, 1942–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bai N., Aida T., Yanagisawa M., Katou S., Sakimura K., Mishina M., Tanaka K. (2013) NMDA receptor subunits have different roles in NMDA-induced neurotoxicity in the retina. Mol. Brain 6, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seki M., Lipton S. A. (2008) Targeting excitotoxic/free radical signaling pathways for therapeutic intervention in glaucoma. Prog. Brain Res. 173, 495–510 [DOI] [PubMed] [Google Scholar]
- 8. Calkins D. J. (2012) Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog. Retin. Eye Res. 31, 702–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goebel D. J., Aurelia J. L., Tai Q., Jojich L., Poosch M. S. (1998) Immunocytochemical localization of the NMDA-R2A receptor subunit in the cat retina. Brain Res. 808, 141–154 [DOI] [PubMed] [Google Scholar]
- 10. Pourcho R. G., Qin P., Goebel D. J. (2001) Cellular and subcellular distribution of NMDA receptor subunit NR2B in the retina. J. Comp. Neurol. 433, 75–85 [DOI] [PubMed] [Google Scholar]
- 11. Levy D. I., Lipton S. A. (1990) Comparison of delayed administration of competitive and uncompetitive antagonists in preventing NMDA receptor-mediated neuronal death. Neurology 40, 852–855 [DOI] [PubMed] [Google Scholar]
- 12. Siliprandi R., Canella R., Carmignoto G., Schiavo N., Zanellato A., Zanoni R., Vantini G. (1992) N-Methyl-d-aspartate-induced neurotoxicity in the adult rat retina. Vis. Neurosci. 8, 567–573 [DOI] [PubMed] [Google Scholar]
- 13. Parsons M. P., Raymond L. A. (2014) Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 82, 279–293 [DOI] [PubMed] [Google Scholar]
- 14. Kornau H. C., Schenker L. T., Kennedy M. B., Seeburg P. H. (1995) Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 269, 1737–1740 [DOI] [PubMed] [Google Scholar]
- 15. Martel M. A., Ryan T. J., Bell K. F., Fowler J. H., McMahon A., Al-Mubarak B., Komiyama N. H., Horsburgh K., Kind P. C., Grant S. G., Wyllie D. J., Hardingham G. E. (2012) The subtype of GluN2 C terminal domain determines the response to excitotoxic insults. Neuron 74, 543–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hardingham G. E., Fukunaga Y., Bading H. (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 5, 405–414 [DOI] [PubMed] [Google Scholar]
- 17. Zhou X., Hollern D., Liao J., Andrechek E., Wang H. (2013) NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis. 4, e560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang J., Diamond J. S. (2009) Subunit- and pathway-specific localization of NMDA receptors and scaffolding proteins at ganglion cell synapses in rat retina. J. Neurosci. 29, 4274–4286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sagdullaev B. T., McCall M. A., Lukasiewicz P. D. (2006) Presynaptic inhibition modulates spillover, creating distinct dynamic response ranges of sensory output. Neuron 50, 923–935 [DOI] [PubMed] [Google Scholar]
- 20. Aarts M., Liu Y., Liu L., Besshoh S., Arundine M., Gurd J. W., Wang Y. T., Salter M. W., Tymianski M. (2002) Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 298, 846–850 [DOI] [PubMed] [Google Scholar]
- 21. Li T., Saro D., Spaller M. R. (2004) Thermodynamic profiling of conformationally constrained cyclic ligands for the PDZ domain. Bioorg. Med. Chem. Lett. 14, 1385–1388 [DOI] [PubMed] [Google Scholar]
- 22. LeBlanc B. W., Iwata M., Mallon A. P., Rupasinghe C. N., Goebel D. J., Marshall J., Spaller M. R., Saab C. Y. (2010) A cyclic peptide targeted against PSD-95 blocks central sensitization and attenuates thermal hyperalgesia. Neuroscience 167, 490–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Piserchio A., Salinas G. D., Li T., Marshall J., Spaller M. R., Mierke D. F. (2004) Targeting specific PDZ domains of PSD-95; structural basis for enhanced affinity and enzymatic stability of a cyclic peptide. Chem. Biol. 11, 469–473 [DOI] [PubMed] [Google Scholar]
- 24. Cao C., Rioult-Pedotti M. S., Migani P., Yu C. J., Tiwari R., Parang K., Spaller M. R., Goebel D. J., Marshall J. (2013) Impairment of TrkB-PSD-95 signaling in Angelman syndrome. PLoS Biol. 11, e1001478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bartlett T. E., Wang Y. T. (2013) The intersections of NMDAR-dependent synaptic plasticity and cell survival. Neuropharmacology 74, 59–68 [DOI] [PubMed] [Google Scholar]
- 26. Goebel D. J., Winkler B. S. (2006) Blockade of PARP activity attenuates poly(ADP-ribosyl)ation but offers only partial neuroprotection against NMDA-induced cell death in the rat retina. J. Neurochem. 98, 1732–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Patra C. R., Rupasinghe C. N., Dutta S. K., Bhattacharya S., Wang E., Spaller M. R., Mukhopadhyay D. (2012) Chemically modified peptides targeting the PDZ domain of GIPC as a therapeutic approach for cancer. ACS Chem. Biol. 7, 770–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee I., Pecinova A., Pecina P., Neel B. G., Araki T., Kucherlapati R., Roberts A. E., Hüttemann M. (2010) A suggested role for mitochondria in Noonan syndrome. Biochim. Biophys. Acta 1802, 275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goebel D. J. (2009) Selective blockade of CaMKII-α inhibits NMDA-induced caspase-3-dependent cell death but does not arrest PARP-1 activation or loss of plasma membrane selectivity in rat retinal neurons. Brain Res. 1256, 190–204 [DOI] [PubMed] [Google Scholar]
- 30. Saro D., Klosi E., Paredes A., Spaller M. R. (2004) Thermodynamic analysis of a hydrophobic binding site: probing the PDZ domain with nonproteinogenic peptide ligands. Org. Lett. 6, 3429–3432 [DOI] [PubMed] [Google Scholar]
- 31. Saro D., Li T., Rupasinghe C., Paredes A., Caspers N., Spaller M. R. (2007) A thermodynamic ligand binding study of the third PDZ domain (PDZ3) from the mammalian neuronal protein PSD-95. Biochemistry 46, 6340–6352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soriano F. X., Martel M. A., Papadia S., Vaslin A., Baxter P., Rickman C., Forder J., Tymianski M., Duncan R., Aarts M., Clarke P., Wyllie D. J., Hardingham G. E. (2008) Specific targeting of pro-death NMDA receptor signals with differing reliance on the NR2B PDZ ligand. J. Neurosci. 28, 10696–10710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buckingham B. P., Inman D. M., Lambert W., Oglesby E., Calkins D. J., Steele M. R., Vetter M. L., Marsh-Armstrong N., Horner P. J. (2008) Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J. Neurosci. 28, 2735–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schmidt N., Mishra A., Lai G. H., Wong G. C. (2010) Arginine-rich cell-penetrating peptides. FEBS Lett. 584, 1806–1813 [DOI] [PubMed] [Google Scholar]
- 35. Qiu J., Tan Y. W., Hagenston A. M., Martel M. A., Kneisel N., Skehel P. A., Wyllie D. J., Bading H., Hardingham G. E. (2013) Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 4, 2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stout A. K., Raphael H. M., Kanterewicz B. I., Klann E., Reynolds I. J. (1998) Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat. Neurosci. 1, 366–373 [DOI] [PubMed] [Google Scholar]
- 37. Kadenbach B., Arnold S., Lee I., Hüttemann M. (2004) The possible role of cytochrome c oxidase in stress-induced apoptosis and degenerative diseases. Biochim. Biophys. Acta 1655, 400–408 [DOI] [PubMed] [Google Scholar]
- 38. Liu S. S. (1999) Cooperation of a “reactive oxygen cycle” with the Q cycle and the proton cycle in the respiratory chain–superoxide generating and cycling mechanisms in mitochondria. J. Bioenerg. Biomembr. 31, 367–376 [DOI] [PubMed] [Google Scholar]
- 39. Kolarow R., Brigadski T., Lessmann V. (2007) Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase II signaling and proceeds via delayed fusion pore opening. J. Neurosci. 27, 10350–10364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Minichiello L. (2009) TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 10, 850–860 [DOI] [PubMed] [Google Scholar]
- 41. Reichardt L. F. (2006) Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1545–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Figurov A., Pozzo-Miller L. D., Olafsson P., Wang T., Lu B. (1996) Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381, 706–709 [DOI] [PubMed] [Google Scholar]
- 43. Ji Y., Lu Y., Yang F., Shen W., Tang T. T., Feng L., Duan S., Lu B. (2010) Acute and gradual increases in BDNF concentration elicit distinct signaling and functions in neurons. Nat. Neurosci. 13, 302–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kang H., Schuman E. M. (1995) Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 267, 1658–1662 [DOI] [PubMed] [Google Scholar]
- 45. Messaoudi E., Bârdsen K., Srebro B., Bramham C. R. (1998) Acute intrahippocampal infusion of BDNF induces lasting potentiation of synaptic transmission in the rat dentate gyrus. J. Neurophysiol. 79, 496–499 [DOI] [PubMed] [Google Scholar]
- 46. Kang H., Welcher A. A., Shelton D., Schuman E. M. (1997) Neurotrophins and time: different roles for TrkB signaling in hippocampal long term potentiation. Neuron 19, 653–664 [DOI] [PubMed] [Google Scholar]
- 47. Almeida R. D., Manadas B. J., Melo C. V., Gomes J. R., Mendes C. S., Grãos M. M., Carvalho R. F., Carvalho A. P., Duarte C. B. (2005) Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 12, 1329–1343 [DOI] [PubMed] [Google Scholar]
- 48. Bell K. F., Bent R. J., Meese-Tamuri S., Ali A., Forder J. P., Aarts M. M. (2013) Calmodulin kinase IV-dependent CREB activation is required for neuroprotection via NMDA receptor-PSD95 disruption. J. Neurochem. 126, 274–287 [DOI] [PubMed] [Google Scholar]
- 49. Meloni B. P., Brookes L. M., Clark V. W., Cross J. L., Edwards A. B., Anderton R. S., Hopkins R. M., Hoffmann K., Knuckey N. W. (2015) Polyarginine and arginine-rich peptides are neuroprotective in stroke models. J. Cereb. Blood Flow Metab. 35, 993–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vaslin A., Rummel C., Clarke P. G. (2009) Unconjugated Tat carrier peptide protects against excitotoxicity. Neurotox. Res. 15, 123–126 [DOI] [PubMed] [Google Scholar]
- 51. Meloni B. P., Craig A. J., Milech N., Hopkins R. M., Watt P. M., Knuckey N. W. (2014) The neuroprotective efficacy of cell-penetrating peptides Tat, penetratin, Arg-9, and Pep-1 in glutamic acid, kainic acid, and in vitro ischemia injury models using primary cortical neuronal cultures. Cell. Mol. Neurobiol. 34, 173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brustovetsky T., Pellman J. J., Yang X. F., Khanna R., Brustovetsky N. (2014) Collapsin response mediator protein 2 (CRMP2) interacts with N-methyl-d-aspartate (NMDA) receptor and Na+/Ca2+ exchanger and regulates their functional activity. J. Biol. Chem. 289, 7470–7482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jonas E. A. (2009) Molecular participants in mitochondrial cell death channel formation during neuronal ischemia. Exp. Neurol. 218, 203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cheng W. C., Leach K. M., Hardwick J. M. (2008) Mitochondrial death pathways in yeast and mammalian cells. Biochim. Biophys. Acta 1783, 1272–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sanderson T. H., Reynolds C. A., Kumar R., Przyklenk K., Hüttemann M. (2013) Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 47, 9–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hüttemann M., Pecina P., Rainbolt M., Sanderson T. H., Kagan V. E., Samavati L., Doan J. W., Lee I. (2011) The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: From respiration to apoptosis. Mitochondrion 11, 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Grover G. J., Atwal K. S., Sleph P. G., Wang F. L., Monshizadegan H., Monticello T., Green D. W. (2004) Excessive ATP hydrolysis in ischemic myocardium by mitochondrial F1F0-ATPase: effect of selective pharmacological inhibition of mitochondrial ATPase hydrolase activity. Am. J. Physiol. Heart Circ Physiol. 287, H1747–H1755 [DOI] [PubMed] [Google Scholar]
- 58. Guigas B., Detaille D., Chauvin C., Batandier C., De Oliveira F., Fontaine E., Leverve X. (2004) Metformin inhibits mitochondrial permeability transition and cell death: a pharmacological in vitro study. Biochem. J. 382, 877–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Owen M. R., Doran E., Halestrap A. P. (2000) Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 348, 607–614 [PMC free article] [PubMed] [Google Scholar]
- 60. Rigobello M. P., Barzon E., Marin O., Bindoli A. (1995) Effect of polycation peptides on mitochondrial permeability transition. Biochem. Biophys. Res. Commun. 217, 144–149 [DOI] [PubMed] [Google Scholar]
- 61. Costantini P., Chernyak B. V., Petronilli V., Bernardi P. (1996) Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J. Biol. Chem. 271, 6746–6751 [DOI] [PubMed] [Google Scholar]
- 62. Eriksson O., Fontaine E., Bernardi P. (1998) Chemical modification of arginines by 2,3-butanedione and phenylglyoxal causes closure of the mitochondrial permeability transition pore. J. Biol. Chem. 273, 12669–12674 [DOI] [PubMed] [Google Scholar]
- 63. Abramov A. Y., Duchen M. R. (2008) Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity. Biochim. Biophys. Acta 1777, 953–964 [DOI] [PubMed] [Google Scholar]
- 64. Szeto H. H. (2006) Cell-permeable, mitochondrial-targeted, peptide antioxidants. AAPS J. 8, E277–E283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guo X., Sesaki H., Qi X. (2014) Drp 1stabilizeson the mitochondria to trigger necrosis under oxidative stress conditions in vitro an in vivo. Biochem. J. 461, 137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nguyen D., Alavi M. V., Kim K. Y., Kang T., Scott R. T., Noh Y. H., Lindsey J. D., Wissinger B., Ellisman M. H., Weinreb R. N., Perkins G. A., Ju W. K. (2011) A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2, e240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Osborne N. N. (2010) Mitochondria: their role in ganglion cell death and survival in primary open angle glaucoma. Exp. Eye Res. 90, 750–757 [DOI] [PubMed] [Google Scholar]
- 68. Gajavelli S., Sinha V. K., Mazzeo A.T., Spurlock M.S., Lee S. W., Ahmed A. I., Yokobori S., Bullock R.M. (2015) Evidence to support mitochondrial neuroprotection, in severe traumatic brain injury. J. Bioenerg. Biomembr. 47, 133–148 [DOI] [PubMed] [Google Scholar]
- 69. Abeti R., Abramov A. Y. (2015) Mitochondrial Ca2+ in neurodegenerative disorders. Pharmacol. Res. 10.1016/j.phrs.2015.05.007 [DOI] [PubMed] [Google Scholar]