

Background: Enhanced IL1R2 expression has frequently been observed in tumors, but its function is unclear.
Results: Intracellular IL1R2 transcriptionally activates IL-6 and VEGF-A by complexing with c-Fos and promotes the angiogenesis of colon cancer cells.
Conclusion: IL1R2 functions as a transcriptional coactivator to enhance the expression of angiogenic factors.
Significance: IL1R2 activates angiogenic factors and hence may serve as a clinical marker for prognosis or treatment.
Keywords: angiogenesis, c-Fos, colon cancer, interleukin 6 (IL-6), vascular endothelial growth factor (VEGF), interleukin-1 receptor type 2
Abstract
Interleukin-1 receptor type 2 (IL1R2) acts as a decoy receptor of exogenous IL-1; however, its intracellular activity is poorly understood. We previously demonstrated that IL1R2 intracellularly activates the expression of several proinflammatory cytokines and affects cell migration. In this study, we found that intracellular IL1R2 expression was increased in human colorectal cancer cells (CRCs) compared with normal colon cells. We also observed that the mRNA levels of IL1R2 were highly correlated with IL-6 in tumor tissues of CRC patients. By modulating its expression in CRC cells, we verified that enhanced IL1R2 expression transcriptionally activated the expression of IL-6 and VEGF-A. Conditioned medium harvested from IL1R2-overexpressing CRC cells contained higher levels of IL-6 and VEGF-A than that from vector control cells and significantly enhanced the proliferation, migration, and tube formation of cultured endothelial cells. We further demonstrated a positive association of intracellular IL1R2 levels with tumor growth and microvessel density in xenograft mouse models. These results revealed that IL1R2 activates the expression of angiogenic factors. Mechanistically, we revealed that IL1R2 complexes with c-Fos and binds to the AP-1 site at the IL-6 and VEGF-A promoters. Together, these results reveal a novel function of intracellular IL1R2 that acts with c-Fos to enhance the transcription of IL-6 and VEGF-A, which promotes angiogenesis in CRC.
Introduction
Angiogenesis involving the sprouting and growth of new blood vessels from existing ones is a hallmark for tumor metastasis (1, 2). Angiogenesis is closely related to chronic inflammation (3), which is well known for its key roles in the development and progression of a variety of human cancers, including colorectal cancer (CRC)2 (3, 4). In the clinic, the serum levels of IL-6 and VEGF are significantly higher in CRC patients than healthy controls (5). Moreover, patients with advanced clinical stage disease have significantly higher levels of IL-6 and VEGF in the serum than those in early clinical stages (5). Although VEGF is one of the most potent endothelial cell mitogens (6–9), IL-6, a critical inflammatory cytokine, may also be involved in angiogenesis in CRC (7, 10, 11).
In addition to IL-6, IL-1 signaling is also a key player in the regulation of inflammatory diseases (12) and a critical pathway involved in tumor growth, invasiveness, angiogenesis, and tumor-host interactions (13). There are two IL-1 receptors, IL1RI and IL1R2 (12). IL1R1 is involved in the signal transduction of exogenous IL-1 (12, 14), whereas IL1R2 is a decoy receptor that lacks the cytoplasmic signaling domain (15, 16) and blocks IL-1 signaling (17). Therefore, IL1R2 may be similar to the IL-1 receptor antagonist, which acts as a negative regulator of IL-1 signaling (18). However, IL1R2 expression is enhanced in keratinocytes treated with IFN-γ or phorbol 12-myristate 13-acetate (19), suggesting that IL1R2 functions as a proinflammatory factor. Recent studies have also shown that the overexpression of intracellular IL1R2 enhances the expression of IL-6 (17, 20). Therefore, IL1R2 has an apparent dual role in the regulation of IL-1 signaling, i.e. IL1R2 suppresses exogenous IL-1 signaling, and intracellular IL1R2 stimulates the expression of inflammatory cytokines. However, studies on the physiological role and biological function of intracellular IL1R2 are limited.
The involvement of IL1R2 overexpression in tumorigenesis has been revealed by an integrative genomics study showing that elevated IL1R2 was significantly associated with the expression of human epidermal growth factor receptor 2 and 3 tyrosine kinase receptors and with reduced relapse-free survival in breast cancer (21). IL1R2 overexpression has been observed in breast cancer patients with recurrences after tamoxifen treatment (22). Increased IL1R2 expression in ovarian and pancreatic cancer tissues (23–25) clinically supported the involvement of IL1R2 in cancer progression. In addition, IL1R2 is increased in an immune-resistant cancer cell line compared with a susceptible cancer cell line (26) and in multidrug-resistant ovarian carcinoma cells (27). These studies suggest that IL1R2 has oncogenic potential; however, the role of IL1R2 on carcinogenesis is far from clear.
We have previously observed that the expression of intracellular IL1R2 is enhanced in long term arsenic-exposed human urothelial cells (28). Furthermore, we showed that the ectopic expression of IL1R2 activates intracellular IL-1α signaling and increases the transcription of IL-6, IL-8, and collagen and the migration of human urothelial cells (17). Consistent with these results, we observed a dose-dependent increase of intracellular IL1R2, IL-6, and VEGF-A levels, as well as tumorigenesis in human keratinocyte cells exposed long term to sodium arsenite. Our previous findings support the hypothesis that the proinflammatory activity of intracellular IL1R2 induces angiogenesis and hence drives malignant transformation.
To better understand the oncogenic activity of intracellular IL1R2, we preliminarily observed that intracellular IL1R2 expression was higher in a variety of CRC cells compared with normal colon epithelial FHC cells. CRC is considered a prominent global health problem because of its increasing prevalence (29). Because angiogenesis is critical for CRC development and metastasis (2), we conducted experiments to elucidate whether and how intracellular IL1R2 acts as an oncogenic and angiogenic factor in CRC.
Experimental Procedures
Cell Culture
The human CRC cell lines Colo205, DLD-1, H3347, SW620, HCT116, and HT29 were cultured in RPMI 1640 medium (Life Technologies, Inc.). Normal colon epithelial cells, FHCs, were cultured in a 1:1 mixture of DMEM/F12 (Life Technologies, Inc.), and RKO, RKO-E6, and hybrid EA.hy926 human endothelial cells were cultured in DMEM (Life Technologies, Inc.). All cells were grown in medium supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm l-glutamine and incubated at 37 °C in a humidified atmosphere containing 5% CO2, and the cells were verified to be mycoplasma free by PCR analysis. RKO, RKO-E6, DLD-1, Colo205, H3347, SW620, HCT116, and HT29 cells were obtained from Jeou-Yuan Chen (Institute of Biomedical Sciences, Academia Sinica, Taiwan), EA.hy926 cells were from Jing-Jy Cheng (National Research Institute of Chinese Medicine, Ministry of Health and Welfare, Taiwan), and FHC cells were from Yuan-Soon Ho (School of Medical Laboratory Science and Biotechnology, Taipei, Medical University, Taiwan). The human keratinocyte A0, A1, and A2 cell lines were generated from HaCaT cells, kindly provided by N. E. Fusenig (German Cancer Research Center, Heidelberg, Germany), by continuously exposing them to 0, 0.5, and 1 μm sodium arsenite in DMEM supplemented with 10% FBS for 20 passages, respectively (30). The T4R2 cell line, derived from a xenograft of A2 cells, was found to be highly tumorigenic in nude mice.
Clinical Samples
In this study, the mRNAs of 40 CRC tissues were used for quantitative real time PCR (qPCR) assay. Patient tissue specimens that were previously collected at the Veterans General Hospital (Taipei, Taiwan) were used with the approval of the Veterans General Hospital's Institutional Review Board.
Western Blotting Analysis
Western blotting analysis was performed as previously described (31). The following primary antibodies were used: goat anti-IL1R2 (GeneTex), rabbit anti-IL1R2 (GeneTex), anti-IL-6 (Abcam), anti-c-Fos (Abcam), anti-VEGF-A (GeneTex), anti-p-c-Jun (Cell Signaling), anti-c-Jun (Cell Signaling), anti-IL1R2 (Abcam), anti-Myc tag (Cell Signaling), and mouse anti-p-c-Fos (Abcam). Nuclei were isolated from human CRC cells using a Nuclei EZ Prep Nuclei Isolation Kit (Sigma).
Quantitative Real Time Polymerase Chain Reaction
qPCR was performed as described by Ponchel et al. (32). The PCR primers used to amplify the genes of interest are listed in Table 1. Thermocycling was performed in a 20-μl reaction solution containing 2.5 μl of cDNA, 200 nm of each primer, and 6.5 μl of SYBR Green I master mix (Roche) using the Roche LightCycler 480 sequence detection system (Roche Applied Science). The expression levels of the genes of interest were normalized to the internal control, GAPDH.
TABLE 1.
Primer sequences used for qPCR, promoter construction, site-directed mutation, and ChIP assay
| Primers | Forward (5′ → 3′) | Reversed (5′ → 3′) |
|---|---|---|
| For qPCR | ||
| IL1R2 | TGTGCTGGCCCCACTTTC | GCACAGTCAGACCATCTGCTTT |
| IL-6 | TGGCTGCAGGACATGACAA | TGAGGTGCCCATGCTACATTT |
| VEGF-A | CTTGCCTTGCTGCTCTACC | CACACAGGATGGCTTGAAG |
| GAPDH | GGAGTCCCTGCCACACTCA | GCCCCTCCCCTCTTCAAG |
| For promoter construction | ||
| KpnI-IL-6 (−710∼+30)-BgIII | CGGGGTACCACGCGGAAGCAGATTCAGAG | GGAAGATCTTTCTCTTTCGTTCCCGGTGG |
| KpnI-IL-6 (−287∼+30)-BgIII | CGGGGTACCCGCCTCAATGACGACCTAAGCTGCA | GGAAGATCTTTCTCTTTCGTTCCCGGTGG |
| KpnI-IL-6 (−206∼+30)-BgIII | CGGGGTACCCATAAGGTTTCCAATCAGCCCCACC | GGAAGATCTTTCTCTTTCGTTCCCGGTGG |
| KpnI-IL-6 (−123∼+30)-BgIII | CGGGGTACCCCAACCCCCAATAAATATAGGACTG | GGAAGATCTTTCTCTTTCGTTCCCGGTGG |
| XhoI-VEGF-A (−1632∼−12)-HindIII | CCGCTCGAGGAATGGAGCGAGCAGCGTCTTCGAGA | CCCAAGCTTCAAGGCAAGGCTCCAATGCACCCAAG |
| For AP-1 site directed mutationa | ||
| IL-6 mutant (−214∼−118) | GTGGGATTTTCCCATTCTTCTCAATATTAGAGT | ACTCTAATATTGAGAAGAATGGGAAAATCCCAC |
| VEGF-A mutant (−621∼−615) | CGACAGGGGCAAAGTGCTGGACCTGCTTTTGGGG | CCCCAAAAGCAGGTCCAGCACTTTGCCCCTGTCG |
| VEGF-A mutant (−1527∼−1521) | GTCTGTGTGGGTGCTGGAGTGTGTGCGTGT | ACACGCACACACTCCAGCACCCACACAGAC |
| For ChIP assay | ||
| IL-6 | ATGCTAAAGGACGTCACATTGCACAA | TGGCAGTTCCAGGGCTAAGGATTTCC |
| VEGF-A | GCAGACGGCAGTCACTAG | GATTCCAATAGATCTGTG |
a Underlining indicates the mutation site.
Selection of IL1R2-silenced Stable Cell Lines
The IL1R2-silenced stable cell lines HT29-shIL1R2 and SW620-shIL1R2 were generated by transfecting HT29 and SW620 cells with the pGIPZ-sh-control and pGIPZ-sh-IL1R2 plasmid, respectively, using Lipofectamine 2000 (Invitrogen). Three days post-transfection, stable HT29-shV and HT29-shIL1R2 cells were established by culturing them in the presence of 0.7 μg/ml puromycin, whereas the SW620-shV and SW620-shIL1R2 cells were established with 0.25 μg/ml puromycin. The control and shRNA plasmids (pGIPZ-sh-control and pGIPZ-sh-IL1R2) were obtained from the siRNA Core Facility of Academia Sinica (Taiwan).
Construction of the IL1R2 Expression Plasmid and Selection of Stable Cell Lines Overexpressing IL1R2
The cDNA encoding human IL1R2 was amplified from MGC full-length cDNA clones (Genome Research Center, National Yang-Ming University) by PCR. The primers used for PCR contained NheI and BamHI restriction sites. The PCR product was digested with NheI and BamHI and subsequently subcloned into pFLAG-myc-CMV22-IL1R2-IRES-EGFP, which had been digested with the same enzymes. The pIRES-EGFP expression (control) and pIRES-EGFP-IL1R2 (IL1R2 expression) vectors were separately transfected into HCT116 and DLD-1 cells using the FuGENE 6 transfection reagent (Roche). Stable transfectants were selected by incubation in culture medium containing 1 mg/ml G418 (Gibco) for 2 weeks.
Transwell Migration Assay
The Boyden chamber system was used to evaluate cell migration. Gelatin (10 μg/ml) was added to each well of a Transwell plate (Corning-Costar, Corning, NY; 8-μm pore size), and then the membranes were allowed to dry at 37 °C for 1 h. Afterward, 5 × 103 cells suspended in 100 μl of FBS-free medium were seeded in the upper chamber of a Transwell plate. The lower chamber was filled with medium containing 5% FBS. After incubating for 8 h, the cells on the top side of the upper Transwell membrane were removed using cotton swabs. The cells trapped on the bottom side of the membrane were fixed with methanol and stained with a 4′,6-diamidino-2-phenylindole solution (10 μg/ml; Invitrogen) for 20 min. The number of cells from eight different fields on each membrane was counted under a fluorescence microscopy.
Conditioned Medium (CM)
Cells were seeded onto 100-mm dishes at a density of 1 × 106 cells/dish. After incubation for 48 h, the medium was replaced with 5 ml of fresh medium with or without 10% FBS and incubated for another 24 h. Then the CM was harvested, centrifuged at 3,000 rpm for 10 min to remove the cell debris, and stored at −20 °C until use.
ELISA
ELISA kits (eBioscience) were used to measure the amount of IL-6 and VEGF-A secreted into serum-free CM according to the manufacturer's instructions.
Cell Proliferation Assay
The proliferation of human IL1R2-adjusted CRC cells was measured by trypan blue assay. The effect of CM on the proliferation of EA.hy926 cells was assayed by cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide reduction, as previously described (33).
Tube Formation Assay
A 96-well plate was precoated with Matrigel 1 h before seeding EA.hy926 hybrid endothelial cells (5 × 105 cells/well) at 37 °C. Tube formation ability was then assayed by quantifying the complexity of the tubes formed using KURABO Angiogenesis Image Analyzer software (34).
Directed in Vivo Angiogenesis Assay (DIVAA)
In vivo angiogenesis assays were performed using a DIVAA kit (Trevigen) according to the manufacturer's instructions. In brief, silicon angioreactors were filled with basement membrane extracts containing various CM and heparin (1 μmol/liter), and these angioreactors were subcutaneously implanted into both flanks of C57/B6 mice (two reactors per side). After 17 days, the reactors were excised and labeled with FITC-lectin. The fluorescence intensity reflects the number of endothelial cells that entered the angioreactors.
In Vivo Xenograft Tumorigenicity Assay
The use of animals followed the guidelines approved by the Institutional Animal Care and Utilization Committee of the Academia Sinica, Taiwan. HCT116-Vec, HCT116-IL1R2, HT29-shV, and HT29-shIL1R2 cells (1 × 106) were injected subcutaneously into 8–12-week-old, male, BALA/c nude mice that were obtained from the National Laboratory Animal Center (Taiwan). Tumor sizes were measured twice per week. When the tumors attained a volume of 1 cm3, the mice were sacrificed, and the tumors were subjected to immunohistochemical staining for CD31 using an anti-CD31 antibody (Abcam). Using ImageJ software, three images were acquired from randomly selected locations in each slide. Random RGB images were converted to 8-bit images for calculating the integrated density parameters. For antibody neutralization experiments, tumor-bearing mice (HT29-shV or HT29-shIL1R2 cells) were treated with the antibodies of isotype control (R&D Systems), IL-6 (MAB206; R&D Systems), or VEGF-A (MAB293; R&D Systems) by administration of 50 μg of antibody (twice a week) into the tumor site when the tumor volume reached 100 mm3.
IL-6 and VEGF-A Promoter-based Dual Luciferase Assays
The IL-6 (−710∼+30) and VEGF (−1632∼−12) promoters were amplified using specific 5′ and 3′ promoter primers (Table 1). The truncated fragments of IL-6 promoter were amplified using various 5′ deletion primers and the same 3′ primer. The following conditions were used to amplify the promoter regions: 5 min at 94 °C and 35 cycles of 1 min at 94 °C, 1 min at 60 °C, and 3 min at 72 °C. The amplified products were digested with specific restriction enzymes and subsequently cloned into the pGL3-basic firefly luciferase reporter vector. For the promoter activity assay, the reporter constructs were co-transfected with the reference pCMV-Renilla luciferase plasmid into cells with different levels of IL1R2 for 24 h. The cells were harvested and lysed with passive lysis buffer (Promega). The promoter activity was determined by the dual luciferase assay according to the manufacturer's instructions.
Mutation of the AP-1 Binding Site in the IL-6 and VEGF-A Promoters
Mutant IL-6 and VEGF-A promoters were generated and used to construct luciferase reporters using the QuikChange II XL site-directed mutagenesis kit (Stratagene) and mutant primers (Table 1). The PCR conditions were as follows: 95 °C for 1 min; 30 cycles of 95 °C for 30 s, 55 °C for 60 s, 66 °C for 5 min, and a final extension step at 68 °C for 7 min. The reaction mixture was digested with DpnI for 2 h to remove the parental methylated or hemimethylated DNA.
Immunoprecipitation-Western Blot Assay
The immunoprecipitation-Western blot assay was performed as previously described (35). Logarithmically growing cells were resuspended in 1 ml of radioimmune precipitation assay lysis buffer. An aliquot of each cell extract was incubated with the indicated antibody, which was immobilized on protein A/G-Sepharose beads (Sigma), overnight at 4 °C with gentle rotation. The beads were extensively washed four times with wash buffer and boiled in 5× SDS loading buffer. The supernatants were immediately used for Western blot analysis as described above.
Chromatin Immunoprecipitation
The ChIP assay was performed using an EZ ChIP chromatin immunoprecipitation kit (Upstate Biotechnology, Inc., Lake Placid, NY). Briefly, cells were cross-linked by the addition of 1% formaldehyde at room temperature, and the reaction was terminated by the addition of glycine. Subsequently, the cells were lysed in SDS lysis buffer. Genomic DNA was extracted and fragmented into ∼200-bp segments using the EZ-Zyme kit (Upstate Biotechnology, Inc., Lake Placid, NY), and the sheared chromatin was diluted 10 times and precleared with protein G-agarose at 4 °C for 1 h with rotation. The protein G-agarose was pelleted by centrifugation, and an aliquot of 10 μl of the supernatant was saved for the input control. The remaining samples were divided into three groups and incubated with 5 μg of anti-IL1R2, anti-c-Fos, and nonspecific rabbit IgG (Millipore). The immunoprecipitated products were washed sequentially according to the manufacturer's instructions. The chromatin was then eluted from the agarose by incubation with elution buffer, and the DNA-protein cross-links were reversed using a high salt solution containing 200 mm NaCl at 65 °C for at least 5 h. Finally, the precipitated DNA was recovered using the provided spin column and eluted with 50 μl of elution buffer. PCR was performed using Taq DNA polymerase (Roche Applied Science) with 2 μl of the precipitated DNA as the template. The primers used are shown in Table 1.
Statistics
The correlation between IL1R2 expression and IL-6 was analyzed using the χ2 test. Two-way analysis of variance using GraphPad Prism software was adopted to compare the growth of tumor in animal studies. Student's t test was used to determine the significance of the differences between the experimental and control groups. Two-sided p values of less than 0.05 were considered to represent significant differences.
Results
IL1R2 Expression Is Higher in Human CRC Cells than Normal Colon Epithelial Cells and Correlated Well with IL-6 mRNA Levels in Tumor Tissues of CRC Patients
As shown in Fig. 1A, CRC cell lines expressed IL1R2 protein to varying degrees, but their expression levels were in general higher than that of normal colon epithelial FHC cells. The CRC cells with high (SW620 and HT29) and low (DLD-1 and HCT116) abundance of IL1R2 were selected to analyze its cellular distribution. The nucleus and cytosol were biochemically separated using a nuclei isolation kit, and the protein levels were analyzed by Western blotting. As shown in Fig. 1B, IL1R2 was present not only in the cytosol but also in the nucleus. We previously observed that IL1R2 overexpression causes increased IL-6 mRNA levels in human urothelial cells (17); we therefore analyzed the IL-6 mRNA levels in 40 CRC tumor specimens by qPCR. As shown in Fig. 1C, there was a positive correlation between IL1R2 and IL-6 mRNA expression (normalized to GAPDH) in these samples (Pearson coefficient r = 0.86684; p < 0.05), implicating the proinflammatory function of IL1R2 in CRC.
FIGURE 1.
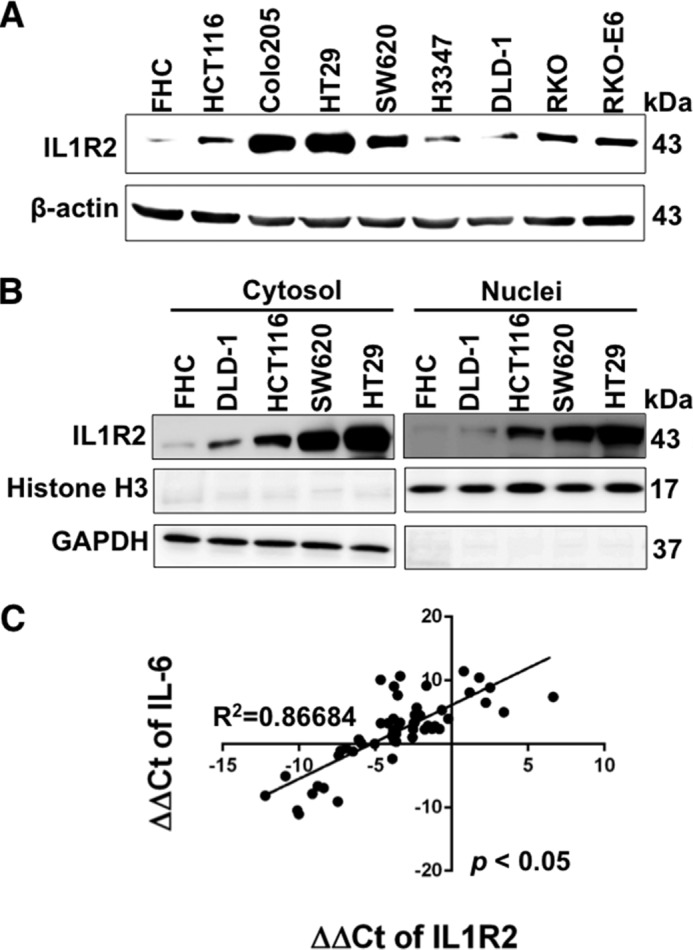
IL1R2 expression is higher in human CRC cells than normal colon epithelial cells and correlates well with IL-6 mRNA levels in tumor tissues of CRC patient. A, the IL1R2 protein levels in normal colon cells (FHC) and eight human CRC cell lines were measured by Western blot analysis. β-Actin served as a loading control. B, the nuclear and cytosolic IL1R2 in FHC and CRC cells was examined by Western blotting. GAPDH served as the loading control for the cytosolic fraction and histone H3 for the nuclear fraction. The Western blots (A and B) were independently repeated at least three times, and representative data are shown. C, correlation between the expression of IL1R2 and IL-6 mRNA (ΔΔCt indicates ΔCt of target gene − ΔCt of GAPDH) in CRC patients. The correlation between IL1R2 and IL-6 was analyzed using the χ2 test.
Intracellular IL1R2 Modulates the Expression of IL-6 and VEGF-A and CRC Cell Migration and Proliferation
Our results showing a positive association of IL1R2 with IL-6 levels in CRC tissue drew us to investigate the roles of proinflammatory IL1R2 on angiogenesis by modulating the expression of IL1R2 using shRNA-mediated knockdown and ectopic IL1R2 expression in human CRC cell lines. Because HT29 and SW620 colon cancer cells express relatively high levels of IL1R2 (Fig. 1A), we generated two stable lines, HT29-shIL1R2 and SW620-shIL1R2 cells, in which the IL1R2 protein levels were significantly reduced compared with cells transfected with the control vector (HT29-shV and SW620-shV; Fig. 2A). IL1R2 silencing resulted in reduced protein (Fig. 2A) and mRNA (data not shown) levels of IL-6 and VEGF-A and also migration (Fig. 2B). Consistently, the proliferation of the HT29-shIL1R2 and SW620-shIL1R2 was significantly reduced compared with HT29-shV and SW620-shV cells (Fig. 2C).
FIGURE 2.

Intracellular IL1R2 modulates the expression of IL-6 and VEGF-A and the migration and proliferation of CRC cells. A, the IL1R2, IL1R1, IL-6, and VEGF-A protein levels in HT29-shV, HT29-shIL1R2, SW620-shV, and SW620-shIL1R2 cells were analyzed by Western blotting. β-Actin served as a loading control. The results shown are representative of three independent experiments with similar results. B, the migration abilities of HT29-shV, HT29-shIL1R2, SW620-shV, and SW620-shIL1R2 cells were analyzed by Transwell migration assays as described under “Experimental Procedures.” C, the proliferation of HT29-shV, HT29-shIL1R2, SW620-shV, and SW620-shIL1R2 cells was analyzed by seeding 2 × 104 cells in each well of 12-well dishes and cultured for 96 h in 10% FCS-containing medium. The cell number was determined using a cell counter after staining with trypan blue. D, the IL1R2, IL-6, IL1R1, and VEGF-A protein levels in HCT116-Vec, HCT116-IL1R2, DLD-1-Vec, and DLD-1-IL1R2 cells were analyzed by Western blotting. β-Actin served as a loading control. The results shown are representative of three independent experiments with similar results. E, the migration abilities of HCT116-Vec, HCT116-IL1R2, DLD-1-Vec, and DLD-1-IL1R2 cells were determined by Transwell migration assays as described under “Experimental Procedures.” F, the proliferation of HCT116-Vec, HCT116-IL1R2, DLD-1-Vec, and DLD-1-IL1R2 cells was determined by trypan blue assay as described in C. The Western blots were independently repeated at least three times, and the representative data are shown. The proliferation and the migration assays were performed in triplicate. The data shown are means ± S.D. of three independent experiments. *, p < 0.05 by Student's t test.
In addition, we generated stable cell lines with ectopic IL1R2 expression using HCT116 and DLD-1 cells (Fig. 2D), in which the IL1R2 levels are relatively low (Fig. 1A). The IL1R2-overexpressing cell lines, HCT116-IL1R2 and DLD-1-IL1R2, showed increased IL1R2, IL-6, and VEGF-A protein levels compared with their vector controls, HCT116-Vec and DLD-1-Vec, respectively (Fig. 2D). The mRNA levels of IL-6 and VEGF-A were also increased in the HCT116-IL1R2 and DLD-1-IL1R2 cells (data not shown). As expected, ectopic expression of IL1R2 enhanced the migration ability of HCT116-IL1R2 and DLD-1-IL1R2 cells (Fig. 2E). In addition, the proliferation of the HCT116-IL1R2 and DLD-1-IL1R2 cells was significantly enhanced compared with those of the HCT116-Vec and DLD-1-Vec cells (Fig. 2F).
Because the protein levels of ILR1 were not changed in IL1R2-silenced (Fig. 2A) or -expressed (Fig. 2D) cells, we may rule out the possibility that the modulation of IL-6 and VEGF-A expression is mediated through IL1R1. Therefore, our present results strongly support that the expression levels of IL1R2 are closely associated with the expression of IL-6 and VEGF-A, migration, and proliferation of human CRC cell lines
The AP-1 Transcription Factor Is Involved in the IL1R2-dependent Increase in IL-6 and VEGF-A Promoter Activity
To understand how IL1R2 activates IL-6 and VEGF-A expression, we constructed luciferase reporters driven by the IL-6 (−710 to + 30 bp) and VEGF-A (−1632 to −12 bp) promoters. These reporters were transfected into various cell lines derived from HCT116 and HT29 cells. The HT29-shIL1R2 cells exhibited lower IL-6 and VEGF-A promoter activity compared with the HT29-shV cells, whereas the HCT116-IL1R2 cells had higher IL-6 and VEGF-A promoter activity than the HCT116-Vec cells (Fig. 3A). These results suggest that IL1R2 is involved in the activation of the IL-6 and VEGF-A promoters.
FIGURE 3.

The AP-1 transcription factor is involved in the IL1R2-dependent increase in IL-6 and VEGF-A promoter activity. A, HT29-shV, HT29-shIL1R2, HCT116-Vec, and HCT116-IL1R2 cells were co-transfected with plasmids containing the firefly luciferase gene driven by the IL-6 or VEGF-A promoter and a Renilla control vector for 24 h. The activities of the IL-6 and VEGF-A promoters were measured as described under “Experimental Procedures.” B, plasmids containing the luciferase reporter driven by serially truncated IL-6 promoters were transiently co-transfected with a Renilla control vector into HT29-shV, HT29-shIL1R2, HCT116-Vec, and HCT116-IL1R2 cells. Dual luciferase assays were performed 24 h post-transfection. C and D, HT29-shV, HT29-shIL1R2, HCT116-Vec, and HCT116-IL1R2 cells were co-transfected with a Renilla control vector and plasmids containing the firefly luciferase gene driven by an AP-1 binding site-mutated IL-6 promoter (C) and VEGF-A promoter mutated at −1527∼−1521 (D, left panels) or mutated at −621∼−615 (D, right panels). The promoter activities of the mutated IL-6 and VEGF-A promoters were determined 24 h post-transfection. The data of relative promoter activity are means ± S.D. of three independent experiments. *, p < 0.05 by Student's t test.
To identify the transcription factors involved in the IL1R2-dependent enhancement of IL-6 promoter activity, a series of truncated IL-6 promoter segments (−710, −287, −206, and −123 bp from the start codon) were placed upstream of the luciferase gene, and these constructs were transfected into HT29-shV, HT29-shIL1R2, HCT116-Vec, and HCT116-IL1R2 cells. As shown in Fig. 3B, the cells expressing higher IL1R2 levels (HCT116-IL1R2 and HT29-shV) exhibited higher IL-6 promoter activity when transfected with constructs starting from −710, −287, and −206 to +30. However, the IL-6 promoter was inactivated when it was truncated at −123, indicating that an essential element for IL-6 promoter activity exists between −206 to −123. Because an activator protein (AP)-1 binding site was located in the −124 to −118 bp region (TGAGTCT) of the IL-6 promoter, we further examined whether the AP-1 transcription factor was involved in the IL1R2-induced activation of the IL-6 by constructing mutated AP-1 (TTCTTCT) reporters. As shown in Fig. 3C, the activity of the mutated IL-6 promoter was significantly reduced in the HT29-shIL1R2 cells. Consistent with this observation, the AP-1-mutated IL-6 promoter was also inactivated in the HCT116-IL1R2 cells (Fig. 3C). Intriguingly, there are two AP-1 binding sites identified in the −621 to −615 bp and −1527 to −1521 bp regions of the VEGF-A promoter. Mutation of the AP-1 binding site in the −621 and −615 bp (TGAGTGA to TGCTGGA) region of the VEGF-A promoter did not affect its activity (Fig. 3D, left panels), whereas mutation in the −1527 to −1521 bp region (TGAGTGA to TGCTGGA) of the VEGF-A promoter abolished the IL1R2-dependent promoter activity (Fig. 3D, right panels). These results reveal that AP-1 transcription factor is essential for increased expression of IL-6 and VEGF-A in IL1R2-overexpressing cells.
IL1R2 Forms a Complex with c-Fos and Activates the IL-6 and VEGF-A Promoters
The AP-1 complex is composed of c-Fos and c-Jun. As shown in Fig. 4A, we observed that the IL1R2 level was positively associated with c-Fos and phosphorylated c-Fos (at Ser 237) but not c-Jun and phosphorylated c-Jun. In addition, the siRNA-mediated silencing of c-Fos further decreased mRNA and protein expression of IL-6 and VEGF-A in HT29-shV and HT29-shIL1R2 cells (Fig. 4B), as well as HCT116-IL1R2 cells (Fig. 4C). These results suggest that c-Fos is likely involved in the activation of IL-6 and VEGF-A transcription in IL1R2-overexpressing cells.
FIGURE 4.

IL1R2 activates c-Fos to enhance the expression of IL-6 and VEGF-A. A, the p-c-Fos (Ser 237), c-Fos, p-c-Jun (Ser-63), and c-Jun protein levels in HT29-shV, HT29-shIL1R2, HCT116-Vec, and HCT116-IL1R2 cells were analyzed by Western blotting. β-Actin served as a loading control. B and C, the mRNA and protein levels of c-Fos, IL-6, and VEGF-A were determined using qPCR and Western blot analysis, respectively, in HT29-shV and HT29-shIL1R2 cells (B) as well as in HCT116-Vec and HCT116-IL1R2-2 cells (C) after c-Fos was knocked down by siRNA transfection. The Western blots were independently repeated at least three times, and the representative data are shown. The data of mRNA levels are means ± S.D. of three independent experiments. *, p < 0.05 by Student's t test.
We therefore performed reciprocal immunoprecipitation-Western blot assays to examine whether IL1R2 interacts with c-Fos. As shown in Fig. 5A, IL1R2 formed a complex with c-Fos in HCT116 and HT29 cells, and their interaction correlated well with IL1R2 levels in cells. To determine the possible domain of IL1R2 interacting with c-Fos, we constructed full-length Myc-tagged and truncated IL1R2 (extracellular domain (1–343) and cytosolic domain (371–398)) and separately transfected them into HCT116 cells. The immunoprecipitation analysis using anti-Myc tag antibody and Western blotting with anti-c-Fos antibody showed that c-Fos failed to interact with IL1R21–343 containing only extracellular domain (Fig. 5B) but interacted with full-length IL1R2 and IL1R2371–398, indicating that the cytosolic domain of IL1R2 is indispensable for its interaction with c-Fos. Intriguingly, ectopic expression of the IL1R2 intracellular domain, IL1R2371–398, was sufficient to enhance the expression of IL-6 and VEGF-A, whereas the extracellular domain of IL1R2 was unable to increase the protein levels of IL-6 and VEGF-A (Fig. 5C). Taken together, the interaction of the intracellular domain of IL1R2 with c-Fos is crucial for IL1R2-enhanced IL-6 and VEGF-A expression.
FIGURE 5.

IL1R2 forms a complex with c-Fos and activates the IL-6 and VEGF-A promoters. A, total lysates of HCT116-Vec and HCT116-IL1R2 cells (left column), as well as HT29-shV and HT29-shIL1R2 cells (right column), were immunoprecipitated using 5 μg of anti-c-Fos (upper panel) or anti-IL1R2 antibodies (lower panel), and Western blotting was performed using the indicated antibodies. B, the extracts of HCT116 cells that had expressed Myc-tagged full-length IL1R2 or its truncated mutant were subjected to immunoprecipitation with 5 μg of anti-Myc tag antibody followed by Western blot with anti-c-Fos or anti-Myc tag antibody. C, the protein lysates of HCT116 cells expressing Myc-tagged full-length IL1R2 or its truncated mutant were harvested and subjected for Western blotting using the indicated antibodies. D, ChIP was performed as follows. Crude lysates from HT29-shV, HT29-shIL1R2, HCT116-Vec, and HCT116-IL1R2 cells were immunoprecipitated using 5 μg of anti-c-Fos, anti-IL1R2, or rabbit IgG and then captured with protein A-agarose beads. The precipitated DNA samples were amplified by PCR to detect a fragment in the IL-6 (upper panel) or VEGF-A (lower panel) promoter containing the AP-1 binding site. The results shown are representative of three independent experiments with similar results. IB, immunoblot; IP, immunoprecipitation.
We further performed ChIP assays to examine whether these proteins modulate the transcription of IL-6 and VEGF-A. As shown in Fig. 5D (upper panel), the binding of c-Fos and IL1R2 to the IL-6 promoter was remarkably suppressed in IL1R2 knockdown HT29 cells, but this binding was elevated in IL1R2-overexpressing HCT116 cells. Similarly, we also observed the binding of IL1R2 to the AP-1 binding sites in VEGF-A promoters (Fig. 5D, lower panel). These results demonstrate that IL1R2 works together with c-Fos to activate the IL-6 and VEGF-A promoters and hence enhances their expression.
Conditioned Medium Harvested from IL1R2-overexpressing Cells is Enriched with IL-6 and VEGF
Because IL-6 and VEGF-A are crucial angiogenic factors, we further evaluated the role of IL1R2 on CRC progression. Because IL-6 and VEGF secreted by tumor cells are critical for angiogenesis (36), we first performed ELISAs to determine the protein levels of IL-6 and VEGF-A in the CM of the aforementioned cell lines. As shown in Fig. 6A, the levels of secreted IL-6 and VEGF-A were reduced in the CM harvested from the IL1R2-silenced HT29-shIL1R2 cells but significantly increased in that from the HCT116-IL1R2 cells ectopically expressing IL1R2 compared with their vector control cells.
FIGURE 6.

CM harvested from IL1R2-overexpressing cells is enriched with IL-6 and VEGF. A, ELISA was used to measure the IL-6 and VEGF-A levels in the serum-free CM harvested from CRC cells with modulated IL1R2 expression. The protein levels are means ± S.D. of three independent experiments. *, p < 0.05 by Student's t test. B and C, the proliferation (B) and migration (C) of EA.hy926 cells were analyzed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (48 h, 10% FCS-containing CM) and Transwell migration assays (8 h, serum-free CM in the upper chamber and 5% FCS-containing medium in the lower chamber), respectively, after incubation in CM (with or without anti-IL-6 (2 μg/ml), anti-VEGF-A (2 μg/ml), or rabbit IgG control (2 μg/ml)) harvested from HT29-shV, HT29-shIL1R2, HCT116-Vec, and HCT116-IL1R2 cells. The data of relative proliferation and migration ability are means ± S.D. of three independent experiments. *, p < 0.05 by Student's t test.
To analyze the effects of CM on the growth and migration of endothelial cells, we performed proliferation and Transwell migration assays using human endothelial cells (EA.hy926). We observed enhanced proliferation (Fig. 6B) and migration (Fig. 6C) of EAhy926 cells when they were cultured in CM harvested from CRC cells with increased expression of IL1R2 (HT29-shV and HCT116-IL1R2). Furthermore, we observed that the proliferation and migration activities of the EA.hy926 cells were significantly decreased by the addition of neutralizing antibodies against IL-6 or VEGF-A to the CM harvested from the HT29-shV and HCT116-IL1R2 cells (Fig. 6, B and C). These results demonstrate that the IL1R2-dependent increase in the secretion of IL-6 and VEGF-A enhances the proliferation and migration of EA.hy926 cells.
Conditioned Medium Harvested from IL1R2-overexpressing Cells Enhances Angiogenicity of Endothelial Cells
To confirm that the CM harvested from CRC cells with enhanced IL1R2 expression promotes endothelial cell angiogenesis, we performed a tube formation assay by culturing EA.hy926 cells with serum-free CM harvested from HCT116-Vec and HCT116-IL1R2 cells and supplemented with 1% fetal calf serum for 24 h. As shown in Fig. 7A, tube formation by the EAhy926 cells was significantly increased by the addition of CM from the HCT116-IL1R2 cells. Furthermore, increased tube formation by the endothelial cells was abolished by the addition of neutralizing antibodies against IL-6 or VEGF-A to the CM harvested from the HCT116-IL1R2 cells (Fig. 7A).
FIGURE 7.

CM harvested from IL1R2-overexpressing cells enhance angiogenicity of endothelial cells. A, CM (1% FCS) was harvested from HCT116-CMV22 and HCT116-IL1R2 cells. EA.hy926 cells (5 × 104) in CM with or without rabbit IgG (2 μg/ml), anti-IL-6 (2 μg/ml), or anti-VEGF-A (2 μg/ml) were seeded in 96-well plates containing Matrigel, and their tube formation ability was examined after 24 h of incubation. The relative tube formation is the mean ± S.D. of three independent experiments. *, p < 0.05 by Student's t test. B, serum-free CM harvested from IL1R2-overexpressing cells enhanced angiogenesis in the DIVAA model. For DIVAA, mice were implanted with silicone tubes containing Matrigel and supplemented with angiogenic factors (100 ng of VEGF and FGF) as positive control or CM (with or without rabbit IgG, anti-IL-6, or anti-VEGF-A antibodies) harvested from HT29-shV, HT29-shIL1R2, HCT116-Vec, and HCT116-IL1R2 cells (n = 6 for each group). Neovascularization in the angioreactors was quantified using a spectrofluorometer 17 days after implantation. The relative invasion is the mean ± S.D. *, p < 0.05 by Student's t test. C, xenograft growth of HT29-shV, HT29-shIL1R2, HCT116-Vec, and HCT116-IL1R2 cells was evaluated in nude mice. Two million cells were subcutaneously inoculated into nude mice (n = 6 for each group). The tumor size (mean ± S.D.) was measured every 3 days. Two-way analysis of variance was performed by GraphPad Prism. **, p < 0.01. D, blood vessels in tumor were visualized by immunohistochemical staining with an antibody against CD31. Tumors formed by HT29-shV, HT29-shIL1R2, HCT116-Vec, and HCT116-IL1R2 cells were fixed, sectioned, and stained. CD31 staining is indicated in brown, and cell nuclei are blue. Total DAB staining was quantified by pixel density analysis using ImageJ software. The data are the averages of six tumors obtained from Fig. 7C (three locations were captured randomly in each tumor). E, the tumor growth of HT29-shV and HT29-shIL1R2 cells in nude mice (n = 5 for each group) was suppressed by intratumoral injection of an IL-6-neutralizing antibody or a VEGF-A-neutralizing antibody (50 μg, twice a week). The IgG antibody was used as control. The data are means ± S.D. Two-way analysis of variance was performed by GraphPad Prism. ***, p < 0.001.
We then performed DIVAA to verify that the CM harvested from CRC cells with enhanced IL1R2 expression stimulates angiogenesis in vivo. As shown in Fig. 7B, the angioreactor containing CM from HT29-shIL1R2 cells had significantly fewer microvascular structures than the angioreactor containing CM from HT29-shV cells. In addition, enhanced microvascular structure formation was suppressed by the addition of neutralizing antibodies against IL-6 or VEGF-A. Furthermore, the CM from the HCT116-IL1R2 cells facilitated the formation of microvascular structures in the angioreactors (Fig. 7B). We also consistently observed that HT29-shIL1R2 xenografts grew significantly slower than HT29-shV xenografts (Fig. 7C), whereas the growth of HCT116-IL1R2 xenografts was significantly faster than that of HCT116-Vec cells (Fig. 7C).
Using immunohistochemical analysis of CD31 expression, we observed that tumors derived from HT29-shV cells had increased microvessel density compared with tumors derived from HT29-shIL1R2 cells, whereas tumors derived from HCT116-IL1R2 cells showed a higher density of microvessels compared with tumors derived from HCT116-Vec cells (Fig. 7D). To further investigate whether IL-6/VEGF-A signaling elevated by IL1R2 was involved in tumor growth in mice, we injected an IL-6 or VEGF-A antibody into HT29-shV tumors. As shown in Fig. 7E, the IL-6 and the VEGF-A antibody significantly decreased the tumor volume of HT29-shV xenografts. Our results clearly demonstrated that intracellular IL1R2 modulates the angiogenesis and growth of human CRC cells through transcriptionally activating the levels of IL-6 and VEGF-A.
IL1R2 Enhances the Expression of IL-6 and VEGF-A and Angiogenic Activity of Keratinocytes
In addition to CRC cells, we noticed that highly tumorigenic human keratinocyte T4R2 cells expressed the highest levels of IL1R2, IL-6, and VEGF-A among the studied HaCaT cell lines (Fig. 8A). Using similar approaches, we silenced the expression of IL1R2 in T4R2 cells and ectopically expressed IL1R2 in the parental HaCaT cells. Our results supported the idea that IL1R2 plays crucial roles in influencing IL-6 and VEGF-A expression (Fig. 8B), which were essential for enhancing angiogenesis using DIVAA assay (Fig. 8C). We also confirmed that the AP-1 site on the promoters of IL-6 and VEGF-A was required for IL1R2-mediated IL-6 and VEGF-A expression (Fig. 8, D and E). Therefore, the novel functions of IL1R2 were observed not only in CRC cells but also in human keratinocytes.
FIGURE 8.
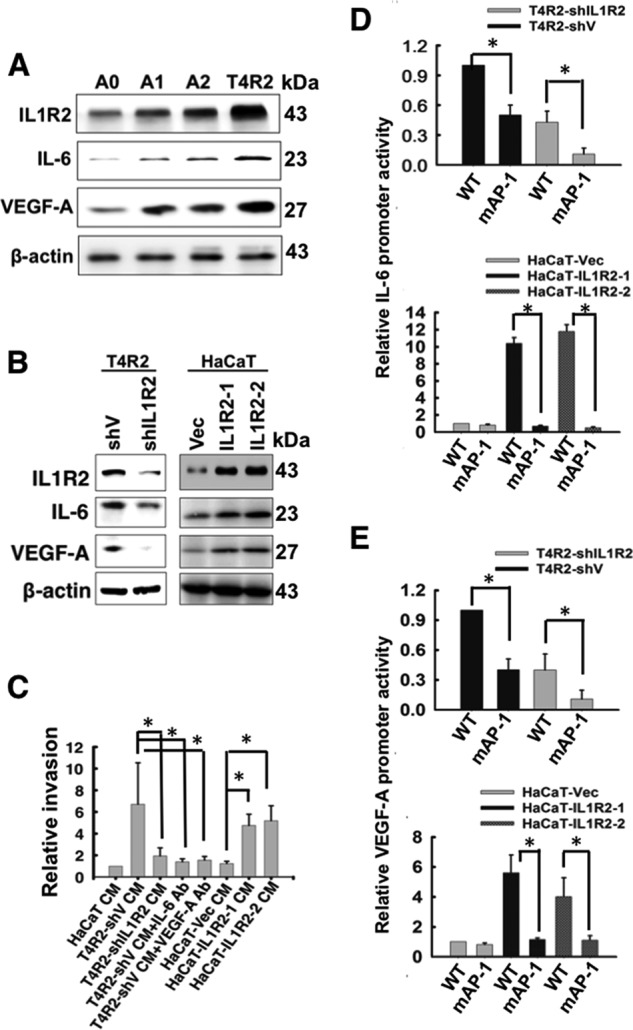
The roles of IL1R2, IL-6, and VEGF-A in A0, A2, and T4R2 cells. A, the protein levels of IL1R2, IL-6, and VEGF-A in A0, A1, A2, and T4R2 cells were analyzed by Western blotting. β-Actin served as a loading control. B, the protein levels of IL1R2, IL-6, and VEGF-A in T4R2-shV, T4R2-shIL1R2, HaCaT-Vec, HaCaT-IL1R2-1, and HaCaT-IL1R2-2 cells were analyzed by Western blotting. C, DIVAA assay was performed as described for Fig. 7B. The CM was harvested from T4R2-shV, T4R2-shIL1R2, HaCaT-Vec, HaCaT-IL1R2-1, and HaCaT-IL1R2-2 cells. D and E, a luciferase assay was used to analyze the activity of wild-type or mutant IL-6 and VEGF-A promoter in T4R2-shV and T4R2-shIL1R2 cells (D) and HaCaT-Vec, HaCaT-IL1R2-1 and HaCaT-IL1R2-2 cells (E). The Western blots were independently repeated at least three times, and the representative data are shown. The relative invasion and promoter activity are means ± S.D. of three independent experiments. *, p < 0.05 by Student's t test.
Discussion
Although IL1R2 acting as a decoy receptor of the IL-1 system (37) might block the signaling of exogenous IL-1 (17), its intracellular activities cannot be overlooked. In this study, we observed a significant correlation between IL1R2 and IL-6 in CRC patients, implying that IL1R2 influences the development of human CRC by affecting key inflammatory and angiogenic factors. Thus, IL1R2 may potentially serve as a novel prognostic and therapeutic target for CRC patients. Further clinical investigation is underway.
A recent study showed that IL1R2 is potentially involved in regulating the transcription of the precursor form of interleukin 1α (pIL-1α) in fibroblasts derived from systemic sclerosis patients (38). In our previous study, we demonstrated that ectopic expression of IL1R2 may activate pIL-1α signaling to enhance IL-6 expression (17). However, the mechanism of IL1R2-mediated pIL-1α activation is not clear. In this study, we demonstrated that the AP-1 binding element on the promoters of IL-6 and VEGF-A is an essential regulatory element responsible for the transcriptional activation of IL-6/VEGF-A by IL1R2. Our present results found that a faction of IL1R2 was localized in the nuclei. We also showed that IL1R2 interacts with and activates c-Fos and binds to the AP-1 segment of IL-6 and VEGF-A promoters. Therefore, this study provides evidence that IL1R2 likely acts as a transcriptional co-activator of c-Fos. Because both IL1R2 and c-Fos bind to IL-6 and VEGF-A promoters, we postulate that IL1R2 and c-Fos may work together to transcriptionally activate IL-6 and VEGF-A genes.
Several c-Fos-interacting proteins have been reported in the literature. For example, c-Fos interacts with CBFA1 to activate the collagenase-3 promoter (39), and the nuclear import of HBXIP (hepatitis B X-interacting protein) relies on the interaction between HBXIP and c-Fos and the phosphorylation of these two proteins in breast cancer cells (40). However, the interaction between c-Fos and these proteins, including IL1R2, and their involvement in transcriptional activation, remain obscure. Our results showing that enhanced IL1R2 expression is associated with increased phosphorylated c-Fos at Ser-237 suggest that the interaction of C-terminal intracellular domain (residues 371–398) of IL1R2 with c-Fos could facilitate c-Fos activation. This novel biological function of IL1R2 also implies that IL1R2 may exert its oncogenic activity via collaboration with c-Fos. However, further investigation is required to understand how IL1R2 interacts with and enhances the transcriptional activity of c-Fos.
Tumor promotion of inflammation and angiogenesis are essential characteristics enabling tumor growth and metastasis (1). IL-6 is involved in a wide spectrum of cellular activities, including cancer cell proliferation (41), cancer apoptosis inhibition (41), and cancer inflammation (42). It has been reported that 60% of colorectal cancer specimens overexpress IL-6 (43). VEGF-A, a crucial factor for angiogenesis (44), is also recognized as a critical indication for the malignancy of human CRC (45–47). However, how IL-6 and VEGF are enhanced in CRC is still unknown. Our present results clearly demonstrated that IL1R2 is a positive regulatory factor affecting the expression of IL-6 and VEGF-A. Furthermore, we show that IL1R2 may act through secreted IL-6 and VEGF-A to activate the stromal populations, particularly to promote angiogenesis. This report is the first to show that IL1R2, a decoy receptor, is actively involved in angiogenesis, which is essential for tumor progression and metastasis (18). Therefore, the proinflammatory activity and oncogenic potential of intracellular IL1R2 warrant attention.
How IL1R2 is activated is currently unclear. IL1R2 was recently identified as a downstream transcriptional target of hedgehog signaling (48). The persistent activation of the Hedgehog signaling pathway is implicated in the development of several human cancers, including basal cell carcinoma (49). In addition, several studies have shown that the signal peptide and flanking amino acid sequences, cell type, and growth conditions influence a signal peptide-containing protein to be nontargeted (50). Therefore, further investigation is required to understand how intracellular IL1R2 is activated in CRC cells.
IL1R2 is mainly involved in inflammation-related diseases. For instance, the hypomethylation of the IL1R2 gene serves as a potential biomarker of autoimmune disease (51), and reduced IL1R2 expression is observed in atherosclerotic lesions compared with normal arteries (52). To our knowledge, this study is the first to report that intracellular IL1R2 is associated with angiogenesis of human CRC. In CRC tissue, accumulated genetic alterations have been found to cause DNA repair defects, enhance growth factor pathways, and activate the tumor microenvironment during CRC progression (53). Although CRC patients at the early stages are curable with recent improvements in therapy, the treatment of patients with late stage CRC remains a challenge (54). The further understanding of molecular mechanisms underlying CRC progression is of clinical importance to develop effective strategies and improve prognoses.
Because IL1R2 is a decoy receptor, it has been well recognized that IL1R2 acts as a negative regulator of exogenous IL-1 signaling (55). However, our results demonstrate the novel function of intracellular IL1R2 as a positive regulator of endogenous signaling that activates the synthesis of IL-6/VEGF-A, which are important proinflammatory cytokines and angiogenic factors. Importantly, the proinflammatory activity of IL1R2 was not restricted to CRC cells but was also identified in tumorigenic human keratinocytes. Because enhanced IL1R2 expression has been observed in a variety of cancers, we may infer the vital functions of IL1R2 on driving tumor progression. Together, these results suggest that IL1R2 is a potential target for cancer prevention, because targeting cancer angiogenesis is an important cancer therapeutic strategy.
Author Contributions
A.-C. M. and T.-C. L. designed the research; A.-C. M., H.-J. L., and C.-W. C. performed the experiments; J.-J. C. contributed analytical tools; C.-C. H., Y.-S. H., and J.-K. J. provided the clinical samples; and A.-C. M. and T.-C. L. analyzed the data and wrote the manuscript. All authors read and approved the manuscript.
Acknowledgment
We thank Prof. Yu Su (Institute of Biopharmaceutical Science, National Yang-Ming University) for valuable suggestions regarding our manuscript.
This work was supported by the Academia Sinica and by Taiwan National Science Council Grants NSC-97-2314-B-0010MY3 and MOST-104-2320-B-001-008-MY3 (to T.-C. L.). The authors declare that they have no conflicts of interest with the contents of this article.
- CRC
- colorectal cancer
- IL1R1
- IL-1 receptor type 1
- IL1R2
- IL-1 receptor type 2
- AP-1
- activator protein-1
- CM
- conditioned medium
- DIVAA
- directed in vivo angiogenesis assay
- qPCR
- quantitative real time PCR.
References
- 1. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 2. Nishida N., Yano H., Nishida T., Kamura T., Kojiro M. (2006) Angiogenesis in cancer. Vasc. Health Risk Manag. 2, 213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jackson J. R., Seed M. P., Kircher C. H., Willoughby D. A., Winkler J. D. (1997) The codependence of angiogenesis and chronic inflammation. FASEB J. 11, 457–465 [PubMed] [Google Scholar]
- 4. Landskron G., De la Fuente M., Thuwajit P., Thuwajit C., Hermoso M. A. (2014) Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eldesoky A., Shouma A., Mosaad Y., Elhawary A. (2011) Clinical relevance of serum vascular endothelial growth factor and interleukin-6 in patients with colorectal cancer. Saudi J. Gastroenterol. 17, 170–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fan Y., Ye J., Shen F., Zhu Y., Yeghiazarians Y., Zhu W., Chen Y., Lawton M. T., Young W. L., Yang G. Y. (2008) Interleukin-6 stimulates circulating blood-derived endothelial progenitor cell angiogenesis in vitro. J. Cereb. Blood Flow Metab. 28, 90–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang S. P., Wu M. S., Shun C. T., Wang H. P., Lin M. T., Kuo M. L., Lin J. T. (2004) Interleukin-6 increases vascular endothelial growth factor and angiogenesis in gastric carcinoma. J. Biomed. Sci. 11, 517–527 [DOI] [PubMed] [Google Scholar]
- 8. Cohen T., Nahari D., Cerem L. W., Neufeld G., Levi B. Z. (1996) Interleukin 6 induces the expression of vascular endothelial growth factor. J. Biol. Chem. 271, 736–741 [DOI] [PubMed] [Google Scholar]
- 9. Hoeben A., Landuyt B., Highley M. S., Wildiers H., Van Oosterom A. T., De Bruijn E. A. (2004) Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 56, 549–580 [DOI] [PubMed] [Google Scholar]
- 10. Wei L. H., Kuo M. L., Chen C. A., Chou C. H., Lai K. B., Lee C. N., Hsieh C. Y. (2003) Interleukin-6 promotes cervical tumor growth by V. E., G. F.-dependent angiogenesis via a STAT3 pathway. Oncogene. 22, 1517–1527 [DOI] [PubMed] [Google Scholar]
- 11. Nagasaki T., Hara M., Nakanishi H., Takahashi H., Sato M., Takeyama H. (2014) Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br. J. Cancer 110, 469–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gabay C., Lamacchia C., Palmer G. (2010) IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 6, 232–241 [DOI] [PubMed] [Google Scholar]
- 13. Apte R. N., Dotan S., Elkabets M., White M. R., Reich E., Carmi Y., Song X., Dvozkin T., Krelin Y., Voronov E. (2006) The involvement of, I. L.-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 25, 387–408 [DOI] [PubMed] [Google Scholar]
- 14. Sims J. E., Acres R. B., Grubin C. E., McMahan C. J., Wignall J. M., March C. J., Dower S. K. (1989) Cloning the interleukin 1 receptor from human T cells. Proc. Natl. Acad. Sci. U.S.A. 86, 8946–8950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McMahan C. J., Slack J. L., Mosley B., Cosman D., Lupton S. D., Brunton L. L., Grubin C. E., Wignall J. M., Jenkins N. A., Brannan C. I. (1991) A novel IL-1 receptor, cloned from B cells by mammalian expression, is expressed in many cell types. EMBO J. 10, 2821–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mantovani A., Locati M., Vecchi A., Sozzani S., Allavena P. (2001) Decoy receptors: a strategy to regulate inflammatory cytokines and chemokines. Trends Immunol. 22, 328–336 [DOI] [PubMed] [Google Scholar]
- 17. Chang S. Y., Su P. F., Lee T. C. (2009) Ectopic expression of interleukin-1 receptor type II enhances cell migration through activation of the pre-interleukin 1α pathway. Cytokine 45, 32–38 [DOI] [PubMed] [Google Scholar]
- 18. Hanahan D., Folkman J. (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86, 353–364 [DOI] [PubMed] [Google Scholar]
- 19. Groves R. W., Giri J., Sims J., Dower S. K., Kupper T. S. (1995) Inducible expression of type 2 IL-1 receptors by cultured human keratinocytes: implications for IL-1-mediated processes in epidermis. J. Immunol. 154, 4065–4072 [PubMed] [Google Scholar]
- 20. Kawaguchi Y., Nishimagi E., Tochimoto A., Kawamoto M., Katsumata Y., Soejima M., Kanno T., Kamatani N., Hara M. (2006) Intracellular IL-1α-binding proteins contribute to biological functions of endogenous IL-1α in systemic sclerosis fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 103, 14501–14506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aceto N., Duss S., MacDonald G., Meyer D. S., Roloff T. C., Hynes N. E., Bentires-Alj M. (2012) Co-expression of HER2 and HER3 receptor tyrosine kinases enhances invasion of breast cells via stimulation of interleukin-8 autocrine secretion. Breast Cancer Res. 14, R131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ma X. J., Wang Z., Ryan P. D., Isakoff S. J., Barmettler A., Fuller A., Muir B., Mohapatra G., Salunga R., Tuggle J. T., Tran Y., Tran D., Tassin A., Amon P., Wang W., Wang W., Enright E., Stecker K., Estepa-Sabal E., Smith B., Younger J., Balis U., Michaelson J., Bhan A., Habin K., Baer T. M., Brugge J., Haber D. A., Erlander M. G., Sgroi D. C. (2004) A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell 5, 607–616 [DOI] [PubMed] [Google Scholar]
- 23. Kondera-Anasz Z., Mielczarek-Palacz A., Switala J. (2003) [Significantly increased interleukin-1A and interleukin-1 soluble type II receptor levels in women with ovarian cancer]. Ginekol. Pol. 74, 761–766 [PubMed] [Google Scholar]
- 24. Laios A., O'Toole S. A., Flavin R., Martin C., Ring M., Gleeson N., D'Arcy T., McGuinness E. P., Sheils O., Sheppard B. L., O'Leary J. J. (2008) An integrative model for recurrence in ovarian cancer. Mol. Cancer 7, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rückert F., Dawelbait G., Winter C., Hartmann A., Denz A., Ammerpohl O., Schroeder M., Schackert H. K., Sipos B., Klöppel G., Kalthoff H., Saeger H. D., Pilarsky C., Grützmann R. (2010) Examination of apoptosis signaling in pancreatic cancer by computational signal transduction analysis. PLoS One 5, e12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin K. Y., Lu D., Hung C. F., Peng S., Huang L., Jie C., Murillo F., Rowley J., Tsai Y. C., He L., Kim D. J., Jaffee E., Pardoll D., Wu T. C. (2007) Ectopic expression of vascular cell adhesion molecule-1 as a new mechanism for tumor immune evasion. Cancer Res. 67, 1832–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Buys T. P., Chari R., Lee E. H., Zhang M., MacAulay C., Lam S., Lam W. L., Ling V. (2007) Genetic changes in the evolution of multidrug resistance for cultured human ovarian cancer cells. Genes Chromosomes Cancer 46, 1069–1079 [DOI] [PubMed] [Google Scholar]
- 28. Su P. F., Hu Y. J., Ho I. C., Cheng Y. M., Lee T. C. (2006) Distinct gene expression profiles in immortalized human urothelial cells exposed to inorganic arsenite and its methylated trivalent metabolites. Environ. Health Perspect. 114, 394–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brenner H., Kloor M., Pox C. P. (2014) Colorectal cancer. Lancet 383, 1490–1502 [DOI] [PubMed] [Google Scholar]
- 30. Chien C. W., Chiang M. C., Ho I. C., Lee T. C. (2004) Association of chromosomal alterations with arsenite-induced tumorigenicity of human HaCaT keratinocytes in nude mice. Environ. Health Perspect. 112, 1704–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Burnette W. N. (1981) “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem. 112, 195–203 [DOI] [PubMed] [Google Scholar]
- 32. Ponchel F., Toomes C., Bransfield K., Leong F. T., Douglas S. H., Field S. L., Bell S. M., Combaret V., Puisieux A., Mighell A. J., Robinson P. A., Inglehearn C. F., Isaacs J. D., Markham A. F. (2003) Real-time PCR based on SYBR-Green I fluorescence: an alternative to the TaqMan assay for a relative quantification of gene rearrangements, gene amplifications and micro gene deletions. BMC Biotechnol. 3, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alley M. C., Scudiero D. A., Monks A., Hursey M. L., Czerwinski M. J., Fine D. L., Abbott B. J., Mayo J. G., Shoemaker R. H., Boyd M. R. (1988) Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 48, 589–601 [PubMed] [Google Scholar]
- 34. Nakagawa K., Shibata A., Yamashita S., Tsuzuki T., Kariya J., Oikawa S., Miyazawa T. (2007) In vivo angiogenesis is suppressed by unsaturated vitamin E, tocotrienol. J. Nutr. 137, 1938–1943 [DOI] [PubMed] [Google Scholar]
- 35. Chen Y. J., Lin Y. P., Chow L. P., Lee T. C. (2011) Proteomic identification of Hsp70 as a new Plk1 substrate in arsenic trioxide-induced mitotically arrested cells. Proteomics 11, 4331–4345 [DOI] [PubMed] [Google Scholar]
- 36. Goto H., Yano S., Matsumori Y., Ogawa H., Blakey D. C., Sone S. (2004) Sensitization of tumor-associated endothelial cell apoptosis by the novel vascular-targeting agent ZD6126 in combination with cisplatin. Clin. Cancer Res. 10, 7671–7676 [DOI] [PubMed] [Google Scholar]
- 37. Rauschmayr T., Groves R. W., Kupper T. S. (1997) Keratinocyte expression of the type 2 interleukin 1 receptor mediates local and specific inhibition of interleukin 1-mediated inflammation. Proc. Natl. Acad. Sci. U.S.A. 94, 5814–5819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ramadas R. A., Li X., Shubitowski D. M., Samineni S., Wills-Karp M., Ewart S. L. (2006) IL-1 Receptor antagonist as a positional candidate gene in a murine model of allergic asthma. Immunogenetics 58, 851–855 [DOI] [PubMed] [Google Scholar]
- 39. D'Alonzo R. C., Selvamurugan N., Karsenty G., Partridge N. C. (2002) Physical interaction of the activator protein-1 factors c-Fos and c-Jun with Cbfa1 for collagenase-3 promoter activation. J. Biol. Chem. 277, 816–822 [DOI] [PubMed] [Google Scholar]
- 40. Zhang Y., Zhao Y., Li H., Li Y., Cai X., Shen Y., Shi H., Li L., Liu Q., Zhang X., Ye L. (2013) The nuclear import of oncoprotein hepatitis B X-interacting protein depends on interacting with c-Fos and phosphorylation of both proteins in breast cancer cells. J. Biol. Chem. 288, 18961–18974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guo Y., Xu F., Lu T., Duan Z., Zhang Z. (2012) Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev. 38, 904–910 [DOI] [PubMed] [Google Scholar]
- 42. Hodge D. R., Hurt E. M., Farrar W. L. (2005) The role of IL-6 and STAT3 in inflammation and cancer. Eur. J. Cancer 41, 2502–2512 [DOI] [PubMed] [Google Scholar]
- 43. Chung Y. C., Chaen Y. L., Hsu C. P. (2006) Clinical significance of tissue expression of interleukin-6 in colorectal carcinoma. Anticancer Res. 26, 3905–3911 [PubMed] [Google Scholar]
- 44. Claesson-Welsh L., Welsh M. (2013) VEGFA and tumour angiogenesis. J. Intern. Med. 273, 114–127 [DOI] [PubMed] [Google Scholar]
- 45. Murukesh N., Dive C., Jayson G. C. (2010) Biomarkers of angiogenesis and their role in the development of VEGF inhibitors. Br. J. Cancer. 102, 8–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Duda D. G. (2012) Molecular biomarkers of response to antiangiogenic therapy for cancer. ISRN Cell Biol. 2012, 587259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Martinetti A., Miceli R., Sottotetti E., Di Bartolomeo M., de Braud F., Gevorgyan A., Dotti K. F., Bajetta E., Campiglio M., Bianchi F., Bregni G., Pietrantonio F. (2014) Circulating biomarkers in advanced colorectal cancer patients randomly assigned to three bevacizumab-based regimens. Cancers (Basel) 6, 1753–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kasper M., Schnidar H., Neill G. W., Hanneder M., Klingler S., Blaas L., Schmid C., Hauser-Kronberger C., Regl G., Philpott M. P., Aberger F. (2006) Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol. Cell. Biol. 26, 6283–6298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schnidar H., Eberl M., Klingler S., Mangelberger D., Kasper M., Hauser-Kronberger C., Regl G., Kroismayr R., Moriggl R., Sibilia M., Aberger F. (2009) Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 69, 1284–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zheng Y., Humphry M., Maguire J. J., Bennett M. R., Clarke M. C. (2013) Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1α, controlling necrosis-induced sterile inflammation. Immunity 38, 285–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin S. Y., Hsieh S. C., Lin Y. C., Lee C. N., Tsai M. H., Lai L. C., Chuang E. Y., Chen P. C., Hung C. C., Chen L. Y., Hsieh W. S., Niu D. M., Su Y. N., Ho H. N. (2012) A whole genome methylation analysis of systemic lupus erythematosus: hypomethylation of the IL10 and IL1R2 promoters is associated with disease activity. Genes Immun. 13, 214–220 [DOI] [PubMed] [Google Scholar]
- 52. Pou J., Martínez-González J., Rebollo A., Rodríguez C., Rodríguez-Calvo R., Martín-Fuentes P., Cenarro A., Civeira F., Laguna J. C., Alegret M. (2011) Type II interleukin-1 receptor expression is reduced in monocytes/macrophages and atherosclerotic lesions. Biochim. Biophys. Acta 1811, 556–563 [DOI] [PubMed] [Google Scholar]
- 53. Fearon E. R. (2011) Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 6, 479–507 [DOI] [PubMed] [Google Scholar]
- 54. Markowitz S. D., Bertagnolli M. M. (2009) Molecular origins of cancer: molecular basis of colorectal cancer. N. Engl. J. Med. 361, 2449–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Neumann D., Kollewe C., Martin M. U., Boraschi D. (2000) The membrane form of the type II IL-1 receptor accounts for inhibitory function. J. Immunol. 165, 3350–3357 [DOI] [PubMed] [Google Scholar]