

Background: The transcription factor ETS1 controls its DNA binding activity through an autoinhibitory module.
Results: The relationship between specific and nonspecific DNA binding, homodimer formation, and autoinhibition was explored.
Conclusion: ETS1 transiently forms dimers as a consequence of interacting with any DNA sequence.
Significance: Relief of autoinhibition by nonspecific DNA interaction leading to subsequent ETS1 dimerization may represent a conserved element of DNA recognition across the ETS family.
Keywords: circular dichroism (CD), dimerization, DNA binding protein, DNA-protein interaction, ETS transcription factor family, protein conformation, protein cross-linking, protein-nucleic acid interaction, autoinhibition
Abstract
ETS1 is the archetype of the ETS transcription factor (TF) family. ETS TFs share a DNA-binding domain, the ETS domain. All ETS TFs recognize a core GGA(A/T) binding site, and thus ETS TFs are found to redundantly regulate the same genes. However, each ETS TF has unique targets as well. One prevailing hypotheses explaining this duality is that protein-protein interactions, including homodimerization, allow each ETS TF to display distinct behavior. The behavior of ETS1 is further regulated by autoinhibition. Autoinhibition apparently modulates ETS1 DNA binding affinity, but the mechanism of this inhibition is not completely understood. We sought to characterize the relationship between DNA binding and ETS1 homodimer formation. We find that ETS1 interrogates DNA and forms dimers even when the DNA does not contain an ETS recognition sequence. Mutational studies also link nonspecific DNA backbone contacts with dimer formation, in addition to providing a new role for the recognition helix of ETS1 in maintaining the autoinhibited state. Finally, in showing that residues in the DNA recognition helix affect autoinhibition, we define a new function of ETS1 autoinhibition: maintenance of a monomeric state in the absence of DNA. The conservation of relevant amino acid residues across all ETS TFs indicates that the mechanisms of nonspecific DNA interrogation and protein oligomer formation elucidated here may be common to all ETS proteins that autoinhibit.
Introduction
Promoter-specific transcription of genes in eukaryotes is controlled by the activities of DNA-bound transcription factors. Many of these act in combination with other transcription factors and are capable of regulating the activities of several different genes. These features are key to coordinated, differential regulation of various gene families. The effectiveness of a given transcription factor across the spectrum of its regulated target genes varies. The variation in transcription factor activity is primarily modulated by differences in the affinity of the factor for its specific binding sites. Transcription factor affinity for its DNA binding sites depends on the sequence of the site, its oligomeric state, identity of its DNA binding partners, and/or the activity of autoregulatory elements within the transcription factor. We are interested in exploring how binding site sequence affects oligomeric form and autoinhibitory function together to regulate the DNA binding activities of transcription factors. To this end, because there is an exact correspondence between DNA binding affinity and transcriptional regulatory activity of ETS family members (1), we are investigating the form and function of ETS1, the prototypical ETS protein.
ETS proteins are found in metazoan species ranging from invertebrates to humans. Each member of this diverse set of proteins shares a conserved sequence of ∼85 amino acids known as the ETS domain (2). This domain directs the sequence-specific DNA binding of these proteins. The ETS domain contains a “winged-helix-turn-helix” motif that is comprised of a four-stranded antiparallel β-sheet scaffold attached to a three-α-helix bundle. All ETS members bind a DNA site that contains an invariant core consensus GGA(A/T) sequence (3). Residues in the recognition α-helix H3 of the ETS domain make direct contact with the major groove surface of the guanine and adenine bases in the conserved core sequence. Protein residues located in the turns between α-helices H2 and H3 and between β-strands 3 and 4 contact other regions of the core site and/or the DNA phosphate backbone (4). Although no base-specifying contacts are made to any DNA base outside of the core, the binding site preferences of ETS family members are modulated by the DNA sequence outside of the core consensus (5, 6).
Full-length ETS1 is not known to bind DNA as a monomer; ETS1 seemingly binds DNA only as a homodimer or as a heterodimer with one of several different partner proteins, including, among others, RUNX1, Pax-5, Pit-1, and NF-κB (7). As a consequence of its interaction with these partners, ETS1 regulates a variety of genes related to functions such as embryonic development, angiogenesis, erythropoiesis, and tumor invasion (7). Interestingly, an ETS1 splice variant that lacks a region N-terminal to the ETS domain does not autoinhibit and can bind DNA as a monomer (1).
The ETS1 homodimer binds DNA sequences comprised of two ETS binding sites (EBS)2 facing each other on opposite strands (4). The head to head arrangement of two EBSs is widespread among ETS1-regulated promoters but is notably found in two particularly well studied cases: the promoter for MMP3, which encodes a matrix metalloprotease, and the promoter for human p53 (8). Both EBSs in head to head sites are required for binding and transcriptional regulations by full-length ETS1; deletion of or mutations in either EBS inhibits ETS1 binding and, thereby, promoter transactivation. These findings indicate that, on a head to head site, two monomers of the ETS1 homodimer bind cooperatively to the two EBSs (4, 9).
In addition to providing additional DNA specifying contacts, formation of either homo- or heterodimeric complexes is apparently required, at least in part, to relieve autoinhibition of ETS1 DNA binding (1, 10, 11). ETS1 autoinhibition is mediated by the packing of helices within the N-terminal inhibitory domain and C-terminal inhibitory domain onto helix H1 of the ETS domain. These inhibitory helices flank the ETS domain and lower the DNA affinity of ETS1, apparently by decreasing the overall flexibility of the three α-helices in the winged helix motif, although the precise mechanistic implications of the change in flexibility are as yet unknown. The function of ETS1 autoinhibition is thought to be to provide a “graded control” of ETS1 DNA binding affinity (5). Most structural studies of ETS1 autoinhibition have focused on a partially inhibited fragment, and to date no plausible model for monomeric relief of autoinhibition has been proposed. On the other hand, there has been much work involving autoinhibition and homodimer formation, culminating in several x-ray crystallography studies (4, 12) of ETS1 homodimers bound to the MMP3 sequence.
Despite extensive investigations aimed at delineating how ETS1 DNA binding is regulated, a precise mechanism has not been articulated. Several questions remain: 1) How is ETS1 dimerization regulated? 2) Can full-length ETS1 bind as a monomer in some DNA contexts? 3) Are DNA binding and protein oligomer formation linked, and if so, how? 4) How do ETS1-DNA interactions lead to relief of autoinhibition? In this study we show that ETS1 forms dimers in the presence of either specific EBS-containing DNA or nonspecific DNA that does not contain an EBS core consensus sequence, but not in the absence of DNA. We have also found that ETS1 does not form dimers when the ability to contact a particular DNA phosphate is blocked. Additionally, we found that an ETS1 splice variant lacking the autoinhibitory module self-associates even in the absence of DNA, suggesting that ETS1 autoinhibition modulates DNA binding activity by preventing oligomerization of the ETS domain. Finally, we show that α-helix H3 mutant ETS1 protein also self-associates in the absence of DNA. This result shows that this DNA binding α-helix H3 also has a role in maintaining autoinhibited state. Autoinhibition was previously thought to only involve allosteric hindrance of a separate region of the ETS domain.
Experimental Procedures
Proteins
Plasmids containing the coding sequence for human ETS1Δ280 and ETS1Δ335 were a kind gift from Dr. Matthias Wilmanns (EMBL, Hamburg, Germany). ETS1 derivatives bearing the R394A/Y395A, L337A, and/or the R378C mutations were constructed by site-directed mutagenesis. The primers used for mutagenesis were purchased from Integrated DNA Technologies (Coralville, IA). The R394A and Y395A mutations were made in one reaction with the following primers: 5′-GAGAAACTGAGCCGTGGCCTAGCCGCCTATTACGACAAAAACATCATCC-3′ (forward) and 5′-GGATGATGTTTTTGTCGTAATAGGCGGCTAGGCCACGGCTCAGTTTCTC-3′ (reverse). The R378C primers are 5′-GGCCAGGAGATGGGGAAAGTGCAAAAACAAACCTAAGATGAATTATG-3′ (forward) and 5′-CATAATTCATCTTAGGTTTGTTTTTGCACTTTCCCCATCTCCTGGCC-3′ (reverse). The L337A primers are 5′-GGCAGTGGACCAATCCAGGCGTGGCAGTTTCTTCTGG-3′ (forward) 5′-CCAGAAGAAACTGCCA±CGCCTGGATTGGTCCACTGCC-3′ (reverse). The R394A/Y395A and R378C primers work on both ETS1Δ280 and ETS1Δ335. The L337A primers are specific to ETS1Δ280. The altered bases are underlined.
Purification of unmodified ETS1Δ280, as well as the R394A/Y395A and L337A mutant proteins, was performed essentially as described in Ref. 4. Purification of R378C mutants was also identical except for the addition of 1 mm DTT to the lysis and storage buffers. Purification of ETS1Δ335 was identical to that in Ref. 4, except instead of concentrating in a centrifugal concentrator, purified protein was concentrated by selective ammonium sulfate precipitation.
DNA Purification
Complementary 60-base oligonucleotides encoding desired ETS1 binding site sequence embedded within their naturally occurring flanking DNA were obtained from Integrated DNA Technologies. The sequences of the top strand of the wild type sites are: MMP3, 5′-TCAGTTTTCTCCTCTACCAAGACAGGAAGCACTTCCTGGAGATTAATCACTGTGTTGCCT-3′; and p53, 5′-CATCAGTTAAAATGTCATTTTTTAGGAAGGCTTTCCGTAATATCACAC CCTAACGTTTTC-3′. The EBSs present are underlined.
Equivalent amounts of each pair of the complementary strands were mixed, heated to 95 °C for 5 min, and slowly cooled over 4 h to anneal the two strands. Double-stranded DNA was separated from the individual single strands by electrophoresis on 8% polyacrylamide gels in 1× TBE (89 mm Tris, pH 8.9, 89 mm borate, 1 mm EDTA) and eluted from them by a crush and soak method. Eluted DNA was separated from contaminants by size exclusion chromatography. Following gel purification, the DNA fragments were radioactively labeled at their 5′ ends by incubating the DNA with γ-[32P]ATP (6000 Ci/mmol) (PerkinElmer Life Sciences) in the presence of T4 polynucleotide kinase (Epicenter, Inc., Madison, WI).
EMSA
Labeled DNA was incubated with the specified concentrations of ETS1 protein in binding buffer (40 μg/ml BSA, 0.01% Triton X-100, 100 mm KCl, 20 mm Tris, pH 7.5, 1 mm EDTA, and 5% glycerol) for 20 min at 4 °C. The protein-DNA complexes were resolved on 5% polyacrylamide gels at 4 °C. The electrophoresis buffer was 1× TBE. The amounts of protein-DNA complexes present on dried gels were quantified using a Storm imager (GE Healthcare). The values of the dissociation constant (KD) were determined by nonlinear squares fitting of the EMSA data to a hyperbolic equation using Prism 4.0 software (GraphPad Software Inc., La Jolla, CA). Each dissociation constant was determined from at least four replicate measurements.
DNase I Footprinting
Single-stranded DNA containing the desired binding sites was gel-purified as described above and radioactively labeled at their 5′ ends by incubating the DNA with γ-[32P]ATP (6000 Ci/mmol) (PerkinElmer Life Sciences) in the presence of T4 polynucleotide kinase (Epicenter, Inc.). Following ethanol precipitation, the labeled DNA was dissolved in TEN (10 mm Tris, pH 7.5, 1 mm EDTA, 50 mm NaCl), and a 1.1 molar excess of the complementary strand was added. The mixture was incubated at 95 °C for 5 min and then slowly cooled over 4 h to anneal the two strands.
DNase I cleavage was performed as follows. In a 20-μl reaction, 1.5 pmol of end-labeled DNA was incubated with increasing amounts of the desired ETS1 protein in buffer containing 40 μg/ml BSA, 0.01% Triton X-100, 100 mm KCl, 20 mm Tris, pH 7.5, 5 mm MgCl2 and 5% glycerol. Protein-DNA complexes were incubated at 4 °C for 20 min and subsequently brought to 25 °C prior to the addition of 3 μg/ml DNase I, an amount sufficient to generate, on average, one cleavage per DNA molecule in 3 min of additional incubation. The cleavage reactions were terminated by precipitation with ethanol and sec-butanol dehydration, and the DNA was dissolved in 90% formamide solution containing tracking dyes. The products along with chemical sequencing reactions (13) derived from the same templates were resolved on 6% acrylamide gels containing 7 m urea in 1× TBE. Cleavage products were visualized using a Storm imager (GE Healthcare).
Protein Cross-linking
Experiments were performed essentially as described in Ref. 14. Briefly, in a 10-μl reaction, 2 pmol of the desired ETS1 protein was incubated with 2–5× molar excess DNA for 20 min at 4 °C in EMSA binding buffer. Subsequently, bismaleimidoethane (BMOE; Thermo Scientific) was added to a final concentration of 10 μm, and samples were incubated for an additional 2 h at 4 °C. Reactions were quenched by adding 10 μm DTT and incubating for 15 min at 37 °C. Subsequent to adding loading dye, the samples were boiled for 5 min, and the reaction products were fractionated on a 15% Tris-Tricine SDS-PAGE gel.
After electrophoretic separation, the reaction products were transferred to nitrocellulose, and the membranes were blocked for 1 h in 2% BSA or 5% milk in TBST (20 mm Tris, pH 7.5, 0.001% Tween 20, 150 mm NaCl) at room temperature and then incubated for 1 h in primary antibody (ETS-1, C-20: sc-350; Santa Cruz Biotechnology, Dallas, TX) at room temperature at a 1:10,000 dilution. The membranes were then rinsed and incubated in goat anti-rabbit secondary antibody (Thermo Scientific, Pierce) conjugated to horseradish peroxidase at a 1:10,000 dilution for 1 h at room temperature. Reaction products were visualized using an ECL detection kit (Bio-Rad). Imaging and band analysis were performed on a Bio-Rad Gel-Doc.
Partial Proteolysis
In a 10 μl reaction, purified ETS1Δ280 (10 μg, ∼500 pmol) was incubated in the presence or absence of 2-fold molar excess of either MMP3 WT or M1 DNA for 20 min at 4 °C in buffer containing 0.01% Triton X-100, 100 mm KCl, 20 mm Tris, pH 7.5, 1 mm EDTA, 5% glycerol. 100 ng of chymotrypsin (Worthington Biochemical Corporation, Lakewood, NJ) was added, and the reaction was allowed to proceed at 4 °C for 3 h, with one-third of the reaction sampled every hour. Proteolysis was quenched by addition of loading dye, after which samples were boiled, and the reaction products were resolved on 15% Tris-Tricine SDS-PAGE. Reaction products were visualized by staining with Coomassie Blue. For mass spectrometry, proteolytic fragments were excised from an SDS-PAGE gel and sent to the Ohio State University Campus Chemical Instrument Center for MALDI-TOF analysis.
Circular Dichroism Spectroscopy
The conformational changes in ETS1Δ280 induced by DNA were monitored by CD spectroscopy. Spectra were recorded at 22 °C from 300 to 190 nm using a JASCO-715 in a 0.4-cm-path length cuvette. Data were collected every 0.1 nm, averaged over 10 scans, and corrected for baseline. The spectra of 13.5 μm protein or a mixture of 13.5 μm protein and equimolar amounts of double-stranded MMP3 WT or M1 DNA dissolved in 1× PBS (3.8 mm NaH2PO4 plus 16.2 mm Na2H2PO4) and 500 mm NaCl buffer were recorded. Prior to CD measurements protein, and DNA were equilibrated, either separately or together, in buffer at 4 °C for 20 min. Spectra were converted to molar ellipticity and plotted.
Results
ETS1 Forms Unstable Dimeric Complexes on Sites Containing a Single EBS
To begin analyzing the effect of DNA sequence, oligomeric state, and autoinhibition on ETS1 DNA binding affinity, we determined the affinity of this protein for the ETS1 homodimer binding site from the promoter of the DNA damage sensing protein p53 and for the wild type and mutants of the binding site in the promoter for MMP3 (stromelysin-1) an extracellular matrix remodeling metalloprotease. The MMP3 and p53 promoters each contain two head to head EBSs (see Table 1 for sequences). The sequences of the two core EBS in each of these promoters are identical, but the sequences flanking these regions differ between them. Unless otherwise noted, for all assays described below, we used ETS1Δ280, which bears a deletion of the N-terminal PNT and transactivation domains present in full-length ETS1. These domains do not contribute to DNA binding, dimerization, or autoinhibition (15); ETS1Δ280 contains all residues required for ETS1 to autoinhibit, dimerize, and bind its specific DNA sites (15–18).
TABLE 1.
Affinity of ETS1Δ280 for wild-type and mutant ETS1 binding sites
The sequences and affinity (dissociation constants, KD) ± standard deviation of ETS1Δ280 for naturally occurring and mutant binding sites were determined by EMSA (see “Experimental Procedures”). EBSs present in each sequence are underlined. NS indicates “nonspecific,” where ETS1Δ280 formed no identifiable complex with the DNA site.
| DNA | Core sequence | KD |
|---|---|---|
| nm | ||
| MMP3 WT | ACAGGAAGCACTTCCTGG | 0.20 ± 0.058 |
| MMP3 M1 | ACAAAAAGCACTTCCTGG | NS |
| MMP3 M2 | ACAGGAAGCACTTTTTGG | NS |
| MMP3 + 4 | AGGAAGCACGCACTTCCT | NS |
| p53 | TTAGGAAGGCTTTCCGTA | 6.2 ± 3.6 |
The affinity of ETS1Δ280 for the p53 and MMP3 DNA sites was initially determined using an electrophoretic mobility shift assay (Fig. 1). We find that when increasing concentrations of ETS1Δ280 are added to radiolabeled DNA containing either the MMP3 or p53 EBS, ETS1Δ280 forms a single complex with each DNA. The complexes formed on these DNAs are larger than those formed when MMP3 is bound by ETS1-p42, a splice variant previously reported to only bind as a monomer. ETS1-p42 does not bind to the p53 site (data not shown) (19). This observation suggests that ETS1-DNA complexes formed with these DNAs each contain two ETS1 molecules (Fig. 1, A and C). Consistent with the suggestion that the identity of the bases that flank the EBS core consensus sequence are important in determining ETS1 DNA affinity, we find that despite having identical core sequences, the dissociation constants (KD) of the complexes between ETS1Δ280 and MMP3 or p53 differ by 30-fold (Table 1).
FIGURE 1.
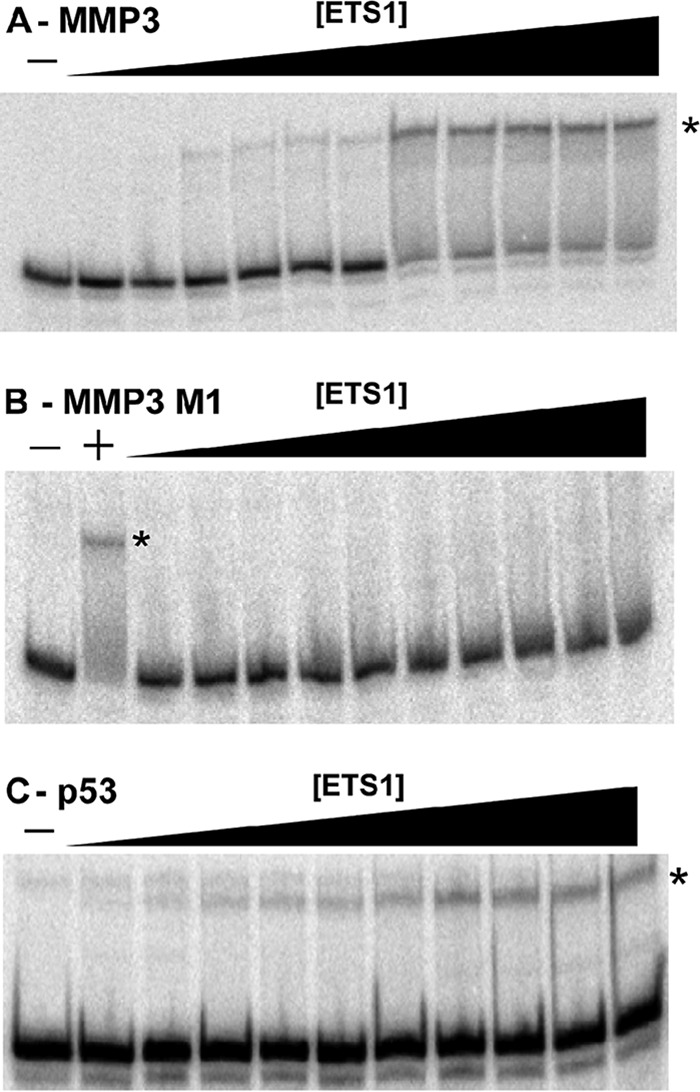
ETS1Δ280-DNA complex formation. Increasing concentrations of ETS1Δ280 were mixed with radiolabeled binding site containing, either MMP3 WT (A), MMP3 M1 (B), or p53 (C) (see Table 1 for sequences), and complexes were resolved via native PAGE as described under “Experimental Procedures.” Shown is a PhosphorImager scan of the gel. Protein-DNA complexes are denoted with asterisks. − indicates no protein added. The + in B denotes a positive control, which is ETS1Δ280 incubated with MMP3 WT DNA.
Mutating either EBS in the MMP3 promoter site prevents ETS1Δ280 from forming a specific protein-DNA complex that is detectable by EMSA, even though each of these sites (MMP3 M1 and MMP3 M2) still contains an intact EBS (Fig. 1B). This result is consistent with previous findings showing that ETS1Δ280 DNA binding to MMP3 DNA requires the core consensus sequence of both of the two head to head EBS sites be intact (20). This finding suggests that two ETS1 monomers bind cooperatively to these two EBSs. Consistent with this idea, ETS1 is also incapable of forming an EMSA-detectable complex with an MMP3 variant in which the two intact core consensus sequences are separated by an additional four bases (MMP3 + 4) (1).
In light of the ability of other ETS family members (21) and the ETS1 splice variant (ETS1-p42) to bind to a single EBS and the ability of ETS1-p51 to bind single EBSs in partnership with heterologous proteins, we find the inability of ETS1Δ280 to form EMSA-detectable complexes with the MMP3 M1, M2, and +4 variant sites somewhat surprising. Considering that rapid dissociation during electrophoresis can prevent detection of protein-DNA complexes by EMSA (22), we hypothesized that ETS1 forms weak and/or transient complexes with these DNAs. To test this idea, we examined ETS1-DNA complex formation using DNase I footprinting.
Adding ETS1Δ280 to wild type MMP3 completely protects both EBS core sequences from digestion by DNase I (Fig. 2A). In addition to the ETS1-mediated protections, we find that the presence of DNA-bound ETS1Δ280 enhances DNase I cleavage at A/T base pairs located six residues upstream and downstream of each of the two EBS regions. A footprint of the p53 binding site showed similar pattern of protection and enhancements (Fig. 2C). In both cases, ETS1-mediated protection and enhancements at both EBSs in both p53 and MMP3 DNA sites arose simultaneously. These findings are consistent with the idea that two ETS1 molecules bind cooperatively to each of these DNA sites, with one monomer associated with each of the two head to head EBS present on these DNAs.
FIGURE 2.
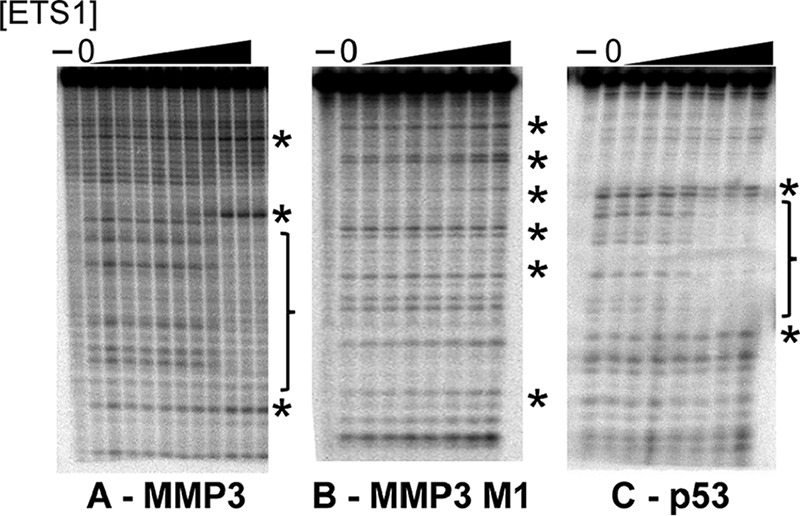
DNase I footprints of ETS1Δ280-DNA complexes. Radiolabeled DNA bearing MMP3 wild type (A), MMP3 M1 (B), or p53 (C) were incubated with increasing amounts of ETS1Δ280. Complexes were then subjected to DNase I cleavage, and the resulting DNA fragments were resolved via denaturing PAGE. Shown is a PhosphorImager scan of the gel. In each panel, the lane labeled with a − contains uncleaved DNA, and the lane labeled with 0 contains DNA incubated with DNase I in the absence of protein. Positions of bases protected from cleavage by ETS1Δ280 binding are bracketed. Enhancements of cleavage are noted with asterisks.
Added ETS1Δ280 is unable to protect either the mutated or wild type EBS core sequences in the MMP3 M1 site from digestion by DNase I (Fig. 2B). However, careful inspection of the footprint patterns reveals that in the presence of ETS1Δ280, regions of enhanced DNase I cleavage are seen in the MMP3 M1 mutant site at positions identical to those seen in the wild type MMP3-ETS1 footprint. Importantly, a nearly identical pattern of protection and enhancement was observed previously (21, 23) in the complex of ETS family member PU.1 with its DNA site and in many other ETS family protein-DNA complexes. Thus, this hypersensitivity is a universal hallmark of sequence-specific ETS-DNA complexes (23–28). Together, our findings suggest that although the M1 mutation prevents formation of an ETS1-DNA complex that is detectable by EMSA (Fig. 1B), ETS1Δ280 apparently does form a complex with DNA containing only a single EBS (Fig. 2B).
Both Specific and Nonspecific DNA Stimulates ETS1 Dimerization
The similarity in the positions of the enhanced DNase I cleavages of the ETS1Δ280-DNA complexes with MMP3 wild type and M1 mutant sites suggests that, despite bearing a different number of EBS core sequences, ETS1Δ280 binds to each of these sites as a dimer. To test this idea, we used cross-linking to determine the oligomeric state of ETS1Δ280 that forms in the presence of each of these DNAs. For these experiments, we created an ETS1Δ280 R378C variant, which in a DNA-bound ETS1Δ280 places two cysteine residues (one from each monomer) within ∼13 Å of each other. Therefore, these residues can be cross-linked using a sulfhydryl-specific bifunctional cross-linker, bismaleimidoethane, or BMOE. The R378C mutation lowers the affinity of ETS1 for the MMP3 site by 9-fold, binding it with a KD of 1.8 nm on MMP3 WT, as compared to 0.20 nm for the wild type protein, but did not affect other characteristics of ETS1Δ280-DNA complexes (data not shown).
ETS1Δ280 R378C does not form a covalent dimer when incubated without BMOE and/or DNA. Similarly, no covalently cross-linked dimers are formed when ETS1Δ280 R378C is incubated with BMOE but without DNA (Fig. 3, A and B). When ETS1Δ280 R378C is incubated in the presence of cross-linker and a 2–5-fold molar excess of DNA that contains either MMP3 or p53 EBSs, ∼50 and 38%, respectively, of ETS1Δ280 forms cross-linked dimers. Because under the conditions of this experiment, all of the ETS1Δ280 R378C is bound to DNA, the amount of dimer formed in these experiments is a consequence of specific binding of ETS1Δ280 R378C to the two head to head EBS in these DNAs. When ETS1Δ280 R378C is incubated with MMP3 mutants in which two head to head EBS in MMP3 are separated (MMP3 + 4) or one or the other EBS is disrupted (MMP3 M1 or MMP3 M2), ∼15–20% of this protein forms cross-linked dimers. Despite the reduction in amount of ETS1Δ280 dimer formed with these mutant sites, the amount of ETS1Δ280 dimers formed in the presence of these mutant binding sites is much greater than that formed without DNA. Therefore the findings in Fig. 3 indicate that that the ETS1Δ280-DNA complexes formed on sites bearing only a single EBS contain two ETS1Δ280 monomers. Hence, these results show that DNA induces dimer formation by ETS1Δ280. Significantly, we find that incubating ETS1Δ280 R378C with poly(dI-dC) also stimulates formation of cross-linked ETS1Δ280 R378C dimers. The amount of ETS1Δ280 R378C dimers formed in the presence of poly(dI-dC) is indistinguishable from that formed in the presence of the MMP3 M1, M2, or +4 mutant sites. This latter finding indicates that the association of ETS1Δ280 with nonspecific DNA is sufficient to induce dimer formation.
FIGURE 3.
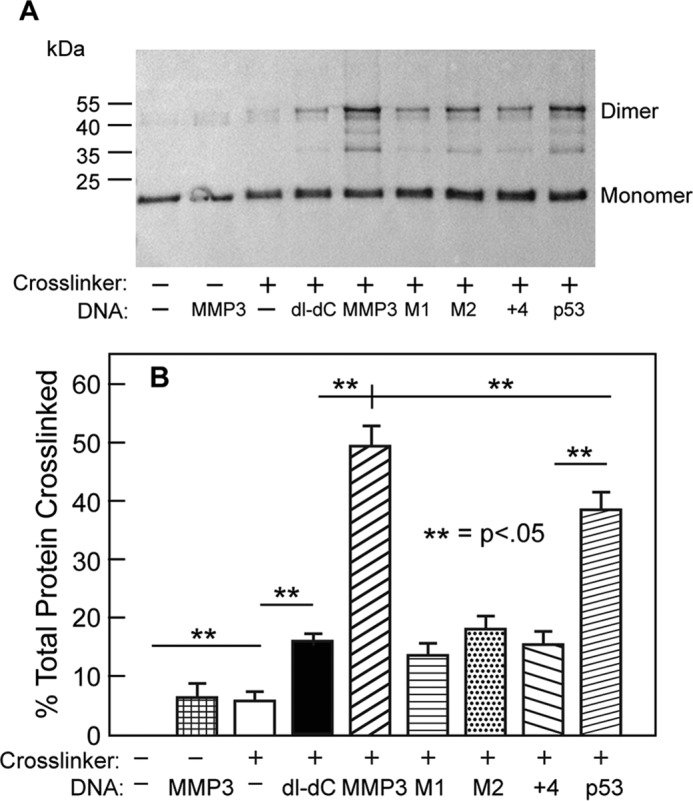
ETS1Δ280 R378C dimer formation. A, shown is an immunoblot of ETS1Δ280 R378C cross-linked by BMOE in the absence or presence of the indicated DNA sequence. dI-dC refers to poly(dI-dC), a repeating inosine-cytosine oligonucleotide used as a nonspecific binding template. The positions of protein monomers and dimers are indicated. Intermediate bands between monomers and dimers are thought to result from incomplete synthesis of ETS1 protein when purified from E. coli. B, quantification of results seen in A. The error bars represent standard deviation derived from four or more replicate experiments.
DNA Alters ETS1 Conformation
Our results show that the presence of DNA catalyzes ETS1Δ280 dimer formation (Fig. 3). It is unclear how DNA stimulates ETS1Δ280 oligomerization. We suggest that DNA triggers dimer formation by stimulating a change in ETS1 conformation and that this conformational change is the same regardless of the DNA sequence being interrogated. To test these ideas, we used partial proteolysis to probe for DNA-induced alterations in ETS1Δ280 conformation. When ETS1 is incubated with chymotrypsin in the absence of DNA, we observed the accumulation of two major proteolytic products (Fig. 4A). When ETS1Δ280 is incubated with chymotrypsin in the presence of DNA, we observed the accumulation of only one of these two products. The proteolysis pattern does not depend on the identity of the DNA added; identical patterns are seen regardless of the sequence of added DNA (Fig. 4A, bottom panel). These findings indicate that the association of ETS1Δ280 with any DNA is sufficient to induce a conformational change. Mass spectrometric analysis of these proteolytic fragments indicates that the conformational change associated with DNA binding that causes these differential cleavage patterns is a result of increased sensitivity of ETS1Δ280 to chymotrypsin in residues 356–361. This region is located in helix H1, a region previously implicated in maintenance of autoinhibition (4, 29). Together with the results in Fig. 3, these results indicate that the conformational change in the region near helix H1 leads to formation of ETS1Δ280 dimers. These data also suggest that the identical changes in ETS1Δ280 conformation accompany the association of ETS1Δ280 with either specific or nonspecific DNA sequences.
FIGURE 4.
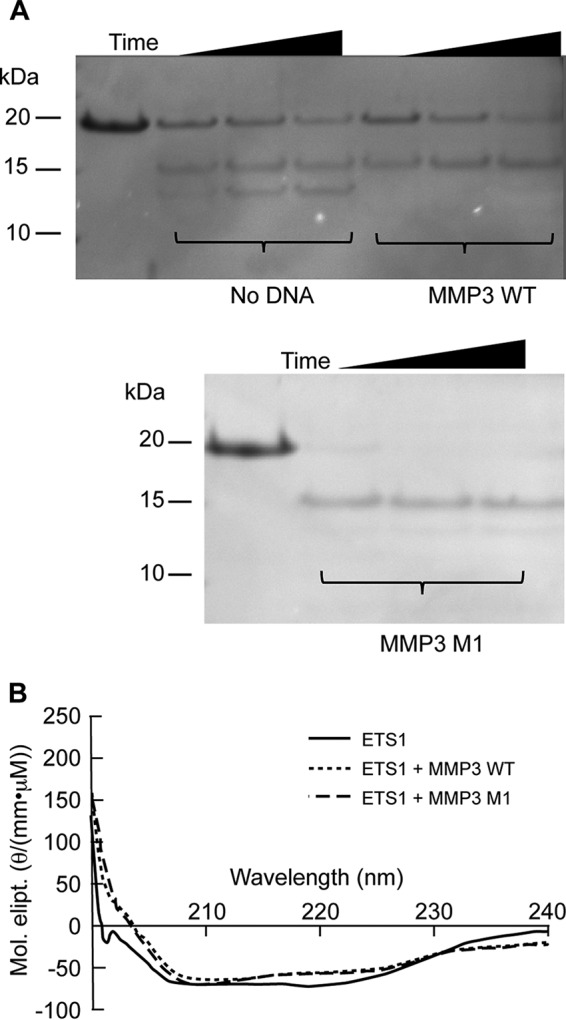
Analysis of ETS1Δ280 conformational states. A, partial proteolysis of ETS1Δ280 in the absence or presence various DNA sequences. Shown is a Coomassie-stained SDS-PAGE of the products of a limited, time-dependent proteolytic cleavage of ETS1Δ280 by chymotrypsin in the absence or presence of the indicated DNA. The electrophoretic mobilities of molecular weight standards are indicated to the left of the gel. B, circular dichroism spectra of ETS1 in the absence or presence of DNA. Similar to A, ETS1Δ280 was saturated with either specific (MMP3 wild type) or nonspecific (MMP3 M1) DNA. The reaction mixture CD spectra was then measured and converted to molar ellipticity (Mol. elipt.). The spectra for ETS1 alone is represented with a solid line, ETS1 in the presence of MMP3 WT is represented with a dotted line, and ETS1 in the presence of MMP3 M1 is represented with a dashed line.
To further test this idea, we used circular dichroism to monitor the changes in ETS1Δ280 secondary structure that accompanies formation of its complex with DNA. Consistent with structural studies (4), the CD spectrum of ETS1Δ280 obtained in the absence of DNA indicates that this protein contains both α helix and β-sheet (Fig. 4B). Adding saturating concentrations of MMP3 DNA causes a decrease in the CD intensity between 210 and 225 nm, a change that is consistent with a reduction in the amount of α-helix present in the protein (30). Relief of autoinhibition upon ETS1 complex formation has previously been found to be associated with unfolding of α-helix HI-1 (residues 304–310) in the N-terminal autoinhibitory region (4). Interestingly, adding an equivalent amount of MMP3 M1 to ETS1Δ280 induces an identical change in CD intensity, as does MMP3. Together, the results in Fig. 4 demonstrate that DNA induces a conformational change in ETS1Δ280 that is prerequisite for its dimerization and subsequent DNA binding.
Residues in the ETS1 DNA Recognition Helix Have a Role in Preventing ETS1 Dimerization
Our results show that regardless of whether it contains an EBS or not, DNA can induce a conformational change in ETS1Δ280 that leads to dimer formation (Figs. 3 and 4). This finding suggests that sequence-specific DNA binding is not required for DNA-stimulated ETS1Δ280 dimer formation. To test this idea and to explore the how DNA stimulates ETS1Δ280 dimerization, we changed both arginine 394 and tyrosine 395 to alanine and examined the effect these changes have on ETS1Δ280 DNA binding and dimerization. These changes remove the base-specifying contacts made by ETS1Δ280 to the first guanine and the first adenine, respectively, of the GGA(A/T) EBS core sequence (4). As shown by the inability of ETS1Δ280 R394A/Y395A to form complexes with either wild type MMP3 or p53 sites in an EMSA, these mutations prevent sequence-specific DNA binding by ETS1Δ280 (data not shown).
We introduced an R378C mutation into ETS1Δ280 R394A/Y395A and used BMOE-mediated cross-linking to probe whether a loss of specific DNA binding determinants affects the ability of DNA to stimulate ETS1Δ280 dimerization. Surprisingly, we find that ETS1Δ280 R378C/R394A/Y395A is capable of forming a dimer in the absence of any added DNA (Fig. 5, A and B). Moreover, the amount of dimer formed in the absence of DNA is identical to that formed in the presence of either MMP3 M1, MMP3 M2, p53, or poly(dI-dC). In fact, with the exception of added wild type MMP3, the presence of DNA did not significantly alter the amount of ETS1Δ280 R378C/R394A/Y395A that forms cross-linked dimers. These results indicate that the R394A/Y395A mutations mimic the effect of the DNA-induced conformational change that leads to ETS1Δ280 dimerization.
FIGURE 5.
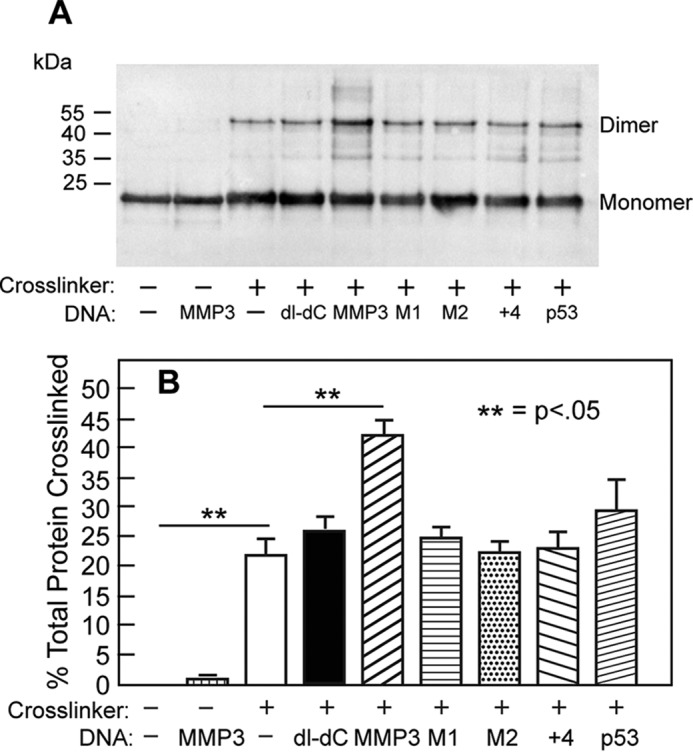
Analysis of the association state of an ETS1Δ280 helix H3 mutant. A, shown is an immunoblot of ETS1Δ280 R378C/R394A/Y395A cross-linked by BMOE in the absence or presence of the indicated DNA sequences. The positions of protein monomers and dimers are indicated. B, quantification of blot in A. The error bars represent the standard deviation derived from four or more replicate experiments.
Taken together, the results in Figs. 3–5 suggest that ETS1Δ280 association with either specific or nonspecific DNA both causes conformational change(s) that lead to relief of autoinhibition and allows dimerization. This suggestion indicates that the same conformational change both relieves autoinhibition and stimulates ETS1Δ280 dimerization. Structural studies indicate that Leu337-mediated packing of helix H1 between the ETS1 recognition helix and the autoinhibitory module plays a role in coupling relief of autoinhibition to DNA binding (31). To explore whether the autoinhibition module also regulates dimerization, we examined the effect of an L337A mutation on ETS1Δ280 dimerization by monitoring BMOE-mediated cross-linking of ETS1Δ280 L337A/R378C. We find that this protein does not dimerize, either in the absence or in the presence of specific or nonspecific DNA (Fig. 6A). This finding argues that relief of autoinhibition and ETS1Δ280 dimerization are linked. Interestingly, the L337A mutation does not prevent a DNA-inducible conformational change. Similar to the wild type ETS1, added DNA causes a reduction in CD intensity in the range of 210–225 nm (Fig. 6B). However, the overall CD change observed with ETS1-L337A differs from that seen with wild type ETS. The precise role of Leu337 in regulating ETS1 dimer formation is as yet unknown.
FIGURE 6.
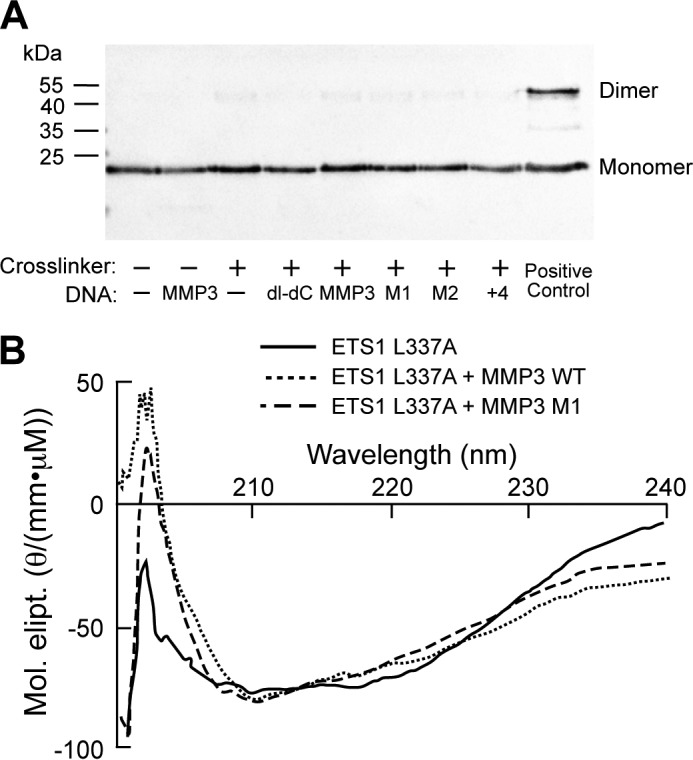
Analysis of the association state and conformational state of an ETS1 helix H1 mutant. A, shown is an immunoblot of ETS1Δ280 L337A/R378C cross-linked by BMOE in the absence or presence of the indicated DNA sequence. The positions of protein monomers and dimers are indicated. ETS1Δ280 R378C incubated with MMP3 WT and BMOE serves as a positive control. B, circular dichroism spectra of ETS1 L337A in the absence or presence of DNA, plotted as molar ellipticity (Mol. elipt.). The spectrum in the absence of DNA is represented with a solid line, the spectrum in the presence of MMP3 is represented with a dotted line, and the spectrum in the presence of MMP3 M1 is represented with a dashed line.
Association State of an ETS1 Splice Variant
Previous investigations showed that an ETS1 splice variant lacking the N-terminal autoinhibitory module encoded by exon VII is able to bind DNA as a monomer (1). Together with our current findings, this observation indicates that DNA-induced dimerization and relief of autoinhibition are linked. To test this idea, we examined the DNA binding and dimerization properties of the ETS1 splice variant, ETS1-p42, which lacks the N-terminal inhibitory domain that forms part of the ETS1 autoinhibitory module (4, 19).
We incorporated the R378C mutation into an N-terminal deletion, ETS1Δ335, a protein that recapitulates the DNA binding of ETS1-p42, and tested the ability of this protein to dimerize in the absence of DNA. We did this via a BMOE cross-linking assay identical to the one performed with full-length ETS1. In these experiments, when incubated with BMOE but without DNA, we find that this protein forms a cross-linked complex twice the size of monomeric ETS1Δ335 (Fig. 7A). This result implies that a lack of autoinhibition, a characteristic inherent to the ETS1-p42 splice variant, results in free ETS1Δ335 molecules dimerizing in the absence of DNA. Additionally, in a native EMSA, ETS1Δ335 R378C forms two different sized protein-DNA complexes, whereas ETS1Δ335 wild type only produces one complex (Fig. 7B). Together, these results show that an ETS1 splice variant lacking autoinhibition self-associates in the absence of DNA.
FIGURE 7.
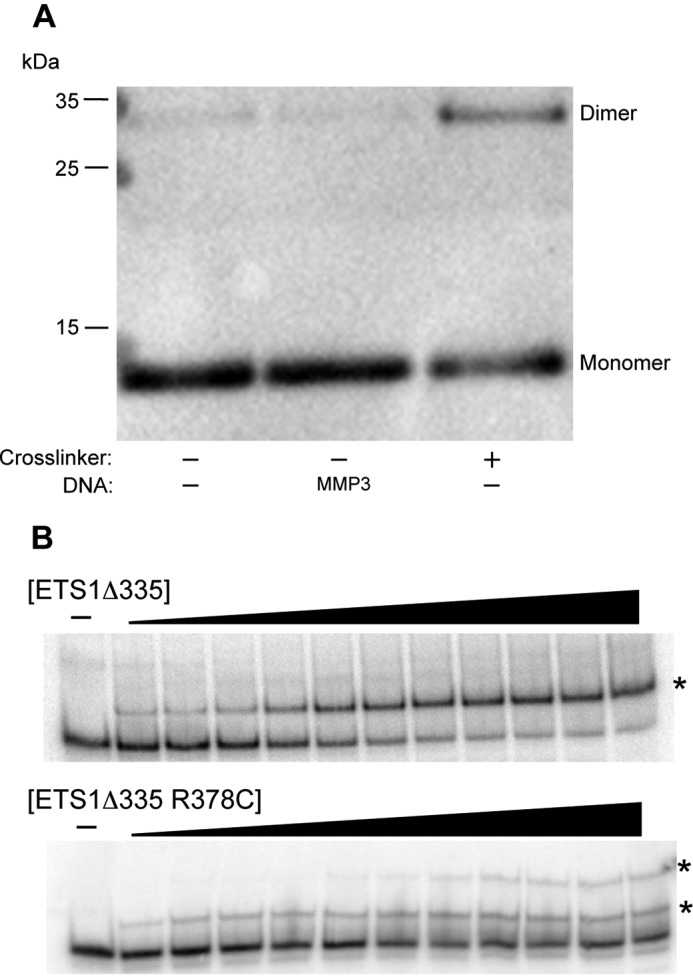
Association state of ETS1Δ335, an ETS1 splice variant lacking the capability for autoinhibition. A, shown is an immunoblot of ETS1Δ335 R378C cross-linked by BMOE in the absence of DNA. The positions of protein monomers and dimers are indicated. B, comparison of the electrophoretic mobilities of complexes formed by ETS1Δ335 and ETS1Δ335 R378C with radiolabeled MMP3 DNA. Positions of the protein-DNA complexes are indicated with asterisks.
Discussion
Despite sharing nearly identical preferred binding site sequences (3), each ETS protein family member binds to and regulates transcription from its own unique set of promoter targets (32). Two prevailing, nonmutually exclusive hypotheses have been proposed to resolve this paradox: 1) indirect readout of different noncontacted bases in each site may differentially modulate the strength of ETS1-DNA interactions by changing the geometry of the protein-DNA interface (5, 31), and 2) the type and strength of protein-protein interactions may be unique to each ETS family member/binding site and thereby influence binding site affinity (7). The effect of varying noncontacted bases on the affinity of ETS1 for artificial DNA sites has been reported previously (31), but prior to our work, the importance of these bases to ETS1 binding site discrimination among natural sites had not demonstrated. Work on other ETS proteins has been reported, however (6). Our finding that ETS1 affinities for its binding sites in the p53 and MMP3 promoters vary by 30-fold (Table 1 and Fig. 1), despite having identical EBSs, demonstrates that indirect readout plays in important role in the ability of ETS1 to distinguish between naturally occurring EBSs. However, the finding that the DNase I footprints of the p53- and MMP3-ETS1 complexes are identical (Fig. 2) indicates that noncontacted bases do not affect the conformation of the protein-DNA complex. This observation suggests that noncontacted bases may affect ETS1 DNA affinity by modulating either the rate of ETS1-DNA complex formation or breakdown.
Previous investigations concluded that relief of autoinhibition mediated by the N-terminal autoinhibitory region of ETS1 occurs as a consequence of hetero- or homodimer formation (1, 4). Results of kinetic analysis of ETS1 binding to specific DNA have also been interpreted as indicating that ETS1 dimer interactions occur only after two monomers encounter two head to head sites (1). Hence current models of ETS1 DNA binding to head to head sites posit that each ETS monomer independently binds each of the two EBS and subsequently interact. Therefore the cooperative binding of two monomers is a consequence of conformational changes in the N-terminal autoinhibitory regions in each of the two bound monomers following DNA binding. According to this idea, these changes relieve autoinhibition and allow the N-terminal regions in each of the two polypeptides to contact each other. The interaction between these regions stabilizes the ETS1 dimer-DNA complex (4).
As opposed to current models, which suggest that specific DNA binding is a prerequisite for ETS1 dimer formation, our observations indicate that association of ETS1 with nonspecific DNA is sufficient for relief of autoinhibition and ETS1 dimerization. In support of this idea, we find that 1) mutations that prevent ETS1 from recognizing specific DNA sequences do not block DNA-stimulated ETS1 dimer formation (Fig. 5); 2) any DNA, regardless of whether or not it contains an EBS, is sufficient to catalyze both conformational changes in the N-terminal autoinhibitory region of ETS1 and ETS1 dimer formation (Figs. 3 and 4, respectively); and 3) mutation of Leu337, a DNA phosphate backbone-contacting residue that couples relief of autoinhibition to DNA binding, renders ETS1 incapable of forming dimers under any condition (Fig. 6). We note that, similar to our findings here, a recent study of ETV6, another ETS protein, showed that specific and nonspecific DNA both induce identical conformational changes in this protein, changes that lead to relief of autoinhibition of ETV6 (33). Therefore, the use of nonspecific DNA to relieve autoinhibition is not limited to ETS1 and may be a common feature of autoinhibited ETS family members.
One puzzling result is that we find that as compared with nonspecific DNA, added MMP3 wild type DNA increases the amount of cross-linked dimer formed by the α-helix H3 mutant protein, whereas added p53 does not (Fig. 5). Both promoter sequences contain the canonical head to head ETS1 dimer site. To explain this observation, we suggest that the loss of the ability of ETS1 R394A/Y395A to recognize the p53 sequence is due to its loss of autoinhibition, allowing its behavior to mirror that of the splice variant ETS1-p42. Similar to ETS1 R394A/Y395A, ETS1-p42 does not display autoinhibition. This protein binds to the MMP3 site but not to the p53 site (data not shown). This suggestion raises the idea that autoinhibition may be required by ETS1 for binding to certain DNA sequences. This idea may also explain the in vivo differences in promoter targeting between the ETS1-p51 and ETS1-p42 splice variants (34). Whether or not the inability to autoinhibit is related to the abilities of the two variants to differentially bend DNA (20) remains to be seen.
Our finding that nonspecific DNA or DNA that contains only a single EBS can catalyze ETS1Δ280 dimerization (Fig. 3) seemingly contradicts previous studies suggesting that ETS1Δ280 only dimerizes on DNA containing two appropriately juxtaposed EBS (1, 4, 31). However, no studies of ETS1Δ280 dimerization that we know of have probed the effect of nonspecific DNA on this process. Moreover, the methods used in previous studies are only able to detect the formation of kinetically stable ETS1Δ280 dimers, whereas our BMOE-mediated cross-linking is sensitive to the transient formation of specific ETS1Δ280 dimers.
Superficially, our finding that ETS1Δ280 forms cross-linked dimers in the presence of the MMP3 M1 differs from the results in Ref. 1, which indicated that only one monomer of ETS1Δ280 can be cross-linked to this DNA. This apparent discrepancy can be resolved in that the cross-linking methods in these studies probe different aspects of ETS1Δ280 complex formation. Our BMOE-mediated cross-linking method probes the interaction between two ETS1Δ280 protein monomers on MMP3 M1, whereas the photo cross-linker used by (1) reports the strong interaction of a monomer of ETS1Δ280 with the single EBS DNA in MMP3 M1. Therefore, together these results indicate that one monomer of ETS1Δ280 strongly interacts with the one intact EBS in MMP3 M1, and this DNA-bound monomer dimerizes with a nonspecifically bound ETS1Δ280 molecule. This suggestion is consistent with the results of our DNase I footprinting experiments showing that regions of enhanced DNase I cleavage are seen in both half-sites of the ETS1Δ280-MMP3 M1 complex at positions identical to those seen in the wild type MMP3-ETS1 footprint (Fig. 2).
Autoinhibition lowers the DNA binding affinity of ETS1 for specific DNA. The lowered DNA affinity is thought to be a consequence of the thermodynamic cost of unfolding N-terminal autoinhibitory region (5, 31). However, the findings that 1) nonspecific DNA can induce unfolding of this region (Fig. 4); 2) ETS1Δ280 bearing mutations in the DNA contacting residues in helix H3 constitutively form dimers in the absence and presence of DNA (Fig. 5); and 3) ETS1Δ335, an ETS1 variant that lacks the N-terminal autoinhibitory domain, also dimerizes in the absence of DNA (Fig. 7), argue for a new role for ETS1 autoinhibition: prevention of ETS1 dimerization in the absence of DNA. Because full-length ETS1 only binds head to head sites as dimers, this new function of the autoinhibitory module suggests that the lowered DNA affinity of ETS1 may also, in whole or part, be caused by blocking ETS1 dimer formation. These results may also explain the results seen in an x-ray crystallography study on a partially inhibited fragment of ETS1, termed Δ300 (15). The propensity of this fragment to dimerize was ascribed to an effect of crystal packing. Our findings show that this study may have been an early indication of the role of autoinhibition in preventing dimer formation.
Regardless, our findings equate a loss of autoinhibition with ETS1 dimerization. Our finding that ETS1 R394A/Y395A forms dimers in the absence of DNA (Fig. 5) implicates the DNA-binding helix H3 in maintenance of autoinhibition, a role not previously described. Despite lacking the ability to recognize EBS DNA, this protein does recognize the presence of DNA. Because its DNA binding surface is disrupted, the nature of ETS1 R394A/Y395A-DNA complexes is unclear. However, Ref. 33 showed that the ETS family member ETV6 uses the same protein surface to bind both specific and nonspecific DNA. Thus, we envision that, identical to unmutated ETS1Δ280, the ETS1Δ280 R394A/Y395A also recognizes DNA using its H3 helix. Therefore, our results link nonspecific ETS1-DNA complex formation to relief of autoinhibition, which subsequently allows dimerization. Consistent with this idea, ETS1Δ280 lacking the Leu337 residue, a mutation that uncouples DNA binding from autoinhibition (31), is incapable of forming cross-linked dimers both in the absence and presence of DNA (Fig. 6). Leu337 sits at the N-terminal end of helix H1. The H1 helix macrodipole is thought to play a large role in ETS1 DNA binding. Because the helix H1 macrodipole is well conserved across the ETS family and Leu337 itself is completely conserved (31), we surmise that this mechanism may exist in all ETS proteins that autoinhibit, which include, in addition to ETV6, ETS2, ETV4, ETV5, and the ESE and TCF subfamilies (5).
We suggest that autoinhibition/prevention of ETS1 oligomerization is essential for ETS1 gene regulatory activities. If ETS1 associates prematurely with any available ETS1 partner, including itself, this protein would unavailable to nonspecifically interrogate DNA and associate with other nonspecifically scanning ETS1 partners. Keeping ETS1 primed for DNA binding is advantageous in that it prevents functional transcription factors from being unnecessarily sequestered from interacting with DNA.
Furthermore, the ability to nonspecifically dimerize and interact with DNA provides ETS1 with the ability to scan large segments of DNA for both a binding site and a potential binding partner. Without this ability, the likelihood of ETS1 encountering a binding site by random walk would be astronomically low.
Because unfolding of the N-terminal inhibitory region flanking the ETS domain and subsequent dimer formation is a consequence of ETS1 interaction with any DNA, regardless of whether it contains an EBS, we propose a new model for ETS1 DNA binding and complex formation. We suggest that as ETS1 searches for its site on nonspecific DNA, this encounter unmasks its oligomerization region. In this conformation, the “scanning” ETS1 molecule is able to interact with available partner proteins that are simultaneously scanning—whether it is another ETS1 molecule or a heterologous partner like RUNX1—to form transient dimers that then search together for a dimer binding site. The dimer may be stabilized if such a site is found or rapidly dissociate if not. This model explains the inability of ETS1Δ280 to form EMSA-detectable complexes on DNAs containing only one EBS while retaining the ability to form cross-linked dimers in the presence of those same DNA sequences. The inability of the Leu337 mutant to forms dimers in the absence or presence of DNA leads us to suspect that the backbone contacts studied previously (31) serve as the register for ETS1 scanning. It also reinforces the notion put forth in Ref. 31 that helix H1 serves as a link between DNA binding and complex formation.
Author Contributions
D. S. and G. B. K. designed the study and wrote the paper. D. S. and C. S. performed the experiments. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank members of the Koudelka lab for critical comments on this work and Jim Stamos for help with figure preparation. We also thank Dr. Arpad Soyogi and Dr. Liwen Zhang at the Ohio State University Campus Chemical Instrument Center for assistance with mass spectrometry analysis.
This work was supported by funds from the College of Arts and Sciences, University at Buffalo (to G. B. K.). The authors declare that they have no conflicts of interest with the contents of this article.
- EBS
- ETS binding site
- BMOE
- bismaleimidoethane
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
References
- 1. Baillat D., Bègue A., Stéhelin D., Aumercier M. (2002) ETS-1 transcription factor binds cooperatively to the palindromic head to head ETS-binding sites of the stromelysin-1 promoter by counteracting autoinhibition. J. Biol. Chem. 277, 29386–29398 [DOI] [PubMed] [Google Scholar]
- 2. Sharrocks A. D. (2001) The ETS-domain transcription factor family. Nat. Rev. Mol. Cell Biol. 2, 827–837 [DOI] [PubMed] [Google Scholar]
- 3. Wei G. H., Badis G., Berger M. F., Kivioja T., Palin K., Enge M., Bonke M., Jolma A., Varjosalo M., Gehrke A. R., Yan J., Talukder S., Turunen M., Taipale M., Stunnenberg H. G., Ukkonen E., Hughes T. R., Bulyk M. L., Taipale J. (2010) Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J.. 29, 2147–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lamber E. P., Vanhille L., Textor L. C., Kachalova G. S., Sieweke M. H., Wilmanns M. (2008) Regulation of the transcription factor Ets-1 by DNA-mediated homo-dimerization. EMBO J. 27, 2006–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hollenhorst P. C., McIntosh L. P., Graves B. J. (2011) Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 80, 437–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Poon G. M., Macgregor R. B. Jr. (2003) Base coupling in sequence-specific site recognition by the ETS domain of murine PU.1. J. Mol. Biol. 328, 805–819 [DOI] [PubMed] [Google Scholar]
- 7. Dittmer J. (2003) The biology of the Ets1 proto-oncogene. Mol. Cancer 2, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baillat D., Laitem C., Leprivier G., Margerin C., Aumercier M. (2009) Ets-1 binds cooperatively to the palindromic Ets-binding sites in the p53 promoter. Biochem. Biophys. Res. Commun. 378, 213–217 [DOI] [PubMed] [Google Scholar]
- 9. Baillat D., Leprivier G., Régnier D., Vintonenko N., Bègue A., Stéhelin D., Aumercier M. (2006) Stromelysin-1 expression is activated in vivo by Ets-1 through palindromic head-to-head Ets binding sites present in the promoter. Oncogene 25, 5764–5776 [DOI] [PubMed] [Google Scholar]
- 10. Kim W. Y., Sieweke M., Ogawa E., Wee H. J., Englmeier U., Graf T., Ito Y. (1999) Mutual activation of Ets-1 and AML1 DNA binding by direct interaction of their autoinhibitory domains. EMBO J.. 18, 1609–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gu T. L., Goetz T. L., Graves B. J., Speck N. A. (2000) Auto-inhibition and partner proteins, core-binding factor beta (CBFbeta) and Ets-1, modulate DNA binding by CBFalpha2 (AML1). Mol. Cell. Biol. 20, 91–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Babayeva N. D., Wilder P. J., Shiina M., Mino K., Desler M., Ogata K., Rizzino A., Tahirov T. H. (2010) Structural basis of Ets1 cooperative binding to palindromic sequences on stromelysin-1 promoter DNA. Cell Cycle 9, 3054–3062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maxam A. M., Gilbert W. (1980) Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 65, 499–560 [DOI] [PubMed] [Google Scholar]
- 14. Sharma S., Gollnick P. (2014) Modulating TRAP-mediated transcription termination by AT during transcription of the leader region of the Bacillus subtilis trp operon. Nucleic Acids Res. 42, 5543–5555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garvie C. W., Pufall M. A., Graves B. J., Wolberger C. (2002) Structural analysis of the autoinhibition of Ets-1 and its role in protein partnerships. J. Biol. Chem. 277, 45529–45536 [DOI] [PubMed] [Google Scholar]
- 16. Lee G. M., Donaldson L. W., Pufall M. A., Kang H. S., Pot I., Graves B. J., McIntosh L. P. (2005) The structural and dynamic basis of Ets-1 DNA binding autoinhibition. J. Biol. Chem. 280, 7088–7099 [DOI] [PubMed] [Google Scholar]
- 17. Lee G. M., Pufall M. A., Meeker C. A., Kang H. S., Graves B. J., McIntosh L. P. (2008) The affinity of Ets-1 for DNA is modulated by phosphorylation through transient interactions of an unstructured region. J. Mol. Biol. 382, 1014–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pufall M. A., Lee G. M., Nelson M. L., Kang H. S., Velyvis A., Kay L. E., McIntosh L. P., Graves B. J. (2005) Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science 309, 142–145 [DOI] [PubMed] [Google Scholar]
- 19. Lionneton F., Lelièvre E., Baillat D., Stehelin D., Soncin F. (2003) Characterization and functional analysis of the p42Ets-1 variant of the mouse Ets-1 transcription factor. Oncogene 22, 9156–9164 [DOI] [PubMed] [Google Scholar]
- 20. Leprivier G., Baillat D., Begue A., Hartmann B., Aumercier M. (2009) Ets-1 p51 and p42 isoforms differentially modulate Stromelysin-1 promoter according to induced DNA bend orientation. Nucleic Acids Res. 37, 4341–4352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Poon G. M. (2012) DNA binding regulates the self-association of the ETS domain of PU.1 in a sequence-dependent manner. Biochemistry 51, 4096–4107 [DOI] [PubMed] [Google Scholar]
- 22. Hellman L. M., Fried M. G. (2007) Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2, 1849–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Poon G. M. (2012) Sequence discrimination by DNA-binding domain of ETS family transcription factor PU.1 is linked to specific hydration of protein-DNA interface. J. Biol. Chem. 287, 18297–18307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rao E., Dang W., Tian G., Sen R. (1997) A three-protein-DNA complex on a B cell-specific domain of the immunoglobulin mu heavy chain gene enhancer. J. Biol. Chem. 272, 6722–6732 [DOI] [PubMed] [Google Scholar]
- 25. Nye J. A., Petersen J. M., Gunther C. V., Jonsen M. D., Graves B. J. (1992) Interaction of murine ets-1 with GGA-binding sites establishes the ETS domain as a new DNA-binding motif. Genes Dev. 6, 975–990 [DOI] [PubMed] [Google Scholar]
- 26. Gross P., Arrowsmith C. H., Macgregor R. B. Jr. (1998) Hydroxyl radical footprinting of DNA complexes of the ets domain of PU.1 and its comparison to the crystal structure. Biochemistry 37, 5129–5135 [DOI] [PubMed] [Google Scholar]
- 27. Graves B. J., Gillespie M. E., McIntosh L. P. (1996) DNA binding by the ETS domain. Nature 384, 322. [DOI] [PubMed] [Google Scholar]
- 28. Gunther C. V., Graves B. J. (1994) Identification of ETS domain proteins in murine T lymphocytes that interact with the Moloney murine leukemia virus enhancer. Mol. Cell. Biol. 14, 7569–7580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petersen J. M., Skalicky J. J., Donaldson L. W., McIntosh L. P., Alber T., Graves B. J. (1995) Modulation of transcription factor Ets-1 DNA binding: DNA-induced unfolding of an alpha helix. Science 269, 1866–1869 [DOI] [PubMed] [Google Scholar]
- 30. Greenfield N. J. (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1, 2876–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang H., McIntosh L. P., Graves B. J. (2002) Inhibitory module of Ets-1 allosterically regulates DNA binding through a dipole-facilitated phosphate contact. J. Biol. Chem. 277, 2225–2233 [DOI] [PubMed] [Google Scholar]
- 32. Hollenhorst P. C., Shah A. A., Hopkins C., Graves B. J. (2007) Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes Dev. 21, 1882–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. De S., Chan A. C., Coyne H. J. 3rd, Bhachech N., Hermsdorf U., Okon M., Murphy M. E., Graves B. J., McIntosh L. P. (2014) Steric mechanism of auto-inhibitory regulation of specific and non-specific DNA binding by the ETS transcriptional repressor ETV6. J. Mol. Biol. 426, 1390–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li R., Pei H., Papas T. (1999) The p42 variant of ETS1 protein rescues defective Fas-induced apoptosis in colon carcinoma cells. Proc. Natl. Acad. Sci. U.S.A. 96, 3876–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]