

Abstract
SOX10 is required for melanocyte development and maintenance, and has been linked to melanoma initiation and progression. However, the molecular mechanisms by which SOX10 guides the appropriate gene expression programs necessary to promote the melanocyte lineage are not fully understood. Here we employ genetic and epigenomic analysis approaches to uncover novel genomic targets and previously unappreciated molecular roles of SOX10 in melanocytes. Through global analysis of SOX10-binding sites and epigenetic characteristics of chromatin states, we uncover an extensive catalog of SOX10 targets genome-wide. Our findings reveal that SOX10 predominantly engages ‘open’ chromatin regions and binds to distal regulatory elements, including novel and previously known melanocyte enhancers. Integrated chromatin occupancy and transcriptome analysis suggest a role for SOX10 in both transcriptional activation and repression to regulate functionally distinct classes of genes. We demonstrate that distinct epigenetic signatures and cis-regulatory sequence motifs predicted to bind putative co-regulatory transcription factors define SOX10-activated and SOX10-repressed target genes. Collectively, these findings uncover a central role of SOX10 as a global regulator of gene expression in the melanocyte lineage by targeting diverse regulatory pathways.
Introduction
Control of gene expression is one of the principal mechanisms by which organisms regulate fundamental cellular processes such as proliferation, survival and differentiation. This involves a complex crosstalk between cis-regulatory sequences, various trans-acting factors, and intrinsic and environmental cues in a cell type-specific manner. In eukaryotes, transcription factor binding to regulatory DNA sequences in the context of chromatin represents a key event in the molecular processes leading up to gene expression. SOX10 is a member of the SRY-related HMG-box containing (SOX) family of DNA binding transcription factors that play key regulatory roles in embryonic development and determination of cell fate (1). SOX10 is essential for the development and physiology of neural crest-derived cell populations including melanocytes, where it is required for proliferation, survival and melanogenesis (2,3). SOX10 mediates these cellular processes by directly activating a number of downstream target genes, including Mitf, Dct, Tyr and Tyrp1 (4–8). The importance of SOX10 in melanocyte/neural crest development is evidenced by the association of SOX10 mutations with the human developmental disorders Waardenburg Syndrome Type 4C, Waardenburg Syndrome Type 2E, and peripheral demyelinating neuropathy, central demyelination, WS and Hirschsprung disease (PCWH), all of which include hypopigmentation and deafness caused by defects in melanocyte development (OMIM # 609136, 611584, 613266). SOX10 is also critical for initiation and maintenance of melanoma, the highly aggressive malignancy of melanocytes, as it shows widespread expression in melanoma tumors and is important for melanoma tissue proliferation (9,10). While we understand some aspects of the transcriptional role of SOX10 in melanocytes, there has not been an exhaustive attempt to identify the targets of SOX10 on a genome-wide scale. The handful of known SOX10 targets provides a very limited view of how SOX10 guides the appropriate gene expression programs necessary to promote the melanocyte lineage.
SOX proteins, including SOX10, are thought to activate target genes through direct DNA contacts via specific sequence motifs involving A/TA/TCAAA/TG (11). Moreover, some SOX proteins are also known to be involved in gene repression by competing with transcriptional activators for binding to regulatory regions and by recruiting co-repressors (12). The versatility of SOX proteins to promote both gene activation and repression suggests as yet undiscovered regulatory roles for SOX10 in melanocytes. Although SOX10 is known to interact with an extensive set of factors, including the RNA pol II transcription machinery, chromatin remodeling complexes, histone deacetylases and cell cycle regulators (1), it is not known whether SOX10 is involved in transcriptional repression. In support of the idea that SOX10 might be involved in both transcriptional activation and repression of genes, we previously observed that overexpression of SOX10 specifically within the melanocyte lineage in vivo is sufficient to upregulate the melanocyte enzyme TYRP1 and increase pigment production within the melanocyte stem cell (McSC) population (3). Unexpectedly, overexpression of SOX10 resulted in the decreased expression of KIT (3), a tyrosine kinase receptor protein that is essential for the survival, growth, differentiation and migration of melanocytes (13). Moreover, in Sox10 null mouse embryos, the expression of the SOX10 target genes Dct, Mitf and Tyr was abolished but a small population of Kit+ cells was retained (14). These data suggest SOX10 may act to both activate and repress downstream targets; however, further analysis is required to fully determine the extent to which SOX10 affects genome-wide transcription in melanocytes.
We propose that high-throughput analysis of transcription factor binding and chromatin states using chromatin immunoprecipitation sequencing (ChIP-Seq) combined with functional genetic and genomic experiments provides the ideal approach to revealing the mechanistic relationship between cis-regulatory elements, transcription factor binding, the epigenetic landscape and gene expression changes. Using in vitro and in vivo models, we present data showing an inverse relationship of Kit and Sox10 expression levels in melanocytes, which would correlate with SOX10 negatively regulating Kit. Via global analysis of SOX10-binding sites and chromatin states, we identify putative cis-regulatory elements that reveal a wide repertoire of SOX10 genomic targets. Furthermore, by integrating epigenomic and transcriptome data we uncover key insights into the functions and mechanisms of SOX10-mediated gene regulation in melanocytes.
Results
SOX10 and Kit expression in the melanocyte lineage
To further investigate the inverse correlation of Sox10 and Kit expression in the melanocyte lineage (3,14), we analyzed Kit gene expression in melan-Ink4a-Arf-1 (melan-a) (15) and melan-Sox10-1 melanocytes, the latter of which are Sox10 haploinsufficient (and referred to here as Sox10LacZ/+ cells; see Methods) and show reduced expression of Sox10 mRNA (Fig. 1A) and SOX10 protein (data not shown). By RT-PCR assays, Kit expression is increased in Sox10LacZ/+ cells (Fig. 1A), and immunohistochemistry shows that a significant percentage of Sox10LacZ/+ melanocytes exhibit high KIT+ immunoreactivity (29.4% high), unlike the syngeneic melan-a SOX10+ melanocytes which exhibit very low to no detectable KIT+ staining (0% high) (Fig. 1B; x2 test P = 5 × 10−9).
Figure 1.

SOX10 and KIT expression are inversely correlated in melanocytes. (A) Quantitative RT-PCR for Sox10 and Kit gene expression in melan-a (control) and Sox10LacZ/+ melanocyte cell lines demonstrates a 50% reduction in Sox10 expression, and a greater than 4-fold increase in Kit gene expression in association with Sox10 haploinsufficiency. Error bars indicate standard deviation of the fold-change corrected for the coefficient of variance for both target and endogenous control expression values. (B) Immunolabeling for KIT protein in vitro reveals that select Sox10LacZ/+ cells exhibit positive KIT expression (green) that is not apparent in control melan-a cells. Individual cells are indicated by nuclear staining for SOX10 (red). (C) Immunolabeling for KIT protein in wild type C57BL/6J mouse hair follicles at postnatal day 2 demonstrates that melanocyte stem cells (arrows) exhibit high KIT fluorescence intensity (green) while hair bulb melanocytes (arrowheads) produce low KIT fluorescence intensity. Hair follicles are outlined by dotted white lines, and melanocyte stem cells and melanocytes are visualized in the right panel by positive staining for the melanocyte marker, DCT (red). (D–G) Double immunolabeling for the melanocyte marker DCT (red) and KIT (green) in mid-anagen hair follicles from wild type, Tg(Dct-Sox10)/0, Sox10LacZ/+ and KitLacZ/+ adult mice demonstrates variability in KIT fluorescence intensity within the hair bulb melanocytes based on genotype. Dotted red lines mark the boundaries of positive DCT staining and indicate the ROI used to calculate KIT fluorescence pixel intensity within hair bulb melanocytes in (H). Hair follicle bulbs are outlined by dotted white lines. (H) Box and whisker plot of KIT mean fluorescence intensity per pixel exhibited by DCT+ hair bulb melanocytes and defined by individual ROIs (error bars indicate min to max, box defines the 25–75th percentile and line marks the median). Examples of ROIs are indicated by the dotted red lines in (D–G). A minimum of 125 ROIs were assessed per genotype and statistical significance determined by the Kolmogorov–Smirnov test (*P < 0.0001). (I) Quantification of the average number of MITF+ nuclei contained within the ROIs used to calculate KIT fluorescence intensity in (H) demonstrates that similar numbers of nuclei per ROI are observed across genotypes. Error bars indicate the standard deviation. (J) Immunolabeling of hair bulb melanocytes demonstrates that tamoxifen-induced Sox10 knockout in adult Sox10fl/fl;Tyr::CreERT2 mice results in the production of a population of PAX3+/SOX10− hair bulb melanocytes (arrows). Triple labeling for KIT further demonstrates that these PAX3+/SOX10− hair bulb melanocytes exhibit notably higher KIT fluorescence intensity than hair bulb melanocytes that retain SOX10 expression (arrowheads). (K) Box and whisker plot of the mean KIT fluorescence intensity per pixel for individual hair bulb melanocytes. Each hair bulb melanocyte was defined by a fixed diameter ROI (10 μm) that was used to encircle each PAX3+ melanocyte nuclei. A minimum of 45 ROIs was assessed per category, and statistical significance determined by the Kolmogorov–Smirnov test (*P < 0.0001). (L–N) Individually, Tg(Dct-Sox10)/0 and KitLacZ/+ genetic alterations produce mice with small white belly spots. Together, overexpression of Sox10 in combination with Kit haploinsufficiency (Tg(Dct-Sox10)/0;KitLacZ/+) results in more extensive congenital white spotting that is present on both the dorsum and ventrum.
To explore the relationship between Sox10 and KIT in vivo, we examined postnatal melanocyte and McSCs within the mouse hair follicle. By immunolabeling hair follicles from wild type C57BL/6 animals at postnatal day 2, we observed that KIT fluorescence intensity appears high in the McSC population and is significantly reduced in the bulb melanocytes (Fig. 1C). This is inversely correlated with data from a previous study, which showed that Sox10 is expressed at relatively low levels in McSCs and at high levels in the more differentiated hair bulb melanocytes that reside within the hair follicle (16). Further, we quantitatively examined changes in KIT protein levels in response to genetically altered Sox10 expression by measuring mean fluorescence intensity per pixel in the DCT+ hair bulb melanocytes found in mid-anagen hairs of adult animals (Fig. 1D–F). Using DCT to define the anatomical boundaries of the hair bulb melanocytes (region of interest, ROI) allowed us to distinguish melanocyte-specific KIT expression from the intense KIT immunoreactivity produced by hair matrix keratinocytes (17). When comparing the distribution of mean fluorescence intensity per hair bulb across genotypes, we found that KIT intensity within the melanocyte ROI is significantly increased with Sox10 haploinsufficiency (Sox10LacZ/+) and significantly decreased with hemizygous Sox10 overexpression (Tg(Dct-Sox10)/0) in comparison to wild type (Fig. 1D–F and H). Haploinsufficiency for Kit (Kittm1Alf/+ (18), referred to here as KitLacZ/+) serves as a control for decreased KIT expression and accordingly shows the lowest KIT intensity per pixel within the melanocyte ROI (Fig. 1G and H). We confirmed that these differences in KIT intensity between genotypes are not simply due to pigment-mediated fluorescence quenching, as H2O2 treatment significantly reduces the light absorbance properties of melanin pigment (19) yet does not change the fluorescence intensity of KIT immunolabeling (Supplementary Material, Fig. S1). Similarities in the number of melanocytes per bulb in each of these genotypes further indicates that these intensity observations are likely due to molecular changes in individual melanocytes rather than a reflection of differences in melanocyte cell number per hair bulb (Fig. 1I).
We also assessed the immediate changes in KIT expression that occur in response to Sox10 reduction in vivo. For this we utilized Sox10fl/fl;Tyr::CreERT2 transgenic mice in which a tamoxifen-inducible, Cre recombinase-estrogen receptor ligand binding domain fusion transgene is under the control of the Tyr promoter to drive homozygous deletion of Sox10 in melanocytes (3). We reported previously that subsequent to tamoxifen-induced Sox10 knockout, a transient population of PAX3+/SOX10− melanocytes is observed in the hair bulb at mid-anagen [Fig. 1J; (3)]. We performed a similar experiment here and treated adult Sox10fl/fl; Tyr::CreERT2/+ mice with systemic tamoxifen for 4 days after initiating the hair cycle by plucking the hairs on their lower back. Seven days after plucking we immunolabeled mid-anagen hairs using specific antibodies to PAX3, SOX10 and KIT. When measuring mean KIT fluorescence intensity per pixel in a defined region encircling each PAX3+ nucleus, we found that KIT intensity is noticeably higher in the PAX3+/SOX10− melanocytes than in those melanocytes that retain SOX10 expression (Fig. 1J and K).
To examine a possible genetic interaction between Sox10 and Kit in vivo, we crossed mice carrying one copy of the Dct-Sox10 transgene (Tg(Dct-Sox10)/0) with KitLacZ/+ mice to determine if overexpression of Sox10 exacerbates haploinsufficiency for Kit. Individually, Tg(Dct-Sox10)/0 and KitLacZ/+ mice both exhibit mild to moderately sized white belly spots at birth (Fig. 1L and M), while Tg(Dct-Sox10)/0;KitLacZ/+ mice exhibit more severe ventral and dorsal white spotting (Fig. 1N). This synergistic effect in the hypopigmentation phenotype is consistent with the participation of SOX10 in the regulation of Kit. Collectively, these observations demonstrate that KIT expression inversely correlates with SOX10 in vitro and in vivo, both physiologically when McSC progeny differentiate into melanocytes in the hair bulb, and in response to stable and transient changes in SOX10 levels. Our findings are consistent with a novel model where SOX10 plays a role in repressing the expression of Kit along with additional targets in melanocytes.
Genome-wide analysis of SOX10 chromatin occupancy in melanocytes
Given the finding that SOX10 is potentially involved in the negative regulation of Kit, we wanted to determine the extent of SOX10-mediated transcriptional regulation on a genome-wide scale and uncover the full range of its genomic targets. Thus, we performed global analysis of SOX10 transcription factor binding using ChIP-Seq on chromatin derived from melan-a melanocytes. We obtained ChIP-Seq data from two independent biological replicates, and only profiles that showed reproducible overlap between both samples were used for downstream analyses (strategy outlined in Supplementary Material, Fig. S2A). Analysis of SOX10 ChIP-Seq sequence tags using MACS (Model-based Analysis of ChIP-Seq) identified 4085 SOX10-binding sites (peaks) genome-wide that replicated across experiments (Supplementary Material, Table S1). To assess the validity of the SOX10 ChIP-Seq peaks we first performed de novo motif analysis using the MEME-ChIP algorithm (20). MEME-ChIP analysis identified two highly enriched de novo sequence motifs that show significant similarity to a known SOX10 motif (MA0442.1, JASPAR database). The first motif, Motif A (motif enrichment E-value = 1.60e-190), is a single seven-nucleotide consensus sequence with a motif match P-value of 2.28e-06 to a known SOX10 motif in the database (Fig. 2A). This suggests that Motif A represents the monomeric binding site of SOX10 in vivo. The second motif, Motif B (motif enrichment E-value = 6.10e-60), involves an inverted spacing of two consensus sequences that also significantly resemble the SOX10-binding site MA0442.1 (motif match P = 1.01e-04), which is consistent with SOX10 binding to DNA in vivo as a head-to-head dimer and in keeping with previous observations in other cell types in vitro (23,24) and in vivo (25). The total SOX10-binding peaks contained 34%, 29% and 57% of Motifs A, B and MA0442.1, respectively. Another assessment of SOX10 ChIP-seq validation was to use central motif enrichment analysis of the identified consensus sequences using Centrimo (21), an algorithm that analyzes the positional distribution of enriched motifs relative to ChIP-seq peak summits. Centrimo results show that both Motif A and Motif B are centrally enriched near the summits of SOX10 ChIP-seq peaks with motif enrichment E-values of 1.7e-125 and 9.1e-148, respectively (Fig. 2B). Similarly, SOX10 motif MA0442.1 exhibits a central motif enrichment with E-value of 1.7e-92, which is indicative of a strong central enrichment. Taken together, these results demonstrate that SOX10 binds to DNA as both a monomer and homodimer in vivo and its chromatin occupancy is mediated via its direct DNA binding activity. In addition, de novo motif analysis of the SOX10 ChIP-Seq regions revealed the presence of potential binding sites for a series of transcription factors that could occupy SOX10-regulated genomic sites and thus potentially regulate its target genes (Supplementary Material, Fig. S3).
Figure 2.

ChIP-Seq analysis of SOX10 chromatin occupancy in melanocytes. (A) Letter logo representations of SOX10 consensus sequences identified using MEME-ChIP (20). De novo motif analysis was performed using sequences within a [−150 bp, +150 bp] window of SOX10 ChIP-Seq summits. Consensus sequences showing significant similarity to known SOX10 motifs are shown. Letter logo size indicates nucleotide frequency and corresponding E-values represent the significance of motif enrichments compared with the genomic background. (B) Graph generated from Centrimo (21) analysis showing the distribution of occurrence probabilities of the consensus sequences enriched under SOX10 ChIP-Seq peaks relative to SOX10-binding summits, i.e. position of best site in sequence. Central motif enrichment E-values are: E-value = 9.1e-148 for Motif B; E-value = 1.7e-125 for Motif A; the distribution of a previously known SOX10 motif (MA0442.1) is shown for comparison (Central motif enrichment E-value = 1.7e-92). (C) Functional GO enrichment analysis of genes associated with SOX10 peaks using GREAT (22).
To evaluate the biological relevance of sequences enriched under SOX10 ChIP-seq peaks we performed evolutionary conservation analysis, a feature known to be associated with regulatory potential, using the PhastCons tool in the Cistrome Integrative Analysis Pipeline (26). Examination of the distribution of PhastCons scores for sequences in a [−1500 bp, +1500 bp] window from SOX10 ChIP-seq summits shows that conservation is highest near the summits of SOX10-binding sites as compared with typical adjacent genomic regions among both 19 placental mammals and 30 vertebrate species (Supplementary Material, Fig. S2B), thus supporting regulatory function. To examine whether the corresponding orthologous human sequences in these highly conserved SOX10-binding sites overlap any SNPs or mutations previously associated with human traits and diseases, we used UCSC genome browser's LiftOver tool to convert the 4085 SOX10 ChIP-Seq peak coordinates in mouse (NCBI37/mm9 assembly) to the corresponding coordinates in the human genome (GRCh37/hg19 assembly) and intersected them with all SNPs contained in the GWAS catalog (27). Of the 4085 SOX10 ChIP-Seq peaks in mouse, 3717 peaks were successfully converted to human genome coordinates and contain 58 SNPs previously identified in published GWAS studies (GWAS catalog; Supplementary Material, Table S2), suggesting potential relevance of SOX10 to the phenotypes associated with these SNPs. Of note was SNP rs12203592, which is known to alter activity of an IRF4 enhancer (28) and is associated with several pigmentation-related phenotypes, including hair color, freckling and tanning (29–31).
Given the established role of SOX10 in the regulation of melanocyte development, maintenance and function, we predicted that genomic sites specifically binding to SOX10 in melanocytes would be enriched near genes relevant to melanocyte biology. Thus, we mapped SOX10 peaks to the nearest transcription start sites (TSSs) of mouse genes (NCBI Build 37 or UCSC mm9) using the Genomic Regions Enrichment of Annotations Tool (GREAT) algorithm (22), and identified 4332 genes associated with the 4085 SOX10 peaks (Supplementary Material, Table S3). GREAT analysis revealed that SOX10 localizes to regulatory regions of genes whose functions are linked to gene ontology (GO) biological process terms directly related to melanocyte development, pigmentation and differentiation (Fig. 2C; Supplementary Material, Table S4). In addition to these known biological roles associated with SOX10 genomic binding, our results suggest previously uncharacterized putative functions for SOX10 in regulating the expression of genes associated with actin cytoskeleton/filament-related processes, negative regulation of transferase activity and negative regulation of kinase activity (Fig. 2C).
Next, we investigated the localization of SOX10 peaks relative to gene regulatory features such as promoters [within a (−2.5 kb, +2.5 kb) window flanking each TSS], exons, introns and intergenic regions. Our analysis showed that the majority of SOX10-binding sites in melanocytes are either intronic or intergenic (Fig. 3A). Interestingly, only a small fraction (<15%) of the SOX10-binding sites are found within 5 kb of any TSS, while the majority (>80%) are located 50–500 kb from the closest TSS (Fig. 3B). This indicates that SOX10 predominantly functions through distal regulatory elements such as enhancers. We examined whether this distal site bias of SOX10 localization is due to higher affinity of binding at distal sites than TSS proximal regions using a strategy similar to that described by Schnetz et al. (32). We classified SOX10 peaks as TSS binding [within a (−2.5 kb, +2.5 kb) window flanking each TSS], gene body binding or distal intergenic binding, and compared their ChIP-Seq enrichment signals. We found that SOX10-binding enrichment is higher at distal sites than gene body sites or TSSs (Fig. 3C). In addition, SOX10 consensus motifs are more enriched in distal SOX10 peaks (E-value < 0) than in TSS SOX10 peaks (E-value > 1) (Supplementary Material, Fig. S2C).
Figure 3.

Genomic distribution of SOX10-binding sites in melanocytes. (A) Pie chart showing the distribution of SOX10 peaks across gene features (promoters or within [−2.5 kb, +2.5 kb] window around TSS, exons, introns and intergenic regions). (B) Genomic distribution of SOX10 peaks with respect to TSS of genes associated with SOX10 ChIP-Seq peaks in melanocytes. Peak and gene associations were performed using GREAT (22) by applying the basal plus extension settings. GREAT assigns biological meaning to genomic regions by analyzing the annotations of nearby genes and regulatory elements wherein genomic distances are divided into bins relative to TSS as [0, 5 kb], [5 kb, 50 kb], [50 kb, 500 kb], [500 kb, infinity]. Values on the Y-axis represent percent of peaks found in a given genomic region. Values displayed on bar graphs represent number of genes associated with peaks in each genomic window. (C) Top, schematic showing genomic categories for classifying SOX10 ChIP-Seq peaks. TSS SOX10 peaks = peaks within [−2.5 kb, +2.5 kb] of TSS; Distal intergenic SOX10 peaks = peaks >2.5 kb away on either sides of TSS; gene body SOX10 peaks = peaks not in TSS or gene body. Bottom, line plot showing average ChIP-seq signal intensities (read counts on y-axis) for SOX10 peaks located at TSS, gene body and distal intergenic site for a [−3000 bp, +3000 bp] window with respect to SOX10 summits (x-axis).
Relationship of SOX10 chromatin occupancy with melanocyte enhancers and other epigenetic features
Previously, via large-scale genomic profiling of the histone acetyltransferase EP300 and the histone modification H3K4me1 as generic chromatin signatures of enhancers, we identified 2489 putative melanocyte enhancer elements that exhibit predicted binding sites for transcription factors such as SOX10 and MITF, and enhanced transcription in vitro and in vivo (33). Since we found that SOX10 primarily binds to distal intergenic regions, likely reflecting targeting of enhancers, we assessed the genomic overlap of our SOX10 peaks with this previously published melanocyte enhancer dataset (33). Our analysis showed that over 50% of the melanocyte enhancers overlap SOX10-occupied genomic regions (Fig. 4A), further substantiating that SOX10 is a bona fide enhancer-binding transcription factor in melanocytes.
Figure 4.

Analysis of the relationship between SOX10 genomic binding and epigenetic chromatin properties in melanocytes. (A) Venn diagram showing overlap of SOX10 peaks with putative melanocyte enhancer elements. (B–D) UCSC genome browser tracks showing genomic interaction of SOX10 with distal enhancer elements at Kit, Tyr, and Sox10 loci, respectively, in melanocytes. Values on the y-axis represent input-normalized read counts or intensities of ChIP-Seq data. For each locus shown, arrows indicate replicated SOX10 peaks having statistically significant binding enrichment over genomic background (input DNA). Green rectangular blocks below the genome browser tracks indicate putative melanocyte enhancer elements (33). Numbers 1 through 9 below the Sox10 genome browser track correspond to approximate locations of Sox10 multispecies conserved sequences or Sox10-MCS (24). (E) Relationship of SOX10 cistrome with EP300 chromatin occupancy and histone modification (H3K4me1, H3K4me2 and H3K4me3) regions identified by ChIP-Seq. Values in each square represent percent overlap of a peak or chromatin region on the left with another peak or chromatin region on the top by at least 1 bp. All ChIP-Seq regions represent replicated peaks from at least two independent experiments. SOX10 (n = 4085); EP300 [n = 3539; (33)]; H3K4me1 [n = 75956; (33)]; Enhancers [n = 2489; (33)]; H3K27ac (n = 52 553); H3K27me3 (n = 37 270). (F) Distribution of SOX10, H3K4me1 (33), EP300 (33), H3K27ac and H3K27me3 ChIP-Seq reads across all SOX10-occupied regions in melanocytes. For each factor average ChIP-Seq reads are plotted within [−3 kb, +3 kb] window around SOX10 peak centers (summits).
Given our in vitro and in vivo data consistent with SOX10 playing a role in the negative regulation of Kit (Fig. 1), we hypothesized that SOX10 directly binds to Kit regulatory elements. Indeed, examination of SOX10 ChIP-Seq at the Kit locus revealed multiple SOX10-binding regulatory elements positioned at −227, −109 and +109 kb with respect to the Kit TSS (Fig. 4B). Interestingly, the −109 kb SOX10-binding element overlaps a previously identified enhancer. Although not significantly enriched based on the P-value cutoff we applied for the detection of SOX10 ChIP-Seq peaks, there is also SOX10 binding to the Kit promoter. These SOX10-binding sites at the Kit locus were confirmed using ChIP/qRT-PCR analysis of chromatin derived from independent experiments (Supplementary Material, Fig. S4). Overall, these findings suggest that SOX10 binding to Kit cis-regulatory elements might be involved in the negative regulation of Kit expression in melanocytes.
ChIP-Seq analysis also revealed SOX10 binding to numerous previously identified regulatory regions of pigmentation genes, thus providing validation of the functionality of our set of SOX10-binding sites. These regions include: a regulatory region at the 5′-region of the Tyr gene TSS (Fig. 4C) that overlaps the approximate site of a previously identified putative melanocyte enhancer element (33), as well as previously identified regions where SOX10 directly binds and activates transcription at Tyrp1 (7), Dct (34), Mef2c (35) and Erbb3 (36) (Supplementary Material, Fig. S5). Although SOX10 ChIP-Seq peaks do not overlap a previously reported SOX10 site within Mitf (5,37), our in vivo ChIP-Seq analysis identified multiple SOX10-binding sites at the Mitf locus (Supplementary Material, Fig. S5). ChIP-Seq analysis also identified several SOX10-binding sites at regulatory regions in the 5′-region of the Sox10 locus itself (Fig. 4D) that were identified previously as multispecies conserved sequences (Sox10-MCS) (24), Importantly, one of these SOX10-binding sites within the Sox10 locus overlaps an element (Sox10-MCS7) whose deletion causes a Waardenburg-Shah syndrome-like phenotype (38) suggesting a direct link between SOX10 transcription factor binding and functional consequences. Taken together, these findings establish that distal enhancer binding is a key molecular feature of SOX10 transcriptional activity and function in melanocytes.
Patterns of histone modifications can distinguish between active and inactive/poised chromatin states, which in turn correlate with the expression status of neighboring genes. For instance, the level of histone H3 acetylation on lysine residue 27 (H3K27ac) is strongly correlated with active chromatin, whereas tri-methylation of the same histone residue (H3K27me3) is associated with chromatin regions repressed by polycomb protein complexes (39–41). Likewise, a combination of histone modification marks can identify specific classes of enhancers, namely active enhancers marked by both H3K4me1 and H3K27ac, and inactive/poised enhancers marked by the presence of H3K4me1 and the absence of H3K27ac marks (40,41). To expand our understanding of the epigenetic landscape of SOX10 chromatin occupancy, we generated genome-wide profiles of H3K27ac and H3K27me3 using ChIP-Seq in melan-a cells (Supplementary Material, Table S1). Then we calculated the genome-wide overlap of SOX10 peaks with H3K27ac, H3K27me3 and the previously generated genomic melanocyte datasets [H3K4me1, EP300 and melanocyte enhancers (33)]. Of the 4085 SOX10 peaks detected in melanocytes, 99% overlap with H3K4me1, 42% with EP300 and 51.2% with H3K27ac (Fig. 4E; Supplementary Material, Table S1). In contrast, the overlap of SOX10 peaks with H3K27me3 is very low (∼0.8% of the time; Fig. 4E) suggesting that SOX10 binding is not frequently associated with polycomb protein-repressed chromatin regions in proliferating melanocytes. In addition, as shown in Figure 4E and Supplementary Material, Table S1, ∼52% of melanocyte enhancers (33) overlap SOX10 peaks. Remarkably, only 0.5% of melanocyte enhancer elements overlap with H3K27me3 regions in contrast to a 57.8% overlap with H3K27ac regions (Fig. 4E; Supplementary Material, Table S1). The relationship of SOX10 chromatin occupancy with the various chromatin features is also evident from examination of the cumulative enrichment profiles of H3K4me1, EP300, H3K27ac and H3K27me3 wherein all but H3K27me3 show obvious enrichment around SOX10-binding sites (Fig. 4F). Specifically, there is a bimodal distribution of both H3K4me1 and H3K27ac around the SOX10 peak summit, and EP300 exhibits an enrichment peak coinciding with the SOX10-binding profile. In contrast, H3K27me3 is generally not enriched at these genomic regions. We also determined the fraction of SOX10-binding sites that reside within the accessible (‘open’) chromatin in melan-a cells using DNase I hypersensitive sites sequencing (DNase-Seq), another approach used to identify active regulatory elements in the genome (42). We found that SOX10 DNA binding occurs almost exclusively within the ‘open’ chromatin region, with a 97.99% overlap between SOX10 peaks and DNase-Seq sites (data not shown). In summary, integration of global SOX10-binding profile, melanocyte enhancer datasets and epigenetic signatures characterizing various chromatin states suggest that SOX10 engages ‘open’ chromatin, mainly binds to distal regulatory sites including a majority of melanocyte enhancer loci, and frequently overlaps with active chromatin regions bearing the H3K27ac mark.
Identification of functional target genes of SOX10 in melanocytes
Although a biological role for SOX10 in melanocytes and melanoma has been well documented (2,3,9,10,43), the molecular functions of SOX10 DNA binding, as it relates to activating or repressing transcription, have not been fully evaluated in the melanocyte lineage. To identify the functional downstream targets of SOX10, we quantitatively assessed the consequences of Sox10 haploinsufficiency on global gene expression in melanocytes using microarray analysis of RNA extracted from Sox10LacZ/+ cells and melan-a cells. Expression analysis of microarray data identified a total of 1549 transcripts, or 1438 unique genes as differentially regulated (P < 0.05) between Sox10LacZ/+ and melan-a cells (Fig. 5A). Of the 1438 genes, 614 genes are downregulated and 824 genes are upregulated in Sox10LacZ/+ cells (Fig. 5B; Supplementary Material, Table S5). This simultaneous upregulation and downregulation of genes following reduction of Sox10 suggests that SOX10 may play a role in both activation and repression of melanocyte genes. Biological pathway enrichment analysis of downregulated and upregulated genes using Ingenuity Pathway Analysis software (IPA®, QIAGEN, www.qiagen.com/ingenuity) shows that downregulated genes in Sox10LacZ/+ cells (genes presumably activated by SOX10) are significantly enriched in pathways regulating melanocyte development and pigmentation, in keeping with the known biological roles of Sox10 in melanocytes (Supplementary Material, Table S6). In addition, these SOX10-activated genes are significantly enriched in a number of biological pathways not described previously as regulated by SOX10, including Clathrin-mediated Endocytosis Signaling, Micropinocytosis Signaling, Integrin Signaling and Focal Adhesion Kinase (FAK) Signaling. On the other hand, Sox10LacZ/+- upregulated genes (genes presumably repressed by SOX10) are highly enriched in pathways such as Embryonic Stem Cell Pluripotency and Regulation of the Epithelial-Mesenchymal Transition Pathway (EMT), the latter of which includes genes involved in TGFb, WNT and PDGF signaling (Supplementary Material, Table S6). Taken together, these results support our hypothesis that SOX10 may play dual roles in the transcriptional regulation of genes expressed in melanocytes: activating genes required for melanocyte proliferation, survival and pigmentation, and potentially repressing genes that promote pluripotency/self-renewal and EMT/invasion.
Figure 5.

Identification of SOX10 target genes in melanocytes. (A) Volcano plot showing all differentially regulated transcripts or probe sets in Sox10LacZ/+ versus melan-a RNA samples assayed by microarray on the Affymetrix GeneChip® Mouse Gene 1.0 ST array. Significance probability values (y-axis) are plotted against the Log2 (expression fold changes) (Sox10LacZ/+/melan-a) on x-axis. Red = upregulated; Green = downregulated. Horizontal blue line represents unadjusted significance P-value cutoff of 0.05. (B) Summary of significantly upregulated or downregulated transcripts (genes) in Sox10LacZ/+ versus control melan-a RNA samples at FDR (B–H) adjusted P < 0.05. (C) Venn diagram showing the overlap of SOX10 ChIP-Seq associated genes (genes that have at least one SOX10-binding site associated with them) and genes whose expression is significantly altered in Sox10LacZ/+ versus control melan-a cells. (D) IPA Pathway enrichment analysis of SOX10-activated and SOX10-repressed target genes, i.e. genes having at least one associated SOX10 peak and showing significant differential expression change in Sox10LacZ/+ versus melan-a cells. Upper panel, SOX10-activated target genes; lower panel, SOX10-repressed target genes. Negative Log10 (B–H corrected P-values) represent significance of enrichment for pathway members shown.
Next, we took an integrated analysis approach to identify the functional target genes of SOX10 DNA binding activity by correlating SOX10-binding profile with gene expression changes related to Sox10 haploinsufficiency. A gene was considered to be a functional target of SOX10 if it shows significant differential expression in Sox10LacZ/+ versus melan-a cells and has at least one SOX10 ChIP-Seq peak associated with it. Of the 4332 genes associated with SOX10 ChIP-Seq, our integrated analysis identified 510 genes that also had significant expression changes (hereafter referred to as ‘SOX10 target genes,’ Fig. 5C). Two hundred and forty-nine of them are significantly downregulated in Sox10LacZ/+ cells (hereafter called ‘SOX10-activated genes’), and the remaining 251 are upregulated in Sox10LacZ/+ cells (hereafter referred to as ‘SOX10-repressed genes’). Thus, there were approximately equal numbers of SOX10-activated genes and SOX10-repressed genes. Biological pathway analysis of SOX10-activated genes reveals enrichment of these genes in pathways related to melanocyte development and pigmentation, in agreement with the known role of SOX10 in melanocytes (Fig. 5D, upper panel; Supplementary Material, Table S7). In addition, SOX10-activated genes are linked to pathway terms such as Integrin Signaling, FAK Signaling and Micropinocytosis Signaling. On the other hand, SOX10-repressed genes show significant enrichment in pathways that are critical regulators of early developmental processes and cancer metastasis, including Human ES cell pluripotency, Mouse ES cell pluripotency and Regulation of the EMT pathway (Fig. 5D; lower panel; Supplementary Material, Table S7). In summary, these results suggest SOX10 shapes the melanocyte gene expression program both by promoting activation of pro-proliferation and pro-pigmentation genes, and also by repressing genes that promote pluripotency/self-renewal and EMT/invasion, cellular phenotypes incompatible with differentiated melanocytes and common traits of early developmental processes and cancer metastasis.
Epigenetic and cis-regulatory mechanisms of transcriptional regulation by SOX10
Having identified a set of 510 functional SOX10 target genes, we sought to identify the molecular mechanisms that distinguish the transcriptional upregulation and downregulation of these genes in response to changes in SOX10 cellular levels. In addition to the underlying DNA sequence, chromatin landscape is a key determinant of eukaryotic gene expression, and specific chromatin histone modification patterns are known to be correlated with the transcriptional status of the genome (39–41). We hypothesized that the magnitude of SOX10-dependent transcriptional outcome is a function of both the chromatin context and the underlying cis-regulatory sequences at these 510 target genes. We first observed that independent of genotype, SOX10-activated genes are expressed at higher levels on average than SOX10-repressed genes in melanocytes (Fig. 6A). To find out whether SOX10-activated and SOX10-repressed genes exhibit distinctive epigenetic statuses, we ranked all functional SOX10 target genes according to their expression fold change in Sox10LacZ/+ cells from low (SOX10-activated genes) to high (SOX10-repressed genes). Then we plotted the ChIP-Seq enrichment profiles of H3K4me1 (33), EP300 (33), H3K27ac and H3K27me3 over a [−3 kb, +3 kb] window relative to the TSS of the ranked gene list (Fig. 6B). This analysis revealed that SOX10-activated and SOX10-repressed genes do not exhibit clear differences in the enrichment of the general enhancer marks H3K4me1 and EP300 (Fig. 6B; two heat maps on the left), but H3K27ac and H3K27me3 enrichment profiles are distinctly different between SOX10-activated and SOX10-repressed genes (Fig. 6B; two heat maps on the right). Overall SOX10-activated genes show high levels of H3K27ac and low levels of H3K27me3, while most SOX10-repressed genes exhibit a silenced chromatin landscape (low H3K27ac and high H3K27me3). Examples of these varied histone modification patterns are shown for a SOX10-repressed gene (Itpr2) and a SOX10-activated gene (Dct) in melan-a cells (Fig. 6). The Dct locus displays a higher frequency of H3K27ac, and remarkably no H3K27me3 surrounding its SOX10-binding sites (Fig. 6C; upper panel) as compared with the Itpr2 locus, which shows very high frequency of H3K27me3 (Fig. 6C, lower panel). The presence of some histone acetylation despite high H3K27me3 at the Itpr2 locus indicates that SOX10-repressed genes are not simply genes already silent in melanocytes, but could be expressed genes maintained in a poised state by SOX10. In summary, these observations indicate that SOX10-activated and SOX10-repressed genes are distinguishable based on their epigenetic status.
Figure 6.

SOX10-activated genes and SOX10-repressed genes exhibit distinct regulatory mechanisms. (A) Average expression level of all SOX10-activated and SOX10-repressed genes. (B) Heat map showing the enrichment profiles of EP300 (33), H3K4me1 (33), H3K27ac and H3K27me3 within [−3 kb, +3 kb] window around the TSS of functional SOX10 target genes. Functional SOX10 target genes were ordered according to relative expression changes. Each row on the heat maps represents one gene. SOX10-activated target genes are at the top of the heat maps while SOX10-repressed target genes are at the bottom of the heat maps. On the heat maps, red color = strong enrichment; yellow color = weak enrichment and white color = no enrichment. (C) UCSC genome browser showing SOX10 ChIP-Seq profile and histone modification marks (H3K27ac and H3K27me3) at SOX10-activated (Dct locus) and SOX10-repressed (Itpr2 locus) targets. Values on the y-axis represent input-normalized read counts or intensities of ChIP-Seq data. For each locus shown, rectangular blocks under the genome browser tracks indicate ChIP-Seq peak boundaries for H3K27ac (in red) and H3K27me3 (in black). (D) Cis-regulatory sequences associated with SOX10-activated and SOX10-repressed targets in melanocytes identified using the BETA tool of the Cistrome Analysis Pipeline (26). Letter logos show transcription factor consensus motifs enriched in SOX10-activated and SOX10-repressed targets. Corresponding P-values represent the significance of motif enrichments compared with the background (non-targets) sequences.
Lastly, we sought to investigate whether the underlying cis-regulatory DNA motifs determine SOX10-mediated transcriptional outcomes. We performed integrated cis-regulatory motif enrichment analysis on the 510 SOX10-activated and SOX10-repressed targets using an integrative tool in the Cistrome Analysis Pipeline (26), which combines transcription factor binding and gene expression data to identify putative transcription factor motifs involved in transcriptional activation and repression. Surprisingly, this analysis revealed that SOX10 consensus motifs are enriched across both SOX10-activated and SOX10-repressed targets with very similar occurrence probabilities (Fig. 6D), suggesting that SOX10 DNA binding motif alone does not distinguish between SOX10-activated and SOX10-repressed genes. However, closer examination of the composition of enriched sequence motifs across the categories of SOX10-activated and SOX10-repressed targets identified distinct secondary consensus sequence motifs. Specifically, SOX10-activated targets show significant enrichment of consensus binding sequences corresponding to the CCAAT/enhancer-binding protein (C/EBP) beta (CEBPB) and the basic leucine zipper (bZIP) domain proteins BHLHB2 (basic helix-loop-helix domain containing, class B, 2) and CREB3L2 (cAMP responsive element binding protein 3-like 2) (Fig. 6D; left panel). This suggests that transcription factors such as CEBPB, CREB3L2, and BHLHB2, a protein family that includes the melanocyte master transcription factor MITF, cooperate with SOX10 to execute melanocyte-specific gene activation. On the other hand, the most significantly enriched sequence motifs among the SOX10-repressed targets include those corresponding to the v-ets avian erythroblastosis virus E26 oncogene homolog 1 (ETS1) and the E2F transcription factor 1 (E2F1) proteins (Fig. 6D; right panel). This includes the Kit locus, as in silico motif analysis of the sequences under SOX10 peaks at the Kit locus found significant matches to one or more ETS1-binding motifs at each SOX10 peak (P < 0.0001, log-likelihood ratio test) as well as one E2F1 binding motif at the SOX10 peak located −227 kb upstream of Kit (Supplementary Material, Fig. S6). Taken together, these findings suggest that SOX10’s participation in the regulation of functionally distinct classes of genes in melanocytes may be defined by the cis-regulatory sequences predicted to bind co-regulatory transcription factors.
Discussion
In this study, we used a combination of genetic and functional genomic analysis approaches to portray a comprehensive and unbiased molecular profile of SOX10-mediated transcriptional activity in proliferating melanocytes. By leveraging the power of high-throughput genomic analysis techniques, we identified putative enhancer elements that mediate transcriptional regulation of Kit by SOX10. We extended these analyses and have discovered previously uncharted genomic targets of SOX10 that included distal enhancers, thus expanding the repertoire of SOX10 targets in melanocytes. Through integrated analysis of transcription factor binding profile, chromatin states and functional expression data, we reveal results consistent with SOX10 playing a role in transcriptional activation and repression of functionally distinct classes of genes via separate mechanisms to shape gene expression in melanocytes. Our study has discovered many new aspects of SOX10-mediated transcriptional regulation and suggests a broad molecular role for SOX10 as a master modulator of multiple genome regulatory programs in the melanocyte lineage.
The relationship between SOX10 and KIT expression in vitro and in vivo
Melanocyte development and maintenance requires a delicate balance between self-renewal, differentiation and proliferation. Although SOX10 and KIT share multiple roles in these various aspects of melanocyte lineage development and maintenance, it is not clear if and how they might interact in these processes. Spurred by previous observations from our laboratory suggesting inverse correlation of SOX10 and KIT expression in the melanocyte lineage (3,14), we conducted in vitro and in vivo experiments revealing that both stable and transient reduction of Sox10 results in increased KIT levels. Conversely, overexpression of Sox10 results in reduction of KIT and exacerbates the spotting phenotype observed in Kit haploinsufficient animals, implying that Sox10 and Kit functionally interact. Collectively, these findings provide evidence consistent with SOX10 playing a role in repression of Kit expression in melanocytes.
Via global analysis of SOX10 genomic localization, we identified multiple SOX10-binding cis-regulatory elements at the Kit locus that may mediate negative regulation of Kit expression by SOX10 in melanocytes. Interestingly, previous studies in other cell types have also reported multiple tissue-specific enhancer elements at the Kit locus (44,45). Particularly, our observations are analogous to findings by Jing et al. (44) identifying GATA1-binding enhancer elements that mediate transcriptional repression of Kit in mature differentiated erythrocytes. Jing et al. demonstrated that a dynamic exchange of GATA1 for GATA2 triggers loss of an upstream enhancer–promoter interaction, and equally facilitates the formation of a downstream repressive chromatin loop resulting in transcriptional repression of Kit (44). It is possible that SOX10-binding enhancer elements within the Kit locus may serve a similar function to the GATA-binding Kit enhancers in erythrocytes. Additionally, the genomic positions of SOX10-binding enhancer elements at the Kit locus identified by the current study are distinct from the GATA-binding Kit enhancers in erythrocytes reported by Jing et al. (44). This suggests that rather than GATA factors, in melanocytes, SOX10 and potentially other SOX proteins may mediate the critical enhancer–promoter interactions and/or the establishment of repressive chromatin loops at the Kit locus in order to confer lineage-specific control of Kit expression.
SOX10 binds to a broad range of genomic targets in melanocytes
Beyond Kit regulation, our study also uncovers an extensive catalog of SOX10 genomic targets in proliferating melanocytes, suggesting that SOX10 potentially regulates the expression of a wider repertoire of genes in melanocytes than appreciated previously. De novo motif analysis of the sequences enriched under SOX10-binding peaks revealed two configurations of SOX10 DNA binding consensus sequences in vivo that is indicative of SOX10 binding to DNA both as a monomer and dimer. This is consistent with previous observations suggesting that SOX10 can bind to DNA both as a monomer and homodimer in vitro (23,24) and in vivo (25). Although SOX10 binds to promoter regions, the majority of SOX10-binding sites occur at distal sites (>5 kb away from the closest TSS), consistent with a model where SOX10 binds to distal enhancer sites and forms chromatin loops to promoters of its target genes. This is further supported by the observations that SOX10 shows higher binding enrichment at distal sites than TSS proximal regions, SOX10-binding sites almost exclusively coincide with DNase I hypersensitive sites and SOX10 peaks overlap a majority of the previously identified putative melanocyte enhancers (33). Binding to distal enhancer elements has been reported for other transcription factors (32,46). Our identification of SOX10-binding sites and the associated distal enhancers across the genome suggests that SOX10 potentially regulates the expression of a wider repertoire of genes in melanocytes than appreciated previously. It is important to point out that our dataset describing genome-wide SOX10 chromatin occupancy potentially includes genomic regions that do not affect gene expression and regions that are functional only in specific cellular contexts and species (47,48). Thus it will be of interest to perform additional future comparative experiments analyzing SOX10-binding patterns in other mammalian melanocytes, such as humans, or other cell types regulated by SOX10 (49).
SOX10 plays a role in transcriptional activation and repression of functionally distinct classes of genes
Encouraged by the discovery of Kit as a potential negative transcriptional target of SOX10, we performed global analysis of gene expression in melanocytes to systematically assess the transcriptional changes associated with Sox10 haploinsufficiency. Our analysis reveals that reduction of Sox10 results in simultaneous downregulation and upregulation of functionally distinct classes of genes, highlighting the complexity of transcriptional regulation by SOX10. In addition to known roles of SOX10 in melanocytes, our analysis identifies a number of pathways not described previously as activated by SOX10, such as Clathrin-mediated Endocytosis Signaling, Micropinocytosis Signaling, Integrin Signaling and FAK Signaling. Importantly, our study uncovers novel aspects of SOX10-mediated gene regulation, such as the negative regulation of genes involved in pluripotency (self-renewal) and EMT, both of which are associated with early developmental processes and cancer metastasis. These findings indicate that SOX10’s ability to promote melanocyte differentiation, proliferation and survival may involve SOX10-mediated repression of alternative pathways incompatible with normal melanocyte state. Our study directly implicates SOX10 in the transcriptional regulation of two distinct classes of genes with opposing biological roles: proliferation and EMT/invasion. This is consistent with observations made in melanoma cells in which depletion of SOX10 leads to cell cycle arrest, induction of senescence and suppression of melanomagenesis (9). Furthermore, SOX10 expression is elevated in highly proliferating melanoma cells as compared with invasive cells, which exhibit low SOX10 levels and high expression of genes associated with tumor metastasis (50,51). Thus, the distinct gene expression patterns and the associated phenotypes observed in melanomas might be a consequence of the dual transcriptional roles associated with SOX10 activity.
Transcriptional activation and repression by SOX10 involves distinct regulatory mechanisms
Transcriptional outcome is influenced both by epigenetic chromatin states and by the combinatorial interactions of transcription factors with specific cis-regulatory sequence motifs. Integration of data from multiple layers of gene regulation, including transcription factor binding, chromatin states and functional transcriptome profiling enabled us to identify the regulatory mechanisms and transcriptional targets of SOX10 in melanocytes. In comparing genes associated with SOX10 ChIP-Seq and SOX10-responsive genes, we find that ∼35.5% of SOX10-responsive genes have SOX10-binding sites associated with them, and are therefore functional SOX10 targets. Genes with significant expression changes but no associated SOX10-binding sites may represent downstream or indirect targets of SOX10, while binding sites with no assigned responsive genes may reflect the possibility of functional redundancy with other SOX10 protein or non-functional binding interactions of SOX10 with the genome. In addition, given the complexity of eukaryotic genomes and the lack of data on the 3D architecture of melanocyte chromatin, the computational approach we employed for assigning peaks-to-genes may have inherently excluded genes that are bona fide targets of SOX10. It is also possible that under the cellular context we performed our studies, i.e. proliferating melanocytes under steady state conditions, only a fraction of SOX10-binding sites are actively engaged in transcriptional regulatory function while the rest may represent poised binding sites that are mobilized depending on alternative cellular contexts. Nonetheless, our integrative genomic analysis suggests that SOX10 plays a role in transcriptional activation and repression of functionally distinct classes of genes via separate regulatory mechanisms.
On average, SOX10-activated genes show high levels of H3K27ac and low levels of H3K27me3 near their TSSs. This suggests that factors that influence histone acetylation may also play a role in the target specificity of SOX10 and the associated transcriptional outcomes. Related to this, we find that SOX10 chromatin occupancy is generally highly correlated with H3K27ac but weakly with EP300 binding. There are two possible explanations for this discrepancy. First, our previous EP300 ChIP-Seq sequencing was not saturated (the sequencing depth was insufficient) to identify all the EP300 peaks in melanocytes. Second, since the homologous protein CBP can also deposit the H3K27ac mark, EP300 chromatin profile alone may not sufficiently capture all the acetylation marks that overlap SOX10 peaks. In contrast, SOX10-repressed genes exhibit low levels of H3K27ac and high levels of H3K27me3. In summary, our data suggest that the histone modification landscape at functional SOX10 target genes is consistent with these genes being either activated or repressed, depending on the cellular levels of SOX10. The lack of significant overlap of SOX10 binding with H3K27me3 may suggest a possible direct involvement of SOX10 in the repression of a subset of its target genes including Kit. Likewise, it is possible that SOX10 chromatin occupancy at repressed loci is associated with other chromatin modification marks linked to gene repression (e.g. H3K9me3). Future studies are needed to directly test these alternative hypotheses.
In this study, we also uncovered putative cis-regulatory sequence motifs that define SOX10-activated and SOX10-repressed targets. Our analysis indicates that although SOX10 DNA binding is associated with transcriptional activation and repression, SOX10 binding alone cannot distinguish SOX10-activated and SOX10-repressed targets as both groups show very similar occurrence probabilities of SOX10-binding sequence motifs. This suggests that additional factors shape the transcriptional outcomes associated with SOX10 DNA binding activity. Indeed, we identify distinct putative co-regulatory transcription factor motifs that define SOX10-activated and SOX10-repressed target genes. These include the enrichment of binding consensus motifs corresponding to CEBPB and the bHLHZIP proteins CREB3L2 and BHLHB2 at SOX10-activated targets. Of note, the bHLHZIP protein family includes the melanocyte master transcription factor MITF. In fact, the bHLHZIP sequence motif identified in this study resembles a variation of an MITF-binding consensus sequence (33). These observations suggest possible collaboration between these cofactors and SOX10 in executing melanocyte-specific gene activation. At SOX10-repressed targets, we find enrichment of transcription factor binding motifs corresponding to ETS1 and E2F1. This suggests a molecular mechanism where the repressive role of SOX10 is achieved through SOX10 competing for ETS1 and E2F1 binding sites. Alternatively, it is possible that SOX10 interaction with these transcription factors leads to the formation of repressive transcriptional complexes. The potential interplay between SOX10 and ETS1 is particularly interesting in light of recent findings showing that one of the most frequent somatic mutations in melanoma involving the telomerase reverse transcriptase (TERT) promoter generates binding motifs for ETS transcription factors (52,53). It is also possible that the Rb proteins and cyclin-dependent kinase signaling pathway could play a role in repression at these E2F1- and SOX10-bound sites, as the Rb/E2F1 pathway is important for normal melanocyte development and its deregulation is associated with melanoma (54,55). Other SOX family proteins could also mediate transcriptional effects downstream of SOX10. Of note, in Sox10LacZ/+ cells Sox11 expression increases more than 4-fold and Sox6 expression decreases more than 6-fold (Supplementary Material, Table S5). Both SOX11 and SOX6 have been shown to regulate multiple developmental processes in a variety of cell types, and are capable of both transcriptional activation and repression (56–59). Since coexpression of SOX proteins in the same cell type can lead to either synergistic or antagonistic effects (1), it is possible that these two SOX proteins could be acting downstream of SOX10 to mediate some of the significant gene expression changes. Future studies involving all these factors will be important to define the detailed biochemical mechanisms by which SOX10 performs this negative regulation of gene expression.
Lastly, melanocytes are heterogeneous in their embryonic origin and anatomical locations such as skin, hair, eye and ear, suggesting their functional diversity. The findings in the current study are only derived from analysis of melan-a cells, an immortal line of pigmented melanocytes derived from normal epidermal melanoblasts of mouse embryos (15). Thus while our data provides interesting findings, additional analyses that include various cell states and subtypes of melanocytes will be needed in order to fully understand the functional diversity of melanocytes.
In summary, our integrated genome-wide analysis approach reveals key molecular functions of SOX10 in melanocytes. SOX10 predominantly binds to distal enhancer elements, and integrated analyses suggest a role for SOX10 in transcriptional activation and repression of functionally distinct classes of genes. Our study provides insight into the potential mechanisms that distinguish the dual transcriptional regulatory role of SOX10, such as the epigenetic status of target genes and the underlying cis-regulatory elements predicted to bind putative secondary transcription factor partners of SOX10. A global hypothesis that emerges from these observations is that SOX10 functions as a lineage-specific master modulator or pioneer transcription factor by marking regulatory sites of its target genes for subsequent recruitment of secondary co-regulatory factors that determine the distinct transcriptional outcomes associated with SOX10 DNA binding (Fig. 7). The identification of SOX10-binding sites across the genome, its functional targets and putative co-regulatory transcription factors in the current study establish the foundation for further investigation into how SOX10 shapes gene expression programs in melanocytes and melanoma, and these future studies could lead to the identification of novel therapeutics for the highly aggressive and often fatal cancer melanoma.
Figure 7.
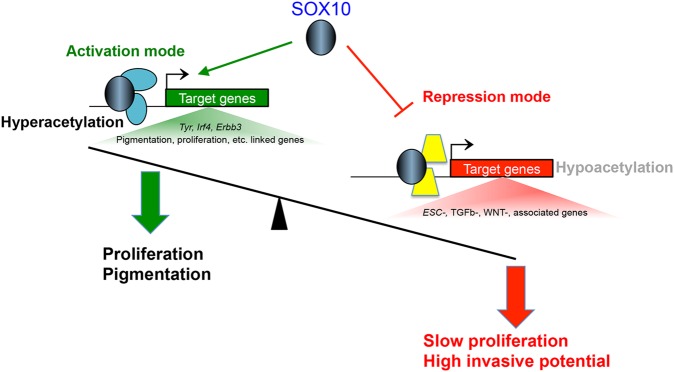
A working model for SOX10-mediated transcriptional regulation in melanocytes. SOX10 DNA binding mediates transcriptional activation and repression of genes linked with functionally distinct pathways. SOX10 activates genes that are required for melanocyte proliferation and pigmentation, and represses genes that promote alternative pathways such as stem cell maintenance and EMT/ invasion. SOX10-activated and SOX10-repressed genes have distinct epigenetic histone modifications as well as co-regulatory transcription partners of SOX10, which determine the transcriptional outcome associated with SOX10 DNA binding activity.
Materials and Methods
Ethics statement
Animal care and experimental animal procedures were performed in accordance with the National Institutes of Health Institutional Animal Care and Use Committee (IACUC) guidelines.
Animals and genotyping
C57BL/6J mice were originally obtained from JAX and maintained by intercross. TYR::CreERT2 (Tg(Tyr-cre/ERT2)13Bos), KitLacZ (Kittm1Alf) (18), Sox10fl (Sox10tm7.1(Sox10)Weg) and Sox10LacZ (Sox10tm1Weg) mice were rederived on and maintained by outcross to C57BL/6J (60–62). The Tg(Dct-Sox10) line (Tg(Dct-Sox10)CF1-10Pav) was maintained through a combination of outcrossing to C57BL/6J and by intercross (63). Ink4a-Arf null mice (B6.129-Cdkn2atm1RDP) were rederived on C57BL/6J and maintained by intercross. Mice were genotyped using DNA isolated from tail tips and PCR analysis. The Sox10LacZ allele was detected using PCR primers for β-galactosidase, 5′-GATCCGCGCTGGCTACCGGC-3′ and 5′-GGATACTGACGAAACGCCTGCC-3′, and the TYR::CreERT2 allele was detected using PCR primers for Cre recombinase, 5′-TCCGCCGCATAACCAGTGAA-3′ and 5′- CGGAAATGGTTTCCCGCAGA, under standard PCR conditions—35 cycles of 45 s at 94°C, 45 s at 65°C and 60 s at 72°C. The Ink4a-Arf null allele was detected using PCR primers, 5′-GTGATCCCTCTACTTTTTCTTCTGACTT -3′ and 5′- GAGACTAGTGAGACGTGCTACTTCCA-3′, under standard PCR conditions—35 cycles of 30 s at 94°C, 30 s at 60°C and 45 s at 72°C. Presence of the Tg(Dct-Sox10) transgene was determined by amplifying across the junction between the Dct promoter and the Sox10 cDNA, using primers 5′-AGCAGTATGGCTGGAGCACT-3′; 5′-TCCAGTCGTAGCCGCTGAGCA-3′. Tg(Dct-Sox10) amplicons were produced using Phusion Hot Start DNA polymerase, GC buffer, 3% DMSO, and the following cycling conditions—35 cycles of 10 s at 98°C, 20 s at 55°C and 20 s at 72°C. Primers and cycling conditions for the Sox10fl allele were described previously (60).
Induction of CRE activity
Tamoxifen (T5648, Sigma) was dissolved in corn oil and 2 mg/animal was administered systemically by intraperitoneal injection for the number of days indicated.
Hair cycle synchronization and immunohistochemistry
Under isoflurane anesthesia, hairs were hand plucked on the lower back of adult animals and were allowed to regenerate for 7 days to reach mid-anagen. Animals were euthanized and the skin from the plucked region was immersed in 2% formaldehyde for 30 min on ice. Skins were cryoprotected in 10% sucrose overnight, embedded in NEG-50 (Thermo Scientific), frozen and sectioned with a cryostat (10 μm). Sections for immunolabeling were first rinsed in PBS with 0.1% Tween 20. For nuclear antigens, sections were permeabilized by treating with 1% Triton X-100 for 15 min. Sections were blocked for 2 h in 1% bovine serum albumin (also containing 100 µg/ml mouse IgG when immunolabeling for PAX3) and incubated with primary antibody overnight at 4°C. Primary antibodies included those against DCT (1:300; TRP2, Santa Cruz Bio, sc-10451), SOX10 (1:75; Santa Cruz Bio, sc-17342), PAX3 (1:75, Developmental Studies Hybridoma Bank, University of Iowa) and c-KIT (1:100; ACK4, Cedarlane, CL8936AP). After washing, sections were incubated in the appropriate secondary antibodies (1:5000; Alexafluor 488, 568 or 633, Invitrogen) for 2 h at room temperature. Fluorescence microscopy was performed on a Zeiss Observer.D1 compound microscope. Images were obtained with an Axiocam Hrc camera using ZEN software and processed with Adobe Photoshop. Quantitation of cellular phenotypes within immunolabeled tissue was performed on every fourth section of sequentially obtained skin sections. KIT mean fluorescence intensity was determined using ZEN software. When determining the intensity of the entire population of melanocytes within each hair bulb, a ROI was drawn exactly around the limits of DCT+ immunolabeling within the red fluorescence channel. This ROI was then applied to the green fluorescence channel to assess KIT immunolabeling. The mean fluorescence intensity of KIT immunolabeling was measured by averaging the fluorescence intensity per pixel within the defined ROI. Similarly, when determining the intensity of individual melanocytes, a circular ROI with a 10 μm diameter was centered on individual PAX3+ nuclei within the red fluorescence channel, and this ROI was applied to the green fluorescence channel to assess KIT mean fluorescence intensity.
Tissue culture, RNA isolation and immunocytochemistry
Three Sox10 haploinsufficient immortal melanocyte cell lines were derived as described previously (15) from 3-day-old Sox10LacZ/+; Ink4a-Arf null mice. They were designated melan-Sox10-1, −2 and −3. The present work used melan-Sox10-1 cells, herein referred to as Sox10LacZ/+ cells. Syngeneic control cells were melan-Ink4a-Arf-1 (15). Established cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (Gibco), 200 nM 12-O-tetradecanoylphorbol 13-acetate (Sigma-Aldrich, St Louis, MO, USA) and 200 pM cholera toxin (Sigma-Aldrich, St Louis, MO, USA) at 37°C in humidified air with 10% CO2 according to the published protocol (15). For RNA isolation, cells were lysed in Trizol reagent and RNA was isolated using RNAeasy Mini Kit (Qiagen). Cells for immunolabeling were grown on glass chamber slides, rinsed in PBS, fixed in 2% formaldehyde for 10 min and permeabilized by treating with 0.5% Triton X-100 for 2 min. The antibodies and their concentrations used for immunolabeling are the same as mentioned above.
Microarray and data analysis
Total RNA (10 µg) isolated from three independent biological samples of melan-a or Sox10LacZ/+ cells was submitted to the NHGRI Microarray Core Facility for subsequent steps of gene expression analysis (cDNA synthesis, amplification, labeling) and hybridization to the Mouse Gene 1.0 ST Array (Affymetrix, Santa Clara, CA, USA). Raw probe intensities from the microarray hybridization were normalized and analyzed by implementing the robust multi-chip average (RMA) algorithm followed by log2 transformation using Partek Genomic Suite v6.6 software (Partek, Inc.; St. Louis, MO, USA). Differentially expressed genes were identified using Analysis of Variance (ANOVA) using a false discovery rate adjusted P-value cutoff of 0.05 (Benjamini–Hochberg method).
Quantitative RT-PCR
RNA from melan-a and Sox10LacZ/+ cell lines was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (ABI). Quantitative real time PCR assays were performed using the Taqman Fast Universal PCR Master Mix or the SYBR® Green PCR Master Mix (ABI/Life Technologies). PCR primers and corresponding gene expression assays used in this study are listed in Supplementary Material, Table S8.
Chromatin immunoprecipitation (ChIP) and high-throughput sequencing (ChIP-Seq)
ChIP-Seq was performed according to previously described protocols (33,64), with slight modifications, in an immortalized melanocyte cell line (melan-a) originally derived from Ink4a-Arf null mice (15). Briefly, ∼2 × 107 cells were harvested and crosslinked at room temperature with 1% formaldehyde for 10 min. Crosslinking reaction was quenched with the addition of glycine to a final concentration of 125 mm for at least 15 min. Crosslinked cells were harvested, snap frozen immediately and stored at −80°C until further use. Cells harvested for ChIP assay were lysed on ice with 1 ml low-salt ChIP buffer (150 mm NaCl, 50 mm Tris–HCl (pH 7.5), 5 mm EDTA, 0.5% NP-40, 1.0%Triton X-100) and sonicated using a Qsonica Sonicator Q500 (Newtown, CT 0647) with the following settings: 90% amplitude; 30 s on/30 s off cycles; total sonication time of 35 min. Soluble chromatin was obtained by centrifugation of sonicated samples and retaining the supernatant (lysate). An aliquot from the lysate was retained for input sample (total genome) and ChIP reaction was carried out by incubating 200 µl of the lysate (∼2 × 106 cells) with each of antibodies to SOX10 (sc-17342X; Santa Cruz Biotechnology; Santa Cruz, CA), H3K27ac (ab4729; Abcam, Massachusetts, US), H3K27me3 (ab6002; Abcam, Massachusetts, US) or a non-specific IgG. Immunoprecipitated chromatin was recovered using recombinant protein G beads or magnetic Dynabeads® (Invitrogen). Input and ChIP samples were reverse crosslinked and DNA was purified using the phenol-chloroform extraction method. Aliquots of input and ChIP DNA obtained from two independent experiments per antibody were submitted for ChIP library construction and high-throughput sequencing, single-end 36-mers on the Illumina Genome Analyzer II for SOX10 ChIP-Seq and single-end 50-mers on HiSeq2500 for H3K27ac and H3K27me3 ChIP-Seqs. Illumina raw sequence reads that passed quality control filtering were mapped to the mouse reference genome NCBI Build 37/UCSC version 9 (mm9) using the Burrows–Wheeler Aligner (BWA) tool (65) by considering only uniquely mapped reads and a maximum duplicate tags at the same position of one read.
ChIP-qPCR assays
Chromatin was prepared from Ink4a-Arf null melanocytes as described above. DNA obtained from input, specific antibody and non-specific IgG ChIPs was analyzed by quantitative PCR (SYBR Green Dye Reagent; Invitrogen) to determine binding enrichment at specific loci. ChIP-qPCR primers were designed using the Primer Express software (Invitrogen) and are listed in Supplementary Material, Table S8. Raw qPCR data were analyzed using the standard curve method as described in the Applied Biosystems qRT-PCR data analysis protocol. ChIP enrichment signals are expressed as percent of input (% input) after non-specific IgG signal subtraction.
Bioinformatic analyses of ChIP-Seq data
Data handling, processing and analysis of ChIP-Seq data were performed using combinations of the UNIX terminal and the Cistrome Integrative Analysis Pipeline [(26); http://cistrome.org/ap/, date last accessed, July 2015]. ChIP-Seq peaks for each experimental replicate were called independently using the Model-based Analysis for ChIP-Seq (MACS v1.4.2) algorithm (66) by applying default settings and a significant P-value threshold of 10−5, except for H3K27me3 where we applied the broad peak parameters as follows: macs14 -t chip.bam -control = input.bam -name = chip_output -format = BAM -gsize = mm -tsize = 50 -bw = 300 -mfold = 10.30 -nolambda -nomodel -shiftsize = 150 -pvalue = 1.00e-05. Only peaks that were called in both biological replicates using the strategy described in Supplementary Material, Figure S2A were considered for subsequent analyses.
Distribution of SOX10 peaks relative to TSS of genes and overlap with genomic regions of interest
Mapping of SOX10 ChIP-Seq peaks to the nearest TSS of the mouse genome (UCSC knownGene) and their distribution profiles across the mouse genome (mm9) were performed using GREAT [Version 2.0.2; (22)]. Analyses involving overlap of SOX10 peaks with genomic features such as proximal promoters (within 2.5 kb of TSS), gene bodies, distal intergenic regions, UTRs, exons and introns were performed as follows: mouse genome annotation files were retrieved from the UCSC genome browser's knownGene track (mm9) and BEDtools (67) was used to calculate genomic intersections. Each SOX10 ChIP-Seq peak was assigned to only one genomic feature using the uninformed hierarchy of promoters > exons > introns > intergenic regions. We performed a similar analysis to calculate SOX10 peak overlap with melanocyte enhancer elements (33), EP300 (33), H3K4me1 (33), H3K27ac, H3K27me3 and DNase I HS sites. A positive overlap was recorded if there is an overlap of at least one base pair between two genomic regions.
Functional enrichment analysis of ChIP-Seq and transcriptome data
Motif discovery
Overrepresented motif analysis of sequences enriched under ChIP-seq peaks was performed using the de novo motif finder MEME-ChIP [(20); http://meme.nbcr.net/meme/tools/meme-chip, date last accessed, July 2015]. A 300 bp sequence surrounding each of the ChIP-Seq peak summits (extending 150 bp on each side) was supplied to MEME-ChIP as input and analyzed with default settings for motif width and significance thresholds.
Evolutionary sequence conservation analysis
The PhastCons average conservation profiles of sequences within 3 kb of ChIP-Seq summits among placental mammals and vertebrates were generated using the Cistrome Integrative Analysis Pipeline [(26); http://cistrome.org/ap/].
Gene ontology (GO) and biological pathway enrichment analyses
Gene ontology analysis of genes associated with ChIP-Seq peaks was generated using GREAT [Version 2.0.2; (22)]. Gene-to-peak association was inferred using the GREAT settings: basal plus extension: 5.0 kb upstream and 1.0 kb downstream, plus up to 1000.0 kb max extension and including curated regulatory domains. Multiple ChIP-Seq peaks for a single gene were allowed in the peak-to-gene assignment rule. The following default significance thresholds were considered: binomial fold enrichment of ≥2; region-based binomial FDR ≤ 0.05 and gene-based hyper-geometric FDR ≤ 0.05. Biological pathway enrichment analysis was performed using the IPA software package (Version 1.0 (1.0); https://analysis.ingenuity.com, date last accessed, July 2015) by applying default parameters and the Benjamini–Hochberg procedure for multiple testing.
Integration of ChIP-Seq and expression data, and motif enrichment analysis
Aggregation plots and heat maps for ChIP-Seq data were generated using either the R programing language or via packages available in the Cistrome Analysis Pipeline [(26); http://cistrome.org/ap/]. Integration of ChIP-Seq and expression data, and motif enrichment analysis were performed using the BETA software package available open source as part of the Cistrome Analysis Pipeline. All microarray and ChIP-Seq data are deposited in NCBI's Gene Expression Omnibus database, GEO accession #GSE69950.
Supplementary Material
Funding
This work was supported by the National Human Genome Research Institute Intramural Research Program at the National Institutes of Health (T.D.F., D.E.W.-C., D.L., S.K.L., D.E.G. and W.J.P.), by the National Institutes of Health (K99/R00 Pathway to Independence Award to M.L.H., GM071648 and NS062972 to A.S.M and D.U.G., 1R21NS084336 and R01DA036865 to L.S., A.S. and G.E.C.); by the Duke Institute for Brain Sciences (L.S., A.S. and G.E.C.) and by the Wellcome Trust (064583 to D.C.B. and E.V.S.).
Supplementary Material
Acknowledgements
The authors would like to thank Lynn Plowright (St George's, University of London, London, UK) for establishing the Sox10 mutant melanocyte lines; the NHGRI Bioinformatics and Scientific Programming Core for Bioinformatics support; Abdel Elkhalhoun and the NHGRI Microarray Core Facility for microarray experiments; The NIH Intramural Sequencing Center for ChIP-Seq library preparation and high-throughput sequencing; and Laura Baxter for critical reading of the manuscript.
Conflict of Interest statement. None declared.
References
- 1.Wegner M. (2010) All purpose Sox: the many roles of sox proteins in gene expression. Int. J. Biochem. Cell Biol., 42, 381–390. [DOI] [PubMed] [Google Scholar]
- 2.Harris M.L., Baxter L.L., Loftus S.K., Pavan W.J. (2010) Sox proteins in melanocyte development and melanoma. Pig. Cell Mel. Res., 23, 496–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris M.L., Buac K., Shakhova O., Hakami R.M., Wegner M., Sommer L., Pavan W.J. (2013) A dual role for SOX10 in the maintenance of the postnatal melanocyte lineage and the differentiation of melanocyte stem cell progenitors. PLoS Genet., 9, e1003644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee M., Goodall J., Verastegui C., Ballotti R., Goding C.R. (2000) Direct regulation of the Microphthalmia promoter by Sox10 links Waardenburg-Shah syndrome (WS4)-associated hypopigmentation and deafness to WS2. J. Biol. Chem., 275, 37978–37983. [DOI] [PubMed] [Google Scholar]
- 5.Potterf S.B., Furumura M., Dunn K.J., Arnheiter H., Pavan W.J. (2000) Transcription factor hierarchy in Waardenburg syndrome: regulation of MITF expression by SOX10 and PAX3. Hum. Genet., 107, 1–6. [DOI] [PubMed] [Google Scholar]
- 6.Potterf S.B., Mollaaghababa R., Hou L., Southard-Smith E.M., Hornyak T.J., Arnheiter H., Pavan W.J. (2001) Analysis of SOX10 function in neural crest-derived melanocyte development: SOX10-dependent transcriptional control of dopachrome tautomerase. Dev. Biol., 237, 245–257. [DOI] [PubMed] [Google Scholar]
- 7.Murisier F., Guichard S., Beermann F. (2006) A conserved transcriptional enhancer that specifies Tyrp1 expression to melanocytes. Dev. Biol., 298, 644–655. [DOI] [PubMed] [Google Scholar]
- 8.Murisier F., Guichard S., Beermann F. (2007) The tyrosinase enhancer is activated by Sox10 and Mitf in mouse melanocytes. Pigment Cell Res., 20, 173–184. [DOI] [PubMed] [Google Scholar]
- 9.Cronin J.C., Watkins-Chow D.E., Incao A., Hasskamp J.H., Schönewolf N., Aoude L.G., Hayward N.K., Bastian B.C., Dummer R., Loftus S.K. et al. (2013) SOX10 ablation arrests cell cycle, induces senescence, and suppresses melanomagenesis. Cancer Res., 73, 5709–5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shakhova O., Zingg D., Schaefer S.M., Hari L., Civenni G., Blunschi J., Claudinot S., Okoniewski M., Beermann F., Mihic-Probst D. et al. (2012) Sox10 promotes the formation and maintenance of giant congenital naevi and melanoma. Nat. Cell Biol., 14, 882–890. [DOI] [PubMed] [Google Scholar]
- 11.Harley V.R., Lovell-Badge R., Goodfellow P.N. (1994) Definition of a consensus DNA binding site for SRY. Nucleic Acids Res., 22, 1500–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stolt C.C., Lommes P., Hillgärtner S., Wegner M. (2008) The transcription factor Sox5 modulates Sox10 function during melanocyte development. Nucleic Acids Res., 36, 5427–5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida H., Kunisada T., Grimm T., Nishimura E.K., Nishioka E., Nishikawa S.I. (2001) Review: melanocyte migration and survival controlled by SCF/c-kit expression. J. Investig. Dermatol. Symp. Proc., 6, 1–5. [DOI] [PubMed] [Google Scholar]
- 14.Loftus S.K., Baxter L.L., Buac K., Watkins-Chow D.E., Larson D.M., Pavan W.J. (2009) Comparison of melanoblast expression patterns identifies distinct classes of genes. Pig. Cell Mel. Res., 22, 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett D.C., Cooper P.J., Hart I.R. (1987) A line of non-tumorigenic mouse melanocytes, syngeneic with the B16 melanoma and requiring a tumour promoter for growth. Int. J. Cancer, 39, 414–418. [DOI] [PubMed] [Google Scholar]
- 16.Osawa M., Egawa G., Mak S.-S., Moriyama M., Freter R., Yonetani S., Beermann F., Nishikawa S.-I. (2005) Molecular characterization of melanocyte stem cells in their niche. Development, 132, 5589–5599. [DOI] [PubMed] [Google Scholar]
- 17.Peters E.M.J., Maurer M., Botchkarev V.A., Jensen K.D., Welker P., Scott G.A., Paus R. (2003) Kit is expressed by epithelial cells in vivo. J. Invest. Dermatol., 121, 976–984. [DOI] [PubMed] [Google Scholar]
- 18.Bernex F., De Sepulveda P., Kress C., Elbaz C., Delouis C., Panthier J.J. (1996) Spatial and temporal patterns of c-kit-expressing cells in WlacZ/+ and WlacZ/WlacZ mouse embryos. Development, 122, 3023–3033. [DOI] [PubMed] [Google Scholar]
- 19.Korytowski W., Sarna T. (1990) Bleaching of melanin pigments. Role of copper ions and hydrogen peroxide in autooxidation and photooxidation of synthetic dopa-melanin. J. Biol. Chem., 265, 12410–12416. [PubMed] [Google Scholar]
- 20.Machanick P., Bailey T.L. (2011) MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics, 27, 1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bailey T.L., Machanick P. (2012) Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res., 40, e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol., 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peirano R.I., Wegner M. (2000) The glial transcription factor Sox10 binds to DNA both as monomer and dimer with different functional consequences. Nucleic Acids Res., 28, 3047–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antonellis A., Huynh J.L., Lee-Lin S.-Q., Vinton R.M., Renaud G., Loftus S.K., Elliot G., Wolfsberg T.G., Green E.D., McCallion A.S. et al. (2008) Identification of neural crest and glial enhancers at the mouse Sox10 locus through transgenesis in zebrafish. PLoS Genet., 4, e1000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srinivasan R., Sun G., Keles S., Jones E.A., Jang S.-W., Krueger C., Moran J.J., Svaren J. (2012) Genome-wide analysis of EGR2/SOX10 binding in myelinating peripheral nerve. Nucleic Acids Res., 40, 6449–6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu T., Ortiz J.A., Taing L., Meyer C.A., Lee B., Zhang Y., Shin H., Wong S.S., Ma J., Lei Y. et al. (2011) Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol., 12, R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci., 106, 9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Praetorius C., Grill C., Stacey S.N., Metcalf A.M., Gorkin D.U., Robinson K.C., Van Otterloo E., Kim R.S.Q., Bergsteinsdóttir K., Ogmundsdóttir M.H. et al. (2013) A polymorphism in IRF4 affects human pigmentation through a tyrosinase-dependent MITF/TFAP2A pathway. Cell, 155, 1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han J., Kraft P., Nan H., Guo Q., Chen C., Qureshi A., Hankinson S.E., Hu F.B., Duffy D.L., Zhao Z.Z. et al. (2008) A genome-wide association study identifies novel alleles associated with hair color and skin pigmentation. PLoS Genet., 4, e1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eriksson N., Macpherson J.M., Tung J.Y., Hon L.S., Naughton B., Saxonov S., Avey L., Wojcicki A., Pe'er I., Mountain J. (2010) Web-based, participant-driven studies yield novel genetic associations for common traits. PLoS Genet., 6, e1000993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang M., Song F., Liang L., Nan H., Zhang J., Liu H., Wang L.-E., Wei Q., Lee J.E., Amos C.I. et al. (2013) Genome-wide association studies identify several new loci associated with pigmentation traits and skin cancer risk in European Americans. Hum. Mol. Genet., 22, 2948–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schnetz M.P., Bartels C.F., Shastri K., Balasubramanian D., Zentner G.E., Balaji R., Zhang X., Song L., Wang Z., Laframboise T. et al. (2009) Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res., 19, 590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gorkin D.U., Lee D., Reed X., Fletez-Brant C., Bessling S.L., Loftus S.K., Beer M.A., Pavan W.J., McCallion A.S. (2012) Integration of ChIP-seq and machine learning reveals enhancers and a predictive regulatory sequence vocabulary in melanocytes. Genome Res., 22, 2290–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiao Z., Mollaaghababa R., Pavan W.J., Antonellis A., Green E.D., Hornyak T.J. (2004) Direct interaction of Sox10 with the promoter of murine Dopachrome Tautomerase (Dct) and synergistic activation of Dct expression with Mitf. Pigment Cell Res., 17, 352–362. [DOI] [PubMed] [Google Scholar]
- 35.Agarwal P., Verzi M.P., Nguyen T., Hu J., Ehlers M.L., McCulley D.J., Xu S.-M., Dodou E., Anderson J.P., Wei M.L. et al. (2011) The MADS box transcription factor MEF2C regulates melanocyte development and is a direct transcriptional target and partner of SOX10. Development, 138, 2555–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prasad M.K., Reed X., Gorkin D.U., Cronin J.C., McAdow A.R., Chain K., Hodonsky C.J., Jones E.A., Svaren J., Antonellis A. et al. (2011) SOX10 directly modulates ERBB3 transcription via an intronic neural crest enhancer. BMC Dev. Biol., 11, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ludwig A., Rehberg S., Wegner M. (2004) Melanocyte-specific expression of dopachrome tautomerase is dependent on synergistic gene activation by the Sox10 and Mitf transcription factors. FEBS Lett., 556, 236–244. [DOI] [PubMed] [Google Scholar]
- 38.Antonellis A., Bennett W.R., Menheniott T.R., Prasad A.B., Lee-Lin S.-Q., Green E.D., Paisley D., Kelsh R.N., Pavan W.J., Ward A., NISC Comparative Sequencing Program, (2006) Deletion of long-range sequences at Sox10 compromises developmental expression in a mouse model of Waardenburg-Shah (WS4) syndrome. Hum. Mol. Genet., 15, 259–271. [DOI] [PubMed] [Google Scholar]
- 39.Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A. et al. (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci., 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S.A., Flynn R.A., Wysocka J. (2011) A unique chromatin signature uncovers early developmental enhancers in humans. Nature, 470, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zentner G.E., Tesar P.J., Scacheri P.C. (2011) Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res., 21, 1273–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song L., Crawford G.E. (2010) DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc., 2010, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun C., Wang L., Huang S., Heynen G.J.J.E., Prahallad A., Robert C., Haanen J., Blank C., Wesseling J., Willems S.M. et al. (2014) Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature, 508, 118–122. [DOI] [PubMed] [Google Scholar]
- 44.Jing H., Vakoc C.R., Ying L., Mandat S., Wang H., Zheng X., Blobel G.A. (2008) Exchange of GATA factors mediates transitions in looped chromatin organization at a developmentally regulated gene locus. Molecular Cell, 29, 232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berrozpe G., Agosti V., Tucker C., Blanpain C., Manova K., Besmer P. (2006) A distant upstream locus control region is critical for expression of the Kit receptor gene in mast cells. Mol. Cell Biol., 26, 5850–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rada-Iglesias A., Bajpai R., Prescott S., Brugmann S.A., Swigut T., Wysocka J. (2012) Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell, 11, 633–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blow M.J., McCulley D.J., Li Z., Zhang T., Akiyama J.A., Holt A., Plajzer-Frick I., Shoukry M., Wright C., Chen F. et al. (2010) ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet., 42, 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmidt D., Wilson M.D., Ballester B., Schwalie P.C., Brown G.D., Marshall A., Kutter C., Watt S., Martinez-Jimenez C.P., Mackay S. et al. (2010) Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science, 328, 1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lopez-Anido C., Sun G., Koenning M., Srinivasan R., Hung H.A., Emery B., Keles S., Svaren J. (2015) Differential Sox10 genomic occupancy in myelinating glia. Glia, 10.1002/glia.22855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoek K.S., Schlegel N.C., Brafford P., Sucker A., Ugurel S., Kumar R., Weber B.L., Nathanson K.L., Phillips D.J., Herlyn M. et al. (2006) Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res., 19, 290–302. [DOI] [PubMed] [Google Scholar]
- 51.Hoek K.S., Eichhoff O.M., Schlegel N.C., Döbbeling U., Kobert N., Schaerer L., Hemmi S., Dummer R. (2008) In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res., 68, 650–656. [DOI] [PubMed] [Google Scholar]
- 52.Horn S., Figl A., Rachakonda P.S., Fischer C., Sucker A., Gast A., Kadel S., Moll I., Nagore E., Hemminki K. et al. (2013) TERT promoter mutations in familial and sporadic melanoma. Science, 339, 959–961. [DOI] [PubMed] [Google Scholar]
- 53.Huang F.W., Hodis E., Xu M.J., Kryukov G.V., Chin L., Garraway L.A. (2013) Highly recurrent TERT promoter mutations in human melanoma. Science, 339, 957–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Halaban R., Cheng E., Smicun Y., Germino J. (2000) Deregulated E2F transcriptional activity in autonomously growing melanoma cells. J. Exp. Med., 191, 1005–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pützer B.M., Steder M., Alla V. (2010) Predicting and preventing melanoma invasiveness: advances in clarifying E2F1 function. Expert Rev. Anticancer Ther., 10, 1707–1720. [DOI] [PubMed] [Google Scholar]
- 56.Hong C.-S., Saint-Jeannet J.-P. (2005) Sox proteins and neural crest development. Semin. Cell Dev. Biol., 16, 694–703. [DOI] [PubMed] [Google Scholar]
- 57.Lefebvre V. (2010) The SoxD transcription factors—--Sox5, Sox6, and Sox13—--are key cell fate modulators. Int. J. Biochem. Cell Biol., 42, 429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Penzo-Méndez A.I. (2010) Critical roles for SoxC transcription factors in development and cancer. Int. J. Biochem. Cell Biol., 42, 425–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hagiwara N. (2011) Sox6, jack of all trades: a versatile regulatory protein in vertebrate development. Dev. Dyn., 240, 1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Finzsch M., Schreiner S., Kichko T., Reeh P., Tamm E.R., Bösl M.R., Meijer D., Wegner M. (2010) Sox10 is required for Schwann cell identity and progression beyond the immature Schwann cell stage. J. Cell Biol., 189, 701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Britsch S., Goerich D.E., Riethmacher D., Peirano R.I., Rossner M., Nave K.A., Birchmeier C., Wegner M. (2001) The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev., 15, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bosenberg M., Muthusamy V., Curley D.P., Wang Z., Hobbs C., Nelson B., Nogueira C., Horner J.W., Depinho R., Chin L. (2006) Characterization of melanocyte-specific inducible Cre recombinase transgenic mice. Genesis, 44, 262–267. [DOI] [PubMed] [Google Scholar]
- 63.Hakami R.M., Hou L., Baxter L.L., Loftus S.K., Southard-Smith E.M., Incao A., Cheng J., Pavan W.J. (2006) Genetic evidence does not support direct regulation of EDNRB by SOX10 in migratory neural crest and the melanocyte lineage. Mech. Dev., 123, 124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee T.I., Johnstone S.E., Young R.A. (2006) Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protoc., 1, 729–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol., 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Quinlan A.R., Hall I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.