

Background: Aberrant activation of Wnt/β-catenin signaling is found in ovarian cancer, even when mutations are absent.
Results: The bioactive lipid lysophosphatidic acid induces β1-integrin clustering, nuclear β-catenin translocation, and activation of Tcf/Lef-regulated gene expression.
Conclusion: Microenvironmental lipids modulate β-catenin signaling.
Significance: Understanding the mechanisms of Wnt/β-catenin activation provides a basis for therapeutic targeting of the pathway in ovarian cancer.
Keywords: β-catenin (β-catenin), integrin, lysophospholipid, ovarian cancer, Wnt signaling, LPA
Abstract
During tumor progression, epithelial ovarian cancer (EOC) cells undergo epithelial-to-mesenchymal transition (EMT), which influences metastatic success. Mutation-dependent activation of Wnt/β-catenin signaling has been implicated in gain of mesenchymal phenotype and loss of differentiation in several solid tumors; however, similar mutations are rare in most EOC histotypes. Nevertheless, evidence for activated Wnt/β-catenin signaling in EOC has been reported, and immunohistochemical analysis of human EOC tumors demonstrates nuclear staining in all histotypes. This study addresses the hypothesis that the bioactive lipid lysophosphatidic acid (LPA), prevalent in the EOC microenvironment, functions to regulate EMT in EOC. Our results demonstrate that LPA induces loss of junctional β-catenin, stimulates clustering of β1 integrins, and enhances the conformationally active population of surface β1 integrins. Furthermore, LPA treatment initiates nuclear translocation of β-catenin and transcriptional activation of Wnt/β-catenin target genes resulting in gain of mesenchymal marker expression. Together these data suggest that LPA initiates EMT in ovarian tumors through β1-integrin-dependent activation of Wnt/β-catenin signaling, providing a novel mechanism for mutation-independent activation of this pathway in EOC progression.
Introduction
Ovarian cancer is a heterogeneous group of cancers subdivided into four types based on cellular phenotype as follows: germ cell tumors, stromal tumors, primary peritoneal tumors, and epithelial tumors (1, 2). Epithelial tumors represent 90% of all occurrences of ovarian cancer (epithelial ovarian cancer or EOC3); of these, high-grade serous EOC is the most commonly occurring (75% of cases and 90% of deaths (2, 3)). The lack of specific targeted therapies and acquisition of resistance to current standard therapies (combination platinum- and taxol-based compounds), coupled with diagnosis at advanced stage, makes ovarian cancer the deadliest gynecological malignancy and fifth leading cause of cancer-related deaths among American women (4–6). Improved understanding of the pathobiology and mechanisms underlying metastasis of EOC will aid in the development of more efficacious treatments for women with this disease.
The etiology of EOC remains unclear; however, two major hypotheses predict that epithelial ovarian cancers arise from genetic aberrations in either the ovarian surface epithelium or the fallopian tube epithelium (3, 7–11). Fallopian tube epithelial cells are derived from the intermediate mesoderm and display classic epithelial markers such as epithelial cadherin (E-cadherin). Although normal ovarian surface epithelium characteristically exhibits epithelial morphology, the mesodermally derived tissue expresses mesenchymal markers (e.g. vimentin, neural (N)-cadherin), and ovarian surface epithelium cells in culture often deposit extracellular matrix components characteristic of mesenchyme (collagen types I and III) (12, 13). Typically, gain of mesenchymal phenotype (epithelial-to-mesenchymal transition, or EMT) is a hallmark tumor dissemination and progression in many carcinomas (14, 15); however, ovarian carcinomas exhibit an early gain of epithelial phenotype or mesenchymal-to-epithelial transition. This gain of an epithelial gene profile aids in disseminating cells in multicellular aggregate (spheroid) formation and anoikis resistance (16). Later in progression, the cells revert to a mesenchymal phenotype (EMT), and it is proposed that this phenotypic plasticity contributes to decreased response to conventional therapeutics (17–20). Furthermore, recent work has demonstrated a role for the mesenchymal phenotype in mesothelial cell invasion at metastatic sites in the peritoneal cavity (21). Understanding the mechanism(s) regulating this phenotypic plasticity may enable reversal of acquired resistance to chemotherapeutic agents.
Acquisition of mesenchymal phenotype via EMT can be initiated by extracellular matrix context, disruption of adhesion, and soluble signaling factors (22, 23). Of these factors, lysophosphatidic acid (LPA) has emerged as a potentially important mediator of EMT in ovarian carcinoma due in part to its high concentration in the ovarian cancer microenvironment (up to 80 μm) (24, 25). LPA modulates mitogenic activity, motility, escape from anoikis, and survival effects through a class of heterotrimeric G-protein-coupled receptors, LPA1 to LPA5 (26, 27). One mechanism by which LPA modulates motility and facilitates the gain of a mesenchymal phenotype in EOC is disruption of E-cadherin-based cell-cell adhesions (28–31). Of particular interest is the fate of adherens junction-associated and/or cytoplasmic β-catenin following disruption of cell-cell adhesions. In intact epithelial tissues, β-catenin displays junctional localization and is associated with the cytoplasmic domain of E-cadherin. Junction disruption can induce pathways that subsequently target β-catenin for proteosome-mediated degradation or for nuclear translocation and transcriptional regulation (32). As previous work has demonstrated the intersection of cytoplasmic and nuclear β-catenin pools (33), this study investigated the consequences of LPA-induced disruption of E-cadherin-mediated cell junctions on subcellular β-catenin localization and Tcf/Lef/β-catenin transcriptional activity. These results provide additional support for the role of ligand-independent β-catenin activity in serous epithelial ovarian carcinomas.
Experimental Procedures
Cell Culture
OVCA429 and OVCA433 cell lines were generously provided by Dr. Robert Bast, Jr. (M.D. Anderson Cancer Center, Houston, TX), and were maintained in MEM (Gibco Invitrogen), 10% fetal bovine serum (Gibco Invitrogen), penicillin/streptomycin (Gibco Invitrogen), amphotericin B (Cellgro by Mediatech), nonessential amino acids (Cellgro by Mediatech, Herndon, VA), and sodium pyruvate (Cellgro by Mediatech) at 37 °C in 5% CO2. Multicellular aggregates were formed in 96-well plates coated with 50 μl of 0.5% agarose in serum-free media by seeding 5000 cells per well in serum-free media and incubating overnight at 37 °C in 5% CO2. Aggregate formation was confirmed by light microscopic visualization.
Lysophosphatidic Acid
LPA was purchased from Cayman Chemical (Ann Arbor, MI) and was prepared for use by dehydrating the lyophilized lipid under a tissue culture hood, on ice, overnight. LPA was resuspended in 1% BSA in PBS at a final concentration of 2 mm, allowed to dissolve on a rotator at 4 °C overnight, and then aliquoted. Aliquots in use were stored at −20 °C and at −80 °C for long term storage. Where indicated, cells were pretreated for 15 min prior to addition of LPA with the LPA receptor inhibitor, Ki16425, or the Rho signaling inhibitor, Y27632, both of which were purchased from Cayman Chemical (Ann Arbor, MI).
Antibodies
Mouse monoclonal anti-active β-catenin (clone 8E7) recognizes β-catenin that was de-phosphorylated on Ser-37 and/or Thr-41 and was purchased from Upstate Biotechnology (Lake Placid, NY). Purified mouse anti-β-catenin monoclonal antibody (clone 14/β-catenin), mouse anti-E-cadherin (clone 36/E-cadherin), and anti-active β1 integrin (clone HUTS21) were purchased from BD Transduction Laboratories. Anti-β1 integrin MAB1959 (function-blocking), anti-β1 integrin MAB2250, and a mouse monoclonal antibody recognizing FAK phosphorylated at tyrosine 397 (clone 18) were purchased from EMD Millipore (Billerica, MA). Anti-HDAC1 was purchased from Thermo Fisher Scientific (Rockford, IL). Anti-proliferating cell nuclear antigen (clone PC10) was purchase from Dako (Glostrup, Denmark). Mouse anti-E-cadherin (clone HECD-1) was purchased from Zymed Laboratories Inc. Monoclonal mouse anti-vinculin (clone VIN-11–5) was purchased from Abcam (Cambridge, MA). Monoclonal mouse anti-vimentin (clone VIM-13.2), polyclonal mouse anti-IgG, peroxidase-conjugated anti-mouse IgG, and peroxidase-conjugated anti-rabbit IgG were purchased from Sigma. Rhodamine phalloidin, Alexa Fluor 488 goat anti-mouse IgG (H+L) was purchased from Molecular Probes (Eugene, OR). Rat anti-Tcf was purchased from Kamiya Biomedical (Tukwila, WA). Polyclonal anti-mouse IgG was purchased from Chemicon (Temecula, CA). Rabbit polyclonal anti-FAK was purchased from Santa Cruz Biotechnology (Dallas, TX).
Immunofluorescence
Cells were plated on 22-mm2 glass coverslips coated by passive adsorption with type I collagen (from rat tail, BD Biosciences) and placed in 6-well tissue culture plates and were treated with LPA at the concentrations and times described in the figure legends. Following treatment, cells were gently washed in phosphate-buffered saline (PBS), fixed in paraformaldehyde (4%), and permeabilized with 0.3% Triton X-100 at room temperature, washed in PBS, and blocked in PBS, 1% bovine serum albumin (BSA), or PBS, 2.5% goat serum, followed by the addition of primary antibody diluted in PBS, 1% BSA, or PBS, 2.5% goat serum at 37 °C. After two washes in PBS, coverslips were incubated with Alexa Fluor 488- or Alexa Fluor 594-conjugated goat anti-mouse or goat anti-rabbit IgG (1:500 dilution) in the dark at room temperature. Coverslips were washed twice in PBS and once in distilled water, fixed using gelvatol, and visualized using fluorescence microscopy (Nikon Microphot FXA or Leica Microsystems DM5500B). Semi-quantitative analysis of surface-associated (junctional) β-catenin staining was determined by counting a minimum of 12 fields per treatment and scoring as positive the number of cells with two remaining fluorescent cell-cell borders (30).
Immunohistochemistry
Immunohistochemical analysis wasperformed on a human tumor tissue microarray prepared at the Robert H. Lurie Comprehensive Cancer Center at Northwestern University with Institutional Review Board approval. Tumor specimens were cut 3–5 μm thick (1 mm in diameter) and deparaffinized. Antigen retrieval was accomplished by heat induction at 99 °C in an antigen retrieval solution (10 mm Tris, 1 mm EDTA, pH 9.0) for ∼1 h. Immunohistochemical staining with anti-β-catenin (BD Transduction Laboratories; 1:50) was performed according to standard procedures and as described previously (31). A total of 105 tissues of varying histotypes were evaluated (41 serous, 26 endometrioid, 3 malignant mixed Mullerian tumors, 6 mucinous, clear cell, 19 borderline, and 1 untyped). A determination of nuclear β-catenin positivity or negativity was determined based on the presence of β-catenin staining coinciding with hematoxylin-stained nuclei (34).
Cell Fractionation
Cells were fractionated following a 2-h incubation with LPA (40 μm) or LiCl (control, 40 μm) as described previously (35). Briefly, cells were washed twice with cold PBS and lysed with a cold hypotonic lysis buffer (10.0 mm NaCl, 20.0 mm HEPES, pH 7.9, 1.0 mm EDTA, 2.0 mm MgCl2, 20.0 mm β-glycerophosphate, 1.0 mm Na3VO4, 1.0 mm PMSF, 1.0 mm DTT, 200 mm sucrose, 0.5% Nonidet P-40, and 10 μg/ml each of aprotinin, pepstatin A, and leupeptin). Lysate was collected by scraping, passed through a 26-gauge syringe, and centrifuged at 16,000 × g for 1 min at 4 °C after a 10-min incubation on ice. The cytoplasmic fraction was collected (supernatant), and the pellet was washed twice with hypotonic lysis buffer before treatment with nuclear extraction buffer (420.0 mm NaCl, 20.0 mm HEPES, pH 7.9, 1.0 mm EDTA, 2.0 mm MgCl2, 20.0 mm β-glycerophosphate, 1.0 mm Na3VO4, 1.0 mm PMSF, 1.0 mm DTT, 25% glycerol, and 10 μg/ml each of aprotinin, pepstatin A, and leupeptin). Following a 10–15-min incubation and 5-min centrifugation (16,000 × g at 4 °C), the nuclear fraction was collected. Protein concentration was measured using a detergent-compatible protein assay kit (Bio-Rad). Western blot analysis was performed as described below. Control blots were probed for HDAC1 and β-actin to assess nuclear and cytoplasmic fractions, respectively.
Immunoprecipitation
The β-catenin/Tcf co-immunoprecipitation protocol was adopted from chromatin immunoprecipitation protocols described previously (36, 37). Cells were grown to 80% confluence and then serum-starved overnight. Appropriate samples were pretreated with 40 μm Ki16425 (LPA receptor inhibitor, Cayman Chemical, Ann Arbor, MI) or DMSO control, followed by addition of 40 μm LPA for 2 h. Nontreated cells, cells treated with Ki16425 alone, and cells treated with 40 μm LiCl served as controls. Cells were collected in a hypotonic lysis buffer (20 mm HEPES, pH 7.9, 25% glycerol, 420 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA), incubated on ice for 20 min, and then centrifuged at 13,000 rpm for 10 min at 4 °C. Nuclei isolated from the previous step were disrupted by resuspension in a “breaking” buffer (50 mm Tris-HCl, pH 8.0, 1 mm EDTA, 150 mm NaCl, 1% SDS, 2% Triton X-100) and then passed through a 26-gauge syringe eight times. The suspension was centrifuged at 13,000 rpm for 10 min at 4 °C, diluted in 1 ml of Triton buffer (50 mm Tris-HCl, pH 8.0, 1 mm EDTA, 150 mm NaCl, 0.1% Triton X-100), and cleared of nonspecific binding by incubation with 20 μl of protein A/G beads (Sigma) at 4 °C overnight. After centrifugation and collection of supernatant, protein concentration was measured using a kit (Bio-Rad). Five hundred micrograms of total protein were added to 1 ml of Triton buffer, 20 μl of protein A/G beads, and 5 μg of anti-β-catenin antibody and then incubated on a rotator overnight at 4 °C. Beads were washed five times in Triton buffer and then resuspended in 2× sample buffer. Samples were boiled and then analyzed for Tcf expression by Western blot as described below. Antibody was removed from the PVDF membrane by washing in stripping buffer (50 mm Tris, pH 6.8, 1% SDS, 150 mm NaCl, 100 mm β-mercaptoethanol, 0.02% sodium azide) and then re-probed for β-catenin as an assay control. Experiments were repeated in triplicate. Western blots were quantified using MultiGauge version 2 (FUJIFILM, Tokyo, Japan).
Flow Cytometry Analysis
Cells were serum-starved overnight, followed by pretreatment of appropriate samples with the LPA receptor inhibitor, Ki16425 (Cayman Chemical, Ann Arbor, MI), for 30 min at 37 °C and LPA treatment for 2 h at 37 °C. Cells were trypsinized, resuspended in serum-free medium, and then incubated with anti-active β1 integrin (HUTS21 clone, BD Transduction Laboratories) for 1 h. Following three washings in PBS, cells were incubated and protected from light with anti-mouse IgG-Alexa Fluor 488 for 30 min at ambient temperature. Excess antibody was removed by washing, and cells were resuspended in PBS and then analyzed with the CyAn ADP analyzer (Beckman Coulter, Brea, CA). All assays were performed three times.
β1 Integrin Cross-linking
To evaluate membrane localization of β1 integrins as individual heterodimers or as clusters, a cross-linking mechanism was utilized to capture integrin distribution following LPA treatment. Analysis of β1 integrin clustering was performed as described previously (49). Cells were serum-starved overnight, trypsinized, and resuspended in fresh serum-free medium in 1.5-ml Eppendorf tubes. Suspensions were incubated with 40 μm LPA for 40 min at room temperature. Control cells were incubated with 1% BSA in PBS (LPA vehicle). To cross-link surface-expressed β1 integrins, anti-β1 integrin (MAB1959) was added for a 40-min incubation on ice. Cells were washed, resuspended in serum-free medium, and then incubated with polyclonal anti-mouse IgG (Chemicon) at 37 °C for 30 min. Cell suspensions were affixed to glass 22-mm2 coverslips by cytology centrifugation using a Cytopro 7620 centrifuge (Wescor, Logan, UT) with acceleration at 2000 rpm for 10 min. Clustering was analyzed by immunofluorescent staining (primary antibody, anti-β1 integrin (MAB2250); secondary antibody, anti-mouse IgG-Alexa Fluor 488), as described below. Fluorescent data were quantified by counting the number of cells with punctate staining patterns indicating clusters, compared with the total number of cells in a given field (Nikon Microphot FXA, ×40 magnification). A minimum of 10 high powered fields were analyzed per condition, and the experiment was repeated in triplicate.
Western Blotting
Samples to be analyzed by Western blotting were loaded onto SDS-polyacrylamide gels, electrophoresed, and then transferred onto polyvinylidene fluoride (PVDF) microporous membranes (Millipore). After blocking nonspecific binding to membranes in 3% BSA in TBST for 1 h at room temperature, membranes were incubated with primary antibodies for 3 h at room temperature or overnight at 4 °C, and then with HRP-conjugated secondary antibodies. Immunoreactivity was determined by SuperSignal West Dura Extended Duration substrate kit (Fisher). Western blots were quantified using MultiGauge version 2 (FUJIFILM, Tokyo, Japan). PVDF membranes were stripped of antibody by washing in a stripping buffer (50 mm Tris, pH 6.8, 1% SDS, 150 mm NaCl, 100 mm β-mercaptoethanol, 0.02% sodium azide) and then re-probed for appropriate assay control(s).
Tcf Reporter Assay
TOPFlash (TCF reporter Plasmid) and FOPFlash (TCF mutant reporter plasmid) were generously provided by Dr. Hans Clevers (Hubrecht Laboratory and Utrecht University, Utrecht, The Netherlands). The Renilla luciferase vector, pRL-CMV, was purchased from Promega (Madison, WI). OVCA433 cells were plated at 40–50% confluence in 6-well plates and transiently co-transfected with a Renilla luciferase reporter construct (pRL-CMV) and either the firefly luciferase TOPFlash TCF reporter plasmid or the FOPFlash TCF mutant reporter plasmid using FuGENE 6 transfection reagent according to the manufacturer's instructions (Roche Diagnostics, Mannheim, Germany). Approximately 18 h after transfection, cells were cultured in low calcium (0.1 mm CaCl2), serum-containing MEM (S-MEM (Invitrogen) for 1 h before the addition of LPA (40 μm) for 2, 4, or 30 h. Cells were then lysed in passive lysis buffer (Promega). Both Renilla and firefly luciferase readings were taken on a Veritas Microplate Luminometer (Turner Biosystems, Sunnyvale, CA) using the reagents and protocol provided in the Dual-Luciferase reporter assay system (Promega). Firefly luciferase readings were first normalized to the reading for the corresponding Renilla luciferase reading to account for transfection efficiency. The adjusted TOPFlash reading was then normalized to the corresponding adjusted FOPFlash reading to account for background reading of the TOPFlash construct.
Primers
Quantitative PCR primers probing VIM, WNT5A, and LRP6 were purchased from SA Biosciences (Frederick, MD). Primers used to probe GAPDH, PTGS2, and SNAI1 were as follows: GAPDH forward, 5′-GAGTCAACGGATTTGGTCGT-3′, and GAPDH reverse, 5′-TTGATTTTGGAGGGATCTCG-3′; PTGS2 forward, 5′-GCCCAGCACTTCACGCATCAG-3′, and PTGS2 reverse, 5′-AGACCAGGCACCAGACCAAAGAC-3′; SNAI1 forward, 5′-TTCCAGCAGCCCTACGACCAG-3′, and SNAI1 reverse, 5′-CGGACTCTTGGTGCTTGTGGA-3′.
Quantitative Real Time PCR
Total RNA was extracted from treated and control cells using TRIzol reagent (Life Technologies, Inc.) according to manufacturer's instructions. cDNA was synthesized from 1 μg of RNA using the RT2 First Strand cDNA synthesis kit (SA Biosciences, Frederick, MD). Amplification was performed using iCycler (Bio-Rad) for 40 cycles, with each cycle consisting of 15 s denaturation at 95.0 °C followed by 1 min of annealing at 60.0 °C. SYBR Green Master Mix and primer sets for RAC1, VIM, LRP6, and WNT5A were purchased from SA Biosciences. GAPDH was used as an internal control in each reaction.
Statistics
p values were determined using the t test function (two sample, unequal variance, one-tailed distribution) using Excel (Microsoft Corp., Redmond, WA).
Results
LPA Initiates EMT and Loss of Junctionally Localized β-Catenin in Ovarian Carcinoma
Previous work by our laboratory and others has demonstrated a potential link between LPA and EMT via LPA-induced activation of RhoA, an initial target in the EMT transduction pathway (38), and via regulation of cell-cell adhesion in EOC (28–31, 39, 40). Ovarian cancers metastasize as both single cells and multicellular aggregates, and it has been proposed that E-cadherin stabilizes aggregate formation and contributes to chemotherapy resistance (12, 16, 21). To assess the potential mechanistic link between LPA and EMT, multicellular aggregates generated from EOC cell lines were treated with LPA and evaluated for changes in mesenchymal (vimentin) and epithelial (E-cadherin) marker expression. E-cadherin cell surface expression was down-regulated in multicellular aggregates (Fig. 1, A–D, green), whereas vimentin expression increased in response to LPA treatment (Fig. 1, A–D, red). Further confirmation of gain of mesenchymal phenotype was demonstrated by evaluation of F-actin stress fiber arrangement and vinculin expression. Reorganization of F-actin stress fibers (Fig. 1, E–H, red) and punctate vinculin staining (Fig. 1, E–H, green) were observed, indicating gain of mesenchymal phenotype in response to LPA treatment.
FIGURE 1.
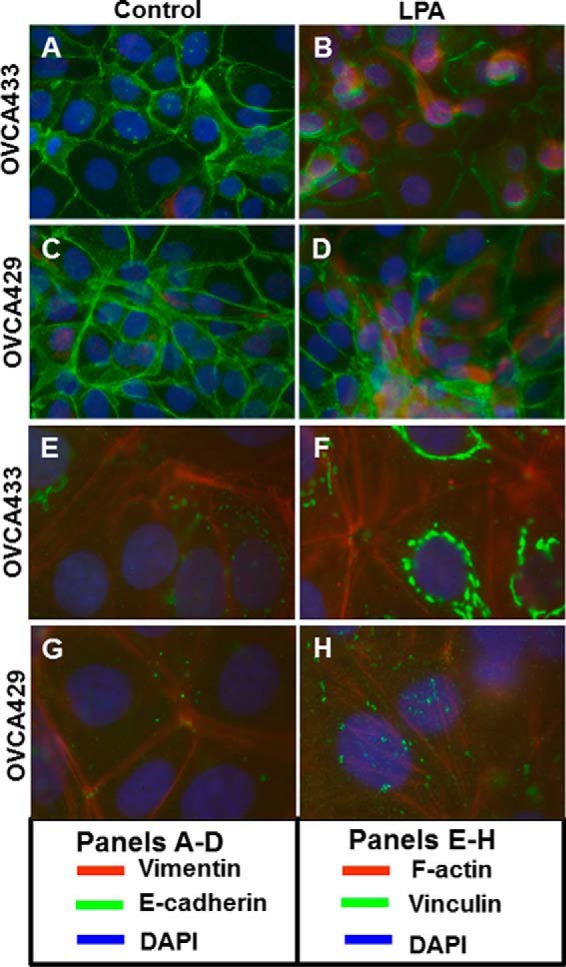
LPA initiates epithelial-to-mesenchymal transition in epithelial ovarian carcinoma cells. Multicellular aggregates formed from OVCA429 or OVCA433 cells were untreated or treated with 40 μm LPA overnight and stained for expression and localization of E-cadherin and vimentin (anti-E-cadherin, green, 1:300; anti-vimentin, red, 1:200) (A–D) or vinculin and F-actin (anti-vinculin, green, 1:100; rhodamine phalloidin labeling, red) (E–H). Nuclei were stained with DAPI (blue). Magnification ×20.
We have previously demonstrated that LPA disrupts E-cadherin junctional integrity in ovarian cancer monolayer cultures (30). To validate LPA-induced adherens junction disruption in multicellular aggregate cultures and to evaluate whether adherens junction disruption alters the subcellular localization of β-catenin, multicellular aggregates of OVCA 433 cells were treated with LPA (40 μm LPA, 2 h) and evaluated for E-cadherin and β-catenin expression by immunofluorescence microscopy. Junctionally localized β-catenin is significantly decreased following LPA treatment compared with control (Fig. 2, C, D, and K) and corresponds temporally to loss of surface-associated E-cadherin (Fig. 2, A and B). Furthermore, β-catenin perinuclear and nuclear accumulation can be observed in response to LPA (Fig. 2D). To determine whether LPA-mediated loss of β-catenin junctional localization is an LPA receptor-dependent event, cells were either treated with LPA (40 μm) alone, pretreated with the pharmacologic LPA receptor inhibitor Ki16425 (40 μm, 30 min), or pretreated with inhibitor and then treated with LPA. LPA treatment led to loss of β-catenin junctional localization with nuclear/perinuclear accumulation of β-catenin (Fig. 2, G and L) compared with untreated control (Fig. 2E), vehicle control (Fig. 2F), and Ki16425 alone (Fig. 2H). This loss of junctional localization was rescued by pretreatment with Ki16425 prior to LPA treatment (Fig. 2, I and L). In positive controls, loss of junctional β-catenin was also observed following treatment with lithium chloride (LiCl, GSK3-β inhibitor; Fig. 2, J and L). Similar results were obtained with OVCA 429 cells (data not shown).
FIGURE 2.

LPA mediates loss of E-cadherin and β-catenin surface expression in an LPA receptor-dependent manner. A–D, OVCA433 cells were untreated (A and C) or treated with 40 μm LPA for 2 h (B and D) and stained for expression of E-cadherin (E-cad) (A and B; anti-E-cadherin, 1:300, green) or β-catenin (β-cat) (C and D; anti-β-catenin, 1:100, red). E–J, cells were pretreated as indicated with the LPA receptor inhibitor, Ki16425 (40 μm; H and I) or DMSO vehicle control (F). Cells were then treated with 40 μm LPA (G and I) for 2 h. Controls included untreated cells (negative control; E) and cells treated with the GSK3-β inhibitor, 40 μm LiCl (positive control; J). Cells were fixed as described above and processed for immunofluorescent staining (anti-β-catenin, green, 1:200). The experiment was repeated in triplicate. K and L, quantitation of junctional β-catenin staining was performed by counting a minimum of 12 fields per treatment and scoring as positive the number of cells with two remaining fluorescent cell-cell borders (30). K, quantitation of data represented by C and D above. L, quantitation of data represented by E–J above. *, p < 0.05
Lysophosphatidic Acid Induces β1 Integrin Activation and Clustering
We have demonstrated that ligand-induced β1 integrin clustering activates pathways leading to enhanced nuclear translocation of β-catenin and activation of β-catenin target genes in ovarian cancer (24). Furthermore, the LPA/LPA receptor interaction has been previously shown to trans-activate a number of surface-expressed signaling receptors (27). To assess the potential for cross-talk between LPA signaling and integrin activation, OVCA433 cells were treated with either 30 or 70 μm LPA for 1 h at 37 °C and then evaluated for β1 integrin activation by flow cytometry analysis using a conformation-specific antibody that detects activated β1 integrin (HUTS21). Both 30 and 70 μm LPA induced β1 integrin activation, and this activation was partially blocked by pretreatment with the LPA receptor inhibitor Ki16425 (Fig. 3, A and B). Disseminating ovarian tumor cells are exposed to LPA in ascites as anchorage-independent cells and multicellular aggregates. To evaluate LPA-induced β1 integrin clustering in anchorage-independent cells, OVCA433 cell suspensions were treated with 20 or 40 μm LPA for 1 h at room temperature. Surface-expressed β1 integrin was cross-linked using a nonactivating anti-β1 integrin antibody, and cells were processed for immunostaining for β1 integrin (41). Representative high powered fields are shown in Fig. 3, C and D. β1 integrin clustering was potentiated by LPA treatment in a dose-dependent manner, with a 1.5-fold increase of the number of β1 integrin clusters (green) following 20 μm LPA treatment and a 2-fold increase of clusters following 40 μm LPA (Fig. 3, C and D).
FIGURE 3.

LPA induces β1 integrin activation and clustering. A and B, LPA activation of β1 integrins. OVCA433 cells were serum-starved, pretreated with the LPA receptor inhibitor Ki16425 (40 μm), where indicated, and then treated with the indicated concentrations of LPA for 1 h. Following incubation with anti-active β1 integrin antibody (1:100, 1 h) and mouse anti-IgG Alexa Fluor 488 (1:500, 30 min), cells were analyzed for surface expression of active β1 integrin by flow cytometry. β1 integrin is activated in response to 30 μm LPA (A, green) and 70 μm LPA (B, purple). Inhibition of LPA receptor with Ki16425 partially blocks β1 integrin activation (A, blue, and B, black). Data from untreated control cells are shown in red. Figure shows a representative histogram from triplicate experiments. C and D, immunofluorescent evaluation of β1 integrin clustering. Suspended cells were treated with LPA (40 μm, 1 h) and then incubated with function-blocking anti-β1-integrin antibody (clone MAB1959, 1:50, 40 min) on ice, followed by mouse anti-IgG (37 °C) to cross-link integrins (30 min). Cell suspensions were cytocentrifuged onto 22-mm2 glass coverslips for immunofluorescent staining with anti-β1-integrin (1:200, clone MAB2250) and mouse anti-IgG-Alexa Fluor 488 (1:500). #, p < 0.1; *, p < 0.05.
To further examine the effect of LPA on β1 integrins and junctional integrity, cells were treated with 30 μm LPA for various time points prior to processing for dual label immunofluorescence microscopy to examine LPA-induced changes in localization of β1 integrin and E-cadherin. Following LPA treatment, a time-dependent aggregation of β1 integrin was observed in both OVCA429 and OVCA433 cells (Fig. 4, A and B). Higher magnification examination of sites of β1 integrin clustering shows recruitment of E-cadherin into clustered integrin complexes (Fig. 4, C and D).
FIGURE 4.

LPA induces β1 integrin aggregation and β1 integrin/E-cadherin co-localization. A and B, OVCA 429 (A) or OVCA433 (B) cells were grown on coverslips coated with type I collagen (10 μg/ml) prior to treatment with LPA (30 μm) for the time periods shown. Cells were stained to evaluate localization of β1 integrin (anti-β1 integrin, 1:50 and Alexa Fluor 594 goat anti-mouse IgG, 1:400, red) or E-cadherin (anti-E-cadherin, 1:100 and Alexa Fluor 488 goat anti-rabbit IgG, 1:400, green). Nuclei are stained with DAPI (blue). Merged images (×40 magnification) show β1 integrin clustering beginning at 4 h. C and D, co-localization of E-cadherin with clustered β1 integrin. OVCA 429 (C) or OVCA433 (D) cells were grown in the absence (left panels) or presence (right panels) of LPA (30 μm) for 24 h prior to analysis by dual label immunofluorescence microscopy. Panels a and g, DAPI; panels b and h, β1 integrin; panels c and i, E-cadherin; panels d–f and j–l, merged images. Panels a–d and g–j, ×20 magnification; panels e and k, ×40 magnification; panels f and l, ×60 magnification. Merged images demonstrate LPA-dependent co-localization of E-cadherin with clustered β1 integrins. Areas of co-localization of E-cadherin with β1 integrin are visible in cells treated with LPA treatment (right panels, C and D) relative to untreated controls (left panels, C and D).
Lysophosphatidic Acid Potentiates Nuclear Accumulation of β-Catenin and Transcriptional Activation
Loss of junctional E-cadherin can target β-catenin to the nucleus or for degradation (33). Although the exact mechanism of β-catenin nuclear transport is unknown, cytoplasmic and perinuclear accumulation of β-catenin is coupled to increased nuclear activation of β-catenin target genes. To evaluate the effect of LPA on nuclear β-catenin accumulation, OVCA429 and OVCA433 cells were treated with LPA; nuclear proteins were isolated by subcellular fractionation as described under “Experimental Procedures,” and fractions were evaluated by Western blot. In control experiments, cells were treated with LiCl (positive control treatment) or left untreated. Results show that LPA treatment potentiates an ∼50% increase in nuclear β-catenin localization in both OVCA429 and OVCA433 cells (Fig. 5, A and B). Mechanisms for activation of Wnt signaling in human EOC tumors exhibit histotype dependence, with endometrioid EOC exhibiting mutations in β-catenin resulting in constitutively active Wnt signaling, although other histotypes do not harbor a significant level of activating mutations in this pathway (42). Immunohistochemical evaluation of a human ovarian tumor tissue microarray demonstrates nuclear localization of β-catenin in each of the four EOC subtypes (Fig. 5, C–F, and Table 1). Levels of nuclear β-catenin in endometrioid tumor samples were consistent with literature values (84.6%). In small cohorts of mucinous and clear cell samples, 66.6 and 62.5% were positive for nuclear β-catenin expression, respectively, although 50.0% of serous tumors exhibited positive nuclear staining. Eight metastatic lesions (seven serous and one clear cell) were also analyzed for nuclear β-catenin with five of the seven serous tumors exhibiting positive staining (71.4%), in addition to positive immunoreactivity in the clear cell metastasis. These data support the hypothesis that factors other than mutational activation may regulate Wnt/β-catenin signaling in human EOC.
FIGURE 5.
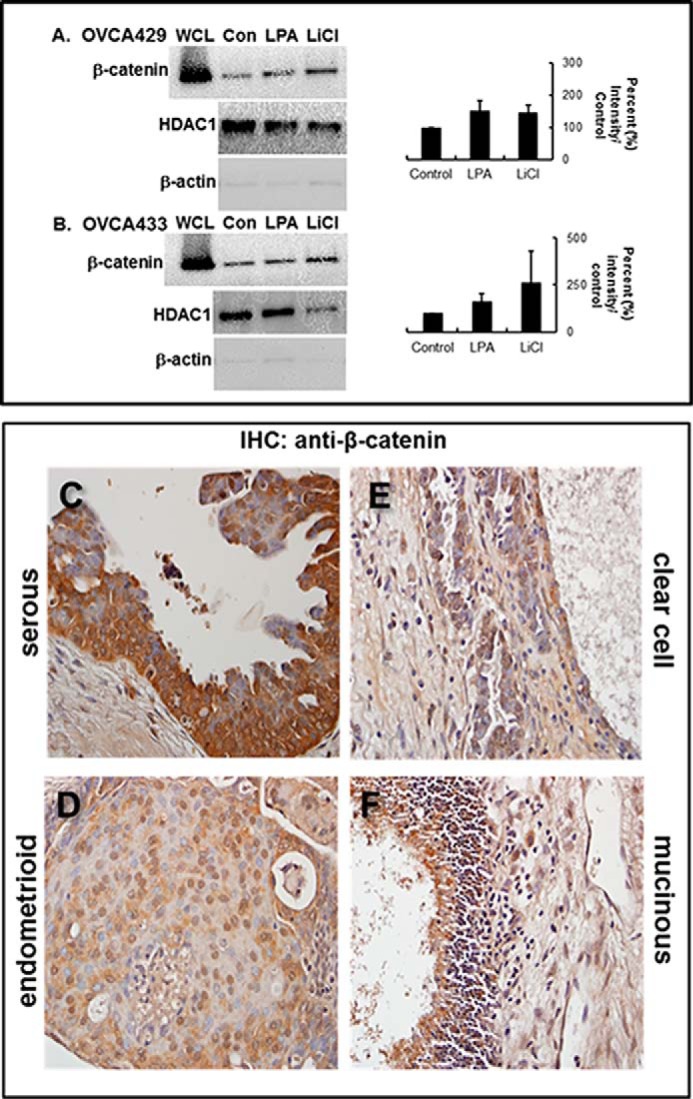
LPA induces LPA receptor-dependent nuclear translocation of β-catenin. A and B, cells were treated as labeled (Con, untreated control; LPA, 40 μm; LiCl, positive control, 40 μm) and then subjected to subcellular fractionation as described under “Experimental Procedures.” Fractions were electrophoresed on 9% SDS-polyacrylamide gels and immunoblotted for β-catenin expression (anti-β-catenin, 1:1000). From analysis of triplicate blots, β-catenin staining in the nuclear fraction of both OVCA429 (A) and OVCA433 (B) cell lines was increased ∼50% in LPA-treated cells compared with control (graphs). Control blots were probed with HDAC1 (middle panels) or β-actin (lower panels) to detect the presence of nuclear or cytoplasmic compartments, respectively. WCL designates unfractionated whole cell lysate. Molecular mass of β-catenin, 92 kDa. C–F, representative samples of each of the four major EOC histotypes were stained by immunohistochemistry (IHC) with anti-β-catenin (BD Transduction Laboratories, 1:50) and a biotinylated secondary antibody (1:200; Vectastain ABC, Vector Laboratories). Slides were finally subjected to 3,3′-diaminobenzidine peroxidase (Vector Laboratories) exposure and hematoxylin staining. β-Catenin staining is present at cell-cell junctions, in the cytoplasm, and in the nucleus. Nuclear β-catenin is found in serous (C), endometrioid (D), clear cell (E), and mucinous (F) tumors.
TABLE 1.
β-Catenin is expressed in human ovarian carcinoma
| Number | Nuclear β-catenin | |
|---|---|---|
| Serous | ||
| Primary | 34 | 17 (50.0%) |
| Metastatic | 7 | 5 (71.4%) |
| Endometrioid | ||
| Primary | 26 | 22 (84.6%) |
| Metastatic | ||
| Mucinous | ||
| Primary | 6 | 4 (66.6%) |
| Metastatic | ||
| Clear cell | ||
| Primary | 8 | 5 (62.5%) |
| Metastatic | 1 | 1 (100.0%) |
The β-catenin protein sequence does not contain a nuclear localization signal domain, and the mechanism of nuclear transport is unclear (43–45). Nevertheless, β-catenin activates transcription by displacing the transcriptional repressor Groucho in Tcf/Lef-binding site-containing gene promoters and subsequently binding the co-activators Tcf/Lef (and potentially other co-factors such as CBP) to initiate transcription (46–48). To assess the potential for LPA regulation of protein/protein interactions between β-catenin and Tcf, nuclear proteins were isolated as described and subjected to immunoprecipitation using anti-β-catenin antibodies. Immunoprecipitates were then probed for β-catenin or Tcf. Results show that Tcf/β-catenin interaction was enhanced in cells treated with LPA relative to untreated or DMSO treated controls (Fig. 6A, inset). Pretreatment with Ki16425 reduced this protein/protein interaction to baseline levels, suggesting that LPA-induced Tcf/β-catenin interaction is LPA receptor-dependent To measure activation of the β-catenin·Tcf·Lef transcriptional complex, cells were transfected with a Renilla luciferase reporter construct and either a TOP (Tcf) reporter construct or a FOP (control) reporter construct and then were treated with LPA for 2, 8, or 24 h. β-Catenin/Tcf/Lef transcriptional activity was increased after a 2-h LPA treatment and was sustained at 8 and 30 h in Fig. 6, B and C. Consequentially, LPA-induced β-catenin/Tcf/Lef transcriptional activity led to up-regulation of five known β-catenin/Tcf/Lef target genes: VIM (vimentin), WNT5A, LRP6, PTGS2 (Cox-2), and SNAI1 (Snail) (Fig. 7A). To evaluate a potential functional link between LPA-induced β1 integrin clustering and transcriptional activation of Wnt/β-catenin targets, cells were treated with LPA, and activation of FAK was evaluated by Western blotting for phospho-Tyr-397 (Fig. 7B, inset). LPA treatment enhanced FAK phosphorylation (Fig. 7B, lane b, inset), and this effect was abrogated by the addition of antibodies that prevent β1 integrin clustering as well as by the LPA receptor inhibitor Ki16427 (Fig. 7B, lanes c and d, inset). As recent studies have demonstrated that LPA can modulate integrin function through Rho/ROCK activation, we also examined the effect of the ROCK inhibitor Y27632 (Fig. 7B, lane e, inset), also resulting in inhibition of FAK phosphorylation. To address the resulting effects on transcription, we examined expression of vimentin, as this β-catenin target gene was most significantly up-regulated following LPA treatment (Fig. 7A). LPA-induced vimentin expression was blocked by inhibition of β1 integrin clustering (Fig. 7C), supporting the hypothesis that LPA-integrin cross-talk and subsequent integrin clustering regulate Wnt/β-catenin signaling and gene expression. Similar results were obtained following inhibition of LPA receptor or Rho/ROCK activity (Fig. 7C).
FIGURE 6.
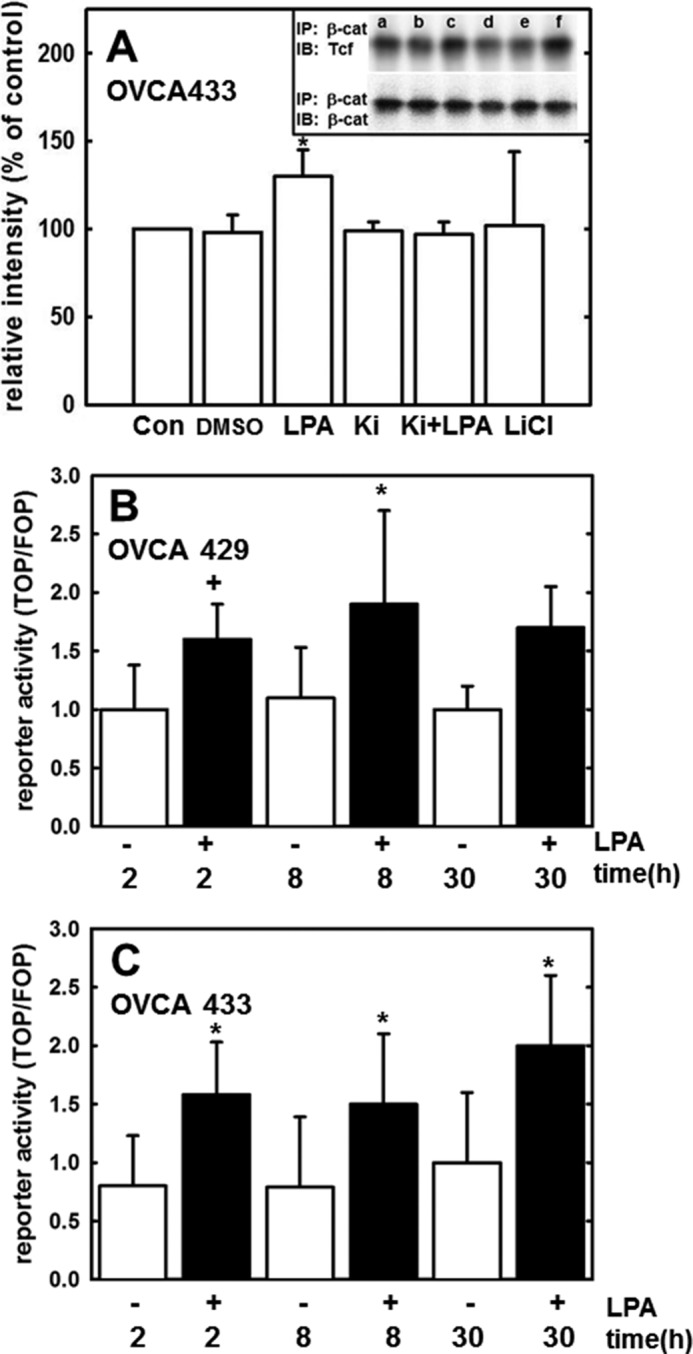
β-Catenin co-localization and activation of Tcf/Lef transcription following LPA treatment. A, inset, cells were treated for 24 h as follows: (Con, lane a) untreated; (DMSO, lane b) DMSO vehicle control; (LPA, lane c) LPA 40 μm; (Ki, lane d) Ki16425; LPA receptor inhibitor, 40 μm (Ki + LPA, lane e) LPA + Ki16425; and (LiCl, lane f) LiCl (GSK3-β inhibitor/positive control, 40 μm). Where indicated, inhibitor was preincubated for 15 min prior to addition of LPA. Following treatment, cells were lysed, and β-catenin was immunoprecipitated (IP) as described under “Experimental Procedures.” Lysates were electrophoresed on 9% SDS-polyacrylamide gels and immunoblotted (IB) with an anti-Tcf antibody (inset, 1:1000) or anti-β-catenin (inset, 1:1000). Band intensity was quantified using FUJIFILM MultiGauge version 3.0 and is represented as relative intensity (percent of control). Experiment was repeated in triplicate. Molecular mass of Tcf, 50 kDa; molecular mass of β-catenin, 92 kDa. *, < 0.05. B and C, OVCA429 and OVCA433 cells, respectively, were transiently co-transfected with either FOP reporter construct/Renilla luciferase reporter construct or TOP reporter construct/Renilla luciferase reporter construct and then treated with 40 μm LPA for 2, 8, or 30 h as indicated. Luciferase reporter activity was measured using a luminometer as described under “Experimental Procedures.” Experiment was conducted in triplicate. *, p < 0.05; +, p = 0.08.
FIGURE 7.
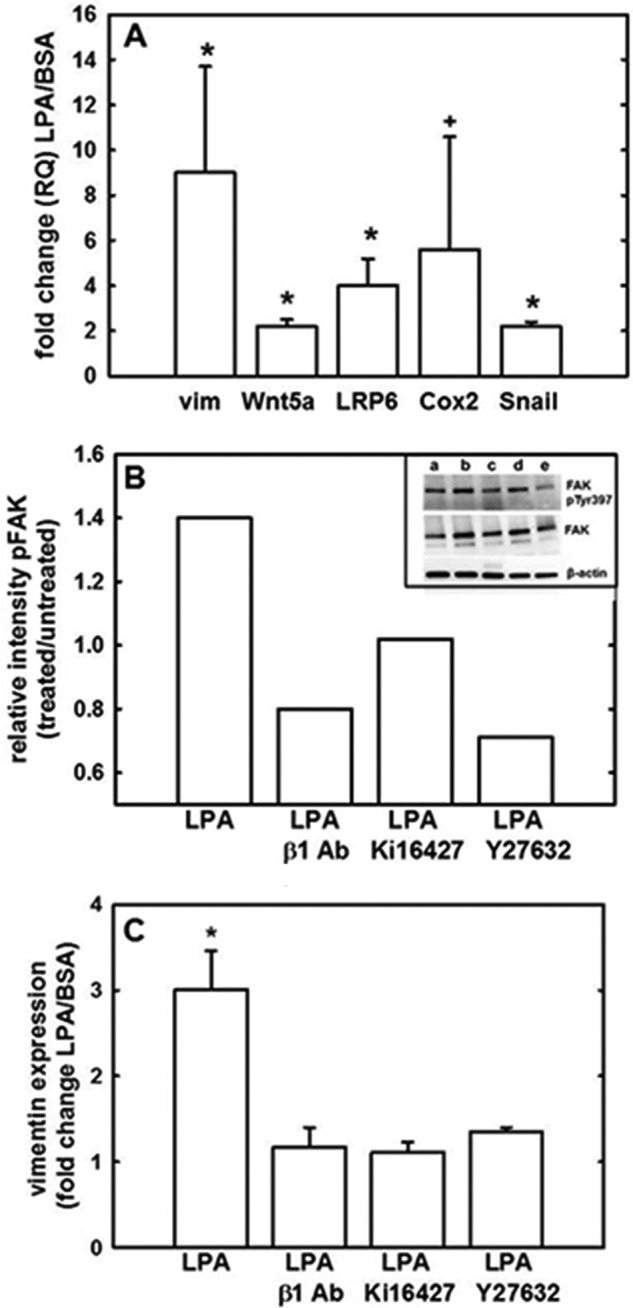
LPA-activated transcription of β-catenin target genes is dependent on β1 integrin clustering. A, LPA induces expression of Wnt/β-catenin target genes. OVCA433 cells were treated with 40 μm LPA or 1% BSA in PBS control, and total RNA was isolated and analyzed for changes in gene expression by quantitative RT-PCR (2(−ΔΔCt) method). Data represent the mean of four independent experiments. *, p < 0.05; +, p < 0.1. B, LPA induces phosphorylation of FAK. Inset, OVCA429 cells were as follows: lane a, untreated or lanes b–e, treated with LPA (40 μm) in the presence of β1 integrin blocking antibodies (1:100) (lane c), the LPA receptor inhibitor Ki16427 (10 μm) (lane d), or the Rho/ROCK inhibitor Y27632 (10 μm) (lane e). Cells were lysed and lysates electrophoresed on 4–20% SDS-polyacrylamide gels (Bio-Rad) and immunoblotted for phospho-Tyr-397 FAK (1:1000), total FAK (1:200), or β-actin (1:5000). Experiment was conducted in duplicate. Band intensity was quantified using FUJIFILM MultiGauge version 3.0 and is represented as relative intensity (treated versus untreated, where untreated is designated as 1). Molecular mass of FAK and phospho-FAK, 125 kDa; molecular mass of β-actin, 40 kDa. C, inhibition of LPA-induced vimentin expression. OVCA429 cells were treated with LPA (40 μm) either alone or in the presence of β1 integrin blocking antibodies (1:100), the LPA receptor inhibitor Ki16427 (10 μm), or the Rho/ROCK inhibitor Y27632 (10 μm). Total RNA was isolated and analyzed for changes in vimentin expression by quantitative RT-PCR (2(−ΔΔCt) method). Data represent the mean of triplicate experiments. *, p < 0.05.
Discussion
LPA regulates a multitude of ovarian tumor cell responses, including proliferation, migration, and invasion (19, 24, 28). LPA is expressed as high as 80 μm in the ascites fluid and serum of patients with ovarian cancer (24, 25, 49–51), underlying the importance of understanding its pathophysiological role in ovarian cancer. Previous studies in cancer cells have shown that LPA induces β-catenin nuclear translocation. For example, in colon cancer cells, LPA activates β-catenin via modulation of glycogen synthase kinase 3β, resulting in enhanced transcription of a β-catenin/TCF reporter gene (52, 53). Similar results were obtained in A431 epidermoid carcinoma cells (33, 54), resulting in loss of junctional E-cadherin and gain of vimentin expression. Data in this study define a role for LPA in activation of β-catenin-regulated transcription and induction of an EMT program in ovarian cancer cells and multicellular aggregates and provide novel mechanistic insight on the role of LPA-induced clustering of β1 integrins in modulation of this signaling pathway.
We have recently shown that LPA modulates loss of epithelial cohesion as a functional result of disrupting cell-cell junctions, through protease-dependent cleavage and altered cadherin trafficking in ovarian carcinoma (30). The fate of β-catenin in response to LPA-mediated adherens junctiondissolution, however, had not been previously reported. Data presented here are consistent with our previous work and demonstrate loss of cell surface-associated β-catenin following LPA treatment. Interestingly, immunofluorescence microscopy results demonstrate that perinuclear accumulation of β-catenin is observed following LPA treatment, corresponding with data that first identified an intersection between the cell-cell adhesion-related and transcription-related cytoplasmic pools of β-catenin (33). The theory that cytoplasmic β-catenin pools intersect nuclear β-catenin pools is further supported by data demonstrating increased nuclear localization, Tcf/Lef reporter activity, and β-catenin target gene transcription in response to LPA treatment.
Although the precise mechanism by which LPA treatment modulates β-catenin translocation is unknown, the current data support the hypothesis that LPA-integrin cross-talk results in β1 integrin activation. Many studies have suggested convergent signaling between growth factors and integrin-mediated adhesion processes (55–57). Several G-protein-coupled agonists, including LPA, have been shown to induce phosphorylation of the integrin effector p125FAK (58). Additionally, LPA-induced migration in fibroblasts is dependent on β1 integrin expression (59), and LPA/LPAR2 interaction activates TGF-β signaling in an integrin-mediated manner (60). Interestingly, LPA treatment also leads to increased ovarian cancer cell adhesion to collagen, as well as enhanced β1 integrin protein expression (29). We have previously reported that β1 integrin aggregation alters β-catenin dynamics via disruption of adherens junctions and inhibition of glycogen synthase kinase 3β activity, leading to enhanced transcriptionally active β-catenin (23). This is consistent with data in the current report showing co-localization of E-cadherin at sites of clustered β1 integrins. Furthermore, three-dimensional collagen culture induces β1 integrin aggregation and down-regulates the expression of dickkopf-1, an inhibitor of canonical Wnt signaling (61, 62). These data support a mechanism whereby LPA-induced β1 integrin aggregation activates β-catenin signaling.
Further support for this mechanism is provided by the observation that the following five pro-metastatic β-catenin target genes are up-regulated following LPA treatment: VIM (vimentin), WNT5A, LRP6, PTGS2 (Cox-2), and SNAI1 (Snail1). Expression of these genes was also up-regulated following multivalent β1 integrin engagement (23) and inhibition of LPA-induced β1 integrin aggregation blocked vimentin transcription. Cox-2 contributes to tumorigenesis by inhibiting apoptosis, increasing growth factor expression to promote angiogenesis, and by enhancing matrix metalloproteinase expression to stimulate invasion (63). Cox-2 protein is expressed in ovarian carcinoma and functions as a downstream effector of LPA-mediated ovarian tumor cell migration and invasion (64). Genes commonly associated with EMT, including vimentin, snail, LRP6, and Wnt5a, were also induced by LPA treatment. Ovarian cancers typically display both epithelial and mesenchymal characteristics, and the mesenchymal marker vimentin is widely expressed in human tumor specimens (16). Recent molecular profiling studies support these findings, showing up-regulation of EMT and EMT-associated invasion programs in primary ovarian tumors and their matched metastases (65, 66). Snail is a key inducer of EMT and functions as a negative regulator of E-cadherin transcription. Several studies have demonstrated that nuclear localization of Snail correlates with tumor progression, with enhanced Snail immunoreactivity in metastatic lesions (67, 68). Snail and other mesenchymal genes were found to be up-regulated in a panel of ovarian cancer cells competent for mesothelial clearance, a surrogate assay that models initial events in intraperitoneal metastasis (21). Furthermore, patients with both primary and metastatic tumors positive for Snail expression showed a significant decrease in overall survival (69). LRP6 functions as a Wnt co-receptor that recruits Axin and Dishevelled to the plasma membrane, thereby disrupting the degradation of β-catenin and facilitating β-catenin nuclear translocation (70). LRP6 is expressed by ovarian carcinoma cell lines and tissues (71). Expression of Wnt5a, a ligand for the Frizzled receptor, is high in ovarian cancer tumors and ascites fluid and is linked to poor overall survival (33, 69, 70). Emerging data suggest that activation of canonical versus noncanonical Wnt signaling by Wnt5a is dependent on the receptor/co-receptor context, and further studies in ovarian cancer cells are warranted (72–75).
In summary, the current data demonstrate that lysophosphatidic acid, expressed in high concentration in ascites fluid of women with ovarian cancer, potentiates β-catenin-dependent Wnt signaling via a mechanism that involves activation and aggregation of β1 integrin. Currently, a number of therapeutic agents that target Wnt signaling, both loss of function and gain of function alterations, are being investigated in the treatment of cancer, bone disease, cardiac and vascular disease, and arthritis (76, 77). Therapeutics under development are designed to target each level of Wnt signaling (extracellular, cytoplasmic, nuclear, and pathway cross-talk) and include small molecule and biotherapeutic agents (78–80). Although not yet extensively evaluated in ovarian cancer, recent preclinical studies support a role for further evaluation of Wnt/β-catenin signaling as a potential therapeutic target (71, 81). Understanding the mechanisms by which microenvironmental factors such as lysophosphatidic acid may contribute to ovarian cancer progression and metastasis via modulation of this pathway may thereby contribute to identification of alternative treatment options for women with ovarian cancer.
Author Contributions
R. J. B. and M. S. S. designed the study and wrote the paper. S. D. W. performed the experiments shown in Fig. 1. Y. L. performed and analyzed the experiments shown in Fig. 4. R. J. B. performed and analyzed the experiments shown in Figs. 2 and 3 and 5–7. All authors reviewed the results and approved the final version of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 CA109545 (to M. S. S.) and RO1 CA086984 (to M. S. S.) from the NCI and Research Supplement to Promote Diversity CA086984-11S1 (to R. J. B.). This work was also supported by the Leo and Ann Albert Charitable Trust (to M. S. S.). The authors declare that they have no conflicts of interest with the contents of this article.
- EOC
- epithelial ovarian cancer
- MEM
- minimal essential medium
- EMT
- epithelial-to-mesenchymal transition
- LPA
- lysophosphatidic acid
- FAK
- focal adhesion kinase.
References
- 1. Cannistra S. A. (2004) Cancer of the ovary. N. Engl. J. Med. 351, 2519–2529 [DOI] [PubMed] [Google Scholar]
- 2. Seidman J. D., Horkayne-Szakaly I., Haiba M., Boice C. R., Kurman R. J., Ronnett B. M. (2004) The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin. Int. J. Gynecol. Pathol. 23, 41–44 [DOI] [PubMed] [Google Scholar]
- 3. Kurman R. J., Shih IeM. (2010) The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am. J. Surg. Pathol. 34, 433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. American Cancer Society (2010) Cancer facts & figures. American Cancer Society, Atlanta, GA [Google Scholar]
- 5. Surveillance Research Program, NCI (2014) SEER Stat Fact Sheets: Ovary cancer. National Cancer Institute, Bethesda [Google Scholar]
- 6. Kim A., Ueda Y., Naka T., Enomoto T. (2012) Therapeutic strategies in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 31, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Auersperg N. (2011) The origin of ovarian carcinomas: a unifying hypothesis. Int. J. Gynecol. Pathol. 30, 12–21 [DOI] [PubMed] [Google Scholar]
- 8. Kim J., Coffey D. M., Creighton C. J., Yu Z., Hawkins S. M., Matzuk M. M. (2012) High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc. Natl. Acad. Sci. U.S.A. 109, 3921–3926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kurman R. J., Shih IeM (2011) Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer–shifting the paradigm. Hum. Pathol. 42, 918–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee Y., Miron A., Drapkin R., Nucci M. R., Medeiros F., Saleemuddin A., Garber J., Birch C., Mou H., Gordon R. W., Cramer D. W., McKeon F. D., Crum C. P. (2007) A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J. Pathol. 211, 26–35 [DOI] [PubMed] [Google Scholar]
- 11. Shih IeM, Kurman R. J. (2004) Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am. J. Pathol. 164, 1511–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Auersperg N., Maines-Bandiera S. L., Kruk P. A. (1994) in Ovarian Cancer III (Sharp F., Mason P., Blacket T., Berek J., eds) pp. 157–169, Chapman Hall, London [Google Scholar]
- 13. Auersperg N., Edelson M. I., Mok S. C., Johnson S. W., Hamilton T. C. (1998) The biology of ovarian cancer. Semin. Oncol. 25, 281–304 [PubMed] [Google Scholar]
- 14. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 15. Hanahan D., Weinberg R. A. (2000) The hallmarks of cancer. Cell 100, 57–70 [DOI] [PubMed] [Google Scholar]
- 16. Hudson L. G., Zeineldin R., Stack M. S. (2008) Phenotypic plasticity of neoplastic ovarian epithelium: unique cadherin profiles in tumor progression. Clin. Exp. Metastasis 25, 643–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ahmed A. A., Etemadmoghadam D., Temple J., Lynch A. G., Riad M., Sharma R., Stewart C., Fereday S., Caldas C., Defazio A., Bowtell D., Brenton J. D. (2010) Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 221, 49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Latifi A., Abubaker K., Castrechini N., Ward A. C., Liongue C., Dobill F., Kumar J., Thompson E. W., Quinn M. A., Findlay J. K., Ahmed N. (2011) Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J. Cell. Biochem. 112, 2850–2864 [DOI] [PubMed] [Google Scholar]
- 19. Marchini S., Fruscio R., Clivio L., Beltrame L., Porcu L., Fuso Nerini I., Cavalieri D., Chiorino G., Cattoretti G., Mangioni C., Milani R., Torri V., Romualdi C., Zambelli A., Romano M., et al. (2013) Resistance to platinum-based chemotherapy is associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Eur. J. Cancer 49, 520–530 [DOI] [PubMed] [Google Scholar]
- 20. Strauss R., Li Z. Y., Liu Y., Beyer I., Persson J., Sova P., Möller T., Pesonen S., Hemminki A., Hamerlik P., Drescher C., Urban N., Bartek J., Lieber A. (2011) Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotypic heterogeneity and plasticity. PLoS One 6, e16186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Davidowitz R. A., Selfors L. M., Iwanicki M. P., Elias K. M., Karst A., Piao H., Ince T. A., Drage M. G., Dering J., Konecny G. E., Matulonis U., Mills G. B., Slamon D. J., Drapkin R., Brugge J. S. (2014) Mesenchymal gene program-expressing ovarian cancer spheroids exhibit enhanced mesothelial clearance. J. Clin. Invest. 124, 2611–2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wells A., Chao Y. L., Grahovac J., Wu Q., Lauffenburger D. A. (2011) Epithelial and mesenchymal phenotypic switchings modulate cell motility in metastasis. Front. Biosci. 16, 815–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burkhalter R. J., Symowicz J., Hudson L. G., Gottardi C. J., Stack M. S. (2011) Integrin regulation of β-catenin signaling in ovarian carcinoma. J. Biol. Chem. 286, 23467–23475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu Y., Gaudette D. C., Boynton J. D., Frankel A., Fang X. J., Sharma A., Hurteau J., Casey G., Goodbody A., Mellors A. (1995) Characterization of an ovarian cancer activating factor in ascites from ovarian cancer patients. Clin. Cancer Res. 1, 1223–1232 [PubMed] [Google Scholar]
- 25. Xu Y., Shen Z., Wiper D. W., Wu M., Morton R. E., Elson P., Kennedy A. W., Belinson J., Markman M., Casey G. (1998) Lysophosphatidic acid as a potential biomarker for ovarian and other gynecologic cancers. JAMA 280, 719–723 [DOI] [PubMed] [Google Scholar]
- 26. Fukushima N., Chun J. (2001) The LPA receptors. Prostaglandins 64, 21–32 [DOI] [PubMed] [Google Scholar]
- 27. Choi J. W., Herr D. R., Noguchi K., Yung Y. C., Lee C. W., Mutoh T., Lin M. E., Teo S. T., Park K. E., Mosley A. N., Chun J. (2010) LPA receptors: subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 50, 157–186 [DOI] [PubMed] [Google Scholar]
- 28. Jourquin J., Yang N., Kam Y., Guess C., Quaranta V. (2006) Dispersal of epithelial cancer cell colonies by lysophosphatidic acid (LPA). J. Cell. Physiol. 206, 337–346 [DOI] [PubMed] [Google Scholar]
- 29. Fishman D. A., Liu Y., Ellerbroek S. M., Stack M. S. (2001) Lysophosphatidic acid promotes matrix metalloproteinase (MMP) activation and MMP-dependent invasion in ovarian cancer cells. Cancer Res. 61, 3194–3199 [PubMed] [Google Scholar]
- 30. Liu Y., Burkhalter R., Symowicz J., Chaffin K., Ellerbroek S., Stack M. S. (2012) Lysophosphatidic acid disrupts junctional integrity and epithelial cohesion in ovarian cancer cells. J. Oncol. 2012, 501492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Symowicz J., Adley B. P., Gleason K. J., Johnson J. J., Ghosh S., Fishman D. A., Hudson L. G., Stack M. S. (2007) Engagement of collagen-binding integrins promotes matrix metalloproteinase-9-dependent E-cadherin ectodomain shedding in ovarian carcinoma cells. Cancer Res. 67, 2030–2039 [DOI] [PubMed] [Google Scholar]
- 32. Barbolina M. V., Burkhalter R. J., Stack M. S. (2011) Diverse mechanisms for activation of Wnt signalling in the ovarian tumour microenvironment. Biochem. J. 437, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kam Y., Quaranta V. (2009) Cadherin-bound β-catenin feeds into the Wnt pathway upon adherens junctions dissociation: evidence for an intersection between β-catenin pools. PLoS One 4, e4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bian Y. S., Osterheld M. C., Bosman F. T., Fontolliet C., Benhattar J. (2000) Nuclear accumulation of β-catenin is a common and early event during neoplastic progression of Barrett esophagus. Am. J. Clin. Pathol. 114, 583–590 [DOI] [PubMed] [Google Scholar]
- 35. Choudhury M., Shukla S. D. (2008) Surrogate alcohols and their metabolites modify histone H3 acetylation: involvement of histone acetyl transferase and histone deacetylase. Alcohol. Clin. Exp. Res. 32, 829–839 [DOI] [PubMed] [Google Scholar]
- 36. Chamorro M. N., Schwartz D. R., Vonica A., Brivanlou A. H., Cho K. R., Varmus H. E. (2005) FGF-20 and DKK1 are transcriptional targets of β-catenin and FGF-20 is implicated in cancer and development. EMBO J. 24, 73–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lowry W. E., Blanpain C., Nowak J. A., Guasch G., Lewis L., Fuchs E. (2005) Defining the impact of β-catenin/Tcf transactivation on epithelial stem cells. Genes Dev. 19, 1596–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thiery J. P. (2002) Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2, 442–454 [DOI] [PubMed] [Google Scholar]
- 39. Do T.-V., Symowicz J. C., Berman D. M., Liotta L. A., Petricoin E. F. 3rd, Stack M. S., Fishman D. A. (2007) Lysophosphatidic acid down-regulates stress fibers and up-regulates pro-matrix metalloproteinase-2 activation in ovarian cancer cells. Mol. Cancer Res. 5, 121–131 [DOI] [PubMed] [Google Scholar]
- 40. Smicun Y., Gil O., Devine K., Fishman D. A. (2007) S1P and LPA have an attachment-dependent regulatory effect on invasion of epithelial ovarian cancer cells. Gynecol. Oncol. 107, 298–309 [DOI] [PubMed] [Google Scholar]
- 41. Gilcrease M. Z., Zhou X., Welch K. (2004) Adhesion-independent α6β4 integrin clustering is mediated by phosphatidylinositol 3-kinase. Cancer Res. 64, 7395–7398 [DOI] [PubMed] [Google Scholar]
- 42. Wu R., Hendrix-Lucas N., Kuick R., Zhai Y., Schwartz D. R., Akyol A., Hanash S., Misek D. E., Katabuchi H., Williams B. O., Fearon E. R., Cho K. R. (2007) Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/β-catenin and PI3K/Pten signaling pathways. Cancer Cell 11, 321–333 [DOI] [PubMed] [Google Scholar]
- 43. Suh E. K., Gumbiner B. M. (2003) Translocation of β-catenin into the nucleus independent of interactions with FG-rich nucleoporins. Exp. Cell Res. 290, 447–456 [DOI] [PubMed] [Google Scholar]
- 44. Krieghoff E., Behrens J., Mayr B. (2006) Nucleo-cytoplasmic distribution of beta-catenin is regulated by retention. J. Cell Sci. 119, 1453–1463 [DOI] [PubMed] [Google Scholar]
- 45. Fagotto F., Gumbiner B. M. (1994) β-Catenin localization during Xenopus embryogenesis: accumulation at tissue and somite boundaries. Development 120, 3667–3679 [DOI] [PubMed] [Google Scholar]
- 46. van Amerongen R., Nusse R. (2009) Towards an integrated view of Wnt signaling in development. Development 136, 3205–3214 [DOI] [PubMed] [Google Scholar]
- 47. Barker N. (2008) The canonical Wnt/β-catenin signalling pathway. Methods Mol. Biol. 468, 5–15 [DOI] [PubMed] [Google Scholar]
- 48. Cavallo R. A., Cox R. T., Moline M. M., Roose J., Polevoy G. A., Clevers H., Peifer M., Bejsovec A. (1998) Drosophila Tcf and Groucho interact to repress Wingless signalling activity. Nature 395, 604–608 [DOI] [PubMed] [Google Scholar]
- 49. Westermann A. M., Havik E., Postma F. R., Beijnen J. H., Dalesio O., Moolenaar W. H., Rodenhuis S. (1998) Malignant effusions contain lysophosphatidic acid (LPA)-like activity. Ann. Oncol. 9, 437–442 [DOI] [PubMed] [Google Scholar]
- 50. Xiao Y. J., Schwartz B., Washington M., Kennedy A., Webster K., Belinson J., Xu Y. (2001) Electrospray ionization mass spectrometry analysis of lysophospholipids in human ascitic fluids: comparison of the lysophospholipid contents in malignant versus nonmalignant ascitic fluids. Anal. Biochem. 290, 302–313 [DOI] [PubMed] [Google Scholar]
- 51. Shen Z., Belinson J., Morton R. E., Xu Y., Xu Y. (1998) Phorbol 12-myristate 13-acetate stimulates lysophosphatidic acid secretion from ovarian and cervical cancer cells but not from breast or leukemia cells. Gynecol. Oncol. 71, 364–368 [DOI] [PubMed] [Google Scholar]
- 52. Guo L., He P., No Y. R., Yun C. C. (2015) Kruppel-like factor 5 incorporates into the β-catenin/TCF complex in response to LPA in colon cancer cells. Cell. Signal. 27, 961–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang M., Zhong W. W., Srivastava N., Slavin A., Yang J., Hoey T., An S. (2005) G protein-coupled lysophosphatidic acid receptors stimulate proliferation of colon cancer cells through the β-catenin pathway. Proc. Natl. Acad. Sci. U.S.A. 102, 6027–6032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jiang Y., Xie X., Li Z., Wang Z., Zhang Y., Ling Z. Q., Ling Z., Pan Y., Wang Z., Chen Y. (2011) Functional cooperation of RKTG with p53 in tumorigenesis and epithelial-mesenchymal transition. Cancer Res. 71, 2959–2968 [DOI] [PubMed] [Google Scholar]
- 55. Alam N., Goel H. L., Zarif M. J., Butterfield J. E., Perkins H. M., Sansoucy B. G., Sawyer T. K., Languino L. R. (2007) The integrin—growth factor receptor duet. J. Cell. Physiol. 213, 649–653 [DOI] [PubMed] [Google Scholar]
- 56. Harburger D. S., Calderwood D. A. (2009) Integrin signalling at a glance. J. Cell Sci. 122, 159–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mitra S. K., Hanson D. A., Schlaepfer D. D. (2005) Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56–68 [DOI] [PubMed] [Google Scholar]
- 58. Rozengurt E. (1995) Convergent signalling in the action of integrins, neuropeptides, growth factors and oncogenes. Cancer Surv. 24, 81–96 [PubMed] [Google Scholar]
- 59. Sakai T., de la Pena J. M., Mosher D. F. (1999) Synergism among lysophosphatidic acid, β1A integrins, and epidermal growth factor or platelet-derived growth factor in mediation of cell migration. J. Biol. Chem. 274, 15480–15486 [DOI] [PubMed] [Google Scholar]
- 60. Xu M. Y., Porte J., Knox A. J., Weinreb P. H., Maher T. M., Violette S. M., McAnulty R. J., Sheppard D., Jenkins G. (2009) Lysophosphatidic acid induces αvβ6 integrin-mediated TGF-β activation via the LPA2 receptor and the small G protein Gα(q). Am. J. Pathol. 174, 1264–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Barbolina M. V., Adley B. P., Ariztia E. V., Liu Y., Stack M. S. (2007) Microenvironmental regulation of membrane type 1 matrix metalloproteinase activity in ovarian carcinoma cells via collagen-induced EGR1 expression. J. Biol. Chem. 282, 4924–4931 [DOI] [PubMed] [Google Scholar]
- 62. Barbolina M. V., Liu Y., Gurler H., Kim M., Kajdacsy-Balla A. A., Rooper L., Shepard J., Weiss M., Shea L. D., Penzes P., Ravosa M. J., Stack M. S. (2013) Matrix rigidity activates Wnt signaling through down-regulation of Dickkopf-1 protein. J. Biol. Chem. 288, 141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dempke W., Rie C., Grothey A., Schmoll H. J. (2001) Cyclooxygenase-2: a novel target for cancer chemotherapy? J. Cancer Res. Clin. Oncol. 127, 411–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Symowicz J., Adley B. P., Woo M. M., Auersperg N., Hudson L. G., Stack M. S. (2005) Cyclooxygenase-2 functions as a downstream mediator of lysophosphatidic acid to promote aggressive behavior in ovarian carcinoma cells. Cancer Res. 65, 2234–2242 [DOI] [PubMed] [Google Scholar]
- 65. Gardi N. L., Deshpande T. U., Kamble S. C., Budhe S. R., Bapat S. A. (2014) Discrete molecular classes of ovarian cancer suggestive of unique mechanisms of transformation and metastases. Clin. Cancer Res. 20, 87–99 [DOI] [PubMed] [Google Scholar]
- 66. Lili L. N., Matyunina L. V., Walker L. D., Wells S. L., Benigno B. B., McDonald J. F. (2013) Molecular profiling supports the role of epithelial-to-mesenchymal transition (EMT) in ovarian cancer metastasis. J. Ovarian Res. 6, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tuhkanen H., Soini Y., Kosma V. M., Anttila M., Sironen R., Hämäläinen K., Kukkonen L., Virtanen I., Mannermaa A. (2009) Nuclear expression of Snail1 in borderline and malignant epithelial ovarian tumours is associated with tumour progression. BMC Cancer 9, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jin H., Yu Y., Zhang T., Zhou X., Zhou J., Jia L., Wu Y., Zhou B. P., Feng Y. (2010) Snail is critical for tumor growth and metastasis of ovarian carcinoma. Int. J. Cancer 126, 2102–2111 [DOI] [PubMed] [Google Scholar]
- 69. Blechschmidt K., Sassen S., Schmalfeldt B., Schuster T., Höfler H., Becker K. F. (2008) The E-cadherin repressor Snail is associated with lower overall survival of ovarian cancer patients. Br. J. Cancer 98, 489–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zeng X., Huang H., Tamai K., Zhang X., Harada Y., Yokota C., Almeida K., Wang J., Doble B., Woodgett J., Wynshaw-Boris A., Hsieh J. C., He X. (2008) Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development 135, 367–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Arend R. C., Londoño-Joshi A. I., Samant R. S., Li Y., Conner M., Hidalgo B., Alvarez R. D., Landen C. N., Straughn J. M., Buchsbaum D. J. (2014) Inhibition of Wnt/β-catenin pathway by niclosamide: a therapeutic target for ovarian cancer. Gynecol. Oncol. 134, 112–120 [DOI] [PubMed] [Google Scholar]
- 72. Ford C. E., Punnia-Moorthy G., Henry C. E., Llamosas E., Nixdorf S., Olivier J., Caduff R., Ward R. L., Heinzelmann-Schwarz V. (2014) The non-canonical Wnt ligand, Wnt5a, is upregulated and associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Gynecol. Oncol. 134, 338–345 [DOI] [PubMed] [Google Scholar]
- 73. McDonald S. L., Silver A. (2009) The opposing roles of Wnt-5a in cancer. Br. J. Cancer 101, 209–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mikels A. J., Nusse R. (2006) Purified Wnt5a protein activates or inhibits β-catenin-TCF signaling depending on receptor context. PLoS Biol. 4, e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ripka S., König A., Buchholz M., Wagner M., Sipos B., Klöppel G., Downward J., Gress T., Michl P. (2007) WNT5A–target of CUTL1 and potent modulator of tumor cell migration and invasion in pancreatic cancer. Carcinogenesis 28, 1178–1187 [DOI] [PubMed] [Google Scholar]
- 76. Luo J., Chen J., Deng Z. L., Luo X., Song W. X., Sharff K. A., Tang N., Haydon R. C., Luu H. H., He T. C. (2007) Wnt signaling and human diseases: what are the therapeutic implications? Lab. Invest. 87, 97–103 [DOI] [PubMed] [Google Scholar]
- 77. Takahashi-Yanaga F., Sasaguri T. (2007) The Wnt/β-catenin signaling pathway as a target in drug discovery. J. Pharmacol. Sci. 104, 293–302 [DOI] [PubMed] [Google Scholar]
- 78. Lepourcelet M., Chen Y. N., France D. S., Wang H., Crews P., Petersen F., Bruseo C., Wood A. W., Shivdasani R. A. (2004) Small-molecule antagonists of the oncogenic Tcf/β-catenin protein complex. Cancer Cell 5, 91–102 [DOI] [PubMed] [Google Scholar]
- 79. Luu H. H., Zhang R., Haydon R. C., Rayburn E., Kang Q., Si W., Park J. K., Wang H., Peng Y., Jiang W., He T. C. (2004) Wnt/β-catenin signaling pathway as a novel cancer drug target. Curr. Cancer Drug Targets 4, 653–671 [DOI] [PubMed] [Google Scholar]
- 80. Zhang B., Abreu J. G., Zhou K., Chen Y., Hu Y., Zhou T., He X., Ma J. X. (2010) Blocking the Wnt pathway, a unifying mechanism for an angiogenic inhibitor in the serine proteinase inhibitor family. Proc. Natl. Acad. Sci. U.S.A. 107, 6900–6905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yan X., Lyu T., Jia N., Yu Y., Hua K., Feng W. (2013) Huaier aqueous extract inhibits ovarian cancer cell motility via the AKT/GSK3β/β-catenin pathway. PLoS One 8, e63731. [DOI] [PMC free article] [PubMed] [Google Scholar]