

Abstract
Environmental factors and susceptible genomes interact to determine the risk of neurodevelopmental disorders. Although few genes and environmental factors have been linked, the intervening cellular and molecular mechanisms connecting a disorder susceptibility gene with environmental factors remain mostly unexplored. Here we focus on the schizophrenia susceptibility gene DTNBP1 and its product dysbindin, a subunit of the BLOC-1 complex, and describe a neuronal pathway modulating copper metabolism via ATP7A. Mutations in ATP7A result in Menkes disease, a disorder of copper metabolism. Dysbindin/BLOC-1 and ATP7A genetically and biochemically interact. Furthermore, disruption of this pathway causes alteration in the transcriptional profile of copper-regulatory and dependent factors in the hippocampus of dysbindin/BLOC-1-null mice. Dysbindin/BLOC-1 loss-of-function alleles do not affect cell and tissue copper content, yet they alter the susceptibility to toxic copper challenges in both mammalian cells and Drosophila. Our results demonstrate that perturbations downstream of the schizophrenia susceptibility gene DTNBP1 confer susceptibility to copper, a metal that in excess is a neurotoxin and whose depletion constitutes a micronutrient deficiency.
Introduction
Environmental factors impinge on pre-existing genetic predispositions to specify pathological neuronal traits. For example, the penetrance of neuropsychiatric phenotypes is dissimilar in individuals with identical genomes, suggesting the participation of environmental factors (1,2). Schizophrenia is one of these instances where there is only a partial phenotypic overlap between monozygotic twins, yet their offspring share a similar risk for schizophrenia (3,4). This has led to the hypothesis that environmental factors strongly modulate the emergence of schizophrenia in subjects with susceptible genomes. Pregnancy and birth complications, paternal age, exposure to xenobiotics, copper, lead, nutritional excesses or deficiencies, oxidative stress and infectious agents have been documented as environmental contributors in epidemiological studies and in genetic animal models of schizophrenia (4–8). The study of gene–environmental factor interactions mostly focuses on gene–environment agent pair correlations in neurodevelopmental disorders. However, the intervening molecular mechanisms linking these genes with environmental factors remain mostly unexplored (9).
Here we focus on the schizophrenia susceptibility gene DTNBP1 and its gene product dysbindin. Dysbindin is part of a larger BLOC-1 complex (biogenesis of lysosome-related complex-1), an octamer to which ∼90% of brain dysbindin belongs (10–13). Genetic polymorphisms in DTNBP1 are risk factors for schizophrenia (14–17), and post-mortem studies show that the vast majority of schizophrenia brains contain reduced dysbindin (18–20). Dysbindin loss-of-function alleles in mice and Drosophila affect synaptic transmission and display schizophrenia endophenotypes, thus indicating that dysbindin and its molecular interactors play fundamental roles in schizophrenia onset/pathogenesis (11,14,15,21–25). To better understand the cellular pathways dysbindin/BLOC-1 is involved in, we took advantage of quantitative proteomics (13). This approach identified proteins involved in response to environmental insults among proteins sensitive to dysbindin/BLOC-1 deficiency. Key molecules identified were the copper transporter ATP7A and its downstream effector, dopamine beta hydroxylase (DBH), a gene product previously implicated in schizophrenia susceptibility (26–28). Loss of function in ATP7A alleles in human and mice results in Menkes disease, an affliction of copper metabolism that reduces DBH activity, and it is dominated by neurodevelopmental phenotypes, which culminate into early childhood neurodegeneration (29–31). ATP7A maintains copper homeostasis through interactions with cytosolic complexes that regulate its subcellular localization, including dysbindin/BLOC-1 (32–37). The association of the dysbindin-containing BLOC-1 complex with ATP7A suggests that genetic defects in dysbindin/BLOC-1 could confer susceptibility to micronutrients and environmental agents such as copper. We tested this concept here, an idea further supported by a plethora of human studies summarized in the ‘copper hypothesis of schizophrenia’ (5).
In this study, we used cell lines, mice and Drosophila carrying diverse loss-of-function alleles or shRNA reagents to understand how disruption in the dysbindin interactome could modulate the susceptibility to environmental agents such as copper. We present evidence of a neuronal dysbindin/BLOC-1-ATP7A-dependent pathway modulating copper metabolism where dysbindin/BLOC-1 resides upstream of ATP7A. However, dysbindin/BLOC-1 disruption does not affect the function of DBH in mouse brain. We characterized this pathway by (i) biochemically identifying an endogenous dysbindin/BLOC-1-ATP7A complex, (ii) demonstrating a concurrent reduction in ATP7A levels in BLOC-1-deficient brains, (iii) defining alterations in the transcriptional profile of ATP7A and related copper-regulatory factors in BLOC-1 deficiencies and (iv) demonstrating an altered susceptibility to toxic copper challenges in dysbindin/BLOC-1 loss of function in cell culture as well as whole organisms. Our results indicate that perturbations downstream of a defect in a schizophrenia susceptibility gene, such as DTNBP1, confer susceptibility to copper, a metal that in excess is a neurotoxin and whose depletion constitutes a micronutrient deficiency. We propose that molecular perturbations downstream of genomic defects associated to schizophrenia risk serve as a convergence point between the genome and the environment to cause neurodevelopmental disorders.
Results
BLOC-1 interacts with copper-sensitive proteins
The role of dysbindin/BLOC-1 in the modulation of cellular copper homeostasis remains largely unexplored. Our focus on copper metabolism stems from our quantitative mass spectrometry data in dysbindin/BLOC-1 downregulated SH-SY5Y human neuroblastoma cells (data not shown). This proteomic study identified two copper-dependent proteins as factors sensitive to dysbindin/BLOC-1 loss of function: ATP7A and DBH. ATP7A encodes a copper transporter, which is upstream of DBH, a copper-binding enzyme necessary for the synthesis of norepinephrine (29,30). DBH requires ATP7A for copper loading and activity, suggesting a novel dysbindin/BLOC-1-ATP7A-DBH copper-dependent pathway. This pathway would be reminiscent of the BLOC-1-ATP7A-tyrosinase route in melanocytes (32). We downregulated dysbindin/BLOC-1 complex subunits to assess whether ATP7A and DBH are sensitive to dysbindin/BLOC-1 expression in SH-SY5Y human neuroblastoma cells. Bloc1s5 (muted) and 6 (pallidin) shRNAs selectively decreased the total cellular content of ATP7A by ∼30–50% and DBH by ∼50–70% (Fig. 1A and A1). To determine whether the DBH dysbindin/BLOC-1 phenotype was solely a consequence of reduced ATP7A content, we downregulated ATP7A using shRNA-mediated knockdown (Fig. 1B and B1). Decreasing ATP7A expression increased the levels of DBH 5-fold, which is in contrast to the effects of dysbindin/BLOC-1 downregulation. Dysbindin/BLOC-1 subunit expression was unchanged by downregulation of ATP7A (ATP7A shRNA, Fig. 1B and B1; ATP7A null fibroblasts, data not shown). These results indicate that dysbindin/BLOC-1 is upstream in a pathway modulating the expression of ATP7A and DBH.
Figure 1.

BLOC-1 is upstream of the ATP7A copper transporter. (A) Neuroblastoma SH-SY5Y cells were shRNA downregulated with a non-targeting hairpin (lane 1) or directed against the BLOC-1 subunits Bloc1s5 and Bloc1s6 (lanes 2–3) for 7 days. The content of ATP7A, DBH and actin was determined by immunoblot. (A1) depicts quantitation of ATP7A and DBH. Each dot represents an independent determination. P-values were determined by Wilcoxon–Mann–Whitney Rank Sum test against actin controls. (B) SH-SY5Y cells were shRNA downregulated with a control non-targeting hairpin or directed against the ATP7A for 7 days. The content of ATP7A, DBH, Bloc1s6 (pallidin) and actin was determined by immunoblot. (B1) depicts quantitation of antigens in B. P-values were calculated by one-way ANOVA with Dunnett multiple comparisons test using actin as control.
We determined whether dysbindin/BLOC-1, ATP7A and DBH biochemically interact as an additional test for a shared cellular pathway between these three factors. Immunoprecipitation of endogenous ATP7A from SH-SY5Y neuroblastoma cells coprecipitated endogenous DBH (Fig. 2). The specificity of the ATP7A-DBH association was determined by out-competition with the ATP7A antigenic peptide and by the absence of transferrin receptor, an abundant membrane protein, from immunoprecipitated ATP7A complexes (Fig. 2 compare lanes 2–3). We did not detect dysbindin/BLOC-1 subunits in these native complexes. Thus, we used in vivo cross-linking with dithiobis-(succinimidylpropionate) (DSP) before cell extract preparation to overcome weak and transient interactions between ATP7A and dysbindin/BLOC-1 (Fig. 2B) (12,38). We detected interactions between ATP7A, DBH and the BLOC-1 subunits Bloc1s6 (pallidin) and Bloc1s8 (dysbindin) after DSP cross-linking. The immunoprecipitation of these factors was abrogated by the excess ATP7A antigenic peptide added during immunoprecipitation (Fig. 2B, compare lanes 2–3). Furthermore, a dysbindin/BLOC-1-ATP7A-DBH association was identified in cell extracts from a FLAG-tagged Bloc1s8-expressing cell line immunoprecipitated with FLAG antibodies (Fig. 2C) (12). These results demonstrate that dysbindin/BLOC-1-ATP7A-DBH forms a complex, further suggesting that dysbindin/BLOC-1 modulates copper metabolism.
Figure 2.

Dysbindin/BLOC-1 forms a complex with ATP7A and DBH. (A) Neuroblastoma SH-SY5Y cell detergent soluble extracts were immunoprecipitated with a monoclonal antibody against ATP7A either in the absence (lane 2) or presence (lane 3) of an ATP7A antigenic peptide. Immuno-isolated complexes were immunoblotted with antibodies against ATP7A and DBH, and transferrin receptor (TrfR). The latter was used as a control membrane protein. (B) SH-SY5Y cells were treated with the cell permeant cross-linker DSP and detergent soluble extracts were immunoprecipitated with a monoclonal antibody against ATP7A either in the absence (lane 2) or presence (lane 3) of an ATP7A antigenic peptide. Immuno-isolated complexes were immunoblotted with antibodies against ATP7A, DBH and the BLOC-1 subunits Bloc1s6 and dysbindin (Bloc1s8). (C) SH-SY5Y cells stably expressing FLAG-tagged dysbindin were treated with the cell permeant cross-linker DSP and detergent soluble extracts were immunoprecipitated with a monoclonal antibody against the FLAG epitope either in the absence (lane 2) or presence (lane 3) of FLAG peptide. Immuno-isolated complexes were immunoblotted with antibodies against ATP7A, DBH, the BLOC-1 subunit Bloc1s6 and FLAG to detect dysbindin (Bloc1s8).
Dysbindin/BLOC-1 deficiency alters transcript levels of copper-sensitive factors
Environmental and genetic alterations of cellular copper content modify the transcription of genes involved in copper cytoplasmic chaperoning and buffering as well as genes encoding copper membrane transporters (30,39–42). We hypothesized that if dysbindin/BLOC-1 modulated cellular copper metabolism, then these copper-sensitive transcripts should also be sensitive to dysbindin/BLOC-1 loss of function. We measured transcripts whose expression is sensitive to copper cellular content and cluster into the GO term GO:0005507, which defines gene products capable of copper ion binding (http://amigo.geneontology.org/amigo/landing) (30) (Fig. 3A). Transcript levels were determined by qRT-PCR in SH-SY5Y neuroblastoma cells treated with control or Bloc1s6 shRNA (Fig. 3B and C); hippocampi from wild-type and dysbindin/BLOC-1 null mice (Fig. 3D and E, Bloc1s8sdy/sdy); and neuroectodermal pigmentary cells (melanocytes) carrying a null allele of Bloc1s5mu/mu and its control, cells rescued by re-expression of wild-type Bloc1s5 cDNA (Fig. 3F) (32). We used as mRNA loading controls the transcripts of the vesicle markers synaptophysin (Fig 3B, SPHY) and vesicle-associated membrane protein 2 (Fig. 3D–F, VAMP2). BLOC-1 loss of function was confirmed measuring Bloc1s5, 6 and 8 transcripts. Transcript levels of the Golgi localized metal transporters ATP7A and ATP7B, plasma membrane copper transporter Crt1 (SLC31A) and the copper chaperone Atox 1 were significantly modified by all dysbindin/BLOC-1 loss-of-function conditions although to a variable extent depending on the cell type. For example, ATP7A mRNA content was increased in the hippocampi of 7-day-old Bloc1s8sdy/sdy mice (Fig. 3D, P7) compared with controls. In contrast, ATP7A transcripts were reduced in 50-day-old adult Bloc1s8sdy/sdy hippocampi (Fig. 3D, P50), BLOC-1-deficient neuroblastoma cells and melanocytes (Fig. 3C and F). Transcript content of metal buffering metallothioneins (Fig. 3; MT1A, MT2A) and the divalent cation metal transporter 1 (Fig. 3, SLC11A2) were variably modified by the absence of BLOC-1 complex depending on the cell or tissue analyzed. These results indicate that BLOC-1 deficiency affects diverse molecules involved in copper homeostasis in melanocytes and brain tissue.
Figure 3.

Dysbindin/BLOC-1 loss of function modifies the expression of transcripts encoding copper metabolism and dependent factors. (A) Putative human copper metabolism interactome as defined by Genemania.org. Green lines depict genetic interactions between factors and pink lines depict protein–protein interactions. Black circles represent inputs into GeneMANIA and gray circles correspond to GeneMANIA proposed hubs. (B–F) Transcript levels were measured by quantitative real-time PCR in samples from control and Bloc1s6 shRNA-treated SH-SY5Y cells (B, loading controls for C), mouse hippocampi from 7 (D) and 50 (E) days postnatal mice, Bloc1s5mu/mu BLOC-1 null or control melanocytes by re-expression of wild-type Bloc1s5 in Bloc1s5mu/mu cells (F). Each dot represents an independent determination from at least three biological replicates except (F) where two biological replicates were made. Wild type and mutants were compared by Wilcoxon–Mann–Whitney Rank Sum test. We denote only not significant changes as NS. All other P-values ≤0.046.
The reduction of ATP7A and the alterations of diverse molecules involved in copper homeostasis in BLOC-1 deficiency suggest that dysbindin/BLOC-1 loss-of-function mutants could behave just as a hypomorphic ATP7A allele. This hypothesis predicts that the transcripts encoding the ATP7A-sensitive enzymes DBH, peptidylglycine alpha-amidating monooxygenase (PAM), lysyl oxidase (LOX) and superoxide dismutase 3 (SOD3) should be similarly affected in both ATP7A- and Bloc1s6 shRNA-treated cells. We focused on these enzymes as their apoenzymes are loaded at the Golgi complex by ATP7A, and their dysfunction has been strongly implicated in the pathogenesis of Menkes disease (30,31,43,44). shRNA downregulation of ATP7A in SH-SY5Y neuroblastoma cells significantly increased DBH transcript content while LOX levels were decreased (Fig. 4A). In contrast, Bloc1s6 shRNA treatment did not affect the transcript content of DBH (Figs. 3C and 4B), and LOX mRNA content was increased instead of reduced (Fig. 4B). We confirmed these Bloc1s6 shRNA enzyme expression patterns in adult Bloc1s8sdy/sdy mouse hippocampi (Fig. 4C). None of these conditions affected PAM and SOD3 (Fig. 4A–C). These results show that dysbindin/BLOC-1 loss-of-function mutants generate molecular phenotypes differing from phenotypes associated to ATP7A loss of function. Therefore, dysbindin/BLOC-1 loss-of-function defects are unlikely to solely be a hypomorphic ATP7A state.
Figure 4.
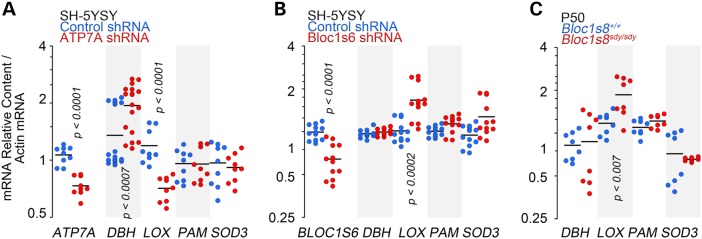
Dysbindin/BLOC-1 and ATP7A loss-of-function transcript phenotypes differ. (A) Transcript levels were measured by quantitative real-time PCR in samples from control and ATP7A shRNA-treated SH-SY5Y cells (A), Bloc1s6 shRNA-treated SH-SY5Y cells (B) and Bloc1s8sdy/sdy mouse hippocampi from 50 days postnatal mice (C). All determinations were normalized to the actin mRNA abundance. Each dot corresponds to an independent determination from at least three biological replicates. Comparisons were made by Wilcoxon–Mann–Whitney Rank Sum test.
The widespread transcript modifications induced by dysbindin/BLOC-1 defects in different cellular systems suggest either that copper homeostasis in dysbindin/BLOC-1 deficiency is compromised or that these transcriptional modifications represent a homeostatic response to maintain a copper metabolism set point. We distinguished between these two hypotheses by measuring the levels of copper and zinc as a control metal in all dysbindin/BLOC-1 loss-of-function models (Fig. 5A and B). Furthermore, we reasoned that a copper deficiency would be reflected in the activity of DBH (Fig. 5C asterisk), a copper-dependent enzyme that catalyzes the conversion of dopamine to norepinephrine (Fig. 5C, DA and NE) (45). Thus, we measured catecholamines in control and adult Bloc1s8sdy/sdy hippocampi. Neither copper content nor the levels of catecholamines were affected by BLOC-1 loss of function (Fig. 5). Thus, we conclude that dysbindin/BLOC-1 deficiency maintains a copper metabolism set point, an adaptive response, in neuronal cells despite of or by modifying the expression of a network of molecules involved in copper homeostasis.
Figure 5.

Copper and catecholamine content are normal in dysbindin/BLOC-1 deficiency. (A and B) Copper and zinc content were determined by inductively coupled plasma mass spectrometry. (C) Catecholamine levels in dysbindin/BLOC-1 null mouse hippocampus (Bloc1s8sdy/sdy) determined by HPLC followed by the coulometric detection. Diagram depicts the catecholamine metabolism step catalyzed by DBH (asterisk). Each dot represents an independent biological replicate or animal. Wild type and loss of function were compared by Wilcoxon–Mann–Whitney Rank Sum test.
Impaired physiological response to excess extracellular copper in dysbindin/BLOC-1 deficiency
While cells are able to maintain copper homeostasis, these mechanisms might not be sufficient to cope with increased extracellular levels of copper. ATP7A resides in the Golgi complex and it translocates to the cell surface when cells are challenged with excess toxic extracellular copper (30,46). BLOC-1 deficiency could compromise this cell surface ATP7A pool impairing responses to toxic copper. Thus, we performed selective cell surface biotinylation of control and Bloc1s6 shRNA-treated cells either after a 2 h acute treatment with copper-chelated media or media with excess extracellular copper (Fig. 6). Surface biotinylated proteins were quantitatively collected from control and Bloc1s6 shRNA-treated cell extracts with streptavidin beads (Fig. 6A). Protein complexes present in streptavidin beads were immunoblotted for ATP7A and transferrin receptor, the latter a BLOC-1- and copper-independent cargo (47). Even though BLOC-1-deficient cells responded to copper excess by mobilizing ATP7A to the cell surface, the surface ATP7A pool remained 50% of that in control shRNA cells (Fig. 6). This reduced ATP7A surface pool was observed in BLOC-1-deficient cells either incubated in copper-depleted media (Fig. 6B, lanes 5–6) or media with excess toxic copper (Fig. 6B, lanes 7–8; and Fig. 6C). In contrast, transferrin receptor surface levels were not modified either by BLOC-1 deficiency, copper challenge or a combination thereof (Fig. 6B–D, TfrR). These results indicate that dysbindin/BLOC-1-deficient cells sense extracellular copper, yet they have an impaired capacity to respond to a toxic challenge with copper.
Figure 6.

ATP7A surface expression is decreased in BLOC-1-deficient cells. (A) SH-SY5Y cells were shRNA downregulated with a control hairpin or one directed against the BLOC-1 subunit Bloc1s6 (lanes 2 and 4) for 7 days. Cell surface was biotinylated at 4°C, and detergent soluble extracts were quantitatively precipitated with neutravidin beads and blotted with streptavidin–peroxidase. Even lanes contain supernatants after bead incubation (odd lanes). (B) It depicts a similar experiment except that cells were incubated for 2 h at 37°C in copper-depleted media or media supplemented with supra-physiological 200 μm copper chloride before surface biotinylation at 4°C. (C) It depicts quantitation of the basal surface levels of ATP7A and transferrin receptor in control and Bloc1s6 shRNA-treated cells. P-value determined with Wilcoxon–Mann–Whitney Rank Sum test. Each dot represents an independent experiment. (D) It depicts quantitation of the basal and copper-induced surface levels of ATP7A and transferrin receptor in control and Bloc1s6 shRNA-treated cells as in (B). P-values were obtained by Kruskal–Wallis Rank Sum test followed by pairwise Wilcoxon–Mann–Whitney Rank Sum tests. Each dot represents an independent experiment.
Dysbindin/BLOC-1 deficiency maintains total cellular copper content despite of or by modifying the expression of copper-regulatory networks (Fig. 3). However, a dysbindin/BLOC-1 defect reduces the pool of ATP7A at the cell surface after toxic copper challenge. These observations predict that cells and organisms lacking dysbindin/BLOC-1 should have differential susceptibilities to environmental challenges of copper homeostasis mechanisms compared with wild type. We used dysbindin/BLOC-1 null melanocytes (Bloc1s5mu/mu) and its control rescued cells to assess cellular susceptibility to toxic concentrations of extracellular copper. Cellular susceptibility to copper was determined measuring the activity of NAD(P)H-dependent oxidoreductase enzymes with the tetrazolium dye (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)) after acute exposure to increasing concentrations of extracellular copper (Fig. 7A and B) (48). BLOC-1 null cells were more susceptible to copper than controls as evidenced by the reduced conversion of MTT to formazan at different supra-physiological copper concentrations (Fig. 7A and B). Decreased MTT metabolization was neither due to differences in cell numbers after copper addition (Fig. 7B) nor to direct effect of copper on MTT (not shown).
Figure 7.

BLOC-1 null cells are susceptible to a copper challenge. Bloc1s5mu/mu BLOC-1 null (red circles) or control melanocytes by re-expression of wild-type Bloc1s5 in Bloc1s5mu/mu cells (blue circles) were incubated in the presence of increasing concentrations of copper chloride for 4 h and the activity of NAD(P)H-dependent oxidoreductases was measured by MTT reduction. Average ± SEM (n = 7). (B) It depicts data in A at 200 μm copper chloride plus cell count assays using crystal violet. Each dot represents an independent experiment. P-values in (A and B) were obtained with one-way ANOVA followed by Bonferroni's all pairs comparison.
Our findings predict that in ATP7A and BLOC-1 gene deficiencies, copper feeding should not be toxic due to impaired ATP7A-dependent intestinal copper absorption (30,49). ATP7A gene dosage modifies Drosophila responses to toxic copper feeding (50). Thus, we used Drosophila BLOC-1 mutants to measure susceptibility to dietary challenges to copper homeostasis mechanisms, because flies assemble a dysbindin/BLOC-1 complex and they reliably recapitulate neuronal and systemic phenotypes associated with dysbindin/BLOC-1 genetic defects in mammals (23–25). To assess whole animal susceptibility to environmental challenges of copper homeostasis mechanisms, we used three BLOC-1 loss-of-function alleles encompassing the orthologs of mammalian Bloc1s1 and 8, blos1 and dysb, respectively. Adult male and female flies were fed with toxic copper concentrations or cisplatin. This compound is an anticancer agent that binds and requires for its biological activity the expression of ATP7A, Atox 1 and SLC31A/Ctr1 (51–54). Importantly, ATP7A, Atox 1 and SLC31A/Ctr1 transcripts are sensitive to BLOC-1 genetic defects (Fig. 3). Wild-type and dysbindin/BLOC-1 mutant flies fed a control glucose diet were viable for 48 h (Fig. 8A and B, No Copper or No Cis-Pt). In contrast, the mortality of wild-type animals progressively increased from 40 to 90% when fed a glucose diet supplemented with copper (Fig. 8A, Plus Copper, blue symbols). Flies carrying two null alleles of blos1, blos1ex2 and blos1ex65, were more resistant than wild-type flies to oral toxic copper irrespective of exposure time and sex (Fig. 8A, Plus Copper, red symbols). Similarly, the blos1ex2 and blos1ex65 alleles also conferred resistance to toxic oral cisplatin, although this phenotype was more pronounced after 24 h of drug exposure (Fig. 8B, Plus Cis-Pt, red symbols). We confirmed these observations analyzing the susceptibility of dysb1 male flies to cisplatin, which were also resistant to this compound (Fig. 8B, Plus Cis-Pt, red symbols). These results indicate that dysbindin/BLOC-1 deficiency confers differential susceptibility to environmental agents that disrupt copper metabolism demonstrating a broader role of dysbindin/BLOC-1 in copper homeostasis.
Figure 8.

Drosophila dysbindin/BLOC-1 loss-of-function alleles confer resistance to copper and cisplatin toxic feeding. Wild-type (w1118, blue symbols) and dysbindin/BLOC-1 loss-of-function mutants [red in the dysbindin-Bloc1s8 ortholog (dysb1) and Bloc1s1 (blos1ex2 and blos1ex65) were fed for 24 or 48 h with glucose solution (no copper] or glucose plus 1 mm copper (A) or 400 μg/ml cisplatin (B), and the number of dead and alive animals was counted. Each dot represents an independent experiment with at least 10 females (F) or males (M). One-way ANOVA followed by Bonferroni's all pairs comparison. *P < 0.0001, **P < 0.0005, #P ≤ 0.05.
Discussion
Here we describe a previously unrecognized dysbindin/BLOC-1-dependent pathway modulating copper metabolism (Fig. 9). We focused on neuronal cells to define a mechanism where dysbindin/BLOC-1 resides upstream of ATP7A. In this study, we immunoprecipitated an endogenous dysbindin/BLOC-1-ATP7A complex and shown a concurrent decrease in total and surface ATP7A protein levels in dysbindin/BLOC-1 loss of function (Fig. 9). Notably, we have described an altered transcriptional profile of not only ATP7A but other copper-regulatory and -dependent factors in the brain and diverse cell systems with impaired dysbindin/BLOC-1 function (Fig. 9). In contrast with other mutations impairing copper-regulatory mechanisms, such as in Menkes disease, dysbindin/BLOC-1 deficiency does not alter levels of catecholamines and copper in the brain (29–31). These normal copper levels in dysbindin/BLOC-1 deficiencies suggest the existence of an adaptive mechanism that occurs despite of or by modifying the expression of transcripts encoding diverse copper-regulatory factors and copper-dependent secretory enzymes. This hypothesis leads to the prediction that dysbindin/BLOC-1 genetic defects would alter susceptibility to environmental challenges with copper and cisplatin, phenotypes that we have documented in mammalian cells and Drosophila carrying loss-of-function alleles in dysbindin/BLOC-1 subunits.
Figure 9.
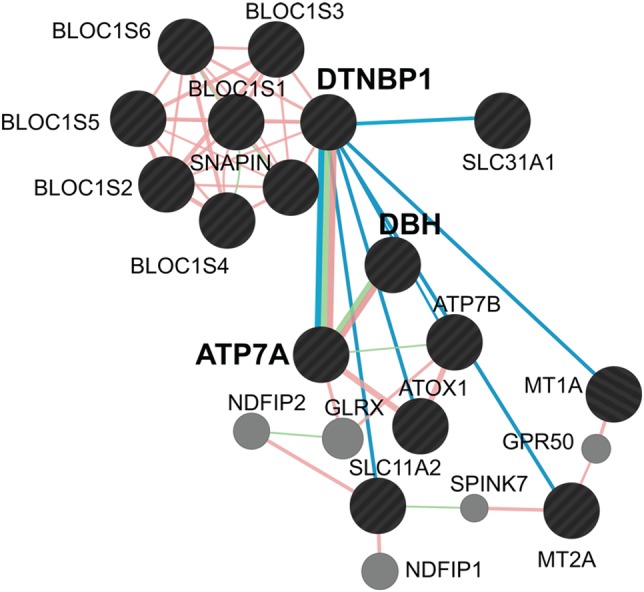
The dysbindin/BLOC-1-copper metabolism interactome. BLOC-1 and copper metabolism interactome as defined by Genemania.org and curated by experimental data herein. Green lines depict genetic interactions between factors and pink lines depict protein–protein interactions. Teal lines depict genetic interactions between the dysbindin null allele Bloc1s8sdy/sdy and transcripts encoding the specified genes. Black circles represent user inputs into GeneMANIA and gray circles correspond to GeneMANIA proposed hubs.
The convergence of dysbindin/BLOC-1, ATP7A and copper homeostasis suggests that changes in micronutrient availability, environmental exposure to metals or metal chelators could influence neurobehavioral phenotypes associated with dysbindin/BLOC-1 null alleles in mice as well as polymorphisms affecting DTNBP1. This model predicts a role for copper in neuronal symptomatology characteristic to schizophrenia. Neuronal copper deficit and overload associate with neurobehavioral phenotypes, some of them overlapping with schizophrenia endophenotypes (5). For example, copper chelators, such as cuprizone and disulfiram, modulate behavior such as impaired social interaction, and cuprizone is a common chemical used to model schizophrenia endophenotypes in rodents (55–57). Disulfiram can cause by itself psychosis in people with low DBH activity (58) or by interaction with other pharmacological agents, including cocaine (59–63). Similarly, disulfiram modifies seeking behaviors for drugs of abuse, a common comorbidity in schizophrenia patients (64,65). Accumulation of copper in tissues also causes neuropsychiatric symptoms. This is exemplified by mutations in human ATP7B, which causes Wilson's disease (29,31). Wilson's disease is a copper storage disorder that manifests as psychiatric pathology, including psychosis, in nearly half of patients and often these behavioral abnormalities precede Wilson's disease diagnosis (66,67). These psychiatric phenotypes in Wilson's disease can be ameliorated by the use of systemic copper chelation (68). Notably, ATP7B transcript levels are susceptible to the Bloc1s8sdy/sdy mutation in hippocampus. Additionally, since the Bloc1s8sdy/sdy mutation also affects the expression of brain metallothioneins and the divalent metal transporter (Slc11a2), it is possible that cellular responses to clearance of other metals, in addition to copper, may also be impaired in dysbindin/BLOC-1 deficiencies and potentially contribute to neurodevelopmental phenotypes.
The susceptibility to metal exposure via a dysbindin/BLOC-1-ATP7A pathway is likely to be developmentally regulated. This assertion is founded on the effects of the dysbindin/BLOC-1 null allele Bloc1s8sdy/sdy on the expression of ATP7A and the metallothionein transcripts. Although the expression of these mRNAs is modified in the hippocampus of young and adult animals, the magnitude and direction of change differ with age. For example, ATP7A mRNA is reduced in Bloc1s8sdy/sdy adult hippocampus, yet it is increased in the hippocampus of 7-day-old Bloc1s8sdy/sdy mice. We chose 7 days as this age corresponds with the peak of hippocampal synaptogenesis in mice (69). We do not know whether this increased ATP7A transcript content observed in Bloc1s8sdy/sdy young hippocampus is solely in neuronal cells. However, it is of interest to speculate that dysbindin/BLOC-1 and ATP7A could be differentially modulated by factors required for synaptogenesis, thus changing ATP7A phenotypes in Bloc1s8sdy/sdy neuronal tissue. In fact, dysbindin/BLOC-1 is necessary for neurite and spine formation (10,70–72). Moreover, copper deficiency induced by ATP7A mutant alleles alters the morphology of dendritic arbors (30). These data stress possible functional connections between the schizophrenia susceptibility factor dysbindin/BLOC-1, copper homeostasis and synaptogenesis.
What metabolic processes could be downstream of a dysbindin/BLOC-1-ATP7A pathway? We propose that dysbindin/BLOC-1 converges on redox metabolism via disregulated copper homeostasis. This concept is supported by the decrease in the levels of peroxiredoxins, enzymes that dispose hydrogen peroxide, that we found in BLOC-1 deficiency in Bloc1s8sdy/sdy hippocampus and Bloc1s6 shRNA-treated cells (12). This peroxiredoxin phenotype coincides with increased reactive oxygen species in Bloc1s6 shRNA-treated cells (12). Impaired responses to oxidative stress and/or decreased antioxidant mechanisms have been documented in schizophrenia affected subjects and animal models of schizophrenia endophenotypes (73–75). In fact, glutathione levels are reduced by 50% in the prefrontal cortex of schizophrenia patients (76,77). The significance of these findings is supported by the ameliorating effects that N-acetylcysteine, a glutathione precursor, possesses in schizophrenia endophenotypes in rodents and humans (75,77,78). Formation of free radicals, such as the one mediated by metals, could be a hub where diverse genetic and environmental schizophrenia susceptibility factors could converge (79).
Our findings have taken advantage of a genetically susceptible model for schizophrenia pathogenesis. Using the DTNBP1 gene product as a start point, we have delineated a pathway (Fig. 9) that includes ATP7A and its substrate copper, ultimately leading to cellular phenotypes that render cells susceptible to environmental insults. We propose that adaptive modifications in downstream mechanisms associated with schizophrenia risk genes are convergence hubs for genomes and environmental factors common to neurodevelopmental disorders.
Materials and Methods
Antibodies used in this study are listed in Supplementary Material, Table S1.
Cell culture
SH-SY5Y (ATCC) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) media supplemented with 10% fetal bovine serum (FBS) and 100 μg/ml penicillin and streptomycin (Hyclone) at 37°C in 10% CO2. SH-SY5Y cell lines stably transfected with 3x-FLAG Dysbindin were previously described (12). Cells were maintained in DMEM media supplemented with 10% FBS, 100 μg/ml penicillin and streptomycin, and 0.2 μg/μl neomycin (catalog no. SV30068, Hyclone) at 37°C in 10% CO2. Immortal melanocytes were a kind gift from Michael Marks (University of Pennsylvania) and were grown in RPMI 1640 media, 10% FBS, 200 nM TPA, 2 mm supplemental glutamine and 100 μg/ml penicillin and streptomycin (Hyclone) at 37°C in 10% CO2 (80).
BLOC-1 and ATP7A knockdowns
For shRNA-mediated knockdowns, shRNA in a pLKO.1 vector for lentiviral infection was obtained from Open Biosystems (Pallidin Clone ID: TRCN0000122781, Item #RHS3979-98828366; Muted Clone ID: TRCN0000128812, Item #RHS3979-98822565; ATP7A Clone ID: TRCN0000043176, Item #RHS3979-9610390). Control shRNA in pLKO.1 was obtained from Addgene (vector 1864). SH-SY5Y cells were treated with lentiviral particles for 7 or 14 days to obtain efficient knockdown. After Day 3 of infection, cells were maintained in DMEM media supplemented with 10% FBS and puromycin (2 μg/ml; Invitrogen).
Mice and qRT-PCR
Mouse strains and procedures are detailed in Larimore et al. 2014 (81). Primer sets are described in Supplementary Material, Table S2.
Immunoprecipitation
To assess interactions between ATP7A, BLOC-1 subunits and DBH, we performed immunoprecipitation experiments either using the ATP7A antibody or the FLAG antibody as previously described. Briefly, untransfected SH-SY5Y cells or SH-SY5Y cells stably transfected with FLAG dysbindin were placed on ice, rinsed twice with phosphate-buffered saline (PBS). The cells were then rinsed twice with PBS and lysed in buffer A (150 mm NaCl, 10 mm HEPES, 1 mm EGTA and 0.1 mm MgCl2, pH 7.4) with 0.5% Triton X-100 and Complete anti-protease (catalog #11245200, Roche), followed by incubation for 30 min on ice. Cells were scraped from the dish, and cell homogenates were centrifuged at 16 100 g for 10 min. The clarified supernatant was recovered, and at least 500 μg of protein extract was applied to 30 μl Dynal magnetic beads (catalog no. 110.31, Invitrogen) coated with antibody (5 μl of ATP7A antibody or 1 μl of FLAG antibody) and incubated for 2 h at 4°C. In some cases, immunoprecipitations were done in the presence of the antigenic 3x-FLAG peptide (340 μm; F4799, Sigma) or antigenic ATP7A peptide (100 μm; custom made from Biosynthesis Inc., Lewisville, TX, USA) as a control. Antigenic ATP7A peptide corresponds to amino acids 42–61 (Uniprot accession number Q04656). The beads were then washed 4–6 times with buffer A with 0.1% Triton X-100. Proteins were eluted from the beads with sample buffer. Samples were resolved by SDS–PAGE and contents analyzed by immunoblot. In some cases, we used cross-linking in intact cells with DSP as previously described (12,37,38). Cells were initially incubated either with 10 mm DSP (Pierce) or as a vehicle control DMSO, diluted in PBS for 2 h on ice. Tris, pH 7.4, was added to the cells for 15 min to quench the DSP reaction. Following this step, cell lysis and immunoprecipitation was performed as described above.
Surface labeling and streptavidin pulldowns
Surface biotinylation and streptavidin pulldowns were performed as described before except for the following modifications (37). After 14-day treatment with scramble or ATP7A-targeted lentiviruses, plates of confluent SH-SY5Y cells were moved to an ice bath and washed two times with warm PBS. Cells were then incubated at 37°C in DMEM media containing either 200 μm bathocuproinedisulfonic acid (BCS) (B1125 Sigma) or 200 μm copper chloride for 90 min. At the end of 90 min, we added an equal volume of DMEM with either 200 μm BCS or 200 μm copper chloride and 100 μg/ml cycloheximide. The cells were then kept at 37°C for an additional 30 min. After this treatment, the cells were treated as described (37). After the strepatividin pulldown, samples were analyzed by SDS–PAGE followed by immunoblot.
MTT assay
Melanocytes were collected and seeded in a 96-well plate at a density of 2.5 × 103cells/well in DMEM + 10% FBS +100 U/ml penicillin, 100 μg/ml streptomycin and 200 nm TPA and incubated overnight. On Day 2, cells were treated with 0–300 μm CuCl2 in Hanks' Balanced Salt Solution (Sigma) + TPA (Sigma). After incubation at 37°C for 1 h, 20 μl 5 mg/ml MTT (Life Technologies) was added to each well, and the plates were incubated for an additional 3.5 h at 37°C. MTT was aspirated, 150 μl DMSO was added and cells were agitated on an orbital shaker for 15 min. The absorbance was read at 595 nm using a microplate reader. Each condition was carried out in quadruplicate, and MTT absorbance was expressed as percentage absorbance of untreated cells.
Catecholamine measurement by HPLC
Levels of dopamine (DA), norepinephrine (NE), serotonin (5-HT) and 3,4-dihydroxyphenylacetic acid (DOPAC) were quantified using high-performance liquid chromatography coupled with coulometric detection using procedures similar to those described previously (64). Briefly, frozen tissue samples were initially prepared by adding 200 μl of ice-cold 0.1 N perchloric acid containing 0.04% sodium metabisulfite and then centrifuged at 16 100 g for 10 min at 4°C. A 50 μl aliquot of each sample was placed into a 200 μl conical sample vial and loaded into a refrigerated autosampler (G1329A, Agilent Technologies, Santa Clara, CA, USA), which injected 15 μl of each sample onto an Ultrasphere ODS 250 × 4.6 mm column, 5 μm (Beckman Coulter, Fullerton, CA, USA) at a constant flow rate of 1.0 ml/min using a mobile phase consisting of 0.1 mm ethylenediaminetetraacetic acid, 0.8 mm sodium octyl sulfate, 20 mm phosphoric acid and 5% acetonitrile (pH 2.6). Separated analytes were detected and quantified using a Coulochem III detector (ESA Inc., Chelmsford, MA, USA), a high sensitivity analytical cell (channel 1, −150 mV; channel 2, +300 mV; model 5011A, ESA Inc.), and a guard cell (+400 mV; model 5020, ESA Inc.). A set of standards containing experimenter-prepared concentrations of DA, NE and DOPAC (50–1000 nm) were analyzed in duplicate along with experimental samples. ChemStation chromatography software (Agilent Technologies) generated chromatograms for each sample analyzed and calculated area under the curve for each peak. Standards were used to generate a standard plot (area under the curve versus analyte concentration) from which the estimated concentration in experimental samples was extrapolated.
ICP-MS determination of copper and zinc levels
Mouse and cell lysates from wild-type and BLOC-1-deficient samples were prepared as follows. Lysis was performed in a buffer with PBS, complete anti-protease and 0.5% ultra-pure Triton-X-100 (Sigma, Catalog number: 93443). The homogenate was then sonicated and kept on ice for 30 min. Cell homogenates were then centrifuged at 16 100 g for 10 min. The clarified supernatant was recovered and analyzed by inductively coupled plasma mass spectrometer (ICP-MS) at the Center for Applied Isotope Studies at the University of Georgia (http://cais.uga.edu/analytical_services/chemical_analysis/services.htm) and at the Barr laboratory at the School of Public Health at Emory University.
Drosophila strains, husbandry and feeding
Flies were raised in a 12 h light:dark cycle at 25°C on standard cornmeal food. Drosophila strains were previously described (25). For copper feeding, adult male and virgin female flies were aged 2–16 days. Flies were starved for 3 h and then fed either a 1 mm CuSO4, 5% glucose solution or a 5% glucose solution placed on a Whatman glass microfiber filter disk. Survival was assayed every 24 h. For cisplatin feeding, adult male and virgin female flies were aged 7–17 days. Flies were starved for 6 h and then fed either a 400 μg/ml cisplatinum, 5% glucose solution or a 5% glucose solution placed on a Whatman glass microfiber filter disk. Survival was assayed every 24 h as described in Podratz et al. (82).
Statistics
Experimental conditions were compared using Synergy Kaleida-Graph, version 4.5.2 (Reading, PA, USA), or StatPlus Mac Built5.6.0pre/Universal (AnalystSoft, Vancouver, Canada).
Supplementary Material
Supplementary material is available at HMG online.
Conflict of Interest statement. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. The authors declare no conflict of interest.
Funding
This work was supported by grants from the National Institutes of Health (GM077569 and NS088503) to V.F., (DA027535, DA034867) to D.W., R01EY004864 to P.M.I., and in part by the Emory University Integrated Cellular Imaging Microscopy Core and Viral Cores of the Emory Neuroscience NINDS Core Facilities grant, P30NS055077, and the HPLC module of the NEI Core Grant for Vision Research, P30EY006360 and HERCULES Center grant NIEHS P30 ES019776. We are indebted to the Faundez lab members for their comments.
Supplementary Material
References
- 1.Zwijnenburg P.J., Meijers-Heijboer H., Boomsma D.I. (2010) Identical but not the same: the value of discordant monozygotic twins in genetic research. Am J. Med. Genet. B Neuropsychiatr. Genet., 153B, 1134–1149. [DOI] [PubMed] [Google Scholar]
- 2.Kato T., Iwamoto K., Kakiuchi C., Kuratomi G., Okazaki Y. (2005) Genetic or epigenetic difference causing discordance between monozygotic twins as a clue to molecular basis of mental disorders. Mol. Psychiatry, 10, 622–630. [DOI] [PubMed] [Google Scholar]
- 3.Gottesman I.I., Bertelsen A. (1989) Confirming unexpressed genotypes for schizophrenia. Risks in the offspring of Fischer's Danish identical and fraternal discordant twins. Arch. Gen. Psychiatry, 46, 867–872. [DOI] [PubMed] [Google Scholar]
- 4.Gejman P.V., Sanders A.R., Kendler K.S. (2011) Genetics of schizophrenia: new findings and challenges. Annu. Rev. Genomics Hum. Genet., 12, 121–144. [DOI] [PubMed] [Google Scholar]
- 5.Bowman M.B., Lewis M.S. (1982) The copper hypothesis of schizophrenia: a review. Neurosci. Biobehav. Rev., 6, 321–328. [DOI] [PubMed] [Google Scholar]
- 6.Abazyan B., Dziedzic J., Hua K., Abazyan S., Yang C., Mori S., Pletnikov M.V., Guilarte T.R. (2014) Chronic exposure of mutant DISC1 mice to lead produces sex-dependent abnormalities consistent with schizophrenia and related mental disorders: a gene-environment interaction study. Schizophr. Bull., 40, 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guilarte T.R., Opler M., Pletnikov M. (2012) Is lead exposure in early life an environmental risk factor for Schizophrenia? Neurobiological connections and testable hypotheses. Neurotoxicology, 33, 560–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamlyn J., Duhig M., McGrath J., Scott J. (2013) Modifiable risk factors for schizophrenia and autism--shared risk factors impacting on brain development. Neurobiol. Dis., 53, 3–9. [DOI] [PubMed] [Google Scholar]
- 9.Uher R. (2014) Gene-environment interactions in severe mental illness. Front. Psychiatry, 5, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghiani C.A., Starcevic M., Rodriguez-Fernandez I.A., Nazarian R., Cheli V.T., Chan L.N., Malvar J.S., de Vellis J., Sabatti C., Dell'angelica E.C. (2009) The dysbindin-containing complex (BLOC-1) in brain: developmental regulation, interaction with SNARE proteins and role in neurite outgrowth. Mol. Psychiatry, 15, 204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghiani C.A., Dell'angelica E.C. (2011) Dysbindin-containing complexes and their proposed functions in brain: from zero to (too) many in a decade. ASN. Neuro., 3, e00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gokhale A., Larimore J., Werner E., So L., Moreno-De-Luca A., Lese-Martin C., Lupashin V.V., Smith Y., Faundez V. (2012) Quantitative proteomic and genetic analyses of the schizophrenia susceptibility factor dysbindin identify novel roles of the biogenesis of lysosome-related organelles complex 1. J. Neurosci., 32, 3697–3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mullin A.P., Gokhale A., Moreno-De-Luca A., Sanyal S., Waddington J.L., Faundez V. (2013) Neurodevelopmental disorders: mechanisms and boundary definitions from genomes, interactomes and proteomes. Transl. Psychiatry, 3, e329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Talbot K., Ong W.Y., Blake D.J., Tang D., Louneva N., Carlson G.C., Arnold S.E. (2009) Dysbindin-1 and its protein family, with special attention to the potential role of dysbindin-1 in neuronal functions and the pathophysiology of schizophrenia. In Kantrowitz J.A. (ed), Handbook of Neurochemistry and Molecular Neurobiology. Springer Science, New York, Vol. 27, pp. 107–241. [Google Scholar]
- 15.Mullin A.P., Gokhale A., Larimore J., Faundez V. (2011) Cell biology of the BLOC-1 complex subunit dysbindin, a schizophrenia susceptibility gene. Mol. Neurobiol., 44, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gornick M.C., Addington A.M., Sporn A., Gogtay N., Greenstein D., Lenane M., Gochman P., Ordonez A., Balkissoon R., Vakkalanka R. et al. (2005) Dysbindin (DTNBP1, 6p22.3) is associated with childhood-onset psychosis and endophenotypes measured by the Premorbid Adjustment Scale (PAS). J. Autism Dev. Disord., 35, 831–838. [DOI] [PubMed] [Google Scholar]
- 17.Fatjo-Vilas M., Papiol S., Estrada G., Bombin I., Peralta V., Rosa A., Parellada M., Miret S., Martin M., Lazaro L. et al. (2011) Dysbindin-1 gene contributes differentially to early- and adult-onset forms of functional psychosis. Am J. Med. Genet. B Neuropsychiatr. Genet., 156B, 322–333. [DOI] [PubMed] [Google Scholar]
- 18.Talbot K., Eidem W.L., Tinsley C.L., Benson M.A., Thompson E.W., Smith R.J., Hahn C.G., Siegel S.J., Trojanowski J.Q., Gur R.E. et al. (2004) Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J. Clin. Invest., 113, 1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Talbot K., Louneva N., Cohen J.W., Kazi H., Blake D.J., Arnold S.E. (2011) Synaptic dysbindin-1 reductions in schizophrenia occur in an isoform-specific manner indicating their subsynaptic location. PLoS ONE, 6, e16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang J., LeGros R.P., Louneva N., Yeh L., Cohen J.W., Hahn C.G., Blake D.J., Arnold S.E., Talbot K. (2009) Dysbindin-1 in dorsolateral prefrontal cortex of schizophrenia cases is reduced in an isoform-specific manner unrelated to dysbindin-1 mRNA expression. Hum. Mol. Genet., 18, 3851–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shao L., Shuai Y., Wang J., Feng S., Lu B., Li Z., Zhao Y., Wang L., Zhong Y. (2011) Schizophrenia susceptibility gene dysbindin regulates glutamatergic and dopaminergic functions via distinctive mechanisms in Drosophila. Proc. Natl. Acad. Sci. USA, 108, 18831–18836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheli V.T., Daniels R.W., Godoy R., Hoyle D.J., Kandachar V., Starcevic M., Martinez-Agosto J.A., Poole S., DiAntonio A., Lloyd V.K. et al. (2010) Genetic modifiers of abnormal organelle biogenesis in a Drosophila model of BLOC-1 deficiency. Hum. Mol. Genet., 19, 861–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dickman D.K., Davis G.W. (2009) The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science, 326, 1127–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dickman D.K., Tong A., Davis G.W. (2012) Snapin is critical for presynaptic homeostatic plasticity. J. Neurosci., 32, 8716–8724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mullin A.P., Sadanandappa M.K., Ma W., Dickman D.K., VijayRaghavan K., Ramaswami M., Sanyal S., Faundez V. (2015) Gene dosage in the dysbindin schizophrenia susceptibility network differentially affect synaptic function and plasticity. J. Neurosci., 35, 325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cubells J.F., Sun X., Li W., Bonsall R.W., McGrath J.A., Avramopoulos D., Lasseter V.K., Wolyniec P.S., Tang Y.L., Mercer K. et al. (2011) Linkage analysis of plasma dopamine beta-hydroxylase activity in families of patients with schizophrenia. Hum. Genet., 130, 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamamoto K., Cubells J.F., Gelernter J., Benkelfat C., Lalonde P., Bloom D., Lal S., Labelle A., Turecki G., Rouleau G.A. et al. (2003) Dopamine beta-hydroxylase (DBH) gene and schizophrenia phenotypic variability: a genetic association study. Am. J. Med. Genet. B Neuropsychiatr. Genet., 117B, 33–38. [DOI] [PubMed] [Google Scholar]
- 28.Cubells J.F., Kranzler H.R., McCance-Katz E., Anderson G.M., Malison R.T., Price L.H., Gelernter J. (2000) A haplotype at the DBH locus, associated with low plasma dopamine beta-hydroxylase activity, also associates with cocaine-induced paranoia. Mol. Psychiatry, 5, 56–63. [DOI] [PubMed] [Google Scholar]
- 29.Kaler S.G. (2011) ATP7A-related copper transport diseases-emerging concepts and future trends. Nat. Rev. Neurol., 7, 15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zlatic S., Comstra H.S., Gokhale A., Petris M.J., Faundez V. (2015) Molecular basis of neurodegeneration and neurodevelopmental defects in Menkes disease. Neurobiol. Dis., 10.1016/j.nbd.2014.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lutsenko S., Barnes N.L., Bartee M.Y., Dmitriev O.Y. (2007) Function and regulation of human copper-transporting ATPases. Physiol. Rev., 87, 1011–1046. [DOI] [PubMed] [Google Scholar]
- 32.Setty S.R., Tenza D., Sviderskaya E.V., Bennett D.C., Raposo G., Marks M.S. (2008) Cell-specific ATP7A transport sustains copper-dependent tyrosinase activity in melanosomes. Nature, 454, 1142–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirst J., Borner G.H., Antrobus R., Peden A.A., Hodson N.A., Sahlender D.A., Robinson M.S. (2012) Distinct and overlapping roles for AP-1 and GGAs revealed by the ‘knocksideways’ system. Curr. Biol., 22, 1711–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinelli D., Travaglini L., Drouin C.A., Ceballos-Picot I., Rizza T., Bertini E., Carrozzo R., Petrini S., de Lonlay P., El Hachem M. et al. (2013) MEDNIK syndrome: a novel defect of copper metabolism treatable by zinc acetate therapy. Brain, 136, 872–881. [DOI] [PubMed] [Google Scholar]
- 35.Materia S., Cater M.A., Klomp L.W., Mercer J.F., La Fontaine S. (2012) Clusterin and COMMD1 independently regulate degradation of the mammalian copper ATPases ATP7A and ATP7B. J. Biol. Chem., 287, 2485–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phillips-Krawczak C.A., Singla A., Starokadomskyy P., Deng Z., Osborne D.G., Li H., Dick C.J., Gomez T.S., Koenecke M., Zhang J.S. et al. (2015) COMMD1 is linked to the WASH complex and regulates endosomal trafficking of the copper transporter ATP7A. Mol. Biol. Cell., 26, 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryder P.V., Vistein R., Gokhale A., Seaman M.N., Puthenveedu M., Faundez V. (2013) The WASH Complex, an endosomal Arp2/3 Activator, interacts with the Hermansky-Pudlak Syndrome Complex BLOC-1 and its Cargo Phosphatidylinositol-4-kinase Type II Alpha. Mol. Biol. Cell., 24, 2269–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zlatic S.A., Ryder P.V., Salazar G., Faundez V. (2010) Isolation of labile multi-protein complexes by in vivo controlled cellular cross-linking and immuno-magnetic affinity chromatography. J. Vis. Exp., pii, 1855 10.3791/1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Armendariz A.D., Gonzalez M., Loguinov A.V., Vulpe C.D. (2004) Gene expression profiling in chronic copper overload reveals upregulation of Prnp and App. Physiol. Genomics, 20, 45–54. [DOI] [PubMed] [Google Scholar]
- 40.Bellingham S.A., Lahiri D.K., Maloney B., La Fontaine S., Multhaup G., Camakaris J. (2004) Copper depletion down-regulates expression of the Alzheimer's disease amyloid-beta precursor protein gene. J. Biol. Chem., 279, 20378–20386. [DOI] [PubMed] [Google Scholar]
- 41.Camakaris J., Petris M.J., Bailey L., Shen P., Lockhart P., Glover T.W., Barcroft C., Patton J., Mercer J.F. (1995) Gene amplification of the Menkes (MNK; ATP7A) P-type ATPase gene of CHO cells is associated with copper resistance and enhanced copper efflux. Hum. Mol. Genet., 4, 2117–2123. [DOI] [PubMed] [Google Scholar]
- 42.Cankorur-Cetinkaya A., Eraslan S., Kirdar B. (2013) Transcriptional remodelling in response to changing copper levels in the Wilson and Menkes disease model of Saccharomyces cerevisiae. Mol. Biosyst., 9, 2889–2908. [DOI] [PubMed] [Google Scholar]
- 43.Niciu M.J., Ma X.M., El Meskini R., Pachter J.S., Mains R.E., Eipper B.A. (2007) Altered ATP7A expression and other compensatory responses in a murine model of Menkes disease. Neurobiol. Dis., 27, 278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaier E.D., Miller M.B., Ralle M., Aryal D., Wetsel W.C., Mains R.E., Eipper B.A. (2013) Peptidylglycine alpha-amidating monooxygenase heterozygosity alters brain copper handling with region specificity. J. Neurochem., 127, 605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Molinoff P.B., Axelrod J. (1971) Biochemistry of catecholamines. Annu. Rev. Biochem., 40, 465–500. [DOI] [PubMed] [Google Scholar]
- 46.Petris M.J., Mercer J.F., Culvenor J.G., Lockhart P., Gleeson P.A., Camakaris J. (1996) Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. EMBO J., 15, 6084–6095. [PMC free article] [PubMed] [Google Scholar]
- 47.Salazar G., Craige B., Styers M.L., Newell-Litwa K.A., Doucette M.M., Wainer B.H., Falcon-Perez J.M., Dell'Angelica E.C., Peden A.A., Werner E. et al. (2006) BLOC-1 complex deficiency alters the targeting of adaptor protein complex-3 cargoes. Mol. Biol. Cell., 17, 4014–4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y., Peterson D.A., Kimura H., Schubert D. (1997) Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. J. Neurochem., 69, 581–593. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y., Zhu S., Hodgkinson V., Prohaska J.R., Weisman G.A., Gitlin J.D., Petris M.J. (2012) Maternofetal and neonatal copper requirements revealed by enterocyte-specific deletion of the Menkes disease protein. Am. J. Physiol. Gastrointest. Liver. Physiol., 303, G1236–G1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hwang J.E., de Bruyne M., Warr C.G., Burke R. (2014) Copper overload and deficiency both adversely affect the central nervous system of Drosophila. Metallomics, 6, 2223–2229. [DOI] [PubMed] [Google Scholar]
- 51.Dolgova N.V., Nokhrin S., Yu C.H., George G.N., Dmitriev O.Y. (2013) Copper chaperone Atox1 interacts with the metal-binding domain of Wilson's disease protein in cisplatin detoxification. Biochem. J., 454, 147–156. [DOI] [PubMed] [Google Scholar]
- 52.Samimi G., Katano K., Holzer A.K., Safaei R., Howell S.B. (2004) Modulation of the cellular pharmacology of cisplatin and its analogs by the copper exporters ATP7A and ATP7B. Mol. Pharmacol., 66, 25–32. [DOI] [PubMed] [Google Scholar]
- 53.Ishida S., Lee J., Thiele D.J., Herskowitz I. (2002) Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA, 99, 14298–14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Palm-Espling M.E., Andersson C.D., Bjorn E., Linusson A., Wittung-Stafshede P. (2013) Determinants for simultaneous binding of copper and platinum to human chaperone Atox1: hitchhiking not hijacking. PLoS ONE, 8, e70473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu H., Yang H.J., Rose G.M., Li X.M. (2011) Recovery of behavioral changes and compromised white matter in C57BL/6 mice exposed to cuprizone: effects of antipsychotic drugs. Front. Behav. Neurosci., 5, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu H., Yang H.J., Zhang Y., Clough R., Browning R., Li X.M. (2009) Behavioral and neurobiological changes in C57BL/6 mice exposed to cuprizone. Behav. Neurosci., 123, 418–429. [DOI] [PubMed] [Google Scholar]
- 57.Tezuka T., Tamura M., Kondo M.A., Sakaue M., Okada K., Takemoto K., Fukunari A., Miwa K., Ohzeki H., Kano S. et al. (2013) Cuprizone short-term exposure: astrocytic IL-6 activation and behavioral changes relevant to psychosis. Neurobiol. Dis., 59, 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Major L.F., Lerner P., Ballenger J.C., Brown G.L., Goodwin F.K., Lovenberg W. (1979) Dopamine-beta-hydroxylase in the cerebrospinal fluid: relationship to disulfiram-induced psychosis. Biol. Psychiatry, 14, 337–344. [PubMed] [Google Scholar]
- 59.de Melo R.C., Lopes R., Alves J.C. (2014) A case of psychosis in disulfiram treatment for alcoholism. Case. Rep. Psychiatry, 2014, 561092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luykx J.J., Vis R., Tijdink J.K., Dirckx M., Van Hecke J., Vinkers C.H. (2013) Psychotic symptoms after combined metronidazole-disulfiram use. J. Clin. Psychopharmacol., 33, 136–137. [DOI] [PubMed] [Google Scholar]
- 61.Grau-Lopez L., Roncero C., Navarro M.C., Casas M. (2012) Psychosis induced by the interaction between disulfiram and methylphenidate may be dose dependent. Subst. Abuse, 33, 186–188. [DOI] [PubMed] [Google Scholar]
- 62.Caci H., Bayle F. (2007) A case of disulfiram-methylphenidate interaction: implications for treatment. Am. J. Psychiatry, 164, 1759. [DOI] [PubMed] [Google Scholar]
- 63.Mutschler J., Diehl A., Kiefer F. (2009) Pronounced paranoia as a result of cocaine-disulfiram interaction: case report and mode of action. J. Clin. Psychopharmacol., 29, 99–101. [DOI] [PubMed] [Google Scholar]
- 64.Schroeder J.P., Cooper D.A., Schank J.R., Lyle M.A., Gaval-Cruz M., Ogbonmwan Y.E., Pozdeyev N., Freeman K.G., Iuvone P.M., Edwards G.L. et al. (2010) Disulfiram attenuates drug-primed reinstatement of cocaine seeking via inhibition of dopamine beta-hydroxylase. Neuropsychopharmacology, 35, 2440–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cooper D.A., Kimmel H.L., Manvich D.F., Schmidt K.T., Weinshenker D., Howell L.L. (2014) Effects of pharmacologic dopamine beta-hydroxylase inhibition on cocaine-induced reinstatement and dopamine neurochemistry in squirrel monkeys. J. Pharmacol. Exp. Ther., 350, 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dening T.R., Berrios G.E. (1989) Wilson's disease. Psychiatric symptoms in 195 cases. Arch. Gen. Psychiatry, 46, 1126–1134. [DOI] [PubMed] [Google Scholar]
- 67.Zimbrean P.C., Schilsky M.L. (2014) Psychiatric aspects of Wilson disease: a review. Gen. Hosp. Psychiatry, 36, 53–62. [DOI] [PubMed] [Google Scholar]
- 68.Srinivas K., Sinha S., Taly A.B., Prashanth L.K., Arunodaya G.R., Janardhana Reddy Y.C., Khanna S. (2008) Dominant psychiatric manifestations in Wilson's disease: a diagnostic and therapeutic challenge! J. Neurol. Sci., 266, 104–108. [DOI] [PubMed] [Google Scholar]
- 69.Mody M., Cao Y., Cui Z., Tay K.Y., Shyong A., Shimizu E., Pham K., Schultz P., Welsh D., Tsien J.Z. (2001) Genome-wide gene expression profiles of the developing mouse hippocampus. Proc. Natl. Acad. Sci. USA, 98, 8862–8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma X., Fei E., Fu C., Ren H., Wang G. (2011) Dysbindin-1, a schizophrenia-related protein, facilitates neurite outgrowth by promoting the transcriptional activity of p53. Mol. Psychiatry, 16, 1105–1116. [DOI] [PubMed] [Google Scholar]
- 71.Jia J.M., Hu Z., Nordman J., Li Z. (2014) The schizophrenia susceptibility gene dysbindin regulates dendritic spine dynamics. J. Neurosci., 34, 13725–13736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ito H., Morishita R., Shinoda T., Iwamoto I., Sudo K., Okamoto K., Nagata K. (2010) Dysbindin-1, WAVE2 and Abi-1 form a complex that regulates dendritic spine formation. Mol. Psychiatry, 15, 976–986. [DOI] [PubMed] [Google Scholar]
- 73.Fournier M., Ferrari C., Baumann P.S., Polari A., Monin A., Bellier-Teichmann T., Wulff J., Pappan K.L., Cuenod M., Conus P. et al. (2014) Impaired metabolic reactivity to oxidative stress in early psychosis patients. Schizophr. Bull., 40, 973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnson A.W., Jaaro-Peled H., Shahani N., Sedlak T.W., Zoubovsky S., Burruss D., Emiliani F., Sawa A., Gallagher M. (2013) Cognitive and motivational deficits together with prefrontal oxidative stress in a mouse model for neuropsychiatric illness. Proc. Natl. Acad. Sci. USA, 110, 12462–12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cabungcal J.H., Counotte D.S., Lewis E.M., Tejeda H.A., Piantadosi P., Pollock C., Calhoon G.G., Sullivan E.M., Presgraves E., Kil J. et al. (2014) Juvenile antioxidant treatment prevents adult deficits in a developmental model of schizophrenia. Neuron, 83, 1073–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Do K.Q., Trabesinger A.H., Kirsten-Kruger M., Lauer C.J., Dydak U., Hell D., Holsboer F., Boesiger P., Cuenod M. (2000) Schizophrenia: glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur. J. Neurosci., 12, 3721–3728. [DOI] [PubMed] [Google Scholar]
- 77.Yao J.K., Leonard S., Reddy R. (2006) Altered glutathione redox state in schizophrenia. Dis. Markers, 22, 83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Berk M., Malhi G.S., Gray L.J., Dean O.M. (2013) The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol. Sci., 34, 167–177. [DOI] [PubMed] [Google Scholar]
- 79.Steullet P., Cabungcal J.H., Monin A., Dwir D., O'Donnell P., Cuenod M., Do K.Q. (2014) Redox dysregulation, neuroinflammation, and NMDA receptor hypofunction: A ‘central hub’ in schizophrenia pathophysiology? Schizophr. Res., 10.1016/j.schres.2014.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Setty S.R., Tenza D., Truschel S.T., Chou E., Sviderskaya E.V., Theos A.C., Lamoreux M.L., Di Pietro S.M., Starcevic M., Bennett D.C. et al. (2007) BLOC-1 is required for cargo-specific sorting from vacuolar early endosomes toward lysosome-related organelles. Mol. Biol. Cell., 18, 768–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Larimore J., Zlatic S.A., Gokhale A., Tornieri K., Singleton K.S., Mullin A.P., Tang J., Talbot K., Faundez V. (2014) Mutations in the BLOC-1 Subunits dysbindin and muted generate divergent and dosage-dependent phenotypes. J. Biol. Chem., 289, 14291–14300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Podratz J.L., Staff N.P., Froemel D., Wallner A., Wabnig F., Bieber A.J., Tang A., Windebank A.J. (2011) Drosophila melanogaster: a new model to study cisplatin-induced neurotoxicity. Neurobiol. Dis., 43, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.