

Abstract
AIM: Hereditary nonpolyposis colorectal cancer (HNPCC) is an autosomal dominantly-inherited cancer-susceptibility syndrome that confers an increased risk for colorectal cancer and a variety of other tumors at a young age. It has been associated with germline mutations in five mismatch repair (MMR) genes (hMSH2, hMLH1, hPMS1, hPMS2, and hMSH6/GTBP). The great majority of germline mutations were found in hMSH2 and hMLH1. The purpose of this study was to analyze the clinical features of Chinese HNPCC patients and to screen hMSH2 and hMLH1 gene mutations.
METHODS: Twenty-eight independent Chinese families were collected, of which 15 met Amsterdam criteria I and 13 met the Japanese clinical diagnosis criteria. The data were recorded including sex, site of colorectal cancer (CRC), age of diagnosis, history of synchronous and/or metachronous CRC, instance of extracolonic cancers, and histopathology of tumors. Peripheral blood samples were collected from all pedigrees after formal written consents were signed. PCR and denaturing high-performance liquid chromatography (DHPLC) were used to screen the coding regions of hMSH2 and hMLH1 genes. The samples showing abnormal DHPLC profiles were sequenced by a 377 DNA sequencer.
RESULTS: One hundred and seventy malignant neoplasms were found in one hundred and twenty-six patients (multiple cancer in twenty-three), including one hundred and twenty-seven CRCs, fifteen gastric, seven endometrial, and five esophageal cancers. Seventy-seven point eight percent of the patients had CRCs, sharing the features of early occurrence (average age of onset, 45.9 years) and of the right-sided predominance reported in the literature. In Chinese HNPCC patients, gastric cancer occurred more frequently, accounting for 11.9% of all cancers patients and ranking second in the spectrum of HNPCC predisposing cancers. Synchronous CRCs occurred less frequently, only accounting for 3.1% of the total CRCs. Twenty percent of the colorectal patients had metachronous CRCs within 10 years after operation. Eight hMSH2 or hMLH1 gene sequence variations were found in twelve families, including the first Mongolian kindred with a hMSH2 gene mutation.
CONCLUSION: HNPCC is characterized by an early-age onset, proximal predominance of CRC, multiple metachronous CRCs, and an excess of extra-colonic cancers. Frequent gastric cancer occurrence and less synchronous CRCs are the remarkable features in Chinese HNPCC patients. DHPLC is a powerful tool in hMSH2 and hMLH1 gene mutation screening. hMLH1 gene mutations, especially of the first nine exons, have been found more common than hMSH2 gene mutations in Chinese patients. Three of seven mutations have been found to be novel, and the germline G204X nonsense mutation in the third exon of hMSH2 has become the first MMR gene mutation found in Chinese Mongolian people.
INTRODUCTION
Hereditary nonpolyposis colorectal cancer (HNPCC, or Lynch syndrome) is an autosomal dominantly-inherited cancer-susceptibility syndrome. It is estimated that HNPCC may account for 5%-10% of the total colorectal cancers (CRC) worldwide[1]. In Western countries, patients inheriting this predisposition are at a particularly high risk of developing CRC and endometrial cancer at a young age, and also at an increased risk of developing various other types of tumors, such as ovarian, uroepithelial, small intestine, biliary tract, stomach, brain, and skin cancers[2]. Five mismatch repair (MMR) genes (hMSH2, hMLH1, hPMS1, hPMS2, and hMSH6/GTBP[3-8]) have been known to be involved in this cancer susceptibility. Currently, more than 300 different mutations have been described in these genes, which account for approximately 500 HNPCC kindreds in the world[9]. hMSH2 and hMLH1 germline mutations were found to be responsible for more than 90% of the HNPCC families[10] (http://www.nfdht.nl/). Therefore, identification of the mutational incidence and spectrum of hMSH2 and hMLH1 genes is important. Identifying the clinical features of HNPCC in China, which might have some differences from those reported in Western countries, will facilitate its diagnosis and treatment. We described the clinical features and the results of mutation screening of both hMSH2 and hMLH1 genes in 28 HNPCC families registered in our hospital. We think that Chinese HNPCC patients have some unique clinic features and MMR gene defects.
MATERIALS AND METHODS
Clinical Data
Subjects were selected from 28 independent Chinese families from January 1992 to August 2003. Among these families, 15 met the Amsterdam criteria I[11]. The criteria were as follows: (1) Three or more relatives had histologically-verified CRC, one of them was a first degree relative to the other two relatives; (2) At least two successive generations were affected; (3) One or more CRC cases were diagnosed under 50 years of age; and (4) Familial polyposis of the colon was excluded. The Japanese clinical diagnosis criteria for HNPCC[12] were used for the other 13 highly-suspected families that did not fully meet the Amsterdam criteria I. Families that met the following A or B were also clinically diagnosed as having HNPCC: A: a case with three or more CRCs within the first-degree relatives; B: a case with two or more CRCs within the first-degree relatives meeting one of the following criteria: age at onset of CRCs being under 50 years, right colon involvement, synchronous or metachronous multiple CRCs, or associated with synchronous or metachronous extracolorectal malignancies.
Detailed family and medical histories were obtained through interview with the proband, and a home visit to extended family members and an extensive review of medical records if available. Peripheral blood samples were collected from all participants after formal written consents were signed.
Eligible HNPCC families were registered and family members were followed up intensively. All patients were reviewed by telephone or outpatient visit at regular intervals. Data concerning sex, site of CRC, age of diagnosis, history of synchronous and/or metachronous CRC, instance of extracolonic cancers, and histopathology of tumors were documented and thoroughly verified.
DNA extraction and PCR amplification
Genomic DNA was isolated from peripheral blood lymphocytes according to the salting-out procedure[13]. The entire hMLH1 and hMSH2 coding region and the splice junctions were amplified by PCR according to Weber methods[14] with minor modifications.
DHPLC analysis
DHPLC analysis was performed on a Transgenomic WAVE system (Transgenomic Inc.) identical with that described previously[15]. Briefly, PCR products (25 μL) were denatured for 5 min at 95 °C and then gradually reannealed by decreasing sample temperature from 95 °C to 45 °C over a period of 30 min to form homo- and/or heteroduplexes. Crude PCR product (7-10 μL) was loaded on the DHPLC column and eluted with a linear acetonitrile gradient at a flow rate of 0.9 mL/min. Gradient parameters were determined based on size and G-C content of the amplicon. Generally, an analysis took approximately 7 min, including column regeneration and re-equilibration to starting conditions. The column mobile phase consisted of a mixture of 0.1 mol/L triethylammonium acetate pH7.0 (TEAA) with (buffer B) or without (buffer A) 250 mL/L acetonitrile. The temperature for heteroduplex analysis was primarily established by using the DHPLC melting algorithm WAVE MakerTM of the WAVETM instrument. The final temperature and denaturing condition for optimal resolution of homoduplexes and heteroduplexes of each fragment were experimentally determined.
DNA sequencing
PCR products displaying a double DHPLC peak indicating existence of heteroduplex were purified with micron microconcentrator filters (Amicon, Beverly, MA) to remove unwanted reagents from the PCR reaction and to concentrate the final products, which were then sequenced by a 377DNA sequencer. All mutations were sequenced in both directions and confirmed in other family members.
RESULTS
Statistics on patients and tumors
A total of 28 kindreds were studied, all of them met the Japanese clinical diagnosis criteria and 15 of them met the Amsterdam I criteria. There were 9 Lynch syndrome I families, in which only colorectal cancers were found, and 19 Lynch syndrome II families, which were characterized by concurrent extracolonic malignancies.
One hundred and seventy malignant neoplasms were found in 126 patients (multiple cancer in twenty-three), including 127 CRCs; 15 gastric, 7 endometrial, 5 esophageal, 2 skin, 2 pancreatic, 2 lung, 1 breast, 1 cervical, 1 ovarian, 1 hepatic, and 1 biliary cancers; 1 gastric leiomyosarcoma, 1 liposarcoma, 1 bone sarcoma, 1 leukemia, and 1 brain glioblastoma. In the present group, 77.8% of the patients had CRCs and 74.7% of the cancers were colorectal ones. There were 45 metachronous CRCs and 4 synchronous CRCs, accounting for 35.4% and 3.1% of the total CRCs, respectively. Right-sided colon cancers constituted 52.9% of the total tumors, and 70.9% of CRCs. Individuals suffering from gastric cancer amounted to 11.9% of total patients.
The average age of malignant neoplasm onset in all the patients was 47.0 years and the ratio of males to females was 1.2:1. Individuals developed CRCs at an average age of 45.9 years. Sixty-two point seven percent of colorectal tumors developed under 50 years of age, 33.3% under 40 years of age and less than 4% occurred above the age of 70 years. In the 28 pedigrees, the average age of tumor occurrence in the first, second, third, and fourth degree was 59.6, 50.0, 44.0, and 31.8 years, respectively.
Eighty-five percent of the patients received radical operations. The remaining patients received chemotherapy, irradiation, and traditional Chinese medicine treatment. Twenty percent of colorectal patients had metachronous CRCs within 10 years after the first operation and required re-operations.
hMSH2 and hMLH1 mutation results
Thirteen double peak profiles displayed in DHPLC were found among 28 probands of all the pedigrees. Finally, 12 probands were identified with a varying DNA sequence by sequencing, of which 7 developed different mutations and 5 had the same hMSH2 polymorphism (Table 1 and Figure 1). We also examined the relatives of affected probands for the same mutations, and found that cancers and mutations were co-segregated in all affected pedigees.
Table 1.
hMSH2/hMLH1 gene sequence variations identified by sequencing
| Sample No. | Gene/Exon | Point of mutation | Mutation result | Reported previously by |
| 231 | hMSH2/3 | g.2610G > T, GGA→TGA | G204X, Truncated protein (nonsense mutation) | None |
| 10, 12, 15, 16, 28 | hMSH2/10 | g.1661+12 A > G | In intron, polymorphism | Scott et al[16] |
| 26 | hMSH2/14 | g.2211-2 A > C | Truncated protein (splice point mutation) | None |
| 11 | hMLH1/3 | g.265 G > T, GAG→TAG | E89X, Truncated protein (nonsense mutation) | Wang et al[17] |
| 14 | hMLH1/6 | g.545+3 A > G | Truncated protein (splice point mutation) | Pensotti et al[18] |
| 8 | hMLH1/8 | g.655 A > G, ATC→GTC | I219V (missense mutation) | Tomlinson et al[19] |
| 25 | hMLH1/8 | g.677 G > A, CGA→CAA | R226Q (missense mutation) | None |
| 18 | hMLH1/9 | g.790+1 G > A | Truncated protein (splice point mutation) | Cunningham[20] |
The first Mongolian family with hMSH2 gene mutation in China.
G refers to genomic DNA.
Figure 1.
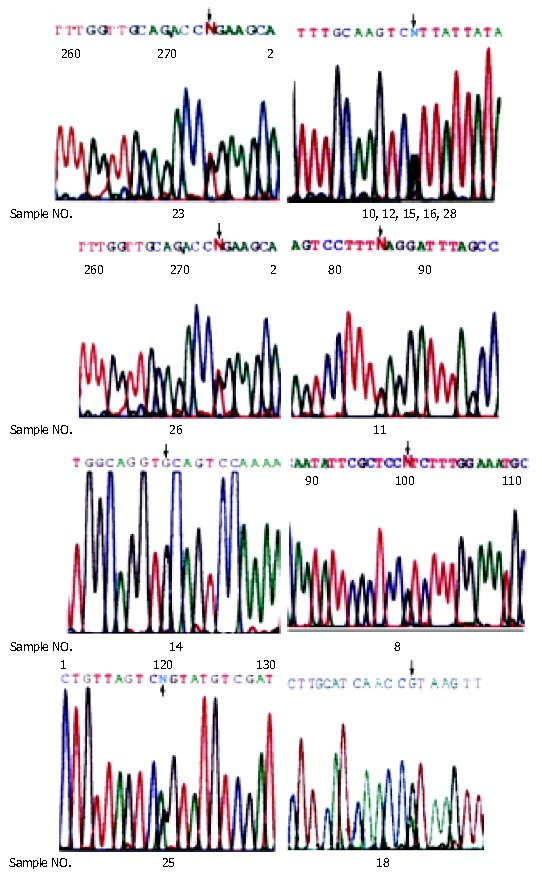
Sequencing graphs of mutations.
DISCUSSION
HNPCC was characterized by an early onset of colorectal cancers (proximal predominance, with 70% proximal to the splenic flexure), multiple synchronous and metachronous CRCs (about 18.1% and 24.2% respectively[21]), and an excess of certain extracolonic cancers[22]. In our study, patients with HNPCC developed CRC at an average age of 45.9 years, much earlier than the general population in China. According to our data, right-sided colon cancers amounted to 52.9% of the total cancers, and 70.9% of CRCs, similar to those reported in Western countries. Moreover, we also found the phenomenon of “generation anticipation”, that is, the later the generation was, the earlier the CRC developed. The fact that the family members tended to be diagnosed early in the follow-up was an explanation for this phenomenon. Another reason for this phenomenon might be that there were carriers who harbored mutated MMR genes but did not become penetrant. Further studies should be carried out.
We found that the following two features were different from those reported in Western countries. Though Chinese patients had a high incidence of metachronous CRCs, synchronous cancer occurred quite rarely, only 3.1% in this study. Our previous studies also had a similar conclusion[20], and the reason for the rare incidence of synchronous cancer in Chinese HNPCC patients remains unclear. The other striking feature was that gastric cancer was the second most common cancer in Chinese HNPCC families, amounting to 11.9% of all cancers patients, much higher than the reported incidence in Western countries[23,24]. Endometrial cancer ranked third, amounting to only 5.6%, and was followed by esophagus carcinoma. In Western countries, however, the second most commonly-seen tumor was endometrial cancer[22,25]. In Japan and Korea, gastric cancer also occurred more frequently[26]. We suppose that these features may represent the ethnic and geographical characteristics that may have some diagnostic significance in China and/or Asia.
The diagnosis of HNPCC depends on the detection of MMR genes. Single-strand conformation polymorphism (SSCP), denaturing gradient gel electrophoresis (DGGE), and direct sequencing have been used in MMR gene defect screening. Using DHPLC, we found a specificity of 92.3%, and recommended it as a hMSH2 and hMLH1 gene mutation screening method. In our study, 5 of 15 families (proband sample NO. 23, 11, 14, 8, 25), who met the Amsterdam I criteria, were found to have mutations and its mutation detection rate is 33.3%. But these criteria could only identify large families where the gene defect was highly penetrant and many small families were inappropriately excluded. In this study, 2 of 13 families (proband sample NO. 26, 18), which did not meet Amsterdam I criteria but fulfilled the Japan criteria, were also found to have mutations. Though the MMR gene defect rate of the latter is comparatively low (only 25%), it has been recommended as the clinical diagnosis criteria in HNPCC patients.
The mutations found in these families were compared with those described already in the human gene mutation database (HGMD) (http://www.uwcm.ac.uk/uwcm/mg/hgmd0.html). To our knowledge, apart from mutation of sample 11 (hMLH1, g.265 G > T,GAG→TAG, E89X) reported before[17], mutations of samples 14, 8, and 18 and the polymorphism of hMSH2 (g.1661+12 A > G) were also reported previously[18-20]. The other three mutations are novel. All the seven mutations resulted in an impaired capacity in MMR, which were consistent with their associations with penetrant tumors in the families. The probability of polymorphism of hMSH2 (g.1661+12 A > G) being pathogenic was very small. Though there were no distinct “hot spot” mutations, we still noticed that hMLH1 gene mutations, especially of the first nine exons, were more common than hMSH2 gene mutations in China. Similarly, Baba[12] and Yuan et al[27] described that hMLH1 gene mutations were more frequently seen in Asia. So, it is worthwhile to initiate MMR gene mutation screening from the first nine exons of hMLH1 gene.
In this study, we identified the first Mongolian family with hMSH2 gene mutation in China. The pedigree is shown in Figure 2. In the large family, the nonsense alteration 610 (genomic DNA) G→T at codon 204 in exon3 of hMSH2 resulted in the substitution of stop codon TGA for glycine codon GGA. This mutation co-segregated with the disease in the family. In five phenotypic normal family members, the same mutated gene was found in the germline. These carriers remain to be followed up intensively.
Figure 2.
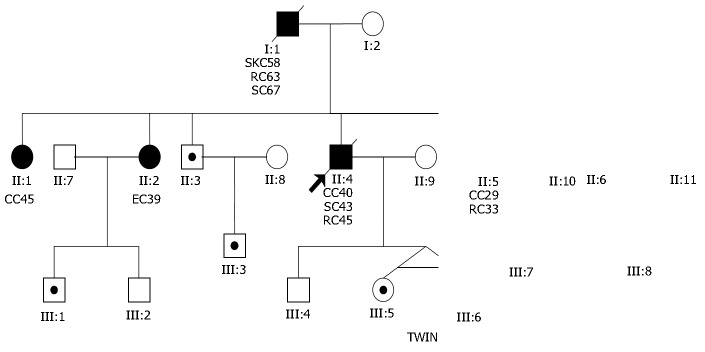
Pedigree of the first Mongolian family with hMSH2 gene mutation in China. ● Cancer patient, ○ Normal person, ⊙ Mutational gene carrier, SKC: skin cancer, RC: rectal cancer, CC: colonic cancer, EC: endometrial cancer, SC: stomach cancer.
Footnotes
Supported by National Natural Science Foundation of China, No. 39970817, and Fund From Ministry of Education of China (the Former National Education Committee)
Edited by Wang XL and Chen ZR Proofread by Xu FM
References
- 1.Stephenson BM, Finan PJ, Gascoyne J, Garbett F, Murday VA, Bishop DT. Frequency of familial colorectal cancer. Br J Surg. 1991;78:1162–1166. doi: 10.1002/bjs.1800781005. [DOI] [PubMed] [Google Scholar]
- 2.Wagner A, Tops C, Wijnen JT, Zwinderman K, van der Meer C, Kets M, Niermeijer MF, Klijn JG, Tibben A, Vasen HF, et al. Genetic testing in hereditary non-polyposis colorectal cancer families with a MSH2, MLH1, or MSH6 mutation. J Med Genet. 2002;39:833–837. doi: 10.1136/jmg.39.11.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1994;77:1 p following 166. [PubMed] [Google Scholar]
- 4.Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, Peltomäki P, Sistonen P, Aaltonen LA, Nyström-Lahti M. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- 5.Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, Kane M, Earabino C, Lipford J, Lindblom A. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–261. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- 6.Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM, Adams MD. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994;263:1625–1629. doi: 10.1126/science.8128251. [DOI] [PubMed] [Google Scholar]
- 7.Nicolaides NC, Papadopoulos N, Liu B, Wei YF, Carter KC, Ruben SM, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature. 1994;371:75–80. doi: 10.1038/371075a0. [DOI] [PubMed] [Google Scholar]
- 8.Akiyama Y, Sato H, Yamada T, Nagasaki H, Tsuchiya A, Abe R, Yuasa Y. Germ-line mutation of the hMSH6/GTBP gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Cancer Res. 1997;57:3920–3923. [PubMed] [Google Scholar]
- 9.Peltomäki P, Gao X, Mecklin JP. Genotype and phenotype in hereditary nonpolyposis colon cancer: a study of families with different vs. shared predisposing mutations. Fam Cancer. 2001;1:9–15. doi: 10.1023/a:1011564720772. [DOI] [PubMed] [Google Scholar]
- 10.Peltomäki P, Vasen HF. Mutations predisposing to hereditary nonpolyposis colorectal cancer: database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology. 1997;113:1146–1158. doi: 10.1053/gast.1997.v113.pm9322509. [DOI] [PubMed] [Google Scholar]
- 11.Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC) Dis Colon Rectum. 1991;34:424–425. doi: 10.1007/BF02053699. [DOI] [PubMed] [Google Scholar]
- 12.Baba S. Hereditary nonpolyposis colorectal cancer: an update. Dis Colon Rectum. 1997;40:S86–S95. doi: 10.1007/BF02062027. [DOI] [PubMed] [Google Scholar]
- 13.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weber TK, Conlon W, Petrelli NJ, Rodriguez-Bigas M, Keitz B, Pazik J, Farrell C, O'Malley L, Oshalim M, Abdo M, et al. Genomic DNA-based hMSH2 and hMLH1 mutation screening in 32 Eastern United States hereditary nonpolyposis colorectal cancer pedigrees. Cancer Res. 1997;57:3798–3803. [PubMed] [Google Scholar]
- 15.Gross E, Arnold N, Goette J, Schwarz-Boeger U, Kiechle M. A comparison of BRCA1 mutation analysis by direct sequencing, SSCP and DHPLC. Hum Genet. 1999;105:72–78. doi: 10.1007/s004399900092. [DOI] [PubMed] [Google Scholar]
- 16.Scott RJ, McPhillips M, Meldrum CJ, Fitzgerald PE, Adams K, Spigelman AD, du Sart D, Tucker K, Kirk J. Hereditary nonpolyposis colorectal cancer in 95 families: differences and similarities between mutation-positive and mutation-negative kindreds. Am J Hum Genet. 2001;68:118–127. doi: 10.1086/316942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao B, Wang ZJ, Xu YF, Wan YL, Li P, Huang YT. Report of 16 kindreds and one kindred with hMLH1 germline mutation. World J Gastroenterol. 2002;8:263–266. doi: 10.3748/wjg.v8.i2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pensotti V, Radice P, Presciuttini S, Calistri D, Gazzoli I, Grimalt Perez A, Mondini P, Buonsanti G, Sala P, Rossetti C, et al. Mean age of tumor onset in hereditary nonpolyposis colorectal cancer (HNPCC) families correlates with the presence of mutations in DNA mismatch repair genes. Genes Chromosomes Cancer. 1997;19:135–142. [PubMed] [Google Scholar]
- 19.Tomlinson IP, Beck NE, Homfray T, Harocopos CJ, Bodmer WF. Germline HNPCC gene variants have little influence on the risk for sporadic colorectal cancer. J Med Genet. 1997;34:39–42. doi: 10.1136/jmg.34.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cunningham JM, Kim CY, Christensen ER, Tester DJ, Parc Y, Burgart LJ, Halling KC, McDonnell SK, Schaid DJ, Walsh Vockley C, et al. The frequency of hereditary defective mismatch repair in a prospective series of unselected colorectal carcinomas. Am J Hum Genet. 2001;69:780–790. doi: 10.1086/323658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fitzgibbons RJ, Lynch HT, Stanislav GV, Watson PA, Lanspa SJ, Marcus JN, Smyrk T, Kriegler MD, Lynch JF. Recognition and treatment of patients with hereditary nonpolyposis colon cancer (Lynch syndromes I and II) Ann Surg. 1987;206:289–295. doi: 10.1097/00000658-198709000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lynch HT, Smyrk T, Lynch J. An update of HNPCC (Lynch syndrome) Cancer Genet Cytogenet. 1997;93:84–99. doi: 10.1016/s0165-4608(96)00290-7. [DOI] [PubMed] [Google Scholar]
- 23.Lynch HT, Richardson JD, Amin M, Lynch JF, Cavalieri RJ, Bronson E, Fusaro RM. Variable gastrointestinal and urologic cancers in a Lynch syndrome II kindred. Dis Colon Rectum. 1991;34:891–895. doi: 10.1007/BF02049703. [DOI] [PubMed] [Google Scholar]
- 24.Hakala T, Mecklin JP, Forss M, Järvinen H, Lehtovirta P. Endometrial carcinoma in the cancer family syndrome. Cancer. 1991;68:1656–1659. doi: 10.1002/1097-0142(19911001)68:7<1656::aid-cncr2820680732>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 25.Froggatt NJ, Green J, Brassett C, Evans DG, Bishop DT, Kolodner R, Maher ER. A common MSH2 mutation in English and North American HNPCC families: origin, phenotypic expression, and sex specific differences in colorectal cancer. J Med Genet. 1999;36:97–102. [PMC free article] [PubMed] [Google Scholar]
- 26.Park YJ, Shin KH, Park JG. Risk of gastric cancer in hereditary nonpolyposis colorectal cancer in Korea. Clin Cancer Res. 2000;6:2994–2998. [PubMed] [Google Scholar]
- 27.Yuan Y, Han HJ, Zheng S, Park JG. Germline mutations of hMLH1 and hMSH2 genes in patients with suspected hereditary nonpolyposis colorectal cancer and sporadic early-onset colorectal cancer. Dis Colon Rectum. 1998;41:434–440. doi: 10.1007/BF02235756. [DOI] [PubMed] [Google Scholar]