

Background: HIV-1 envelope (Env) glycoprotein is targeted to endoplasmic reticulum (ER)-associated protein degradation (ERAD) pathway for degradation after infecting cells.
Results: ER class I α-mannosidase (ERManI) interacts with Env and initiates this degradation process.
Conclusion: ERManI is essential for the Env degradation.
Significance: These findings define a novel endogenous and potential therapeutically applicable antiretroviral mechanism by targeting Env for degradation.
Keywords: endoplasmic reticulum stress (ER stress), endoplasmic-reticulum-associated protein degradation (ERAD), glycoprotein, glycoprotein biosynthesis, human immunodeficiency virus (HIV), protein degradation, unfolded protein response (UPR), viral protein
Abstract
Previously, we reported that the mitochondrial translocator protein (TSPO) induces HIV-1 envelope (Env) degradation via the endoplasmic reticulum (ER)-associated protein degradation (ERAD) pathway, but the mechanism was not clear. Here we investigated how the four ER-associated glycoside hydrolase family 47 (GH47) α-mannosidases, ERManI, and ER-degradation enhancing α-mannosidase-like (EDEM) proteins 1, 2, and 3, are involved in the Env degradation process. Ectopic expression of these four α-mannosidases uncovers that only ERManI inhibits HIV-1 Env expression in a dose-dependent manner. In addition, genetic knock-out of the ERManI gene MAN1B1 using CRISPR/Cas9 technology disrupts the TSPO-mediated Env degradation. Biochemical studies show that HIV-1 Env interacts with ERManI, and between the ERManI cytoplasmic, transmembrane, lumenal stem, and lumenal catalytic domains, the catalytic domain plays a critical role in the Env-ERManI interaction. In addition, functional studies show that inactivation of the catalytic sites by site-directed mutagenesis disrupts the ERManI activity. These studies identify ERManI as a critical GH47 α-mannosidase in the ER-associated protein degradation pathway that initiates the Env degradation and suggests that its catalytic domain and enzymatic activity play an important role in this process.
Introduction
Viral Env3 glycoproteins bind to receptors and mediate the entry of virions into cells to initiate infection. Unlike viral structural and enzymatic proteins, Env is produced through the host secretory pathway, where Env is folded into a natural conformation in the ER and delivered to the cell surface (1). Notably, the efficiency of HIV-1 Env folding is very low: almost 85% Env proteins are retained in the ER and degraded (2–4). The degradation mechanism remained unknown until we recently demonstrated that Env is targeted to the ERAD pathway for degradation (5). ERAD is a host quality control mechanism for protein folding (6). It specifically delivers misfolded proteins to the SEL1L-containing translocon pore complex on the ER membrane and elicits their retro-translocation to the cytoplasm and subsequent degradation by the ubiquitin/proteasome system.
Class I α-mannosidases belong to the carbohydrate-active enZymes (CAZy) GH47 (7), which consists of seven members: ERManI, EDEM1, EDEM2, EDEM3, and Golgi mannosidase IA, IB, and IC (8). Although the enzymatic activity of EDEM1, EDEM2, and EDEM3 has not been demonstrated in vitro, the others specifically cleave the α1,2-linked mannose residues during protein N-glycosylation. In addition, they also play an important role in the ERAD pathway.
N-Glycosylation involves a number of enzymes and chaperones in the ER and requires the dedicated ERAD pathway to server as surveillance system. When nascent glycoprotein precursors enter the ER lumen, they are covalently modified with pre-assembled oligosaccharides on Asn residues in a consensus Asn-X-(Ser/Thr) motif (9). The N-linked oligosaccharides contain 14 sugars consisting of 2 N-acetylglucosamine (GlcNAc), 9 mannose (Man, 4 are α1,2-linked), and 3 terminal glucose (Glc) residues distributed on three extended Man branches A, B, and C (Fig. 1). The sequential removal of the two outermost Glc residues on branch A by glucosidases I and II allows client proteins to interact with ER chaperones calnexin and calreticulin. In conjunction with other chaperones and thiol-disulfide oxidoreductases, precursors are folded and oligomerized into native proteins. During this process, ERManI cleaves the outermost Man residue on branch B on native proteins (Fig. 1). After further removal of the last Glc residue on branch A by glucosidase II, native glycoproteins are released from calnexin/calreticulin and transported to their final destinations. Noticeably, the glycoprotein folding in the ER is error-prone. If glycoproteins display non-native conformation, they are then reglucosylated by the UDP-Glc:unfolded glycoprotein glucosyltransferase and subject to additional rounds of re-engagement with the chaperone machinery until folding is achieved. However, if proteins are terminally misfolded, accumulation of misfolded proteins activates the unfolded protein response. Misfolded proteins are then guided to the ERAD pathway for degradation.
FIGURE 1.
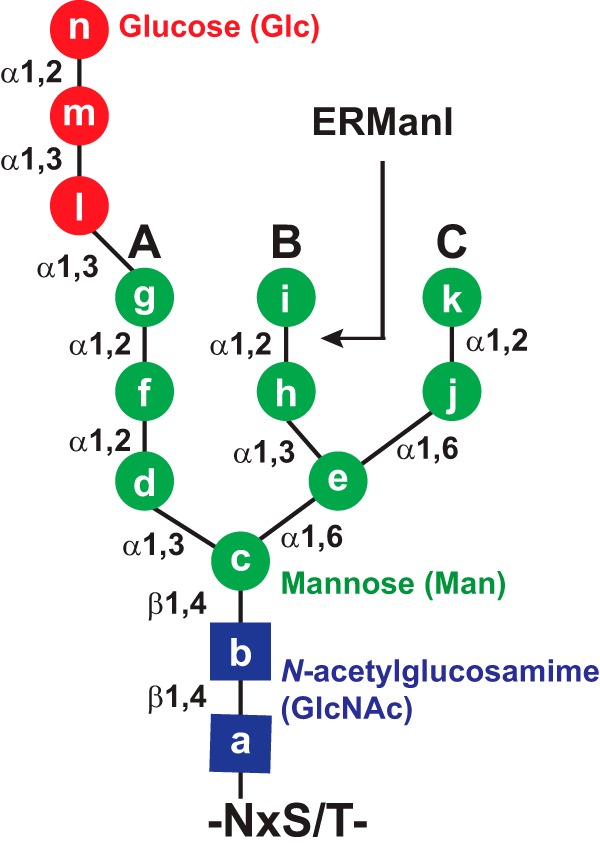
Schematic presentation of the N-linked core oligosaccharide structure. The core is composed of two N-acetylglucosamine (GlcNAc, blue squares), nine mannose (Man, green circles), and three glucose (Glc, red circles) residues. A, B, and C are three oligosaccharide branches. The ERManI preferred cleavage site is indicated.
ERManI and EDEM1 play an indispensable role in ERAD. Genetic knock-out of the ERManI gene MAN1B1 orthologue Mns1p and EDEM1 orthologue Htm1p in Saccharomyces cerevisiae showed a clear involvement of these two genes in this pathway (10, 11). In mammalian cells an inhibition of ERAD is achieved by inhibiting the CAZy GH47 α-mannosidase activity with kifunensine or by small interfering RNA-mediated gene knockdown (12–14). In addition, both ERManI and EDEM1 accelerate misfolded glycoprotein degradation in a dose-dependent manner (13–15). It has been suggested that EDEM1 extracts misfolded proteins from the calnexin/calreticulin cycle (16, 17), and misfolded proteins are targeted to the ER-derived quality control compartment where ERManI is enriched (12, 18). Although ERManI prefers to cleave the outermost Man residue on branch B, it may continue to cleave the other α1,2-linked Man residues on branches A and C under conditions of overexpression (19). Thus, ERManI and possibly the EDEM proteins may catalyze more extensive demannosylation, which constitutes a signal of protein misfolding, resulting in misfolded proteins being degraded via ERAD.
Recently, we reported that the mitochondrial translocator protein TSPO induces HIV-1 Env glycoprotein degradation via ERAD in the human CD4+ T cell line CEM.NKR (NKR), resulting in a potent HIV-1 restriction (5). TSPO associates with the mitochondrial permeability transition pore complex by interacting with one of its component, the voltage-dependent anion channel protein (20). Mitochondrial permeability transition pore establishes the mitochondrial transmembrane potential (Δψm), which allows carrier proteins to exchange small molecules between the mitochondrial matrix and cytoplasm for energy production and controls the integrity of the mitochondrial membrane (21). The goal of this study was to elucidate how HIV-1 Env is degraded via the ERAD pathway, and we identified ERManI as a critical initiator for the Env degradation, resulting in inhibition of HIV-1 replication.
Experimental Procedures
Chemicals and Antibodies
Kifunensine, tunicamycin, anti-HA antibodies, anti-FLAG M2 antibodies, and anti-FLAG M2-agarose beads were purchased from Sigma. Lactacystin and anti-actin antibodies were purchased from Santa Cruz Biotechnology. The enhanced chemiluminescence detection kit was purchased from Amersham Bioscience. Monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase antibodies were purchased from Meridian Life Science. Goat anti-human TSPO antibodies and monoclonal anti-MAN1B1 antibodies (3C2) were purchased from Novus. HIV-1 proteins were detected by antibodies from the NIH AIDS Research and Reference Reagent Program, and their catalogue numbers are 1513 (HIV-1 Gag), 526 (HIV-1 gp41), and 521 (HIV-1 gp120). Horseradish peroxidase-conjugated anti-rabbit, -goat, or -mouse immunoglobulin G secondary antibodies were purchased from Pierce.
Cell Lines
The human 293T cell line was purchased from ATCC. The human CEM-T4 T cell line and HIV-1 luciferase reporter GHOST cells were obtained from the NIH AIDS Research and Reference Reagent Program. The TSPO-KO 293T cell line A3 was reported before (5). The human CEM.NKR T cell line subclones N2-NP and N5-P were described before (22). CEM-T4, N2-NP, and N5-P cells were cultured in RPMI 1640 with 10% fetal bovine serum (HyClone). 293T and GHOST cells were cultured in DMEM with 10% bovine calf serum (HyClone).
Plasmids
The HIV-1 proviral vector pNL4-3 was obtained from the NIH AIDS Research and Reference Reagent Program. The HIV-1 luciferase reporter proviral vector pNL-Luc and the pcDNA3.1-TSPO-V5-His vector were described before (22, 23). Mammalian vectors expressing human ERManI, murine (m) EDEM1, mEDEM2, and mEDEM3 fused with a C-terminal HA tag were kindly provided by the Hosokawa and the Suzuki laboratories. pCMV6-Entry vectors expressing human EDEM1, EDEM2, and EDEM3 with a C-terminal FLAG tag were purchased from OriGene. Vectors expressing human ERManI C-terminal deletion mutants FL-1–240 and FL1–240/ΔDPS were provided by the Sifers laboratory. The full-length human ERManI cDNA was subcloned into the pcDNA3.1 vector by replacing the APOBEC3G cDNA in the pcDNA3.1-A3G-HA-FLAG vector that expresses an in-frame C-terminal tandem arrayed HA-FLAG tag after HindIII/NotI digestion. The human ERManI single-point mutants E330A, R334C, E397K, D463A, C527A, C556A, and E599A were directly created in the pcDNA3.1-ERManI-HA-FLAG vector using QuikChange II Site-Directed Mutagenesis kit (Agilent Technologies). The pcDNA3.3-TOPO vector expressing human codon-optimized Cas9 was obtained from the Church laboratory through Addgene (24). To express MAN1B1 guide RNA (gRNA; see Fig. 5A), a 455-bp gBlock that contained the U6 promoter, 19-bp gRNA, gRNA scaffold, and termination signal sequences was ordered from Integrated DNA Technologies (IDT) and cloned into the pGEM-T Easy vector (Promega) after PCR amplification, according to the Church laboratory protocol (24).
FIGURE 5.

Role of the endogenous ERManI protein in the TSPO inhibitory activity. A, schematic illustration of MAN1B1. Numbers indicate the nucleotide or amino acid positions in the ERManI open reading frame. The intron 3–4 sequence is shown in lowercase, and the exon 4 sequence is shown in uppercase. The 19-bp gRNA target sequence is shown in green, and the protospacer-adjacent motif (PAM) is shown in red. The sense primer ERManI-ko-S and antisense primer ERManI-ko-A sequences that were used to amplify this gene locus are underlined. A 5-bp deletion detected in MAN1B1-KO cells is boxed. B, analysis of the endogenous ERManI protein expression in three 293T clones (B4, E7, F7) isolated after transfection with Cas9 and MAN1B1 gRNA expression vectors by Western blotting. C, analysis of the MAN1B1 gene locus by PCR. An 83-bp DNA fragment was PCR-amplified from the MAN1B1 locus using primers ERManI-ko-S and ERManI-ko-A and analyzed by 10% TBE-polyacrylamide gel. M, marker. D and E, influence of MAN1B1 KO on HIV-1 Env inhibition. WT and E7 cells were transfected with indicated amounts of HIV-1 proviral vector pNL4-3 and TSPO expression vector in the absence (D) or presence (E) of an ERManI expression vector. Viral and cellular protein expressions were analyzed by Western blotting. F, quantification of the Env expression in D and E. The levels of HIV-1 gp120 expression in untransfected cells were set up as 100%, and the others were normalized and are presented as relative values. Error bars represent S.E. from three independent experiments.
Analysis of HIV-1 Infectivity
HIV-1 particles were produced from 293T cells after transfection with pNL-Luc and an ERManI expression vector. After being normalized by p24Gag ELISA, equal amounts of viruses were used to infect GHOST cells. After 48 h of infection, cells were lysed, and viral infectivity was determined by measuring the cellular luciferase activity using a firefly luciferase reporter assay kit from Promega.
Immunoprecipitation
To determine ERManI and HIV-1 Env interaction, 293T cells were transfected with the HIV-1 proviral vector pNL4-3 and ERManI expression vectors that have a FLAG tag. After 48 h, cells were lysed with a buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100, 1 mm EDTA). The cytosolic fraction was rocked with anti-FLAG M2-agarose beads for 4 h at 4 °C. After extensive washing with phosphate-buffered saline, bead-associated proteins were detected by Western blotting.
Knock-out of MAN1B1 in 293T Cells by CRISPR/Cas9
A detailed protocol was described before (5). Briefly, 293T cells were transfected with a Cas9 expression vector and a MAN1B1 gRNA expression vector, and cloned by limiting dilution. Clones were screened for ERManI expression by Western blotting, and ERManI knock-out (KO) clones were identified. The MAN1B1 locus in these KO clones was further analyzed by PCR using ERManI-ko-S and ERManI-ko-A as a primer pair (see Fig. 5A), and sequenced. A verified MAN1B1-KO clone E7 was finally identified.
Quantitation of Protein and DNA Levels
Images from Western blots were quantitated using the ImageJ program. Protein expression levels were calculated and presented as relative values.
Results
TSPO Triggers Env Degradation via ERAD in the Human T Cell Line NKR
The human CD4+ T cell line NKR is non-permissive for HIV-1 replication due to TSPO overexpression, which causes rapid Env turnover by ERAD (5). This is further demonstrated in its permissive clone N5-P and non-permissive clone N2-NP, which were obtained by limiting dilution of NKR cells (22). N2-NP cells expressed significantly higher TSPO levels than N5-P (Fig. 2A), resulting in ∼8-fold more TSPO expression (Fig. 2B). After HIV-1 infection, levels of Env expression were much lower in N2-NP cells than in N5-P cells (Fig. 2C), resulting in ∼10-fold Env reduction (Fig. 2D). In addition, treatment of these infected cells with an ERAD inhibitor kifunensine (KIF) significantly increased the Env expression in N2-NP cells (Fig. 2, C and D); KIF also increased HIV-1 replication in N2-NP cells but not in N5-P cells (Fig. 2E). These results suggest that Env is degraded via ERAD, which is responsible for HIV-1 inhibition in N2-NP cells.
FIGURE 2.

HIV-1 Env is degraded via ERAD in N2-NP cells. A, comparison of the endogenous TSPO expression in N2-NP and N5-P cells. Equal numbers of cells were lysed, and cellular TSPO expression was determined by Western blotting using actin as a loading control. B, quantitation of the TSPO expression from A, as described under “Experimental Procedures.” The levels of TSPO expression in N2-NP cells were set up as 100%, and the levels in N5-P cells were normalized and are presented as relative values. C, inhibition of HIV-1 Env expression by ERAD. N2-NP and N5-P cells were infected with HIV-1 in the presence or absence of 5 μm KIF. HIV-1 Env and Gag expression were determined by Western blotting. D, quantitation of the Env expression from C. The levels of HIV-1 gp120 expression in untreated N5-P cells were set up as 100%, and the others were normalized and are presented as relative values. E, inhibition of HIV-1 replication by ERAD. N2-NP and N5-P cells were infected with HIV-1 in the presence or absence of 5 μm KIF. HIV-1 replication was determined by measuring the Gag protein levels in supernatants of infected cells by p24Gag ELISA. Error bars represent S.E. from three independent experiments.
TSPO Triggers Env Degradation via ERAD in 293T Cells
To explore the mechanism of HIV-1 Env degradation by ERAD, the endogenous TSPO activity was further investigated in 293T cells. A3 is a clonal 293T cell line where the TSPO gene was knocked out by the advantageous “clustered, regularly interspaced, short palindromic repeat (CRISPR)/CRISPR-associated-9 (Cas9)” technology (5). When HIV-1 protein expression was compared in A3 and the wild-type (WT) 293T cells after transfection with the HIV-1 proviral vector pNL4-3, similar levels of Gag (p24, p55) were detected in both cell lines, but much more Env (gp41, gp160) proteins were detected in A3 than WT cells (Fig. 3A, lanes 1 and 10). In fact, a 4–8-fold higher Env expression was detected in A3 cells than in WT 293T cells after comparing serially diluted samples (Fig. 3A, lanes 3, 4, 10). Next these HIV-transfected cells were treated with increasing amounts of KIF, and the Env expression was determined. It was found that the Env expression was increased in a dose-dependent manner in WT cells (Fig. 3B). When levels of the increase were quantified, a maximal 4-fold increase was detected, which almost reached the Env expression levels in A3 cells (Fig. 3C). The same treatment did not increase the Env expression in A3 cells or the Gag expression in both A3 and WT cells (Fig. 3, B and C). These results further confirmed the TSPO activity in 293T cells.
FIGURE 3.

TSPO inhibits HIV-1 Env expression via ERAD in 293T cells. A, the TSPO-knock-out (KO) 293T cell line A3 and wild-type (WT) 293T cells were transfected with HIV-1 proviral clone pNL4-3. After 48 h, transfected cells were lysed, and the A3 cell lysate was serially diluted. Diluted A3 and undiluted WT samples were analyzed by Western blotting using indicated antibodies. B, A3 and WT 293T cells were transfected with pNL4-3 and cultured under treatment with the indicated amounts of KIF. Viral protein expression was analyzed by Western blotting using indicated antibodies. C, quantification of Gag and Env protein expression from B. In each cell line, levels of Gag or Env expression in samples treated with 100 μm KIF were set up as 100%, and the others were normalized to the standards and presented as relative values, respectively.
Identification of ERManI from the ERAD Pathway That Inhibits HIV-1 Env Expression
KIF is an alkaloid that specifically inhibits CAZy GH47 α-mannosidases (25). Results that KIF rescues HIV-1 Env expression in both N2-NP and 293T cells suggest that these enzymes are involved in the Env degradation. Among the seven CAZy GH47 α-mannosidases, ERManI, EDEM1, EDEM2, and EDEM3 have been found to play a role in ERAD. To understand how they are involved in HIV-1 Env degradation, 293T cells were transfected with HIV-1 proviral vector pNL4-3 plus a human ERManI, murine (m) EDEM1, mEDEM2, or mEDEM3 expression vector or a human APOBEC3A (A3A) expression vector, which served as a control. After 48 h of transfection, protein expression was determined by Western blotting. It was found that although all these enzymes were expressed, only ERManI was able to inhibit the Env gp120 and gp41 expression (Fig. 4A, lane 4). Human EDEM proteins share an overall ∼90% amino acid sequence identity with their murine orthologues (26). To confirm the lack of inhibitory activity of these EDEM proteins, human EDEM proteins were ectopically expressed with HIV-1 in 293T cells, and the Env expression was determined. The A3A protein was also used as a control in this experiment. Again, like their murine orthologues and the A3A protein, these human EDEM proteins did not show any inhibitory effect on HIV-1 Env expression (Fig. 4B, lanes 1, 2, and 3).
FIGURE 4.

ERManI inhibits HIV-1 Env expression. A and B, 293T cells were transfected with pNL4-3, and a mammalian vector expressing indicated human or murine (m-) proteins, respectively. After 48 h, protein expression was determined by Western blotting using the indicated antibodies. C, titration of the ERManI anti-Env activity. 293T cells were transfected with pNL4-3 and an ERManI expression vector at indicated ratio, and protein expression was determined by Western blotting. D, 293T cells were transfected with 2.0 μg of pNL4-3 and 1.0 μg of ERManI expression vectors. Cells were treated with 25 μm lactacystin or 5 μm KIF for 4 h, or untreated (control, (Ctrl)). Protein expression was determined by Western blotting using indicated antibodies. E, ERManI reduces HIV-1 infectivity. HIV-1 luciferase reporter viruses were produced after transfection of 293T cells with the HIV-1 proviral vector pNL-Luc and a WT ERManI, its catalytically inactive mutant E330A, or a control (Ctrl) vector. Viruses were normalized by p24Gag ELISA and used to infect GHOST cells. After 48 h, cells were lysed, and viral infectivity was determined by measuring the Luc activity. Error bars represent S.E. from three independent experiments.
To verify the ERManI activity, 293T cells were transfected with fixed amounts of pNL4-3 and serially diluted ERManI expression vector, and levels of Env expression were determined. It was found that ERManI could inhibit HIV-1 Env expression in a dose-dependent manner, suggesting that the Env inhibition is indeed caused by ERManI (Fig. 4C). In addition, these transfected cells were treated with KIF and a proteasomal inhibitor lactacystin. Both KIF and lactacystin were previously found to block the ERManI-mediated degradation of misfolded human α1-antitrypsin (A1AT) genetic variant-null Hong Kong (NHK) (13, 14). As expected, both KIF and lactacystin also rescued the HIV-1 Env expression (Fig. 4D). Moreover, the ERManI activity was further evaluated in a HIV-1 replication assay. HIV-1 reporter viruses were produced from 293T cells after ectopic expression of WT ERManI or its catalytic mutant E330A (see below). After normalization of viral production by the Gag protein levels, equal amounts of HIV-1 were used to infect the GHOST cells, and viral infectivity was determined. It was found that unlike the E330A mutant, WT ERManI significantly reduced the HIV-1 infectivity (Fig. 4E). Taken together, these experiments identified ERManI as a potent CAZy GH47 α-mannosidase that strongly inhibits HIV-1 Env expression via the ERAD pathway.
Knock-out of ERManI Disrupts TSPO Activity
To demonstrate the role of ERManI in TSPO-induced Env degradation, the ERManI gene MAN1B1 was knocked out in 293T cells using the CRISPR/Cas9 technology (24, 27). MAN1B1 is located on human chromosome 9, which has 13 exons. A specific 19-nucleotide gRNA was designed to target the exon 4 and inactivate this gene (Fig. 5A). A clone E7, which did not show any ERManI expression, was identified by Western blotting (Fig. 5B). When an 83-bp DNA fragment was amplified from the targeted locus in E7 cells by PCR, a small deletion was identified (Fig. 5C). After cloning and sequencing the DNA fragment, a 5-bp deletion was found (Fig. 5A). These results demonstrate that MAN1B1 is successfully knocked out in these E7 cells.
Next, HIV-1 protein expression was compared in E7 and WT 293T cells after ectopic expression of TSPO. Cells were transfected with a fixed amount of pNL4-3 and increasing amounts of TSPO expression vector, and the protein expression was determined by Western blotting. It was found that although TSPO could inhibit the Env (gp41, gp120) expression in WT cells in a dose-dependent manner, resulting in a maximal 10-fold reduction of Env expression, this activity was almost completely lost in E7 cells (Fig. 5, D and F). To confirm that MAN1B1 KO was responsible for the loss of the TSPO activity, the TSPO activity was tested again in E7 cells after the ERManI expression was restored by co-transfection with an ERManI expression vector. It was found that TSPO became able to reduce the Env expression in E7 cells in a dose-dependent manner, indicating that the TSPO activity was restored (Fig. 5E, lanes 4–6, and F). In addition, the TSPO activity was stronger in WT cells than in E7 cells, which could result from the endogenous ERManI activity. Taken together, these results demonstrate that ERManI plays an indispensable role in HIV-1 Env degradation via the ERAD pathway.
Mapping of the Critical ERManI Determinants for HIV-1 Env Inhibition
Human ERManI is a 79.5-kDa, type II membrane protein. It has 699 amino acids and consists of an N-terminal cytoplasmic domain (CD), a transmembrane (TM) helix, a lumenal stem domain, and a lumenal catalytic domain (Fig. 6A) (28, 29). The catalytic domain contains seven residues critical for the catalytic activity and protein stability, which includes Glu-330, Arg-334, Glu-397, Asp-463, Cys-527, Cys-556, and Glu-599 (see “Discussion”). To understand how these residues contribute to the HIV-1 Env inhibitory activity, they were targeted for site-directed mutagenesis by generating seven single-point mutants: E330A, R334C, E397K, D463A, C527A, C556A, and E599A. When these mutants were ectopically expressed with HIV-1 provirus in 293T cells, it was found that all these mutants were expressed at similar levels as the WT protein (Fig. 6B). Nevertheless, even though they were expressed, their inhibitory activity on Env expression was decreased to the similar levels as the control protein A3A (Fig. 6B). These results suggest that the catalytic domain is required for the ERManI inhibition of HIV-1 Env expression.
FIGURE 6.

Mapping of critical ERManI determinants for Env inhibition. A, schematic description of the ERManI protein. The cytoplasmic domain (CD), transmembrane (TM) domain, lumenal stem domain, and lumenal catalytic domain are indicated. Numbers are amino acid positions that divide these domains. Three catalytic residues, two conserved cysteine residues, two genetic mutations, and the DPS are indicated. In addition, two catalytic domain deletion mutants, FL-1–240 and FL-1–240/ΔDPS, are also illustrated. B, mapping of critical ERManI residues for Env inhibition. 293T cells were transfected with pNL4-3 plus a vector expressing indicated ERManI mutants at 1:1 ratio. Viral and cellular protein expression was determined by Western blotting. C, mapping of the critical ERManI domain for Env inhibition. 293T cells were transfected with pNL4-3 plus a vector expressing indicated ERManI mutants at 2:1 ratio. Viral and cellular protein expression was determined by Western blotting. D, interaction between ERManI and HIV-1 Env. 293T cells were transfected with pNL4-3 and an indicated ERManI deletion mutant expression vectors. After 48 h, proteins were immunoprecipitated (IP) with anti-FLAG M2-agarose beads. Proteins in cell lysate (Input), and in association with beads (IP) were analyzed by Western blotting. A3A was used as a control in B–D.
To confirm the role of the catalytic domain in HIV-1 Env inhibition, two previously described ERManI catalytic domain deletion mutants (FL-1–240 and FL-1–240/ΔDPS) were employed (30). Both mutants lack the lumenal catalytic domain, and the FL-1–240/ΔDPS mutant has an additional deletion of a highly conserved decapeptide sequence (DPS) in the lumenal stem domain (Fig. 6A). When these mutants were tested for Env inhibition together with the WT ERManI, the ERManI E330 mutant, and the human A3A protein, it was found that only the WT ERManI exhibited the Env inhibitory activity, whereas A3A, FL-1–240, FL-1–240/ΔDPS, and E330A were all inactive (Fig. 6C). These results further confirmed the indispensible role of the catalytic domain in the ERManI activity.
To understand how ERManI inhibits HIV-1 Env expression, the interaction between ERManI and HIV-1 Env was studied. A3A, WT ERManI, FL-1–240, or FL-1–240/ΔDPS was co-expressed with HIV-1 Env in 293T cells, and proteins were immunoprecipitated by an anti-FLAG antibody. It was found that the WT ERManI could pull down the HIV-1 Env precursor gp160, whereas A3A, FL-1–240, or FL-1–240/ΔDPS could not (Fig. 6D). These results demonstrate that ERManI interacts with Env and suggest the lumenal catalytic domain is involved in this interaction.
Discussion
In this report we studied the molecular mechanism of TSPO-induced HIV-1 Env degradation via ERAD and identified ERManI as a critical initiator for the degradation. Env is expressed through the classical secretory pathway, in which it needs to be properly folded in the ER (1). The Env folding involves cross-linking of 20 cysteine residues, which is dependent on heavy N-glycosylation and the most oxidizing redox status in the ER (31). It has been suggested that the oxidative protein folding in the ER is controlled by mitochondria, likely via regulating the ER redox status through releasing reactive oxygen species (32). Intracellular reactive oxygen species is mainly produced by mitochondria as a byproduct from energy production. Indeed, ER contains a specialized subcompartment that is called the mitochondrial-associated ER membrane, which physically connects ER to mitochondria (33). In mammalian cells, mitochondrial-associated ER membrane is supported by a protein complex consisting of voltage-dependent anion channel and several other proteins (34). As introduced earlier, TSPO is a mitochondrial protein (35) that interacts with voltage-dependent anion channel (20). We speculate that TSPO overexpression reduces the oxidative redox status in the ER, likely by blocking the mitochondria-ER communication, to interfere with HIV-1 Env folding. Accumulation of misfolded Env then activates unfolded protein response, resulting in recognition of these misfolded Env proteins by ERManI and their degradation via ERAD.
We found that the catalytic domain of ERManI plays an indispensible role in inhibition of HIV-1 Env expression. The structure of this domain shows an (αα)7-barrel composed of 14 consecutive helices, and Glu-330, Asp-463, and Glu-599 were proposed as potential catalytic residues (36). Mutations of Glu-330, Asp-463, and Glu-599 caused 96.5%, 99.9%, or ∼100% reduction in enzyme efficiency (kcat/Km), respectively (37). In addition, ERManI has two highly conserved cysteine residues Cys-527 and Cys-556, which are also conserved in three other Golgi CAZy GH47 α1,2-mannosidases, IA, IB, and IC, but not in EDEM proteins (36). The formation of a disulfide bond between these residues was demonstrated in the yeast Mns1, which was proposed to stabilize the protein (38). Moreover, R334C and E397K mutations are identified in nonsyndromic autosomal-recessive intellectual disability (NS-ARID) patients (39), and the R334C mutation is also found in the congenital disorders of glycosylation (40). The E397K mutation was found to reduce the ERManI expression, and the R334C mutation was found to reduce the enzyme efficiency by ∼100% (39). We created seven ERManI mutants, E330A, R334C, E397K, D463A, C527A, C556A, and E599A, to inactivate these critical residues, and found that they all lost the Env inhibitory activity (Fig. 6B). In addition, we tested the activity of two previously reported catalytic domain deletion mutants, FL-1–240 and FL-1–240/ΔDPS. Although the FL-1–240 mutant still has the activity to trigger NHK degradation, the FL-1–240/ΔDPS mutant does not (30). Nevertheless, we found that they all lost the Env inhibitory activity (Fig. 6C). Together, these results demonstrate that the catalytic activity and the catalytic domain are required for the ERManI activity. The importance of the catalytic domain was further underscored from our investigation on Env-ERManI interaction. We found that WT ERManI could pull down HIV-1 Env, whereas both FL-1–240 and FL-1–240/ΔDPS mutants could not, suggesting that ERManI interacts with Env, and this interaction is dependent on the catalytic domain (Fig. 6D). Therefore, it is likely that Env cycles between the ER and Golgi and interacts with ERManI in a post-ER compartment, resulting in Env degradation.
Results from this report point out two remarkable differences in ERAD-mediated degradation of HIV-1 Env and misfolded host glycoproteins. First, although ectopic expression of EDEM proteins is able to accelerate the degradation of NHK and/or misfolded β-secretase (15, 41–43), it is unable to inhibit HIV-1 Env expression (Fig. 4, A and B). Second, although the ERManI catalytic domain is not required for NHK degradation, it is required for the Env degradation. Because the FL-1–240 mutant still triggers the NHK degradation but the FL-1–240/ΔDPS mutant does not, it is suggested that instead of the catalytic domain, the conserved DPS in the stem domain is critical for the NHK degradation (30). However, because both FL-1–240 and FL-1–240/ΔDPS mutants fail to inhibit HIV-1 Env expression, it is suggested that the catalytic domain is critical for the Env degradation (Fig. 6C). Thus, although both HIV-1 Env and NHK are degraded via ERAD, different downstream signaling cascades could be involved in their degradation. A further understanding of these differences may identify a specific pathway for inhibition of the Env expression and HIV-1 replication.
Author Contributions
Y.-H. Z. conceived and coordinated the study and wrote the paper. T. Z. designed, performed, and analyzed the experiments shown in Figs. 2–5. D. A. F. designed, performed, and analyzed the experiments shown in Fig. 6. Y.-H. Z. and K. W. M. edited the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Ying Zhang, Changqing Yu, Jiajun Zhou and Matthew A. Wexler for experimental supports. We thank the Hosokawa, Suzuki, and Sifers laboratories for providing expression vectors. We also thank the NIH AIDS Research and Reference Reagent Program for providing the other reagents.
This work was supported, in whole or in part, by National Institutes of Health Grants AI063944 (to Y.-H. Z) and P41GM103390, GM47533, and DK075322 (to K. W. M.). The authors declare that they have no conflicts of interest with the contents of this article.
- Env
- envelope
- ER
- endoplasmic reticulum
- ERAD
- ER-associated protein degradation
- CAZy
- carbohydrate-active enZyme
- ERManI
- endoplasmic reticulum class I α-mannosidase
- GH47
- glycoside hydrolase family 47
- EDEM
- ER-degradation enhancing α-mannosidase-like
- NKR
- CEM.NKR
- TSPO
- mitochondrial translocator protein
- gRNA
- guide RNA
- KIF
- kifunensine
- CRISPR
- clustered, regularly interspaced, short palindromic repeat
- Cas9
- CRISPR-associated-9
- A3A
- APOBEC3A
- NHK
- null Hong Kong
- DPS
- decapeptide sequence.
References
- 1. Checkley M. A., Luttge B. G., Freed E. O. (2011) HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol. 410, 582–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fennie C., Lasky L. A. (1989) Model for intracellular folding of the human immunodeficiency virus type 1 gp120. J. Virol. 63, 639–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hallenberger S., Tucker S. P., Owens R. J., Bernstein H. B., Compans R. W. (1993) Secretion of a truncated form of the human immunodeficiency virus type 1 envelope glycoprotein. Virology 193, 510–514 [DOI] [PubMed] [Google Scholar]
- 4. Willey R. L., Bonifacino J. S., Potts B. J., Martin M. A., Klausner R. D. (1988) Biosynthesis, cleavage, and degradation of the human immunodeficiency virus 1 envelope glycoprotein gp160. Proc. Natl. Acad. Sci. U.S.A. 85, 9580–9584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou T., Dang Y., Zheng Y. H. (2014) The mitochondrial translocator protein, TSPO, inhibits HIV-1 envelope glycoprotein biosynthesis via the endoplasmic reticulum-associated protein degradation pathway. J. Virol. 88, 3474–3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meusser B., Hirsch C., Jarosch E., Sommer T. (2005) ERAD: the long road to destruction. Nat. Cell Biol. 7, 766–772 [DOI] [PubMed] [Google Scholar]
- 7. Henrissat B., Davies G. (1997) Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7, 637–644 [DOI] [PubMed] [Google Scholar]
- 8. Mast S. W., Moremen K. W. (2006) Family 47 α-mannosidases in N-glycan processing. Methods Enzymol. 415, 31–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Helenius A., Aebi M. (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 [DOI] [PubMed] [Google Scholar]
- 10. Jakob C. A., Bodmer D., Spirig U., Battig P., Marcil A., Dignard D., Bergeron J. J., Thomas D. Y., Aebi M. (2001) Htm1p, a mannosidase-like protein, is involved in glycoprotein degradation in yeast. EMBO Rep. 2, 423–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jakob C. A., Burda P., Roth J., Aebi M. (1998) Degradation of misfolded endoplasmic reticulum glycoproteins in Saccharomyces cerevisiae is determined by a specific oligosaccharide structure. J. Cell Biol. 142, 1223–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Avezov E., Frenkel Z., Ehrlich M., Herscovics A., Lederkremer G. Z. (2008) Endoplasmic reticulum (ER) mannosidase I is compartmentalized and required for N-glycan trimming to Man5–6GlcNAc2 in glycoprotein ER-associated degradation. Mol. Biol. Cell 19, 216–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hosokawa N., Tremblay L. O., You Z., Herscovics A., Wada I., Nagata K. (2003) Enhancement of endoplasmic reticulum (ER) degradation of misfolded null Hong Kong α1-antitrypsin by human ER mannosidase I. J. Biol. Chem. 278, 26287–26294 [DOI] [PubMed] [Google Scholar]
- 14. Wu Y., Swulius M. T., Moremen K. W., Sifers R. N. (2003) Elucidation of the molecular logic by which misfolded α1-antitrypsin is preferentially selected for degradation. Proc. Natl. Acad. Sci. U.S.A. 100, 8229–8234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hosokawa N., Wada I., Hasegawa K., Yorihuzi T., Tremblay L. O., Herscovics A., Nagata K. (2001) A novel ER α-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2, 415–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Molinari M., Calanca V., Galli C., Lucca P., Paganetti P. (2003) Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 299, 1397–1400 [DOI] [PubMed] [Google Scholar]
- 17. Oda Y., Hosokawa N., Wada I., Nagata K. (2003) EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299, 1394–1397 [DOI] [PubMed] [Google Scholar]
- 18. Benyair R., Ogen-Shtern N., Mazkereth N., Shai B., Ehrlich M., Lederkremer G. Z. (2015) Mammalian ER mannosidase I resides in quality control vesicles, where it encounters its glycoprotein substrates. Mol. Biol. Cell 26, 172–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Herscovics A., Romero P. A., Tremblay L. O. (2002) The specificity of the yeast and human class I ER α1,2-mannosidases involved in ER quality control is not as strict previously reported. Glycobiology 12, 14G–15G [PubMed] [Google Scholar]
- 20. McEnery M. W., Snowman A. M., Trifiletti R. R., Snyder S. H. (1992) Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. U.S.A. 89, 3170–3174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zamzami N., Kroemer G. (2001) The mitochondrion in apoptosis: how Pandora's box opens. Nat. Rev. Mol. Cell Biol. 2, 67–71 [DOI] [PubMed] [Google Scholar]
- 22. Zhou T., Han Y., Dang Y., Wang X., Zheng Y. H. (2009) A novel HIV-1 restriction factor that is biologically distinct from APOBEC3 cytidine deaminases in a human T cell line CEM.NKR. Retrovirology 6, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dang Y., Wang X., Esselman W. J., Zheng Y. H. (2006) Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J. Virol. 80, 10522–10533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., Church G. M. (2013) RNA-guided human genome engineering via Cas9. Science 339, 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Herscovics A. (2001) Structure and function of Class I α1,2-mannosidases involved in glycoprotein synthesis and endoplasmic reticulum quality control. Biochimie 83, 757–762 [DOI] [PubMed] [Google Scholar]
- 26. Olivari S., Molinari M. (2007) Glycoprotein folding and the role of EDEM1, EDEM2 and EDEM3 in degradation of folding-defective glycoproteins. FEBS Lett. 581, 3658–3664 [DOI] [PubMed] [Google Scholar]
- 27. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gonzalez D. S., Karaveg K., Vandersall-Nairn A. S., Lal A., Moremen K. W. (1999) Identification, expression, and characterization of a cDNA encoding human endoplasmic reticulum mannosidase I, the enzyme that catalyzes the first mannose trimming step in mammalian Asn-linked oligosaccharide biosynthesis. J. Biol. Chem. 274, 21375–21386 [DOI] [PubMed] [Google Scholar]
- 29. Tremblay L. O., Herscovics A. (1999) Cloning and expression of a specific human α1,2-mannosidase that trims Man(9)GlcNAc(2) to Man(8)GlcNAc(2) isomer B during N-glycan biosynthesis. Glycobiology 9, 1073–1078 [DOI] [PubMed] [Google Scholar]
- 30. Iannotti M. J., Figard L., Sokac A. M., Sifers R. N. (2014) A Golgi-localized mannosidase (MAN1B1) plays a non-enzymatic gatekeeper role in protein biosynthetic quality control. J. Biol. Chem. 289, 11844–11858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feige M. J., Hendershot L. M. (2011) Disulfide bonds in ER protein folding and homeostasis. Curr. Opin. Cell Biol. 23, 167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang Y., Song Y., Loscalzo J. (2007) Regulation of the protein disulfide proteome by mitochondria in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 104, 10813–10817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Simmen T., Lynes E. M., Gesson K., Thomas G. (2010) Oxidative protein folding in the endoplasmic reticulum: tight links to the mitochondria-associated membrane (MAM). Biochim. Biophys. Acta 1798, 1465–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kornmann B., Walter P. (2010) ERMES-mediated ER-mitochondria contacts: molecular hubs for the regulation of mitochondrial biology. J. Cell Sci. 123, 1389–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dellisanti C. (2015) TSPO through the crystal looking glass. Nat. Struct. Mol. Biol. 22, 184 [Google Scholar]
- 36. Vallee F., Karaveg K., Herscovics A., Moremen K. W., Howell P. L. (2000) Structural basis for catalysis and inhibition of N-glycan processing class I α1,2-mannosidases. J. Biol. Chem. 275, 41287–41298 [DOI] [PubMed] [Google Scholar]
- 37. Karaveg K., Moremen K. W. (2005) Energetics of substrate binding and catalysis by class 1 (glycosylhydrolase family 47) α-mannosidases involved in N-glycan processing and endoplasmic reticulum quality control. J. Biol. Chem. 280, 29837–29848 [DOI] [PubMed] [Google Scholar]
- 38. Lipari F., Herscovics A. (1996) Role of the cysteine residues in the α1,2-mannosidase involved in N-glycan biosynthesis in Saccharomyces cerevisiae. The conserved Cys-340 and Cys-385 residues form an essential disulfide bond. J. Biol. Chem. 271, 27615–27622 [DOI] [PubMed] [Google Scholar]
- 39. Rafiq M. A., Kuss A. W., Puettmann L., Noor A., Ramiah A., Ali G., Hu H., Kerio N. A., Xiang Y., Garshasbi M., Khan M. A., Ishak G. E., Weksberg R., Ullmann R., Tzschach A., Kahrizi K., Mahmood K., Naeem F., Ayub M., Moremen K. W., Vincent J. B., Ropers H. H., Ansar M., Najmabadi H. (2011) Mutations in the α1,2-mannosidase gene, MAN1B1, cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 89, 176–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rymen D., Peanne R., Millón M. B., Race V., Sturiale L., Garozzo D., Mills P., Clayton P., Asteggiano C. G., Quelhas D., Cansu A., Martins E., Nassogne M. C., Gonçalves-Rocha M., Topaloglu H., Jaeken J., Foulquier F., Matthijs G. (2013) MAN1B1 deficiency: an unexpected CDG-II. PLoS Genet. 9, e1003989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hirao K., Natsuka Y., Tamura T., Wada I., Morito D., Natsuka S., Romero P., Sleno B., Tremblay L. O., Herscovics A., Nagata K., Hosokawa N. (2006) EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 281, 9650–9658 [DOI] [PubMed] [Google Scholar]
- 42. Mast S. W., Diekman K., Karaveg K., Davis A., Sifers R. N., Moremen K. W. (2005) Human EDEM2, a novel homolog of family 47 glycosidases, is involved in ER-associated degradation of glycoproteins. Glycobiology 15, 421–436 [DOI] [PubMed] [Google Scholar]
- 43. Olivari S., Galli C., Alanen H., Ruddock L., Molinari M. (2005) A novel stress-induced EDEM variant regulating endoplasmic reticulum-associated glycoprotein degradation. J. Biol. Chem. 280, 2424–2428 [DOI] [PubMed] [Google Scholar]