

Background: S49 kin− cells, which lack active protein kinase A (PKA), can identify roles of PKA, including at the mitochondria, in which such roles are not well defined.
Results: PKA activation alters expression of mitochondrial proteins, including in the branched-chain amino acid (BCAA) degradation pathway.
Conclusion: Expression of BCAA degradative enzymes is increased by PKA.
Significance: Cellular responses mediated by cAMP/PKA may derive from effects on mitochondrial proteins.
Keywords: cell metabolism, cyclic AMP (cAMP), mitochondria, protein kinase A (PKA), proteomics
Abstract
Cyclic AMP (cAMP), acting via protein kinase A (PKA), regulates many cellular responses, but the role of mitochondria in such responses is poorly understood. To define such roles, we used quantitative proteomic analysis of mitochondria-enriched fractions and performed functional and morphologic studies of wild-type (WT) and kin− (PKA-null) murine S49 lymphoma cells. Basally, 75 proteins significantly differed in abundance between WT and kin− S49 cells. WT, but not kin−, S49 cells incubated with the cAMP analog 8-(4-chlorophenylthio)adenosine cAMP (CPT-cAMP) for 16 h have (a) increased expression of mitochondria-related genes and proteins, including ones in pathways of branched-chain amino acid and fatty acid metabolism and (b) increased maximal capacity of respiration on branched-chain keto acids and fatty acids. CPT-cAMP also regulates the cellular rate of ATP-utilization, as the rates of both ATP-linked respiration and proton efflux are decreased in WT but not kin− cells. CPT-cAMP protected WT S49 cells from glucose or glutamine deprivation, In contrast, CPT-cAMP did not protect kin− cells or WT cells treated with the PKA inhibitor H89 from glutamine deprivation. Under basal conditions, the mitochondrial structure of WT and kin− S49 cells is similar. Treatment with CPT-cAMP produced apoptotic changes (i.e. decreased mitochondrial density and size and loss of cristae) in WT, but not kin− cells. Together, these findings show that cAMP acts via PKA to regulate multiple aspects of mitochondrial function and structure. Mitochondrial perturbation thus likely contributes to cAMP/PKA-mediated cellular responses.
Introduction
The second messenger cAMP is found in virtually every eukaryotic cell. Through compartmentalized signaling and activation of protein kinase A (PKA) and other effectors, cAMP regulates functional activities at discrete cellular locations (1, 2). PKA signaling can be pro-apoptotic in lymphoid cells through activation of a mitochondria-dependent apoptotic pathway (3). Mitochondrial proteins involved in oxidative phosphorylation, ketogenesis, the TCA cycle, and fatty acid oxidation have consensus sequences for phosphorylation by PKA and can be phosphorylated (4). However, the role of PKA in regulating mitochondrial metabolism remains controversial due to difficulty in distinguishing the effects of PKA that occur outside or inside mitochondria, opposing theories regarding the source of cAMP within the mitochondrial matrix, and the poorly understood trafficking of PKA to mitochondria (5). Moreover, even though PKA is a key mechanism for cAMP-mediated regulation of cells, knowledge is limited regarding the mitochondrial proteins that are regulated by PKA.
Murine S49 T lymphoma (CD4+/8+) cells are a useful system to assess cAMP/PKA signaling (6–10). Selective pressure by cAMP analogs has been used to generate clonal variants of WT S49 cells (6, 10, 11). Incubation with cAMP analogs or agents that raise endogenous cAMP induces G1-phase cell-cycle arrest and apoptosis of WT S49 cells (6, 7, 12). The S49 variants that resist cAMP-induced apoptosis have helped reveal the functional role of components in the cAMP/PKA signaling pathway (3, 6, 8, 10).
kin−3 S49 cells are a clonal S49 variant that lack active PKA. The PKA catalytic (C) subunit in kin− cells has improper cis-phosphorylation at Ser338 during translation, rendering the C subunit insoluble; accordingly, kin− S49 cells have no PKA activity (13) yet grow robustly and resist cAMP-induced growth arrest and apoptosis (7, 8, 11). kin− cells thus provide a PKA-null cell system to identify cAMP/PKA-dependent responses.
We previously found (14, 15) that cAMP acts via PKA to alter the expression of many mRNAs in WT, but not kin− S49 cells, indicating that PKA produces substantial transcriptional regulation (14). Changes in mRNA may not, however, predict effects on protein expression as mRNA and protein expression may not highly correlate (16, 17). Due to the role of mitochondria in cAMP/PKA-mediated death of S49 cells (6, 7), here we used proteomic analysis to identify cAMP/PKA-mediated changes in protein expression in a mitochondria-enriched fraction of S49 cells and conducted additional studies to assess cAMP/PKA-regulated control of mitochondrial proteins and mitochondrial function.
Experimental Procedures
Growth of S49 Cells and Treatment with a cAMP Analog
WT and kin− S49 cells were grown in suspension culture in a humidified atmosphere containing 10% CO2 at 37 °C in DMEM with 4.5 g/liter glucose supplemented with 10% heat-inactivated horse serum, 1 mm sodium pyruvate, and 10 mm HEPES (pH 7.4). Cells were incubated for the indicated times with 100 μm CPT-cAMP (Sigma), a cAMP analog that activates PKA (18). Cultures were initiated at a density of 2 × 105; cells were maintained at a density of 1 × 105–2 × 106 cells/ml. Cell viability was determined using a Coulter Z2 Particle analyzer (Beckman Coulter).
Mitochondria Preparation
Mitochondria were prepared according to an adaptation of the method described by Kristián et al. (19). Briefly, 2 × 108 cells were harvested by centrifugation (1000 × g, 5 min, 4 °C), washed with ice-cold PBS, centrifuged again, and resuspended in MSHE (0.21 m mannitol, 0.07 m sucrose, 10 mm HEPES (pH 7.4), 1 mm EGTA) with a protease inhibitor mixture (MSHE-P) (Sigma). The cells were disrupted by 1400 p.s.i. for 10 min by nitrogen cavitation (Parr). The homogenate was centrifuged at 625 × g for 10 min to remove nuclei and unbroken cells. The supernatant was then centrifuged at 15,000 × g for 10 min. The supernatant (containing the endoplasmic reticulum) was removed. The pellet, the mitochondria-enriched fraction, was washed twice by resuspension in MSHE-P with centrifugation at 15,000 × g for 10 min followed by resuspension in MSHE-P.
Proteomic Analysis
Equal (100 μg) aliquots of proteins from WT and kin− S49 cells (0, 6, and 16 h CPT-cAMP treatment) were prepared for isobaric tagging and analyzed by mass spectrometry (MS) as previously described (15) with the following modification; the peptides were labeled with different 4-plex isobaric tagging for relative and absolute quantitation (iTRAQ) reagents (20). Spectrum Mill v3.03 was used to analyze the MS data as described (15) using 3 biological replicates to calculate protein iTRAQ reporter ion intensities. Proteins with five or more unique peptides were selected for quantitative analysis. A minimal total iTRAQ reporter ion intensity (sum of 4 channels compared) of 100 was used to filter out low intensity spectra. Conclusions regarding a change in protein abundance required the following criteria to be fulfilled. 1) The protein had to be quantified in at least two datasets. 2) If the protein was quantified in all three replicates, its abundance ratios had to be ≤0.67 or ≥1.5 in all three replicates. 3) If the protein was quantified in only two datasets, both had to yield abundance ratios of ≤0.67 or ≥1.5. We opted not to use a t test for iTRAQ quantification because that test can be too stringent for identifying proteins with -fold differences that are biologically significant (21). The DAVID 6.7 Bioinformatics tool (david.abcc.ncifcrf.gov) (22) was used to provide gene annotation and gene ontology term enrichment analysis.
Immunoblot Analysis
Immunoblotting was used to verify increased expression of branched-chain amino acid transferase (Bcat2), medium-chain specific acyl-CoA dehydrogenase (Acadm), and short-chain specific acyl-CoA dehydrogenase (Acads) in WT S49 cells incubated with CPT-cAMP. Whole cell lysates prepared from WT and kin− cells incubated with CPT-cAMP for 0–24 h were separated by 10% NuPAGE Bis-Tris gels (Invitrogen) in MOPS running buffer and transferred using an iBlot according to the manufacturer's instructions. Antibodies for Acadm were from Santa Cruz Biotechnology, for Bcat2 and anti-rabbit secondary antibodies were from Cell Signaling Technologies, and for GAPDH antibody were from Abcam. Protein expression was quantitated by densitometry using ImageJ 1.41o software (imagej.nih.gov).
Real-time PCR of Metabolic Genes
Cell pellets were collected and snap-frozen from untreated WT and kin− S49 cells, cells were incubated with CPT-cAMP for 16 h, or WT S49 cells were incubated for 40 min with the PKA inhibitor H89 (20 μm) and then with CPT-cAMP for 0 or 16 h. Pellets were stored at −80 °C until used. RNA was isolated from frozen pellets using Direct-zol RNA MiniPrep Kit (Zymo) according to the manufacturer's instructions and converted to cDNA using SuperScript III Reverse Transcriptase (Invitrogen) using the manufacturer's recommended protocol for random hexamer priming. Real-time PCR reactions contained 1× SYBR Green Master Mix (Eurogentec), 30–60 ng of cDNA, and primers at a final concentration of 0.2 μm. Primer sequences were as follows: Acads, forward 5′-GAC TGG CGA CGG TTA CAC A-3′; reverse 5′-GGC AAA GTC ACG GCA TGT C-3′; Acadm forward 5′-AAC ACA ACA CTC GAA AGC GG-3′; reverse 5′-TTC TGC TGT TCC GTC AAC TCA-3′; Bcat2 forward 5′-ACA GAC CAC ATG CTG ATG GTG-3′; reverse 5′-CTG GGT GTA GCG TGA GGT TC-3′.
Culture of S49 Cells in Media Lacking Glutamine or Glucose
WT and kin− S49 cells were grown in suspension culture in a humidified atmosphere containing 10% CO2 at 37 °C in media for each tested condition. Culture media formulations were as follows: regular (high glucose) media (DMEM with 4.5 g/liter glucose supplemented with 10% heat-inactivated horse serum, 1 mm sodium pyruvate, and 10 mm HEPES (pH 7.4)); minimal glucose media (DMEM without glucose supplemented with 10% heat-inactivated horse serum, 1 mm sodium pyruvate, and 10 mm HEPES (pH 7.4)); glutamine-deficient media (DMEM with 4.5 g/liter glucose and no l-glutamine supplemented with 10% heat-inactivated horse serum, 1 mm sodium pyruvate, and 10 mm HEPES (pH 7.4)). Cultures were initiated at a density of 5 × 105 cells/ml and incubated with CPT-cAMP as described above, with 10 μm forskolin or with H89 (1–20 μm) for 40 min before incubation with CPT-cAMP. Viability was determined using a Coulter Z2 Particle analyzer (Beckman Coulter); further analysis of apoptotic versus necrotic death was by flow cytometry. Freshly isolated cells were pelleted and washed twice in PBS, stained with annexin V-FITC (BD Biosciences) for 30 min at room temperature, diluted to a final volume of 500 μl in PBS and propidium iodide (BD Biosciences) was added just before reading on a FACSAria (BD Biosciences). Data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR). Events were gated to exclude debris; the data shown are the percentages of gated events.
Oxygen Consumption and Proton Production Measurements
WT, kin− and CPT-cAMP-treated cells (1.25 × 105 cells/well) were placed in XF96 microplates pretreated with Cell-Tak (BD Biosciences) and assayed in a Seahorse XF96 Analyzer (24). Plates were spun at 500 × g for 5 min, and growth medium was replaced with unbuffered DMEM (Sigma #D5030) supplemented with 8 mm glucose, 3 mm glutamine, 1 mm pyruvate, and 0.5 mm carnitine. ATP-linked respiration was measured as the respiration rate sensitive to 2 μm oligomycin. Maximal respiration was reported as the difference between the rate of protonophore-stimulated respiration (calculated by using sequential additions of FCCP; final concentrations, 400–800 nm) and the rate of respiration in the presence of 1 μm rotenone and 2 μm antimycin A (non-mitochondrial respiration). The response to inhibitors of specific oxidative pathways was assessed using UK5099 (Tocris) (2 μm) (25), BPTES (3 μm) (26), or etomoxir (20 μm) (27) added to cells 1 h before measurements.
Adherent cells were permeabilized, and ADP-stimulated respiration (State 3 respiration) was measured as previously described (28). Briefly, cells were permeabilized with recombinant perfringolysin O (commercially XF PMP; Seahorse Bioscience) and provided with 4 mm ADP and the following concentrations of respiratory substrates: 10 mm pyruvate with 1 mm malate, 5 mm glutamate with 5 mm malate, 10 mm succinate with 2 μm rotenone, 40 μm palmitoyl carnitine or octanoyl carnitine with 1 mm malate, and 4 mm l-α-ketoisocaproate (the keto acid of leucine) or 4 mm l-α-keto-β-methyl valerate (the keto acid of isoleucine) with 1 mm malate. All bioenergetic experiments were conducted with a minimum of three biological replicates and at least five technical replicates per experimental condition.
Electron Microscopy (EM)
S49 cells were pelleted and prepared for EM according to the method of Niikura et al. (29) before imaging using an FEI spirit transmission electron microscope operated at 120 kV.
Data Analysis
Statistical analyses were performed using GraphPad Prism 6 (La Jolla, CA). We performed one-way analysis of variance followed by Dunnett's multiple comparison test to determine statistical differences for untreated WT S49 cells or cells incubated with CPT-cAMP or forskolin and for WT compared with kin− S49 cells. Where only t tests were applied, we used the Holm-Sidak method, with α = 5.0%, analyzing each comparison without the assumption of a consistent standard deviation.
Results
Quantitative Proteomic Analysis of Mitochondrial Proteins
Fig. 1A illustrates our workflow for quantitative proteomic analysis of mitochondria-enriched fractions of WT and kin− S49 cells. We conducted this analysis in three biological replicates using 4-plex iTRAQ labeling (20) and identified and quantified the proteins with Spectrum Mill. Applying a 1% false discovery rate cutoff for peptide identification (to minimize detection of false positives) and requiring detection of at least 5 unique peptides for each protein, we identified 2013 proteins (supplemental Table 1). Known or predicted mitochondrial localization was determined using the COMPARTMENTS database (30); 1871 of the proteins we detected were listed in COMPARTMENTS, 1305 of which had known or predicted mitochondrial localization based on text mining or protein sequence.
FIGURE 1.

A, diagram of the workflow of mitochondrial proteomic studies. Proteins from mitochondria-enriched fractions of WT and kin− S49 cell were digested in three biological replicates of cells (WT control, WT treated for 6 or 16 h with CPT-cAMP (100 μm), kin− control, and kin− cells treated for 6 or 16 h with 100 μm CPT-cAMP). After digestion, peptides were desalted and labeled with iTRAQ reagents, and the labeled peptides from each sample were pooled and then separated by online two-dimensional LC with (PQD/CAD) pulsed Q dissociation-collision/collision0activated dissociation) acquisition on an LTQ-Orbitrap. Raw data for protein identification and quantitation were obtained using Spectrum Mill. Functional categories of proteins whose level of expression was increased or decreased were assigned according to the results of the DAVID bioinformatics tool. B, location of proteins with PKA-dependent increases within BCAA degradation pathway. Proteins related to degradation of leucine, isoleucine, and valine show PKA-dependent increases in WT S49 cells incubated for 16 h with CPT-cAMP. 1, Bcat2 (BCAA transferase, mitochondrial precursor) mediates the first step in the degradation of each BCAA. 2, Acadm (medium-chain specific acyl-CoA dehydrogenase, mitochondrial precursor) catalyzes a reversible step in the degradation of all three BCAA. Acads (short-chain specific acyl-CoA dehydrogenase, mitochondrial precursor) (3) and Acadsb (short/branched-chain specific acyl-CoA dehydrogenase, mitochondrial precursor) (4) catalyze steps in valine and isoleucine degradation. 5, Hibch (3-hydroxyisobutyryl-CoA hydrolase, mitochondrial precursor) is unique to valine degradation. 6, Acat1 (acetyl-CoA acetyltransferase, mitochondrial precursor) reversibly catalyzes the conversion of acetyl-CoA to acetoacetyl-CoA. 7, Mccc1 (methylcrotonoyl-CoA carboxylase subunit α, mitochondrial precursor is unique to leucine degradation. 8, Hmgcs2 (hydroxymethylglutaryl-CoA synthase, mitochondrial precursor) converts acetoacetyl-CoA to (S)-3-hydroxy-3-methylglutaryl-CoA, which can be converted further to acetoacetate and acetyl-CoA and is, therefore, involved in ketone body oxidation. This pathway map is based on the (human) KEGG (Kyoto Encyclopedia of Genes and Genomes) valine, leucine, and isoleucine degradation pathway.
Differences in Proteins between WT and kin− S49 Cells under Basal Conditions
Under basal conditions, 75 proteins demonstrated differences (36 decreases, 39 increases) in abundance in kin− compared with WT S49 cells (supplemental Table 2). Most of these proteins were not previously identified as regulated by or interacting with PKA. Analysis by the DAVID bioinformatics tool indicated that these proteins include ones involved in formation of intracellular structures, trafficking, oxidation/reduction, and metabolism. Table 1 lists a subset of differentially expressed proteins in the oxidation/reduction and metabolism categories.
TABLE 1.
Subset of proteins with greater or lower abundance in untreatred kin− S49 relative to untreated WT (kin−:WT ratio >1.5 or <0.67)
kin− cells a displayed higher and lower abundance of proteins within DAVID categories related to metabolism and oxidation/reduction. A full listing of basally differential proteins is contained in supplemental Table 2. Data for proteins whose expression increased are shown in bold; proteins with decreased expression are shown in italics.
| Gene symbol | Accession no. | Average ratio | Protein | Unique peptide no. | Spectrum no. | % Protein coverage |
|---|---|---|---|---|---|---|
| Metabolic process | ||||||
| Pck2 | IPI00223060 | 4.87 | Pck2 mitochondrial phosphoenolpyruvate carboxykinase 2 | 25 | 139 | 38 |
| Tars2 | IPI00132419 | 3.91 | Tars2 threonyl-tRNA synthetase, mitochondrial precursor | 12 | 40 | 19 |
| Gm2296 | IPI00851029 | 3.75 | Ubiquitin-conjugating enzyme E2S pseudogene | 6 | 12 | 28 |
| Aup1 | IPI00137725 | 3.25 | Aup1 ancient ubiquitous protein | 6 | 39 | 19 |
| Gart | IPI00230612 | 3.03 | Gart phosphoribosylglycinamide formyltransferase | 12 | 30 | 17 |
| Gls | IPI00671957 | 2.57 | Gls glutaminase isoform 5 | 22 | 242 | 3 |
| Usp39 | IPI00457815 | 2.23 | Usp39 U4/U6.U5 tri-snRNP-associated protein 2 | 7 | 9 | 13 |
| Nfs1 | IPI00311072 | 1.97 | Nfs1 Adult male kidney cDNA, RIKEN full-length enriched library, clone:0610009K17 product:nitrogen fixation gene 1 | 12 | 51 | 31 |
| LOC100047372 | IPI00222546 | 1.91 | Rpl22,LOC100047372 60S ribosomal protein L22 | 8 | 117 | 47 |
| Hk2 | IPI00114342 | 1.59 | Hk2 hexokinase-2 | 55 | 1122 | 44 |
| Gfm1 | IPI00230283 | 1.51 | Gfm1 elongation factor G 1, mitochondrial precursor | 20 | 119 | 27 |
| MPC2 | IPI00131896 | 0.64 | Mitochondrial pyruvate carrier 2 | 7 | 23 | 29 |
| Tmlhe | IPI00129163 | 0.60 | Tmlhe trimethyllysine dioxygenase, mitochondrial precursor | 5 | 13 | 8 |
| Taok3 | IPI00670075 | 0.44 | Taok3 similar to Serine/threonine-protein kinase TAO3 (thousand and one amino acid protein 3) isoform 9 | 5 | 9 | 6 |
| Ssb | IPI00134300 | 0.36 | Ssb lupus La protein homolog | 9 | 18 | 19 |
| Lipid metabolic process | ||||||
| Osbp | IPI00755161 | 1.61 | Osbp oxysterol binding protein | 6 | 8 | 9 |
| Cyb5r3 | IPI00759904 | 1.53 | Cyb5r3 Isoform 2 of NADH-cytochrome b5 reductase 3 | 12 | 56 | 34 |
| Gpam | IPI00387288 | 0.57 | Gpam glycerol-3-phosphate acyltransferase, mitochondrial | 8 | 46 | 12 |
| Fech | IPI00608064 | 0.25 | Fech Fech protein (fragment) | 10 | 36 | 9 |
| Oxidation/reduction | ||||||
| Mthfd2 | IPI00109824 | 3.36 | Mthfd2 bifunctional methylenetetrahydrofolate dehydrogenase/cyclohydrolase, mitochondrial precursor | 20 | 223 | 69 |
| LOC100046934 | IPI00854030 | 2.98 | LOC100046934 similar to amine oxidase (flavin-containing) domain 2 | 8 | 20 | 11 |
| Ndufv3 | IPI00128285 | 0.67 | NADH dehydrogenase (ubiquinone) flavoprotein 3 | 9 | 19 | 22 |
| Ndufa12 | IPI00344004 | 0.54 | Ndufa12 NADH dehydrogenase (ubiquinone) 1α subcomplex, 12 | 7 | 27 | 54 |
kin− cells have lower basal expression of a set of proteins involved in mitochondrial oxidative metabolism including MPC2 (a component of the mitochondrial pyruvate carrier), trimethyllysine dioxygenase (Tmlhe), the first enzyme in the carnitine biosynthesis pathway, glycerol-3-phosphate acyltransferase (Gpam), the first enzyme in the synthesis of glycerolipids, and two components of Complex I of the electron transport chain, Ndufv3 and Ndufa12.
Changes in Protein Expression in Response to Incubation with CPT-cAMP
Incubation of WT S49 cells with CPT-cAMP for 6 h altered the expression of 83 (52 increases, 31 decreases) proteins in the mitochondria-enriched fraction, but none of these changes occurred in CPT-cAMP-incubated kin− cells. The 16-h incubation with CPT-cAMP altered the expression of 110 proteins (67 increases, 43 decreases) in WT cells but produced no changes in protein expression in kin− S49 cells (supplemental Table 3). Thus, under basal conditions and in response to cAMP, PKA regulates the expression of numerous proteins in a mitochondria-enriched fraction of WT S49 cells.
The DAVID Bioinformatics tool identified seven gene ontology classifications (oxidation/reduction, cell cycle/cell cycle process/cell proliferation, BCAA degradation, FA metabolism, and leukocyte (lymphocyte) activation) of proteins whose expression increased in WT S49 cells incubated for 16 h with CPT-cAMP (Table 2). Proteins with decreased abundance after 16 h of CPT-cAMP treatment include ones involved in regulation of the cell cycle and oxidation/reduction.
TABLE 2.
Proteins showing significant changes in CPT-cAMP-treated WT S49 cells, 16h
Proteins whose levels of expression increased or decreased in WT S49 cells incubated with CPT-cAMP for 16 h relative to basal (control) conditions are shown. No protein had altered expression in kin− S49 cells incubated with CPT-cAMP for 16 h. The average ratios of protein abundance (WT 16 h:WT control) from at least two biological replicates are presented. Data for proteins whose expression increased are shown in bold; proteins with decreased expression are shown in italics. The data include the number of unique peptides identified for each protein (Unique_pep), the number of PDQ spectra (Spectrum_num) used for each protein quantitation, the full protein name as determined by Spectrum Mill, and the functional classification assigned by the DAVID bioinformatics software. Gene expression from previously published microarray analysis (14) is represented at the right by a heatmap.
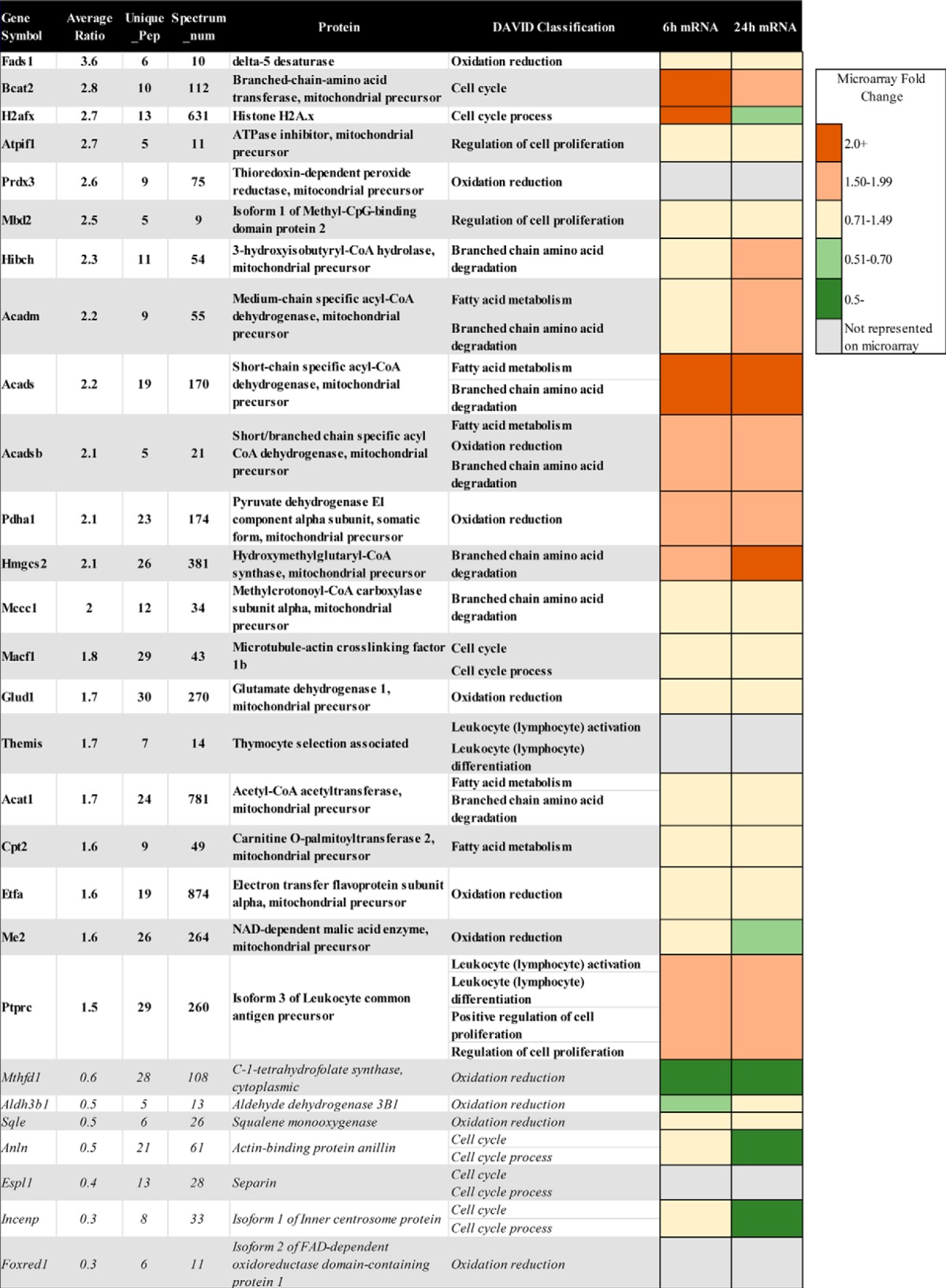
Proteins classified as “oxidation/reduction” have diverse roles. Proteins with decreased abundance in CPT-cAMP-treated WT cells include C-1-tetrahydrofolate synthase (Mthfd1), which is associated with methionine, thymine, and purine synthesis (31) and squalene epoxidase (Sqle), the first oxygenation step in cholesterol biosynthesis (32). Foxred1, an assembly factor specific to Complex I, may have a role in metabolism, as bacterial FOXRED genes appear in operons that regulate degradation of creatine and creatinine, resulting in glycine, a component of the antioxidant glutathione (33). Oxidation/reduction proteins that are increased by incubation with CPT-cAMP include electron transfer flavoprotein subunit α (Etfa), which catalyzes the initial step of FA β-oxidation, and aldehyde dehydrogenase family 3 member B1 (Aldh3b1), which oxidizes long-chain FAs (34) and may protect cells from oxidative stress (35). CPT-cAMP also increases expression of pyruvate dehydrogenase 1 (Pdha1), glutamate dehydrogenase 1 (Glud1), and NAD+-dependent malic acid enzyme (Me2), proteins providing substrates to the TCA cycle.
Incubation of WT S49 cells with CPT-cAMP for 16 h increased the abundance of multiple proteins involved in BCAA, FA, and ketone body catabolism, including medium-chain-specific acyl-CoA dehydrogenase (Acadm) and short-chain-specific acyl-CoA dehydrogenase (Acads), both involved in FA oxidation, short/branched-chain-specific acyl-CoA dehydrogenase (Acadsb), which participates in BCAA and FA oxidation, acetyl-CoA acetyltransferase (Acat1), which plays a role in BCAA and ketone body oxidation, and hydroxymethylglutaryl-CoA synthase (Hmgcs2), involved in ketone body oxidation (Fig. 1B). Carnitine O-palmitoyltransferase 2 (Cpt2), critical in long-chain fatty acid oxidation, was also elevated. CPT-cAMP also increased expression of other proteins more exclusively involved in BCAA degradation, including 3-hydoxyisobutyryl-CoA hydrolase (Hibch), methylcrotonoyl-CoA carboxylase subunit α (Mccc1), and BCAA transferase (Bcat2).
Gene expression data of S49 cells (14) are available for 24 of the 28 proteins identified in Table 2, which shows gene expression at 6 and 24 h of incubation with CPT-cAMP as a heatmap. Comparison of microarray and protein expression data demonstrated that mRNA expression of genes associated with cell cycle or regulation of cell proliferation, oxidation/reduction, and FA and BCAA metabolism correlated with protein abundance at 16 h.
Acads, Acadsb, Hmgcs2, and Bcat2, each involved in BCAA, FA, and ketone body catabolism, increased at 6 h; expression of Hibch and Acadm increased by 24 h. Altered abundance of the corresponding proteins (at 16 h) was as predicted by their gene expression changes. Thus, there is clearly a shift as a result of CPT-cAMP incubation toward expression of proteins involved in oxidative pathways for FA and BCAA. We next sought to validate these proteomic changes with alternative methods.
Increased Expression of Protein and mRNA of BCAA Metabolizing Genes in WT, but Not kin− S49 Cells
Immunoblot analysis confirmed that incubation of WT S49 cells with CPT-cAMP increased the protein expression of Bcat2, Acads, and Acadm; the expression of Bcat2 in kin− S49 cells did not significantly change with CPT-cAMP treatment, and expression of Acads and Acadm was too low to quantify (Fig. 2, A–F).
FIGURE 2.

PKA-dependent increases in protein and gene expression of BCAA degradation pathway members in WT S49 cells. Immunoblotting of whole-cell lysates shows that protein levels of Bcat2, Acads, and Acadm increase in WT S49 cells after treatment with CPT-cAMP. Representative immunoblots of Bcat2, Acads, and Acadm are shown in A, C, and E, respectively. B, quantification of Bcat2 protein in WT and kin− S49 incubated with CPT-cAMP. Data shown are the mean ± S.E. of n = 33, * = p < 0.05 versus 0 h. D, quantification of Acadm protein in WT. Data shown are the mean ± S.E. of n = 4 * = p < 0.05 versus 0 h. F, quantification of Acads protein in WT. Data shown are the mean ± S.E. of n = 4. * = p < 0.05 versus 0 h. G, CPT-cAMP induces mRNA expression of Acads, Acadm, and Bcat2 at 16 h incubation time 2-fold over basal expression. H89 blunts the CPT-cAMP-induced expression of Acads, Acadm, and Bcat2. WT, kin−, and WT S49 cells preincubated with 20 μm H89 were treated with CPT-cAMP, and expression of Acads, Acadm, and Bcat2 at 16 h were determined; the results are expressed as relative to expression at 0 h. Data shown are the mean ± S.E., n = 3, p values from Tukey multiple comparisons test after two-way analysis of variance: ** = p < 0.01 versus WT, *** = p < 0.001. ns, not significant.
We used real-time qPCR analysis to assess the mRNA expression of Acads and Acadm, two genes in the FA oxidation pathway, and Bcat2, a mitochondrial BCAA aminotransferase. The DAVID bioinformatics tool classifies Bcat2 as a cell cycle related protein, but it catalyzes the first step in BCAA catabolism. CPT-cAMP increased the expression (>2-fold at 16 h compared with basal) of mRNA for Acads, Acadm, and Bcat2 (Fig. 2G). None of these significantly changed in kin− S49 cells. Consistent with this finding, treatment of WT cells with the PKA inhibitor H89 (20 μm) blunted the increase by CPT-cAMP in mRNA expression of Acads, Acadm, and Bcat2 (Fig. 2G).
cAMP/PKA Protects WT, but Not kin− S49 Cells from Death in Media That Lacks Glutamine
The lower abundance of proteins related to pyruvate and FA metabolism in kin− than WT S49 cells and the higher abundance of HK, Gls, and Pck2 (Table 1) suggested that kin− cells may have a higher dependence upon glucose or amino acids such as glutamine to meet energetic requirements. CPT-cAMP-stimulated elevations in genes and proteins involved in BCAA degradation in WT but not kin− cells indicate a role for PKA in regulation of proteins involved in BCAA degradation. To metabolically stress the cells, we cultured WT and kin− S49 cells in media lacking added glucose or glutamine. WT cells grown in media with DMEM with no added glucose for 72 h had an ∼40% decrease in viability but the addition of CPT-cAMP mitigated this loss in viability (Fig. 3A). kin− cells were more resistant to glucose withdrawal, and CPT-cAMP had no effect on viable cell number (Fig. 3A), thus implying that the increased survival of WT cells is mediated by PKA.
FIGURE 3.

Effects of cAMP/PKA signaling on survival in low nutrient media. A, CPT-cAMP enhances viability of WT S49 cells in low glucose media after 72 h; viability of kin− S49 cells is unaffected. Data shown are the mean ± S.E., n = 6, *** = p < 0.001. ns, not significant. B, WT and kin− S49 cells in glutamine-free media have ∼50% loss of viability at 36 h. CPT-cAMP protects WT, but not kin− S49 cells, from this loss in viability. Data shown are the mean ± S.E., n = 8, *** = p < 0.001, † = p < 0.001 versus WT with the same treatment. C, protection by CPT-cAMP of WT S49 in glutamine-free media declines over time. D, an ∼50% loss of viability in kin− S49 cells in glutamine-free media occurs as early as 18 h. E, viability of the D- (cAMP-deathless) variant of S49 cell does not decline in glutamine-free media, and the cells do not undergo apoptosis in response to CPT-cAMP. Data in C–E are the mean ± S.E., n = 4, *** = p < 0.001, ** = p < 0.01. F, the percentage of apoptotic cells (high annexin V staining) are highest in kin− S49 cells in glutamine-free media, whereas CPT-cAMP (100 μm) or forskolin (10 μm) lowers the percentage of apoptotic WT S49 cells. Data are the mean ± S.E., n = 4, ** = p < 0.001 versus WT, *** = p < 0.001 versus WT. G, comparisons of live cells (low annexin V/propidium iodide staining) show that the PKA inhibitor H89 suppresses CPT-cAMP protection of WT S49 cells in glutamine-free media. Data are the mean ± S.E., n = 4, *** = p < 0.001. H, necroptosis and apoptosis appear to contribute equally to WT cell death in glutamine-free media, whereas apoptosis is more prevalent in kin− cell death in glutamine-free media. Inhibition of caspases with benzyloxycarbonyl-VAD-fluoromethyl ketone (z-VAD-fmk) reduces early apoptotic events in kin− cells (glutamine-free media). Inhibition of necroptosis with necrostatin appears to accelerate apoptotic cell death in kin− cells in glutamine-free media. Data shown are the mean ± S.D., n = 2, *** = p < 0.001 versus kin− untreated, ** = p < 0.01 versus kin− untreated.
Both WT and kin− S49 cells grown for 36 h in (high glucose) media that lacks glutamine had a >50% loss in viability; the addition of CPT-cAMP to WT, but not kin− cells protected them from this loss in viability (Fig. 3B). Moreover, the two cell types differed in their rates of loss in viability in media that lacked glutamine; WT cells had a gradual decrease in viability, which was slowed (up to 48 h) by the addition of CPT-cAMP (Fig. 3C). By contrast, kin− S49 cells had lower viability, which was detectable within 18 h (Fig. 3D) and appears to reflect an increase in apoptosis. The inability of CPT-cAMP to sustain the increase in the viability of WT S49 cells likely results from its pro-apoptotic action that occurs in these cells by 48 h (7, 11) and by findings with cAMP-deathless (D-) S49 cells, which do not undergo CPT-cAMP-promoted apoptosis (Fig. 3E) (3). CPT-cAMP and forskolin (which increases endogenous cAMP via activation of adenylyl cyclase) protected WT S49 cells from death in media lacking glutamine; the PKA inhibitor H89 eliminated this protection (Fig. 3, F and G). Flow cytometry analysis revealed a higher percentage of early apoptotic events compared with necroptotic events in glutamine-deprived kin− S49 cells, suggesting that growth of kin− cells in glutamine-free media enhances apoptosis. Consistent with this idea, the caspase inhibitor benzyloxycarbonyl-VAD-fluoromethyl ketone reduced apoptosis of kin− S49 cells in this media. Glutamine-deprived WT S49 cells display similar percentages of cells in early apoptosis and necroptosis. (Fig. 3H).
Bioenergetic Measurements in WT and kin− S49 Cells
To determine whether PKA-dependent changes in protein expression and sensitivity to nutrient deprivation manifested in changes in cellular energetics, respiration and proton production rates were measured. Cellular ATP needs are predominantly supplied by oxidative phosphorylation and glycolysis. Mitochondrial ATP production can be estimated by the respiration rate sensitive to the ATP synthase inhibitor oligomycin. Glycolysis, the catabolism of uncharged glucose into anionic lactate, can be indirectly measured by extracellular acidification and expressed quantitatively as the proton production rate (PPR; Refs. 36 and 37). The contribution of respiratory CO2 to medium acidification was broadly similar across groups and unlikely to confound interpretation, as basal respiration (Fig. 4A) and cellular substrate preference (see below) were similar in WT and kin− cells. This renders PPR a reliable indicator of glycolytic turnover for this specific comparison (36, 37).
FIGURE 4.
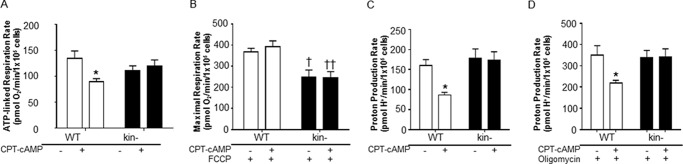
CPT-cAMP/PKA regulates cellular bioenergetics. CPT-cAMP decreases ATP-linked respiration (A), and PKA is necessary to drive ATP synthesis in the presence of the protonophore FCCP (stimulating maximal activity of the electron transport chain) (B). Cells were incubated with 8 mm glucose, 1 mm pyruvate, and 3 mm glutamine in unbuffered DMEM. C, medium acidification can serve as an indirect measure of glycolytic turnover as catabolism of a molecule of uncharged glucose into two molecules of lactate (pKa 3.9) requires the release of two protons at physiological pH. Stimulation of PKA with CPT-cAMP in WT cells decreased proton production. D, a similar effect of CPT-cAMP on WT cells occurs when the mitochondrial ATP synthase is blocked by Oligomycin. Cells were incubated with 8 mm glucose, 1 mm pyruvate, and 3 mm glutamine in unbuffered DMEM. All data are the mean ± S.E. (n≥3), * = p < 0.05 versus control, † = p < 0.05 versus WT control, †† = p < 0.01 versus WT +CPT-cAMP.
CPT-cAMP treatment of WT cells decreased the rate of ATP-linked respiration (Fig. 4A), implying mitochondrial dysfunction, a shift of ATP production to glycolysis, or an overall decrease in the rate of ATP utilization by the cells (37). To determine if CPT-cAMP induces mitochondrial dysfunction, activity of the electron transport chain was disengaged from cellular ATP turnover by the addition of oligomycin and the protonophore FCCP (Fig. 4B). Uncoupler-stimulated rates of respiration were similar between control and CPT-cAMP-treated WT cells, suggesting that PKA activation does not induce a defect in electron transport chain function.
With regard to glycolytic flux, CPT-cAMP treatment decreased endogenous (basal) PPR (Fig. 4C). These data, combined with the findings discussed above (Fig. 4, A and B), strongly suggest that CPT-cAMP diminishes cellular ATP demand in the WT cells. Oligomycin significantly stimulates PPR (Fig. 4D), blocking mitochondrial ATP production and forcing glycolysis to meet the cellular ATP demand. Under these conditions, CPT-cAMP negatively regulated glycolytic turnover (Fig. 4D). Taken together, the results in Fig. 4 imply that CPT-cAMP acts via PKA to regulate the ATP utilization rate as well as the poise between oxidative phosphorylation and glycolysis to meet the energy demand of the cell.
WT and kin− cells had similar rates of ATP-linked respiration (Fig. 4A), suggesting that the absence of PKA does not change the mitochondrial contribution to overall cellular ATP production. In contrast, maximal rates of FCCP-stimulated respiration were decreased in kin− compared with WT cells (Fig. 4B), indicating that the lack of PKA-mediated signaling lowers the cells' respiratory capacity. As expected, respiratory rates in the kin− cells were insensitive to CPT-cAMP (Fig. 4, A and B).
With regard to PPR in kin− cells, rates of glycolytic flux were not significantly different in the basal state (Fig. 4C) or in the presence of oligomycin (Fig. 4D) from WT cells. As expected, there was no change in the glycolytic rate upon CPT-cAMP treatment in kin− cells under either condition (Fig. 4, C and D).
To examine whether PKA can mediate not only global changes in cellular bioenergetics but also the flux through specific respiratory pathways, rates of State 3 (ADP-stimulated, phosphorylating) respiration were measured in permeabilized cells. Unlike with intact cells, this approach allows experimental control over the specific substrates offered to mitochondria, allowing for the direct interrogation of specific metabolic pathways (28).
The proteomic and Western analysis described above suggested that PKA-mediated regulates of expression of proteins involved in BCAA and FA catabolism. To test whether this regulatory control can translate into an altered capacity for carbon flux through these pathways, we measured the oxygen consumption on several substrates, including branched-chain keto acids and fatty acyl carnitines. First, we found that maximal rates of State 3 respiration on a variety of substrates were lower in kin− than WT S49 cells (Fig. 5, A–C), suggesting a generalized decrease in oxidative capacity in kin− cells consistent with the decrease in uncoupler-stimulated respiration seen in intact cells. CPT-cAMP acting via PKA increased the capacity to oxidize branched-chain keto acids of leucine and isoleucine (Fig. 5A) as well as the medium- and long-chain FA conjugates octanoyl carnitine and palmitoyl carnitine (Fig. 5B). Such changes were not seen in kin− cells, indicating that PKA upregulates the capacities for BCAA and FA oxidation. CPT-cAMP did not change the maximal, ADP-stimulated rates of permeabilized cells for pyruvate, glutamate, or succinate (Fig. 5C), thus ruling out a global effect of CPT-cAMP on mitochondrial function, branched-chain keto acids, and FA conjugates.
FIGURE 5.

PKA activity affects BCAA and FA substrate usage. In WT, but not kin−, S49 cells, CPT-cAMP increased the capacity to oxidize the branched chain keto acids for leucine (l-α-ketoisocaproate (KIC)) and isoleucine (l-α-keto-β-methyl valerate (KMV)) (A) as well as the FA conjugates palmitoyl carnitine and octanoyl carnitine (B). C, other substrates showed no effect, and the relative usage of glucose and glutamine for oxidative metabolism was not changed. D, inhibitors were used to determine the extent to which cells oxidize pyruvate (UK5099), glutamine (BPTES), and long-chain FAs (etomoxir) and to assess for differences in substrate oxidation in the mitochondria of WT and kin− S49 cells. Only etomoxir produced a significant change in substrate oxidation between WT S49 control and WT S49 +CPT-cAMP (* = p < 0.05 versus WT control, ** = p < 0.01 versus WT control). All data are the mean ± S.E. (n ≥ 3).
To determine whether the relative contribution of different substrates was altered in whole cells, rates of maximal, uncoupler-stimulated respiration in intact cells were measured in response to UK5099 (which inhibits mitochondrial pyruvate uptake), BPTES (a glutaminase inhibitor), and etomoxir (an inhibitor of the carnitine palmitoyl transferase-1). CPT-treatment did not alter the sensitivity to blocking pyruvate uptake or glutamine oxidation, but blocking of mitochondrial long-chain FA uptake was observed in WT but not kin− cells, suggesting an increased capacity to oxidize endogenous long-chain FAs (Fig. 5D). These results support the idea that CPT-cAMP treatment induces a shift toward use of FAs as an energy substrate in WT cells.
Electron Microscopy and Mitochondrial Morphology of WT and kin− S49 Cells
EM analyses indicated that, basally, the mitochondria of kin− cells resemble those of WT cells (Fig. 6, a and b), having similar volume density, number and size of mitochondria, and cristae abundance (Table 3), consistent with the health and protection from damage by the reactive oxygen species of kin− cells (15). These data also suggest that the decreased rates of maximal respiration in kin− cells (Figs. 4B and 5, A–C) is not the result of lower mitochondrial content in these cells.
FIGURE 6.
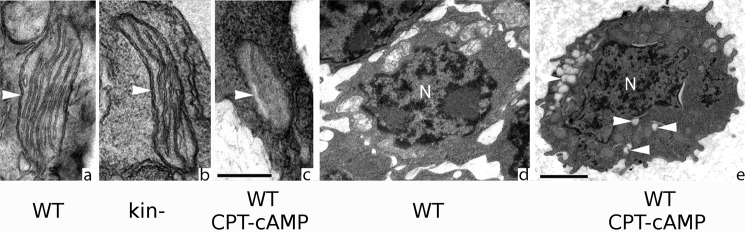
CPT-cAMP treatment changes mitochondrial structure and increases autophagy. Electron microscopy showed medium to large mitochondria typically with long cristae (arrowhead) in WT S49 cells (a). b, similar mitochondrial structure (arrowhead) in kin− S49 cells. c, CPT-cAMP-treated WT S49 cells have smaller mitochondria with fewer cristae (arrowhead) than do untreated WT cells. Scale bar, 500 nm for panels a, b, and c. d and e, lysosomal vesicles (arrowheads) are more prevalent in CPT-cAMP-treated WT cells (e) than in untreated WT cells (d), consistent with measurements (Table 3) that show decreased mitochondria volume density and size, but no change in number, suggesting degradation of a portion of individual mitochondria, likely by fission of the dysfunctional part and its subsequent autophagosomal digestion. Scale bar, 1000 nm and applies to panels d and e.
TABLE 3.
Morphologic characteristics of mitochondria of WT and kin−S49 cells
Measurements derived from EM micrographs of WT S49 cells (untreated or incubated with CPT-cAMP for 16 h) and kin− S49 cells. Quantification of mitochondrial volume density (expressed as %, n =10), mitochondria number per μm2 cytoplasmic area (n =10), and mitochondria size (n =20) is represented by cross-sectional area (μm2; n =10), and cristae abundance is represented by the ratio of total cristae membrane surface area to mitochondrial outer membrane surface area (n =10). CPT-cAMP-treated WT S49 cells had a lower volume density of mitochondria and smaller mitochondria but no change in mitochondria number compared to untreated WT cells. CPT-cAMP-treated WT cells also have mitochondria with fewer cristae. p values in boldface have a value of < 0.01.
| Mean | S.E. | t test vs. control | t test vs. kin− | |
|---|---|---|---|---|
| Mitochondrial volume density (%) | ||||
| WT untreated | 14 | 0.79 | ||
| WT CPT-cAMP | 10 | 0.8 | p = 0.0022 | p = 0.13 |
| kin− | 13 | 1.3 | p = 0.29 | |
| kin− CPT-cAMP | 17 | 2.9 | p = 0.15 | |
| Mitochondrial number/cytoplasmic area (μm2)a | ||||
| WT untreated | 0.14 | 0.017 | ||
| WT CPT-cAMP | 0.12 | 0.038 | ||
| kin− | 0.14 | 0.026 | ||
| kin− CPT-cAMP | 0.12 | 0.011 | ||
| Mitochonrial cross-sectional area (μm2) | ||||
| WT untreated | 0.54 | 0.048 | ||
| WT CPT-cAMP | 0.35 | 0.075 | p = 0.0015 | p = 0.0032 |
| kin− | 0.61 | 0.045 | p = 0.61 | |
| kin− CPT-cAMP | 0.70 | 0.087 | p = 0.37 | |
| Total cristae membrane SA/mito outer membrane SA | ||||
| WT untreated | 1.9 | 0.17 | ||
| WT CPT-cAMP | 0.48 | 0.11 | p = 0.0000036 | p = 0.000010 |
| kin− | 1.6 | 0.14 | p = 0.17 | |
a Error was not significant in any comparison.
CPT-cAMP altered the morphology of WT, but not kin−, S49 cells with evidence of apoptosis by 16 h, including reduction in mitochondrial volume density and size and in the surface area of cristae (Fig. 6, see a and c and d and e; Table 3). These findings are consistent with data showing that treatment of WT cells with CPT-cAMP induces loss of mitochondrial membrane potential, mitochondrial release of cytochrome c, and second mitochondria-derived activator of caspases (SMAC) and increases caspase-3 activity (3, 6).
The decrease in volume density and size of mitochondria but not their number implies that CPT-cAMP may increase autophagy in WT cells. CPT-cAMP treatment increased lysosomal vesicle number (Fig. 6, d and e), which suggests that some mitochondria have been degraded, perhaps by fission of the dysfunctional part and its subsequent digestion by an autophagosome.
Discussion
We studied S49 lymphoma cells and utilized multiple complementary approaches to determine if cAMP/PKA regulates the mitochondrial proteome and influences the structure and function of mitochondria. Prior studies of the mitochondrial proteome (38–40) have not assessed a role for cAMP or PKA nor the contribution of mitochondria to cellular actions of cAMP/PKA. We reasoned that a comparison of WT and kin− cells would be useful for such an assessment.
Under basal conditions, 36 and 39 proteins had decreased and increased expression, respectively, in kin− cells compared with WT S49 cells, which implies that under steady-state conditions PKA both stimulates and inhibits the expression of proteins in the mitochondrial proteome. Consistent with these bidirectional effects of PKA, incubation of WT, but not kin− S49 cells, with CPT-cAMP both decreased and increased protein expression. In CPT-treated WT S49 cells, expression of 31 proteins decreased after 6 h and of 43 proteins after 16 h, whereas 52 and 67 proteins increased after 6 and 16 h, respectively. The DAVID bioinformatics tool predicted that numerous cell cycle-related proteins were among the cAMP/PKA-regulated proteins with increased expression after 16 h, consistent with the G1 phase growth arrest of WT cells in response to increases in intracellular cAMP concentration (6, 8, 10).
Incubation of WT cells with CPT-cAMP prominently increased expression of proteins involved in FA oxidation, ketogenesis, and BCAA degradation, changes not seen in kin− S49 cells treated with CPT-cAMP. Immunoblot analysis confirmed the increases in expression of the BCAA and FA degradation pathway proteins, and real-time PCR showed that these increases result from their increased gene expression. Functional studies revealed a cAMP/PKA-dependent survival response and increased BCAA oxidation in WT S49 cell under glutamine-deprived conditions. Furthermore, analysis of respiratory rates revealed that CPT-cAMP treatment of WT cells results in an increased capacity to oxidize branched-chain α-ketoacids and fatty acyl carnitines, whereas no effect was observed on other oxidizable substrates including succinate, pyruvate, and glutamate. Together, these data imply that the regulation by cAMP/PKA of BCAA and FA degradation is transcriptional; the increase in BCAA and FA oxidation may be part of the pro-apoptotic events promoted by cAMP in WT S49 cells (3, 6, 7, 14).
In agreement with this effect of cAMP/PKA in S49 cells, yeast cAMP-dependent protein kinase type 1 (TPK1), a PKA, regulates BCAA metabolism through the de-repression of genes for BCAA aminotransferase, a homolog of murine Bcat1/2, and ketol-acid reductoisomerase (ILV5), involved in BCAA synthesis (41). Murine Acadm is transcriptionally regulated by PKA (42), but we believe ours is the first demonstration that Bcat2 or Acads is transcriptionally regulated by PKA.
The ability of forskolin to promote survival of WTS49 cells in media lacking glutamine supports the idea that cAMP/PKA mediates the increased transcription of metabolic genes. Actions of cAMP can be spatially limited by phosphodiesterase activity and complexes between PKA and its targets (43, 44). Mitochondria-located membrane adenylyl cyclases have not been identified (5), but a soluble adenylyl cyclase is present in mitochondria and regulated by changes in bicarbonate, Ca2+, and ATP levels (45, 46).
A phosphoproteomic analysis of mouse liver mitochondria revealed changes in phosphorylation of Hmgcs2, but not other proteins, related to BCAA degradation or FA oxidation (4). Assessment of the Jurkat T cell PKA-regulated phosphoproteome (47) did not identify the proteins that we found. An unexpected aspect of PKA deficiency in the kin− S49 cells is their sensitivity to a lack of glutamine, which includes apoptotic death (Fig. 3H). kin− S49 cells rapidly proliferate in complete media and remain viable in low glucose media. K-ras-transformed NIH-3T3 cells display aerobic glycolysis (the Warburg effect) and are sensitive to glucose withdrawal, undergoing apoptosis that can be prevented by PKA stimulation (48) and have decreased proliferation in the absence of glutamine (49). Because S49 cells do not possess a Ras mutation (50), the sensitivity of WT S49 cells to reduced glucose and its alleviation by stimulation of PKA occurs without constitutive Ras activity. Perhaps PKA-mediated up-regulation of alternative mitochondrial pathways for energy metabolism, including BCAA and FA oxidation, enhances survival in the face of glucose or glutamine deprivation. We identified two proteins encoded by mitochondria DNA mitochondrial cytochrome c oxidase subunit 2 (mt-CO2 or COX2) and NADH-ubiquinone oxidoreductase chain 1 (mt-Nd1), but their basal expression is similar in WT and kin− S49 cells, and CPT-cAMP did not increase their expression.
The current mitoproteomic analysis identified 2013 proteins, whereas our previous proteomic study of lysates from WT and kin− S49 cells identified 1056 proteins (15); 469 proteins were in common between these studies. The greater number of proteins identified in this study may result from the use of iTRAQ 4-plex in place of 8-plex (51). One protein, LOC100046934 (similar to amine oxidase (flavin-containing) domain 2) was increased in both studies. Other proteins had significantly high or low kin−:WT ratios but not in at least two experimental sets or lacked statistical significance in the other study. DAVID analysis of proteins in common showed enrichment for nucleotide binding proteins. Other highly enriched categories include protein folding/chaperone function, vesicle, mitochondria, and oxidation/reduction. The mitochondrial proteomic analysis thus revealed more proteins but shared similarities in terms of basal differences between WT and kin− cells, the effect of cAMP/PKA on protein abundance and enriched categories. The relatively small number of “overlapping” proteins in the two datasets may, at least in part, reflect our rather conservative approach for protein identification.
Future phosphoproteomic analyses of WT and kin− S49 cells should enhance understanding of PKA-specific substrates and mechanisms involved in cAMP/PKA-mediated actions (6). Such analyses may not only reveal the PKA-mediated phosphorylation profile but also if other kinases compensate for the lack of PKA in kin− cells. In yeast, deletion of cAMP-dependent protein kinase type 2 or 3 (TPK2 or TPK3) had a prominent impact on the phosphoproteome, whereas deletion of TPK1 had a smaller effect but changed the morphology of the yeast (23).
The current study provides new insights regarding the regulation by cAMP/PKA of mitochondrial function, including stimulated expression of enzymes involved in BCAA and FA oxidation, especially under states of nutritional stress. Our findings raise the possibility that the regulation of mitochondrial metabolism contributes to, and perhaps helps mediate, cellular responses that are altered by cAMP acting via PKA.
Author Contributions
S. S. T. and P. A. I. conceived and coordinated the study. Y. G. and L. Z. performed and analyzed the data in the proteomics experiments. A. W. performed and analyzed the experiments shown in Figs. 2 and 3. A. S. D. and A. N. M. designed, performed, and analyzed the experiments shown in Figs. 4 and 5. G. P. performed and analyzed the electron microscopy shown in Fig. 6. A. W. and P. A. I. wrote the manuscript, but all authors contributed to the writing and approved the final version.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants DK54441 (to S. S. T.) and GM066232, HL107200, and CA189477 (to P. A. I.). This work was also supported by the Howard Hughes Medical Institute (to S. S. T.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables 1–3.
- kin−
- protein kinase A null variant of S49
- CPT-cAMP
- 8-(4-chlorophenylthio)-cAMP
- BCAA
- branched-chain amino acid
- BPTES
- bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide
- FA
- fatty acid
- FCCP
- carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- iTRAQ
- isobaric tagging for relative and absolute quantitation
- LTQ
- linear trap quadrupole
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PPR
- proton production rate.
References
- 1. Dekkers B. G., Racké K., Schmidt M. (2013) Distinct PKA and Epac compartmentalization in airway function and plasticity. Pharmacol. Ther. 137, 248–265 [DOI] [PubMed] [Google Scholar]
- 2. Stangherlin A., Zaccolo M. (2012) Phosphodiesterases and subcellular compartmentalized cAMP signaling in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 302, H379–H390 [DOI] [PubMed] [Google Scholar]
- 3. Zhang L., Zambon A. C., Vranizan K., Pothula K., Conklin B. R., Insel P. A. (2008) Gene expression signatures of cAMP/protein kinase A (PKA)-promoted, mitochondrial-dependent apoptosis: comparative analysis of wild-type and cAMP-deathless S49 lymphoma cells. J. Biol. Chem. 283, 4304–4313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grimsrud P. A., Carson J. J., Hebert A. S., Hubler S. L., Niemi N. M., Bailey D. J., Jochem A., Stapleton D. S., Keller M. P., Westphall M. S., Yandell B. S., Attie A. D., Coon J. J., Pagliarini D. J. (2012) A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab. 16, 672–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Valsecchi F., Ramos-Espiritu L. S., Buck J., Levin L. R., Manfredi G. (2013) cAMP and mitochondria. Physiology 28, 199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Insel P. A., Wilderman A., Zhang L., Keshwani M. M., Zambon A. C. (2014) Cyclic AMP/PKA-promoted apoptosis: insights from studies of S49 lymphoma cells. Horm. Metab. Res. 46, 854–862 [DOI] [PubMed] [Google Scholar]
- 7. Zhang L., Insel P. A. (2004) The Pro-apoptotic protein Bim is a convergence point for cAMP/protein kinase A- and glucocorticoid-promoted apoptosis of lymphoid cells. J. Biol. Chem. 279, 20858–20865 [DOI] [PubMed] [Google Scholar]
- 8. Insel P. A., Bourne H. R., Coffino P., Tomkins G. M. (1975) Cyclic AMP-dependent protein kinase: pivotal role in regulation of enzyme induction and growth. Science 190, 896–898 [DOI] [PubMed] [Google Scholar]
- 9. Richardson M. D., Goka T. J., Barber R., Butcher R. W. (1994) Growth of S49 wild type cells in 3 nm epinephrine increases cyclic AMP phosphodiesterase activity. Life Sci. 54, 863–875 [DOI] [PubMed] [Google Scholar]
- 10. Hochman J., Insel P. A., Bourne H. R., Coffino P., Tomkins G. M. (1975) A structural gene mutation affecting the regulatory subunit of cyclic AMP-dependent protein kinase in mouse lymphoma cells. Proc. Natl. Acad. Sci. U.S.A. 72, 5051–5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Daalen Wetters T., Coffino P. (1987) Cultured S49 mouse T lymphoma cells. Methods Enzymol. 151, 9–19 [DOI] [PubMed] [Google Scholar]
- 12. Insel P. A., Zhang L., Murray F., Yokouchi H., Zambon A. C. (2012) Cyclic AMP is both a pro-apoptotic and anti-apoptotic second messenger. Acta Physiol. (Oxf.) 204, 277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Keshwani M. M., Klammt C., von Daake S., Ma Y., Kornev A. P., Choe S., Insel P. A., Taylor S. S. (2012) Cotranslational cis-phosphorylation of the COOH-terminal tail is a keypriming step in the maturation of cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. U.S.A. 109, E1221–E1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zambon A. C., Zhang L., Minovitsky S., Kanter J. R., Prabhakar S., Salomonis N., Vranizan K., Dubchak I., Conklin B. R., Insel P. A. (2005) Gene expression patterns define key transcriptional events in cell-cycle regulation by cAMP and protein kinase A. Proc. Natl. Acad. Sci. U.S.A. 102, 8561–8566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo Y., Wilderman A., Zhang L., Taylor S. S., Insel P. A. (2012) Quantitative proteomics analysis of the cAMP/protein kinase A signaling pathway. Biochemistry 51, 9323–9332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tian Q., Stepaniants S. B., Mao M., Weng L., Feetham M. C., Doyle M. J., Yi E. C., Dai H., Thorsson V., Eng J., Goodlett D., Berger J. P., Gunter B., Linseley P. S., Stoughton R. B., Aebersold R., Collins S. J., Hanlon W. A., Hood L. E. (2004) Integrated genomic and proteomic analyses of gene expression in mammalian cells. Mol. Cell Proteomics 3, 960–969 [DOI] [PubMed] [Google Scholar]
- 17. Hegde P. S., White I. R., Debouck C. (2003) Interplay of transcriptomics and proteomics. Curr. Opin. Biotechnol. 14, 647–651 [DOI] [PubMed] [Google Scholar]
- 18. Lamb D., Steinberg R. A. (2002) Anti-proliferative effects of 8-chloro-cAMP andother cAMP analogs are unrelated to their effects on protein kinase A regulatory subunit expression. J. Cell Physiol. 192, 216–224 [DOI] [PubMed] [Google Scholar]
- 19. Kristián T., Hopkins I. B., McKenna M. C., Fiskum G. (2006) Isolation of mitochondria with high respiratory control from primary cultures of neurons and astrocytes using nitrogen cavitation. J. Neurosci. Methods 152, 136–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 21. O'Brien R. N., Shen Z., Tachikawa K., Lee P. A., Briggs S. P. (2010) Quantitative proteome analysis of pluripotent cells by iTRAQ mass tagging reveals post-transcriptional regulation of proteins required for ES cell self-renewal. Mol. Cell Proteomics 9, 2238–2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 23. Bodenmiller B., Wanka S., Kraft C., Urban J., Campbell D., Pedrioli P. G., Gerrits B., Picotti P., Lam H., Vitek O., Brusniak M. Y., Roschitzki B., Zhang C., Shokat K. M., Schlapbach R., Colman-Lerner A., Nolan G. P., Nesvizhskii A. I., Peter M., Loewith R., von Mering C., Aebersold R. (2010) Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci. Signal 3, rs4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gerencser A. A., Neilson A., Choi S. W., Edman U., Yadava N., Oh R. J., Ferrick D. A., Nicholls D. G., Brand M. D. (2009) Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal. Chem. 81, 6868–6878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Halestrap A. P. (1975) The mitochondrial pyruvate carrier. Kinetics and specificity for substrates and inhibitors. Biochem. J. 148, 85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robinson M. M., McBryant S. J., Tsukamoto T., Rojas C., Ferraris D. V., Hamilton S. K., Hansen J. C., Curthoys N. P. (2007) Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES) Biochem. J. 406, 407–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lopaschuk G. D., Wall S. R., Olley P. M., Davies N. J. (1988) Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ. Res. 63, 1036–1043 [DOI] [PubMed] [Google Scholar]
- 28. Divakaruni A. S., Rogers G. W., Murphy A. N. (2014) Measuring mitochondrial function in permeabilized cells using the Seahorse XF Analyzer or a Clark-type oxygen electrode. Curr. Protoc. Toxicol. 60, 25.2.1–25.2.16 [DOI] [PubMed] [Google Scholar]
- 29. Niikura Y., Dixit A., Scott R., Perkins G., Kitagawa K. (2007) BUB1 mediation of caspase-independent mitotic apoptosis determines cell fate. J. Cell Biol. 178, 283–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Binder J. X., Pletscher-Frankild S., Tsafou K., Stolte C., O'Donoghue S. I., Schneider R., Jensen L. J. (2014) COMPARTMENTS: unification and visualization of protein subcellular localization evidence. Database (Oxford), 10.1093/database/bau012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Field M. S., Kamynina E., Watkins D., Rosenblatt D. S., Stover P. J. (2015) Human mutations in methylenetetrahydrofolate dehydrogenase 1 impair nuclear de novo thymidylate biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 112, 400–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Laden B. P., Tang Y., Porter T. D. (2000) Cloning, heterologous expression, and enzymological characterization of human squalene monooxygenase. Arch. Biochem. Biophys. 374, 381–388 [DOI] [PubMed] [Google Scholar]
- 33. Lemire B. D. (2015) Evolution of FOXRED1, an FAD-dependent oxidoreductase necessary for NADH:ubiquinone oxidoreductase (Complex I) assembly. Biochim. Biophys. Acta 1847, 451–457 [DOI] [PubMed] [Google Scholar]
- 34. Kitamura T., Naganuma T., Abe K., Nakahara K., Ohno Y., Kihara A. (2013) Substrate specificity, plasma membrane localization, and lipid modification of the aldehyde dehydrogenase ALDH3B1. Biochim. Biophys. Acta 1831, 1395–1401 [DOI] [PubMed] [Google Scholar]
- 35. Marchitti S. A., Brocker C., Orlicky D. J., Vasiliou V. (2010) Molecular characterization, expression analysis, and role of ALDH3B1 in the cellular protection against oxidative stress. Free Radic. Biol. Med. 49, 1432–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mookerjee S. A., Goncalves R. L., Gerencser A. A., Nicholls D. G., Brand M. D. (2015) The contributions of respiration and glycolysis to extracellular acid production. Biochim. Biophys. Acta. 1847, 171–181 [DOI] [PubMed] [Google Scholar]
- 37. Divakaruni A. S., Paradyse A., Ferrick D. A., Murphy A. N., Jastroch M. (2014) Analysis and Interpretation of microplate-based oxygen consumption and pH data. Methods Enzymol. 547, 309–354 [DOI] [PubMed] [Google Scholar]
- 38. Herrmann P. C., Herrmann E. C. (2012) Mitochondrial proteome: toward the detection and profiling of disease associated alterations. Methods Mol. Biol. 823, 265–277 [DOI] [PubMed] [Google Scholar]
- 39. Pagliarini D. J., Rutter J. (2013) Hallmarks of a new era in mitochondrial biochemistry. Genes Dev. 27, 2615–2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Warda M., Kim H. K., Kim N., Ko K. S., Rhee B. D., Han J. (2013) A matter of life, death and diseases: mitochondria from a proteomic perspective. Expert. Rev. Proteomics 10, 97–111 [DOI] [PubMed] [Google Scholar]
- 41. Robertson L. S., Causton H. C., Young R. A., Fink G. R. (2000) The yeast A kinases differentially regulate iron uptake and respiratory function. Proc. Natl. Acad. Sci. U.S.A. 97, 5984–5988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang B., Zhu L., Sui S., Sun C., Jiang H., Ren D. (2014) Cilostazol induces mitochondrial fatty acid β-oxidation in C2C12 myotubes. Biochem. Biophys. Res. Commun. 447, 441–445 [DOI] [PubMed] [Google Scholar]
- 43. Conti M., Mika D., Richter W. (2014) Cyclic AMP compartments and signaling specificity: role of cyclic nucleotide phosphodiesterases. J. Gen. Physiol. 143, 29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Houslay M. D. (2010) Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem. Sci. 35, 91–100 [DOI] [PubMed] [Google Scholar]
- 45. Chen Y., Cann M. J., Litvin T. N., Iourgenko V., Sinclair M. L., Levin L. R., Buck J. (2000) Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science 289, 625–628 [DOI] [PubMed] [Google Scholar]
- 46. Litvin T. N., Kamenetsky M., Zarifyan A., Buck J., Levin L. R. (2003) Kinetic properties of “soluble” adenylyl cyclase. Synergism between calcium and bicarbonate. J. Biol. Chem. 278, 15922–15926 [DOI] [PubMed] [Google Scholar]
- 47. Giansanti P., Stokes M. P., Silva J. C., Scholten A., Heck A. J. (2013) Interrogating cAMP-dependent kinase signaling in Jurkat T cells via a protein kinase A targeted immune-precipitation phosphoproteomics approach. Mol. Cell Proteomics 12, 3350–3359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Palorini R., De Rasmo D., Gaviraghi M., Sala Danna L., Signorile A., Cirulli C., Chiaradonna F., Alberghina L., Papa S. (2013) Oncogenic K-ras expression is associated with derangement of the cAMP/PKA pathway and forskolin-reversible alterations of mitochondrial dynamics and respiration. Oncogene 32, 352–362 [DOI] [PubMed] [Google Scholar]
- 49. Gaglio D., Soldati C., Vanoni M., Alberghina L., Chiaradonna F. (2009) Glutamine deprivation induces abortive s-phase rescued by deoxyribonucleotides in k-ras transformed fibroblasts. PLoS ONE 4, e4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dent P., Wang Y., Gu Y. Z., Wood S. L., Reardon D. B., Mangues R., Pellicer A., Schonbrunn A., Sturgill T. W. (1997) S49 cells endogenously express subtype 2 somatostatin receptors which couple to increase protein tyrosine phosphatase activity in membranes and down-regulate Raf-1 activity in situ. Cell. Signal. 9, 539–549 [DOI] [PubMed] [Google Scholar]
- 51. Pichler P., Köcher T., Holzmann J., Mazanek M., Taus T., Ammerer G., Mechtler K. (2010) Peptide labeling with isobaric tags yields higher identification rates using iTRAQ 4-Plex compared to TMT 6-Plex and iTRAQ 8-Plex on LTQ Orbitrap. Anal. Chem. 82, 6549–6558 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.