

Abstract
Deficiencies in vitamins or other factors (B6, B12, folic acid, betaine) and genetic disorders for the metabolism of the non-protein amino acid-homocysteine (Hcy) lead to hyperhomocysteinemia (HHcy). HHcy is an integral component of several disorders including cardiovascular disease, neurodegeneration, diabetes and alcoholic liver disease. HHcy unleashes mediators of inflammation such as NFκB, IL-1β, IL-6, and IL-8, increases production of intracellular superoxide anion causing oxidative stress and reducing intracellular level of nitric oxide (NO), and induces endoplasmic reticulum (ER) stress which can explain many processes of Hcy-promoted cell injury such as apoptosis, fat accumulation, and inflammation. Animal models have played an important role in determining the biological effects of HHcy. ER stress may also be involved in other liver diseases such as α1-antitrypsin (α1-AT) deficiency and hepatitis C and/or B virus infection. Future research should evaluate the possible potentiative effects of alcohol and hepatic virus infection on ER stress-induced liver injury, study potentially beneficial effects of lowering Hcy and preventing ER stress in alcoholic humans, and examine polymorphism of Hcy metabolizing enzymes as potential risk-factors for the development of HHcy and liver disease.
INTRODUCTION
Homocysteine (Hcy) is a toxic non-protein sulfur containing amino acids in humans. It is formed exclusively upon demethylation of the essential amino acid-methionine. Hcy is metabolized either through remethylation or transsulfuration pathways and is nutritionally regulated. Normal concentrations of total homocysteine in plasma are in the range of 5 to 16 μmol/L and the desired upper limit for Hcy concentration should be 10 μmol/L. An elevated plasma Hcy level is denoted hyperhomocysteinemia (HHcy). Three ranges of HHcy are defined: Moderate (16 to 30 μmol/L), intermediate (31 to 100 μmol/L), and severe (> 100 μmol/L). Individuals who consume a large amount of food rich in animal protein may ingest two to three grams of methionine, resulting in postprandial Hcy concentrations greater than 20 μmol/L. Clinical HHcy was first described more than 40 years ago in children with learning difficulties[1-3], and it has since been estimated that moderate HHcy occurs in 5%-7% of the general population. Evidence now indicates that moderate HHcy is an important and independent risk factor for several disorders, including atherosclerosis, diabetes, fatty liver, immune activation, and neurodegenerations such as Alzheimer’s and Parkinson’s diseases[3-9].
Readers are referred to recent reviews on HHcy and functions of Hcy[10,11]. The main goal of this article is to provide information on major causes of HHcy, potential mechanisms of Hcy toxicity, with emphasis on endoplasmic reticulum (ER) stress mechanism, and animal models for the study of biological effects of HHcy. We would also summarize our ongoing work on ethanol-induced HHcy and liver injury in an intragastric ethanol fed murine model.
HCY METABOLISM AND HHCY
Hcy was formed from methionine after removal of the methyl group on S-adenosylmethionine (SAM) (Figure 1). Hcy metabolism involves reversible formation of S-adenosylhomocysteine (SAH), remethylation to methionine by betaine-homocysteine methyltransferase (BHMT) (liver and kidney restricted), which is vitamin-independent, and by the ubiquitous methionine synthase (MS), which is dependent on vitamin B12 and methylenetetrahydrofolate (MTHF) production via 5, 10-methylenetetrahydrofolate reductase (MTHFR). Hcy can also be converted through transsulfuration to cystathionine for the formation of cysteine and glutathione (GSH). The transsulfuration is catalyzed by cystathionine-β-synthase (CBS) and is dependent on vitamin B6. In addition, Hcy can be converted to Hcy-tRNA. Although it was not incorporated into protein due to editing mechanisms, nitroso-Hcy-tRNA is stable and might play a role in Hcy-induced protein misfolding along with the formation of Hcy-protein-SH mixed disulfides and Hcy thiolactone covalent binding to lysine amino groups[11]. Hcy-t-RNA is edited through the action of methionyl-t-RNA synthetase (ATP consuming) by formation of a thioester thiolactone which could covalently bind to protein amino groups. Thus, homocysteinylation of proteins depends on the formation of thiolactone[12,13].
Figure 1.
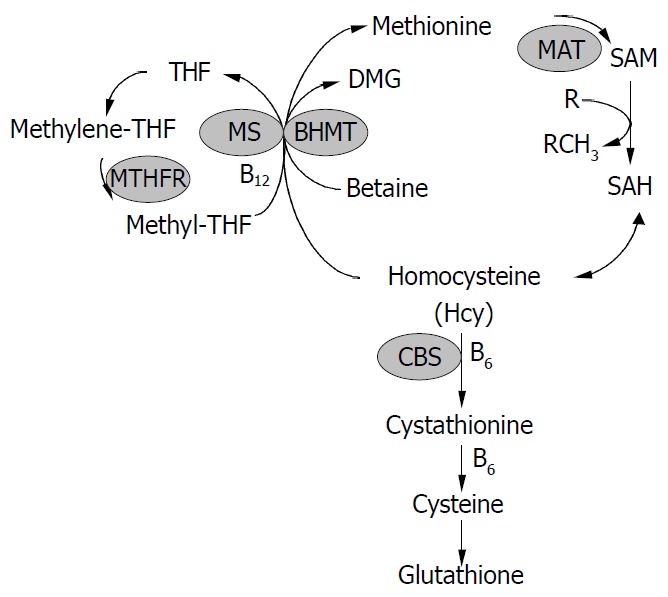
Homocysteine metabolism. Homocysteine has three main metabolic fates: To be remethylated to methionine, to en-ter the cysteine biosynthetic pathway, and to be released into the extracellular medium. CBS, cystathionine -synthase; MS, methionine synthase; THF, tetrahydrofolate; MTHFR, 5, 10-methylenetetrahydrofolate reductase; BHMT, betaine-ho-mocysteine methyltransferase; DMG, dimethylglycine; MAT, methionine adenosyltransferase; SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine.
Tight regulation of Hcy metabolism depends on different affinities of MS, BHMT, and CBS for Hcy. MS and BHMT show low Km values for Hcy (< 0.1 mmol/L), and CBS has high Km values for Hcy (> 1 mmol/L). At low Hcy concentrations, methionine conservation was favored; and at high Hcy concentrations, immediate and long-term drainage of Hcy via the transsulfuration pathway was ensured[14]. SAM could play a key regulatory role by allosterically inhibiting MTHFR and BHMT and activating CBS[15-18]. Thus, SAM may be a regulatory switch in Hcy metabolism: Low SAM favors remethylation and conservation of Hcy for methionine synthesis, whereas high levels favor transsulfuration. High Hcy levels can decrease the SAM/SAH ratio, since most methyltransferases bind to SAH with higher affinity than SAM, elevated SAH inhibits methylation. In vitro, under “physiological” conditions of concentrated 27 000 g postmitochondrial supernatant with 8 mmol/L GSH, 0.3 mmol/L serine, 2 mmol/L betaine, 60 μmol/L methionine, 50 μmol/L methyl THF, 60 μmol/L SAM and 10 μmol/L SAH, transsulfuration accounted for 46% of Hcy metabolism and the remainder was equally contributed to by MS and BHMT. The need to conserve methionine (e.g. low protein diet) resulted in decreased cystathionine production and increased Hcy remethylation. Conversely, in the presence of excess methionine, SAM activated the cystathionine pathway.
HHcy results from increased levels of intracellular Hcy that is readily released into the extracellular medium: Plasma or body fluid. Kidney might be a major site for the removal and metabolism of Hcy primarily through the transsulfuration pathway[19]. Renal impairment often causes HHcy, reflecting a role of kidney in Hcy clearance from plasma. This fact might contribute to the high incidence of vascular complications in patients with chronic renal failure[20]. Genetic abnormalities, age, sex and various nutritional and hormonal determinants contribute to HHcy. However, genetic and nutritional disorders are the major factors. Genetic disorders involve polymorphism in the genes coding for MTHFR and CBS. The most common genetic defect associated with mild HHcy is a point mutation, namely, a C to T substitution at nucleotide 677 (C677→T) in the open reading frame of the gene for MTHFR. This point mutation could cause a substitution of valine for alanine in the functional enzyme[21], resulting in a thermolabile variant of the enzyme with decreased total activity. This is an autosomal recessive mutation, and the frequency of the C677→T polymorphism varied among racial and ethnic groups, with 13% of T/T homozygous and 50% C/T heterozygous among Caucasian and Asian populations, and very low incidence among African-Americans[21-27]. Premature atherosclerosis and thrombotic disease were observed in MTHFR deficiency[23-28]. The most common genetic cause associated with severe HHcy is homozygous CBS deficiency, which resulted in plasma Hcy concentrations of up to 400 μmol/L, compared to normal plasma levels of 10 μmol/L[28-30]. Homozygous CBS deficiency, T833→C and G919→A mutations, were inherited as an autosomal recessive disorder with pleiotropic clinical manifestations, including mental retardation, ectopia lentis, osteoporosis, skeletal abnormalities and hepatic steatosis[28-30]. Patients were usually at higher risk for premature atherosclerosis and thrombotic disease, which is the major cause of death[31-33]. CBS deficiency has a worldwide incidence of 1:344 000 live births, ranging from 1:58 000 to 1:1 000 000 in countries that perform newborn screening[31]. While homozygous CBS deficiency is rare, heterozygous CBS deficiency occurs in approximately 1% of the general population and is associated with premature atherosclerosis and thrombotic disease in phenotypically normal individuals[31-33].
Nutritional disorders that potentially lead to HHcy include deficiencies in vitamin B12, folate and vitamin B6, as the de novo synthesis of methionine methyl groups requires both vitamin B12 and folate cofactors whereas the synthesis of cystathionine requires pyridoxal 5-phosphate (vitamin B6). Although it has been shown that deficiencies of vitamin B12 and folate are related to increased plasma Hcy concentrations[32-35], the relationship of Hcy levels to vitamin B6 status is less clear[36,37]. In addition, excess dietary methionine in normal mice has been shown to induce HHcy[38]. Under normal conditions, several methylation reactions in the liver contribute to the bulk (90%) of SAM utilization and Hcy production via SAH. For example, phosphatidylethanolamine to phosphatidylcholine is mediated by phosphatidylethanolamine N-methyltransferase (PEMT). PEMT-/- mice had 50% decreased plasma Hcy despite being choline and betaine deficient[39]. PEMT null mice exhibited fatty liver and apoptosis but this was not prevented by betaine administration, impaired lipoprotein secretion rather than methyl donor deficiency appeared to be the dominant effect of choline deficiency[40]. The other major source of Hcy is the activity of hepatic guanidinoacetate (GAA) N-methyltransferase (NMT). GAA is produced in the kidney by L-arginine:glycine amidinotransferase. GAA is then converted to creatine in the liver by GAA-NMT, utilizing SAM and generating SAH. Creatine is exported to muscle and also represses the kidney enzyme which produces GAA. GAA supplementation could induce HHcy and creatine feeding lowers Hcy[41].
HCY TOXICITY
Possible cellular mechanisms by which elevated Hcy promotes liver disease are oxidative stress, endoplasmic reticulum (ER) stress and the activation of pro-inflammatory factors (Figure 2). Hcy enhances the production of several pro-inflammatory cytokines. Expression of monocyte chemoattractant protein 1 (MCP-1) was increased in cultured human vascular endothelial cells, smooth muscle cells and monocytes treated with Hcy[42-44]. Hcy has also been shown to increase expression of IL-8[42], a T-lymphocyte and neutrophil chemoattractant, in cultured endothelial cells. Hcy-induced expression of MCP-1 and IL-8 in monocytes and endothelial cells has been shown to occur through activation of NF-κB, a transcription factor involved in mediating downstream inflammatory processes[44,45]. Active NF-κB could stimulate production of cytokines, chemokines, interferons, leukocyte adhesion molecules, hemopoietic growth factors and major histocompatibility (MHC) class I molecules-all of which are thought to influence inflammation[45,46].
Figure 2.
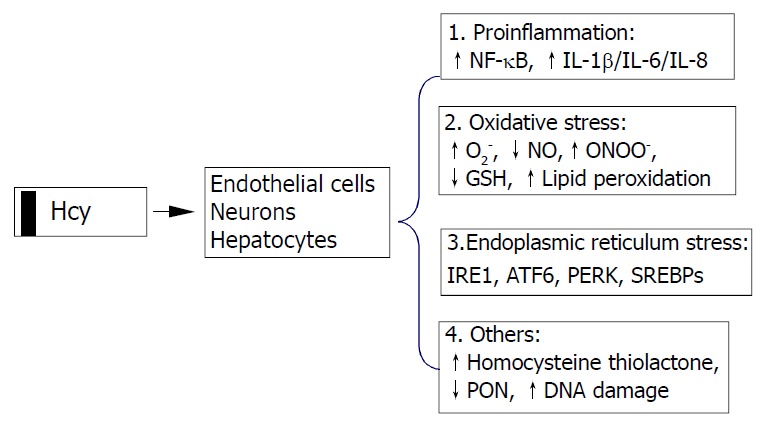
Cellular mechanisms by which homocysteine pro-motes cell injury. Homocysteine causes activation of necrosis factor-κB (NF-κB) and enhances production of cytokines (IL-1β, IL-6, and IL-8) resulting in inflammatory reactions, increases intracellular levels of superoxide anion causing oxidative stress, and induces endoplasmic reticulum (ER) stress by causing misfolding of proteins traversing the ER. Homocysteinyl-tRNA increases production of highly reactive derivative homocys-teine thiolactone which damages enzymes and DNA. IRE1, type 1 ER transmembrane protein kinase; ATF6, the activating tran-scription factor 6; PERK, the PKR like ER kinase; SREBP, sterol regulatory element binding protein, PON, paraoxonase.
Hcy can generate a procoagulant state, which may be related to its proclivity to auto-oxidize, generating H2O2. Various in vitro studies using vascular tissues have implicated Hcy in causing abnormal vascular relaxation responses by enhancing the intracellular production of superoxide anion (O2-)[47-54]. O2- is believed to react with and decrease the availability of endothelial nitric oxide (NO) and yield peroxynitrite, thereby limiting normal vasodilation responses[55,56]. Deceased GSH peroxidase transcription (reduction of peroxides protects NO) may play a role in this process[49,57], since overexpression of GSH peroxidase could restore the NO response[57]. O2- and peroxynitrite are also known to contribute to the oxidative modification of tissues, resulting in the formation of lipid peroxides and nitrosated end products such as 3-nitrotyrosine. The observations that Hcy decreased the expression of a wide range of antioxidant enzymes[57-59] and impaird endothelial NO bioavailability by inhibiting glutathione peroxidase activity raise the possibility that Hcy sensitizes cells to the cytotoxic effects of agents or conditions known to generate ROS. Decreased NO bioavailability has also been shown in vitro to increase the expression of MCP-1, which may enhance intravascular monocyte recruitment and lead to accelerated lesion formation[60].
Intracellular Hcy can be converted by methionyl tRNA synthase into an Hcy-AMP complex, which is subsequently catabolised to Hcy thiolactone, thereby preventing the incorporation of Hcy into nascent polypeptide chains. Hcy thiolactone has unique reactive properties that can lead to the homocysteinylation of lysine residues and free amine groups on numerous cellular proteins, thereby resulting in decreased biological activity and premature degradation[61]. In addition, Hcy thiolactone secreted into the circulation may induce widespread modifications of plasma proteins that could potentially contribute to the development of liver and cardiovascular diseases. Recent studies have demonstrated that Hcy thiolactone decreases paraoxonase activity associated with HDL, thereby rendering HDL less protective against oxidative damage or against toxicity of Hcy thiolactone[62].
HCY-INDUCED ER STRESS
ER is a principal site for protein synthesis and folding, calcium storage and calcium signaling. It also serves as a site of biosynthesis for steroids, cholesterol and other lipids. The physiological roles of the ER include regulation of protein synthesis, folding and targeting and maintenance of cellular calcium homeostasis. The ER has a high concentration of numerous resident chaperone proteins such as glucose regulated protein-78 (GRP78) and GRP94, a high level of calcium and an oxidative environment to carry out these functions efficiently. Proteins that were translocated into the ER lumen underwent post-translational modifications and the folding required for optimal function. Properly folded proteins were allowed to reach their destiny via the secretory pathway, whereas unfolded and misfolded proteins were exported or dislocated from the ER and degraded by cytoplasmic proteasomes[63-68]. ER stress is a condition under which unfolded and misfolded proteins accumulate (Figure 3). ER stress triggers unfolded protein response (UPR), which is an intracellular signaling pathway and is mediated via three ER-resident sensors in mammalian cells: A type-I ER transmembrane protein kinase (IRE-1), the activating transcription factor 6 (ATF-6) and the PKR like ER kinase (PERK). Activation of these three pathways is mediated by GRP78, which is associated with each sensor in the absence of ER stress. As unfolded proteins accumulated in the ER, GRP78 dissociated from and thereby activating IRE-1, ATF-6 and PERK[68-70]. Activation of both IRE-1 and ATF-6 increases the expression of ER-resident chaperones. IRE-1 is a stress-activated transmembrane protein kinase having endoribonuclease activity. Following ER stress, IRE-1 dimerized and was autophosphorylated, thereby allowing IRE-1 to act as an endoribonuclease in the alternative splicing of XBP-1 mRNA. The removal of a 26 base pair intron resulted in a translation frameshift that permits XBP-1 to act as a transcriptional activator of genes containing upstream ER stress response elements (ERSE). Upon ER stress, ATF-6 was transported to the Golgi where the cytosolic transactivation domain of ATF-6 is cleaved from the membrane by specific proteases (S1P and S2P) that also recognize, cleave and activate sterol regulatory element-binding proteins (SREBPs) leading to increased lipids needed for ER membrane synthesis. Following release, the transactivation domain of ATF-6 localized to the nucleus where it interacts with ERSE, thereby activating transcription of numerous UPR-responsive genes, including GRP78, GADD153 (CHOP), XBP-1, ERp72, and Hcy-induced ER protein (Herp). ER stress could also lead to a rapid attenuation in protein synthesis, a cellular process mediated by the transmembrane protein kinase, PERK. Activation of PERK could cause phosphorylation of eukaryotic initiation factor-2α (eIF-2α), which blocks mRNA translation initiation to help relieve the unfolded protein burden on the ER. Recent studies have also demonstrated that PERK-dependent eIF-2 α phosphorylation is required for transcriptional activation of a wide range of UPR-responsive genes[71,72]. The early UPR co-coordinately enhances cell survival by ensuring that the adverse effects of ER stress are dealt with in a timely and efficient manner. However, prolonged UPR following ER stress has severe consequences. It can lead to activation of the tumor necrosis factor receptor associated facter 2 (TRAF2), which activates caspases (e.g. caspase-12 in mice) and JNK resulting in programmed cell death. Over-expression of CHOP, a basic region leucine zipper transcription factor, could also promote cell death[71]. Overproduction of lipids by SREBP can lead to fat accumulation. In addition, ER stress is associated with release of ER Ca2+ stores which can trigger oxidative stress via effects on mitochondria and NF-κB activation leading to inflammatory reactions[73]. NF-κB activation could be blocked by calcium chelators and antioxidants[19]. Increased cytosol calcium also activates calpains which proteolytically cleave Bcl-XL (inactivation) and caspase 12 (activation). ER stress could contribute to the pathogenesis of a number of human diseases, including diabetes, Alzheimer’s disease, Parkinson’s disease and cancer[72].
Figure 3.
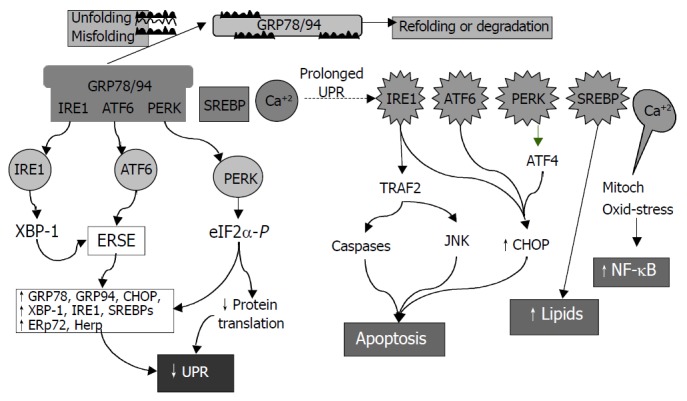
Consequences of endoplasmic reticulum (ER) stress response. In the early phase, unfolded proteins cause dissociation of chaperones such as Bip/GRP78 from ER resident kinases-IRE1 and PERK and transcription factor-ATF6. Activated PERK phos-phorylates eIF2 resulting in translational attenuation. Activated IRE1 and ATF6 up-regulate genes encoding ER chaperone pro-teins such as GRP78/94 leading to increased protein-folding capacity. Overall, the unfolded protein response (UPR) goes down. In the late phase, IRE1 interacts with TRAF2 (tumor necrosis factor receptor associated factor 2) which activates caspases and JNK (cJUN NH2-terminal kinase) leads to apoptosis. ATF6 and PERK upregulate CHOP (C/EBP homologous protein) promoting cell death. SREBP upregulates lipid synthesis. Prolonged UPR leads to Ca2+ release from ER causing production of reactive oxygen intermediates which may lead to activation of NF-κB.
Hcy induced ER stress response has recently received much attention[6,74-79]. Hcy causes ER stress by disrupting disulphide bond formation and causing misfolding of proteins traversing the ER. Elevated levels of intracellular Hcy could increase the expression of several ER stress response genes, including GRP78, GRP94, Herp and RTP[6,58,74,77,78,80-82]. Hcy could induce expression of GADD153[58,78,79] involved in ER stress-induced cell death[83]. Hcy-induced ER stress could cause dysregulation of lipid biosynthesis by activating the SREBPs[6,76-79], ER resident transcription factors are responsible for the induction of genes in the cholesterol/triglyceride biosynthesis and uptake pathways[6]. Hcy-induced cell death was mimicked by other ER stress agents and was dependent on IRE-1 signaling. Activation of IRE-1 by Hcy could lead to a rapid and sustained activation of JNK protein kinases[84,85], a result consistent with the finding that activation of JNK by ER stress involved binding of IRE-1 to TRAF2[86]. Because persistent activation of JNK correlated with cell death[87], these studies could provide further support for a mechanism involving Hcy-induced programmed cell death.
APPROACHES FOR STUDY OF HHCY
Cell and animal models with altered plasma Hcy are among the most useful approaches in determining the biological effects of HHcy. However, cell and transgenic animal models expressing Hcy metabolism-related genes/enzymes are not available. Nevertheless, diet- and, especially, genetic-induced animal models of HHcy have been developed. The gene knockout animals have significantly enhanced the status of Hcy as an independent risk factor for several disorders.
Homozygous and heterozygous CBS-deficient mice were generated in 1995[88]. Homozygous mutants completely lacking CBS were born at the expected frequency from mating of heterozygotes, but they suffered from severe growth retardation and a majority of them died within 5 wk after birth. Histological examination showed that the hepatocytes of homozygotes were enlarged, multinucleated, and filled with microvesicular lipid droplets. Plasma Hcy levels of the homozygotes (203.6 ± 65.3 μmol/L) were 33 times higher than normal (6.1 ± 0.8 μmol/L). The homozygous CBS deficient mice represented a model for severe HHcy. Heterozygous CBS deficient mice had 50% reduction in CBS mRNA and enzyme activity in the liver and had twice normal plasma Hcy levels (13.5 ± 3.2 μmol/L). The CBS knockouts significantly help elucidate the in vivo role of elevated levels of Hcy in the etiology of several HHcy-related disorders and in the cellular mechanisms by which Hcy promote cell injury. The CBS-deficient mice were predisposed to HHcy during dietary folate deficiency, and moderate HHcy was associated with marked impairment of endothelial function in mice[89]. Results from a subsequent study indicated that endothelial dysfunction occurred in HHcy mice even in the absence of folate deficiency[90]. Endothelial dysfunction in CBS (+/-) mice was associated with increased tissue levels of SAH, which suggests that altered SAM-dependent methylation may contribute to vascular dysfunction in HHcy[91]. Further studies with the CBS deficient mice revealed the importance of intracellular redox balance for nitric oxide bioactivity and endothelial function, and the importance of ER stress in abnormal hepatic accumulation of lipid[49,92]. Expression of several genes analyzed by DNA microarray was found to be reproducibly abnormal in the livers of heterozygous and homozygous CBS-deficient mice[93]. These genes encode ribosomal protein S3a and methylthioadenosine phosphorylase, suggesting cellular growth and proliferation perturbations may occur in homozygous CBS-deficient mice liver.
MTHFR-deficient mice have been recently developed to examine the effects of HHcy resulting from genetic deficiencies in the remethylation pathway[94]. MTHFR-deficient mice shared basic phenotypic similarities with CBS-deficient mice. However, they were unique in that they developed mild HHcy and atherosclerosis. Recent study has demonstrated the importance of choline metabolism in HHcy in this model[95]. Comparison study by administrating the alternate choline-derived methyl donor, betaine, to wild-type mice and MTHFR deficient mice revealed that plasma Hcy and liver choline metabolite levels were strongly dependent on the MTHFR genotype. Betaine supplementation decreased Hcy in all three genotypes, restored liver betaine and phosphocholine pools, and prevented severe steatosis in MTHFR-deficient mice. Since there was a significant negative correlation between plasma betaine and Hcy concentrations in humans with cardiovascular disease, the results emphasize the strong interrelationship between Hcy, folate, and choline metabolism. MTHFR-compromised mice with HHcy appeared to be much more sensitive to changes of choline/betaine intake than wild-type animals. HHcy, in the range of that associated with folate deficiency or with homozygosity for the 677T MTHFR variant, may be associated with disturbed choline metabolism.
MS could directly catalyze the remethylation pathway and inactivation of this gene has been attempted recently[96]. Heterozygous MS knockout mice from an outbred background had slightly elevated plasma Hcy (6.1 mol/L) and methionine compared to wild-type mice (4.1 μmol/L) but seemed to be otherwise indistinguishable. Homozygous knockout embryos survived through implantation but died soon thereafter. Nutritional supplementation during pregnancy was unable to rescue embryos that were completely deficient in MS. This study indicated that MS activity was essential for early embryonic development of mice. Although the MS knockout mouse has not provided an immediately obvious animal model of human disease, heterozygotes with 50% reduction of MS activity may be useful. It is likely that MS heterozygote knockouts are more susceptible to dietary deficiencies than wild type mice and thus having merits as a model in which interactions between genetic status and nutritional status can be studied.
The animal models are valuable in vivo tools to further examine potential therapeutic approaches in lowering plasma Hcy while decreasing the prevalence of HHcy-induced disorders. However, the animal models neither have tissue or organ specificity nor exclude potential compensatory pathways of Hcy metabolism. Conditional disruption of Hcy metabolism-related genes and crossing between animal models that are deficient in different genes should be the future directions in the effort of creating animal models for study of HHcy.
ETHANOL-INDUCED HHCY AND LIVER INJURY
The pathogenesis of the pathologic features of alcoholic liver injury, namely steatosis, apoptosis, necrosis, inflammation and fibrosis, is an area of intense interest. Although much progress has been made over the past decade, we still do not have a complete understanding of this process[97]. We recently found that in a murine model of intragastric ethanol there was an upregulation of genes associated with endoplasmic reticulum (ER) stress response, including GRP78 and 94, CHOP and SREBP. The expression of these genes was associated with protein malfolding as well as apoptosis and lipid synthesis[78,79]. Since alcoholism and alcohol-related diseases constitute a severe health problem in the world and ER stress has been linked to Hcy in the pathogenesis of several disorders such as atherosclerosis, Alzheimer’s disease, and liver steatosis, we would direct the readers’ attention to ethanol-induced alterations in Hcy metabolism.
Alcoholic patients have been shown to have elevated plasma Hcy (average two-fold) which rangeed from 10-120 mol/L (normal 5-15 mol/L)[98-100]. Total folate, B12 and B6 levels were normal. However, Hcy levels correlated with folate levels and blood alcohol levels. Well nourished alcoholics exhibited markedly lower levels of serum pyridoxal-phosphate (PLP) and mildly lower red cell folate[37]. Even “social” drinking (30 g/d×6 wk) caused 20% increased Hcy and decreased folate[98,101]. Heavy alcohol consumption is a risk factor for stroke and brain atrophy[101-103]. Rats fed ethanol exhibited a doubling of plasma Hcy despite normal levels of folate, PLP and B12[100]. We have observed a 7 fold increase of plasma Hcy levels (22.3 ± 2.8 μmol/L vs pair-fed control 3.0 ± 0.9 μmol/L) in mice fed ethanol intragastrically for 4 wk[78].
With alcohol feeding of rats intragastrically for 9 wk liver specific MAT1A protein expression did not change, whereas MAT2A increased in conjunction with -40% decreased hepatic levels of methionine and SAM[104]. However, shorter exposure of rats and minipigs to ethanol was not associated with a decrease in methionine or SAM in most studies. Depending on route, ethanol dose and duration, variable changes in SAM and SAH have been described[104-107].
Ethanol feeding has been known to lower MS[108], leading to increased accumulation of 5-methyl THF and to increased BHMT leading to utilization-induced decreased betaine levels[108]. These effects depended on increased blood ethanol. Golden Syrian hamsters with high ADH fed a 360 mL/L ethanol diet did not develop increased blood ethanol levels or changes in Hcy metabolism unless ADH was inhibited[109]. Of note, the SAM levels were maintained by the utilization of betaine. However prolonged ethanol feeding eventually could lead to depletion of SAM. Chronic alcohol could increase choline uptake[110] and mitochondrial oxidation to betaine[111] suggesting compensation for increased demand for betaine. Feeding betaine (0.5%) raised SAM levels which was accentuated in alcohol fed animals (minimal to begin with) and prevented fatty liver[78,105,108]. Raised SAM was initially assumed to contribute to betaine’s ameliorative effect on fatty liver. It may be equally important that the protective role of betaine was due to lowering Hcy directly through BHMT and indirectly by raised SAM leading to activation of CBS.
The mechanism of the ethanol induced decrease in MS is not well understood. Kenyon et al[107] showed that the enzyme was inhibited by high concentrations of acetaldehyde, whereas we have found decreased mRNA. Increase in BHMT activity appeared to be a compensatory phenomenon to maintain methionine and was seen after 2 wk in ethanol fed rats and after 4 wk in ethanol fed mice.
In the chronic (12 mo) ethanol fed micropigs with adequate folate, MS activity decreased by 20% which was associated with slightly decreased serum methionine, 20% increased serum Hcy, and increased hepatic SAH but no change in SAM. These small changes due to ethanol were not associated with increased ALT or fatty liver but were associated with increased scattered apoptosis[112]. Interestingly addition of folate deficient diet to ethanol feeding of the castrated minipig accentuated plasma Hcy and liver injury[116] although ER stress was not considered in this study.
Betaine lowered Hcy and prevented ER stress and alcoholic liver injury in alcohol fed mice[78]. However, we need to be cognizant of other actions of betaine. Feeding rats betaine in drinking water (1.5 g/kg) blunted the TNF response to LPS and decreased concomitant liver injury[114]. Importantly, however, taurine was equally effective. Earlier work has suggested an indirect protective role of choline supplementation, suggesting choline oxidation to betaine could protect against Kupffer cell activation[115-117]. It has been suggested that betaine and taurine serve as organic osmolytes which are critical in regulating Kupffer cell function[118]. Hyperosmotic conditions induce Na+ betaine transporter mRNA while hypoosmotic conditions do the opposite, this occurred in Kupffer cells but not hepatocytes. Betaine or taurine protects the liver against warm ischemia-reperfusion. Recently, betaine pretreatment was shown to protect the hepatocyte from bile acid induced apoptosis. The mechanism is not certain and high concentrations of betaine (mmol/L) were required[119,120]. We observed that increased gene expression of TNF and CD 14 was indicative of the alcohol-induced Kupffer cell activation[78]. However, betaine treatment did not significantly attenuate these changes, suggesting that betaine either acts downstream of alcohol-induced Kupffer cell activation or acts via an independent pathway.
The effect of SAM feeding is of interest since it was reported to decrease fatty liver and mitochondrial abnormalities[112]. SAM might be expected to inhibit re-methylation and promote transsulfuration of Hcy. It is unclear as to what the overall effect on Hcy would be. Severe SAM deficiency in MAT1A knockout did not alter Hcy but was associated with increased expression of BHMT and CBS[121]. SAM could transcriptionally activate MAT1A and suppress MAT2A[122].
Overall chronic ethanol exposure seemed to cause a modest decrease in SAM and increase in SAH along with early-decreased MS and late-increased BHMT. All these changes were accompanied with increased Hcy which occurs despite adequate dietary folate, B12, B6 and choline. Thus there are possible contributions of decreased MS, unknown effects on CBS, and decreased SAM (decreased activation of CBS) leading to HHcy. The decrease in SAM levels was accompanied with increased SAH levels. Since both SAM and SAH activated CBS, it is doubtful that changes in levels of these metabolites exerted a significant regulatory role on transulfuration[11,16]. The increase in BHMT appeared insufficient to lower Hcy due to limitation on the availability of betaine and already-impaired cell function. Although high dietary choline might generate sufficient betaine in rodents, the choline oxidase pathway is normally low in primates. Thus, providing excess dietary betaine rather than choline would seem to be an approach more applicable to the human situation. Since betaine corrects hyperhomocysteinemia, fatty liver injury and ER stress, and homocysteine is known to cause all these changes, it is reasonable to state that an important mechanism by which betaine protects against alcoholic liver disease is the correction of hyperhomocysteinemia and proof of this hypothesis requires further work.
POSSIBLE ROLE OF ER STRESS IN OTHER LIVER DISEASES
ER stress may also be involved in liver injury caused by α1-antitrypsin (α1-AT) deficiency and hepatitis C virus (HCV) or hepatitis B virus (HBV) infection. α1-antitrypsin (α1-AT) deficiency was caused by a point mutation encoding a substitution of lysine for glutamate-342[123]. Aggregated mutant α1-AT was retained in ER rather than secreted in the blood and body fluids where its function is to inhibit neutrophil proteases. Individuals with this deficiency had a markedly increased risk of developing emphysema by a loss of function mechanism by which reduced levels of α1-AT in the lung inhibit connective tissue breakdown by neutrophil elastase, cathepsin G, and proteinase 3. Some individuals with α1-AT deficiency developed liver injury and hepatocellular carcinoma by a gain of function mechanism, i.e., accumulation of aggregated mutant α1-AT within the ER which is toxic to liver cells. However, the exact mechanism by which ER retention of this aggregated mutant protein leads to cellular injury is still unknown. Recent studies have demonstrated that ER retention of mutant α1-AT induces a marked autophagic response in cell culture and transgenic mouse models of α1-AT deficiency as well as in the liver of patients with α1-AT deficiency[124]. The autophagic response is a general mechanism whereby cytosol and intracellular organelles, such as ER, are first sequestered from the rest of the cytoplasm within unique vacuoles and then degraded by fusion with lysosomes to clear the cells of senescent constituents. Under a fasting condition, a marked increase in fat accumulation was observed in α1-AT-containing globules in the liver of α1-AT deficient mice[125], suggesting a malfunction of ER caused by accumulation of mutant α1-AT. Investigations of ER stress markers such as GRP78, CHOP, SREBP, XBP1, and ATF6 are needed to assess the direct involvement of ER stress in α1-AT deficiency-induced liver injury.
Evidence of ER stress in HBV or HCV infection is emerging. HBV codes for three forms of surface protein. The minor and large forms are translated from transcripts specified by the preS1 promoter, while the middle and small forms are translated from transcripts specified by the downstream S promoter. Overexpression of the large surface protein of HBV could lead to a 10-fold activation of the S promoter but not of an unrelated promoter[126]. The large surface protein could also activate the cellular grp78 and grp94 promoters. Neither the middle nor the small surface protein, nor a secretable form of the large surface protein, could activate the S promoter, but agents that induced endoplasmic reticulum (ER) stress had an effect similar to that of the large surface protein, suggesting that HBV may evolve a feedback mechanism, such that ER stress induced by accumulation of the large surface protein increases the synthesis of the middle and small surface proteins, which in combination with the large surface protein can form mixed, secretable particles. HCV-induced ER stress was more evident. HCV replicates from a ribonucleoprotein (RNP) complex that is associated with ER membrane. The replication activities of the HCV subgenomic replicon have been shown to induce ER stress[127]. HCV replicons induce the UPR which is paralleled by the proteolytic cleavage of ATF6. The HCV non-structural protein 5A (NS5A) can bind to and inactivate the cellular double-stranded RNA-activated protein kinase, PKR. NS5A has recently been demonstrated to engage ER-nucleus signal transduction pathway[131]. Expression of NS5A in the ER could induce an ER stress leading to the activation of STAT-3 and NF-κB, which is sensitive to inhibitors of Ca2+ uptake. The NS5A-induced ER stress signaling has also been shown in the context of an HCV subgenomic replicon[128]. Another HCV component, the HCV envelope protein E2, is an ER-bound protein that contains a region of sequence homology with the PKR and its substrate, the eIF2α. E2 could modulate global translation by inhibition of the interferon-induced PKR through its PKR-eIF2α phosphorylation site homology domain (PePHD)[129]. E2 could also bind to and inhibit PERK[129]. At low expression levels, E2 induced ER stress, but at high expression levels, E2 inhibited PERK kinase activity in vitro. Mammalian cells that stably expressed E2 were refractory to the translation-inhibitory effects of ER stress inducers, and E2 relieved general translation inhibition induced by PERK. The PePHD of E2 was required for the rescue of translation that was inhibited by activated PERK. These findings may explain why the virus promotes persistent infection by overcoming the cellular ER stress response. In addition, HCV-induced ER stress resulted in a decline in protein glycosylation. Decreasing protein glycosylation could disrupt the proper protein folding of MHC class I molecules, preventing the assembly of MHC class I molecules. Cells expressing HCV subgenomic replicons had a lower MHC class I cell surface expression[130]. HCV-infected cells may thus be undetectable in the immune system by suppressing MHC class I antigen presentation to cytotoxic T lymphocytes. Therefore, the persistence and pathogenesis of HCV may depend upon the ER stress-mediated interference of MHC class I assembly and cell surface expression. Finally, HCV infection may suppress the degradation of misfolded proteins while stimulating the synthesis of its viral proteins in the ER. In the ER, IRE1-XBP1 pathway directs both protein refolding and degradation in response to ER stress. It was demonstrated that XBP1 expression was elevated in cells carrying HCV subgenomic replicons, but XBP1 transactivating activity was repressed[131]. This prevents the IRE1-XBP1 transcriptional induction of EDEM (ER degradation-enhancing -mannosidase-like protein), which is required for the degradation of misfolded proteins. Consequently, misfolded proteins are stable in cells expressing HCV replicons. Study with a cell line with a defective IRE1-XBP1 pathway showed elevated levels of HCV internal ribosome entry site-mediated translation[131]. This study indicated that the HCV suppression of misfolded protein degradation in the ER not only promoted HCV expression but also contributed to the persistence of the virus in infected hepatocytes.
HCV infection is common in alcoholic patients presenting with liver disease. Heavy alcohol intake would worsen the outcome of HCV infection[132-134], which has directed much attention to the interaction between alcohol and HCV infection. Alcohol plays an important role in HCV infection resulting in increased viral replication, enhanced HCV quasispecies complexity, increased liver-cell death, suppression of immune responses, and iron overload[135]. However, the pathogenic mechanisms underlying the alcohol-HCV interactions are not fully understood. Based on the above evidence that both HCV and homocysteine could induce endoplasmic reticulum (ER) stress response[6,78,127] and that there was a link between alcohol-induced significant elevation of homocysteine level, ER stress, and the pathogenesis of liver injury[78], it is reasonable to hypothesize that a locus of the potentiative interaction between alcohol and HCV in accelerating the progression of liver disease is at the level of ER stress. In the case of both HBV and HCV infection, it is widely recognized that severely immunosuppressed patients may develop a paradoxically severe and rapidly progressive liver disease. This has been seen in the post-OLT setting and in patient with AIDS. Therefore, the loss of immune detection of viral-infected hepatocytes may lead to an unopposed massive overload of hepatocytes with viral proteins triggering ER stress. Future studies should examine the contribution of ER stress to these pathologic conditions.
CONCLUSION
HHcy is an integral component of several disorders including cardiovascular and cerebrovascular diseases, neurodegeneration, liver steatosis, diabetes, and cancer. HHcy can result from deficiencies of vitamin cofactors (B6, B12, folic acid) required for Hcy metabolism and/or from genetic disorders of its metabolism. Hcy unleashes inflammation mediators such as NFκB, IL-1β, IL-6, and IL-8. Hcy increases production of intracellular superoxide anion causing oxidative stress. Hcy-induced misfolding or malfunctioning of numerous intracellular proteins are increasingly important and attract much attention because the Hcy-induced ER stress mechanism can explain many processes of cell injury. Animal model creation and integral investigation of available animal models will certainly play important role in determining precisely the biological effects of HHcy. Our observations with the murine intragastric ethanol fed model have suggested a link between Hcy metabolism, ER stress, and the pathogenesis of alcohol induced liver injury. Figure 4 demonstrates our hypothesis in which ethanol feeding causes HHcy which then induces the ER stress response in parenchymal and nonparenchymal cells in the liver leading to fatty liver, apoptosis and possibly inflammation. The potential beneficial effects of lowering Hcy and preventing ER stress in alcoholic humans needs to be studied. In addition, since a minority of alcoholics develop liver disease and a wide range of Hcy levels are seen in alcoholics, it will be important to examine polymorphism of Hcy metabolizing enzymes as potential risk-factors for the development of HHcy and liver disease.
Figure 4.
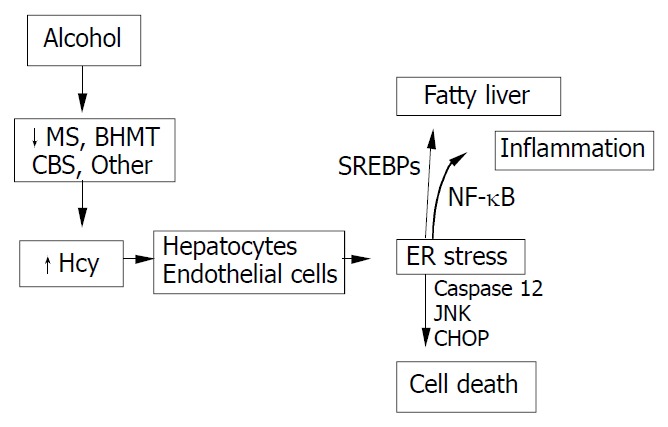
Hypothesis for the role of ethanol-induced HHcy in the pathogenesis of alcoholic liver disease.
Footnotes
Supported by the U.S. National Institute of Alcohol Abuse and Alcoholism, R01 AA014428 and by the Robert E. and May R. Wright Foundation, No. 263
Edited by Wang XL and Xu XQ Proofread by Xu FM
References
- 1.Carson NAJ, Neil DW. Metabolic abnormalities detected in a survey of mentally backward individuals in Northern Ireland. Arch Dis Child. 1962;37:505–513. doi: 10.1136/adc.37.195.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerritsen T, Vaughn JG, Weisman HA. The identification of ho-mocysteine in the urine. Biochem Biophys Res Commun. 1962;9:593–596. doi: 10.1016/0006-291x(62)90114-6. [DOI] [PubMed] [Google Scholar]
- 3.Mudd SH, Finkelstein JD, Irreverre F, Laster L. Homocystinuria: An Enzymatic Defect. Science. 1964;143:1443–1445. doi: 10.1126/science.143.3613.1443. [DOI] [PubMed] [Google Scholar]
- 4.Clarke R, Daly L, Robinson K, Naughten E, Cahalane S, Fowler B, Graham I. Hyperhomocysteinemia: an independent risk factor for vascular disease. N Engl J Med. 1991;324:1149–1155. doi: 10.1056/NEJM199104253241701. [DOI] [PubMed] [Google Scholar]
- 5.Audelin MC, Genest J. Homocysteine and cardiovascular disease in diabetes mellitus. Atherosclerosis. 2001;159:497–511. doi: 10.1016/s0021-9150(01)00531-7. [DOI] [PubMed] [Google Scholar]
- 6.Werstuck GH, Lentz SR, Dayal S, Hossain GS, Sood SK, Shi YY, Zhou J, Maeda N, Krisans SK, Malinow MR, et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Invest. 2001;107:1263–1273. doi: 10.1172/JCI11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schroecksnadel K, Frick B, Wirleitner B, Winkler C, Schennach H, Fuchs D. Moderate hyperhomocysteinemia and immune activation. Curr Pharm Biotechnol. 2004;5:107–118. doi: 10.2174/1389201043489657. [DOI] [PubMed] [Google Scholar]
- 8.Herrmann W, Knapp JP. Hyperhomocysteinemia: a new risk factor for degenerative diseases. Clin Lab. 2002;48:471–481. [PubMed] [Google Scholar]
- 9.Polidori MC, Marvardi M, Cherubini A, Senin U, Mecocci P. Heart disease and vascular risk factors in the cognitively impaired elderly: implications for Alzheimer's dementia. Aging (Milano) 2001;13:231–239. doi: 10.1007/BF03351481. [DOI] [PubMed] [Google Scholar]
- 10.Lawrence de Koning AB, Werstuck GH, Zhou J, Austin RC. Hyperhomocysteinemia and its role in the development of atherosclerosis. Clin Biochem. 2003;36:431–441. doi: 10.1016/s0009-9120(03)00062-6. [DOI] [PubMed] [Google Scholar]
- 11.Medina M, Urdiales JL, Amores-Sánchez MI. Roles of homocysteine in cell metabolism: old and new functions. Eur J Biochem. 2001;268:3871–3882. doi: 10.1046/j.1432-1327.2001.02278.x. [DOI] [PubMed] [Google Scholar]
- 12.Jakubowski H. Molecular basis of homocysteine toxicity in humans. Cell Mol Life Sci. 2004;61:470–487. doi: 10.1007/s00018-003-3204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jakubowski H, Zhang L, Bardeguez A, Aviv A. Homocysteine thiolactone and protein homocysteinylation in human endothelial cells: implications for atherosclerosis. Circ Res. 2000;87:45–51. doi: 10.1161/01.res.87.1.45. [DOI] [PubMed] [Google Scholar]
- 14.Finkelstein JD, Martin JJ, Harris BJ. Methionine metabolism in mammals. The methionine-sparing effect of cystine. J Biol Chem. 1988;263:11750–11754. [PubMed] [Google Scholar]
- 15.Finkelstein JD. Pathways and regulation of homocysteine metabolism in mammals. Semin Thromb Hemost. 2000;26:219–225. doi: 10.1055/s-2000-8466. [DOI] [PubMed] [Google Scholar]
- 16.Finkelstein JD. Homocysteine: a history in progress. Nutr Rev. 2000;58:193–204. doi: 10.1111/j.1753-4887.2000.tb01862.x. [DOI] [PubMed] [Google Scholar]
- 17.Finkelstein JD, Martin JJ. Methionine metabolism in mammals. Distribution of homocysteine between competing pathways. J Biol Chem. 1984;259:9508–9513. [PubMed] [Google Scholar]
- 18.Mato JM, Corrales FJ, Lu SC, Avila MA. S-Adenosylmethionine: a control switch that regulates liver function. FASEB J. 2002;16:15–26. doi: 10.1096/fj.01-0401rev. [DOI] [PubMed] [Google Scholar]
- 19.Bostom AG, Lathrop L. Hyperhomocysteinemia in end-stage renal disease: prevalence, etiology, and potential relationship to arteriosclerotic outcomes. Kidney Int. 1997;52:10–20. doi: 10.1038/ki.1997.298. [DOI] [PubMed] [Google Scholar]
- 20.van Guldener C, Stehouwer CD. Homocysteine metabolism in renal disease. Clin Chem Lab Med. 2003;41:1412–1417. doi: 10.1515/CCLM.2003.217. [DOI] [PubMed] [Google Scholar]
- 21.Mudd SH, Lev HL, Skovby F. Disorders of transsulfuration. In Scriver CR, Beaudet AL, Sly WS, Valle D. (eds), The Metabolic Basis of Inherited Disease. New York: McGraw-Hill; 1995. p. 1279–1327. [Google Scholar]
- 22.Rosenblatt DS. Inherited disorders of folate transport and metabolism. In Scriver CR, Beaudet AL, Sly WS, Valle D. (eds), The Metabolic Basis of Inherited Disease. New York: McGraw-Hill; 1995. p. 3111–3128. [Google Scholar]
- 23.Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Matthews RG, Boers GJ, den Heijer M, Kluijtmans LA, van den Heuvel LP. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;10:111–113. doi: 10.1038/ng0595-111. [DOI] [PubMed] [Google Scholar]
- 24.Ma J, Stampfer MJ, Hennekens CH, Frosst P, Selhub J, Horsford J, Malinow MR, Willett WC, Rozen R. Methylenetetrahydrofolate reductase polymorphism, plasma folate, homocysteine, and risk of myocardial infarction in US physicians. Circulation. 1996;94:2410–2416. doi: 10.1161/01.cir.94.10.2410. [DOI] [PubMed] [Google Scholar]
- 25.Jacques PF, Bostom AG, Williams RR, Ellison RC, Eckfeldt JH, Rosenberg IH, Selhub J, Rozen R. Relation between folate status, a common mutation in methylenetetrahydrofolate reductase, and plasma homocysteine concentrations. Circulation. 1996;93:7–9. doi: 10.1161/01.cir.93.1.7. [DOI] [PubMed] [Google Scholar]
- 26.Jeng YL, Wu MH, Huang HB, Lin WY, You SL, Chu TY, Chen CJ, Sun CA. The methylenetetrahydrofolate reductase 677C→T polymorphism and lung cancer risk in a Chinese population. Anticancer Res. 2003;23:5149–5152. [PubMed] [Google Scholar]
- 27.Woo KS, Qiao M, Chook P, Poon PY, Chan AK, Lau JT, Fung KP, Woo JL. Homocysteine, endothelial dysfunction, and coronary artery disease: emerging strategy for secondary prevention. J Card Surg. 2002;17:432–435. [PubMed] [Google Scholar]
- 28.Mudd SH, Levy HL, Skovby F. Disorders of transsulfation. In: Scriver CR, Beadet AL, Sly WS, Vallee D, editors. The Metabolic Basis for Inherited Disease. New York: McGraw-Hill; 1989. p. 693. [Google Scholar]
- 29.Kalra DK. Homocysteine and cardiovascular disease. Curr Atheroscler Rep. 2004;6:101–106. doi: 10.1007/s11883-004-0097-3. [DOI] [PubMed] [Google Scholar]
- 30.Wi lcke n DE L, Du dma n N PB. H o m o cy st inu ria a nd atherosclerosis. In: Lusis AJ, Rotter JI, Sparkes RS, editors. Mo-lecular genetics of coronary artery disease; candidate genes and process in atherosclerosis. Monograms in human genetics. New York: Karger; 1992. p. 311. [Google Scholar]
- 31.Mudd SH, Levy HL, Kraus JP. Disorders of transsulfuration. In: Scriver CR, Beaudet AL, Sly WS, Eds , editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 2007–2056. [Google Scholar]
- 32.Ueland PM, Refsum H. Plasma homocysteine, a risk factor for vascular disease: plasma levels in health, disease, and drug therapy. J Lab Clin Med. 1989;114:473–501. [PubMed] [Google Scholar]
- 33.Moat SJ, Bao L, Fowler B, Bonham JR, Walter JH, Kraus JP. The molecular basis of cystathionine beta-synthase (CBS) deficiency in UK and US patients with homocystinuria. Hum Mutat. 2004;23:206. doi: 10.1002/humu.9214. [DOI] [PubMed] [Google Scholar]
- 34.Haynes WG. Hyperhomocysteinemia, vascular function and atherosclerosis: effects of vitamins. Cardiovasc Drugs Ther. 2002;16:391–399. doi: 10.1023/a:1022130217463. [DOI] [PubMed] [Google Scholar]
- 35.Billion S, Tribout B, Cadet E, Queinnec C, Rochette J, Wheatley P, Bataille P. Hyperhomocysteinaemia, folate and vitamin B12 in unsupplemented haemodialysis patients: effect of oral therapy with folic acid and vitamin B12. Nephrol Dial Transplant. 2002;17:455–461. doi: 10.1093/ndt/17.3.455. [DOI] [PubMed] [Google Scholar]
- 36.Lakshmi AV, Maniprabha C, Krishna TP. Plasma homocysteine level in relation to folate and vitamin B6 status in apparently normal men. Asia Pac J Clin Nutr. 2001;10:194–196. doi: 10.1046/j.1440-6047.2001.00252.x. [DOI] [PubMed] [Google Scholar]
- 37.Cravo ML, Camilo ME. Hyperhomocysteinemia in chronic alcoholism: relations to folic acid and vitamins B(6) and B(12) status. Nutrition. 2000;16:296–302. doi: 10.1016/s0899-9007(99)00297-x. [DOI] [PubMed] [Google Scholar]
- 38.Troen AM, Lutgens E, Smith DE, Rosenberg IH, Selhub J. The atherogenic effect of excess methionine intake. Proc Natl Acad Sci USA. 2003;100:15089–15094. doi: 10.1073/pnas.2436385100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noga AA, Stead LM, Zhao Y, Brosnan ME, Brosnan JT, Vance DE. Plasma homocysteine is regulated by phospholipid methylation. J Biol Chem. 2003;278:5952–5955. doi: 10.1074/jbc.M212194200. [DOI] [PubMed] [Google Scholar]
- 40.Shin OH, Mar MH, Albright CD, Citarella MT, da Costa KA, Zeisel SH. Methyl-group donors cannot prevent apoptotic death of rat hepatocytes induced by choline-deficiency. J Cell Biochem. 1997;64:196–208. doi: 10.1002/(sici)1097-4644(199702)64:2<196::aid-jcb3>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 41.Stead LM, Au KP, Jacobs RL, Brosnan ME, Brosnan JT. Methylation demand and homocysteine metabolism: effects of dietary provision of creatine and guanidinoacetate. Am J Physiol Endocrinol Metab. 2001;281:E1095–E1100. doi: 10.1152/ajpendo.2001.281.5.E1095. [DOI] [PubMed] [Google Scholar]
- 42.Poddar R, Sivasubramanian N, DiBello PM, Robinson K, Jacobsen DW. Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: implications for vascular disease. Circulation. 2001;103:2717–2723. doi: 10.1161/01.cir.103.22.2717. [DOI] [PubMed] [Google Scholar]
- 43.Wang G, O K. Homocysteine stimulates the expression of monocyte chemoattractant protein-1 receptor (CCR2) in human monocytes: possible involvement of oxygen free radicals. Biochem J. 2001;357:233–240. doi: 10.1042/0264-6021:3570233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang G, Siow YL, O K. Homocysteine induces monocyte chemoattractant protein-1 expression by activating NF-kappaB in THP-1 macrophages. Am J Physiol Heart Circ Physiol. 2001;280:H2840–H2847. doi: 10.1152/ajpheart.2001.280.6.H2840. [DOI] [PubMed] [Google Scholar]
- 45.Collins T, Cybulsky MI. NF-kappaB: pivotal mediator or innocent bystander in atherogenesis. J Clin Invest. 2001;107:255–264. doi: 10.1172/JCI10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hofmann MA, Lalla E, Lu Y, Gleason MR, Wolf BM, Tanji N, Ferran LJ, Kohl B, Rao V, Kisiel W, et al. Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J Clin Invest. 2001;107:675–683. doi: 10.1172/JCI10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loscalzo J. The oxidant stress of hyperhomocyst(e)inemia. J Clin Invest. 1996;98:5–7. doi: 10.1172/JCI118776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tawakol A, Omland T, Gerhard M, Wu JT, Creager MA. Hyperhomocyst(e)inemia is associated with impaired endothelium-dependent vasodilation in humans. Circulation. 1997;95:1119–1121. doi: 10.1161/01.cir.95.5.1119. [DOI] [PubMed] [Google Scholar]
- 49.Weiss N, Heydrick S, Zhang YY, Bierl C, Cap A, Loscalzo J. Cel-lular redox state and endothelial dysfunction in mildly hyperhomocysteinemic cystathionine beta-synthase-deficient mice. Arterioscler Thromb Vasc Biol. 2002;22:34–41. doi: 10.1161/hq1201.100456. [DOI] [PubMed] [Google Scholar]
- 50.Lang D, Kredan MB, Moat SJ, Hussain SA, Powell CA, Bellamy MF, Powers HJ, Lewis MJ. Homocysteine-induced inhibition of endothelium-dependent relaxation in rabbit aorta: role for superoxide anions. Arterioscler Thromb Vasc Biol. 2000;20:422–427. doi: 10.1161/01.atv.20.2.422. [DOI] [PubMed] [Google Scholar]
- 51.Chambers JC, McGregor A, Jean-Marie J, Obeid OA, Kooner JS. Demonstration of rapid onset vascular endothelial dysfunction after hyperhomocysteinemia: an effect reversible with vitamin C therapy. Circulation. 1999;99:1156–1160. doi: 10.1161/01.cir.99.9.1156. [DOI] [PubMed] [Google Scholar]
- 52.Stanger O, Weger M. Interactions of homocysteine, nitric oxide, folate and radicals in the progressively damaged endothelium. Clin Chem Lab Med. 2003;41:1444–1454. doi: 10.1515/CCLM.2003.222. [DOI] [PubMed] [Google Scholar]
- 53.Franken DG, Boers GH, Blom HJ, Trijbels FJ, Kloppenborg PW. Treatment of mild hyperhomocysteinemia in vascular disease patients. Arterioscler Thromb. 1994;14:465–470. doi: 10.1161/01.atv.14.3.465. [DOI] [PubMed] [Google Scholar]
- 54.Mosharov E, Cranford MR, Banerjee R. The quantitatively im-portant relationship between homocysteine metabolism and glu-tathione synthesis by the transsulfuration pathway and its regu-lation by redox changes. Biochemistry. 2000;39:13005–13011. doi: 10.1021/bi001088w. [DOI] [PubMed] [Google Scholar]
- 55.Gryglewski RJ, Palmer RM, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320:454–456. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]
- 56.Heinecke JW, Rosen H, Suzuki LA, Chait A. The role of sulfur-containing amino acids in superoxide production and modification of low density lipoprotein by arterial smooth muscle cells. J Biol Chem. 1987;262:10098–10103. [PubMed] [Google Scholar]
- 57.Upchurch GR, Welch GN, Fabian AJ, Freedman JE, Johnson JL, Keaney JF, Loscalzo J. Homocyst(e)ine decreases bioavailable nitric oxide by a mechanism involving glutathione peroxidase. J Biol Chem. 1997;272:17012–17017. doi: 10.1074/jbc.272.27.17012. [DOI] [PubMed] [Google Scholar]
- 58.Outinen PA, Sood SK, Liaw PC, Sarge KD, Maeda N, Hirsh J, Ribau J, Podor TJ, Weitz JI, Austin RC. Characterization of the stress-inducing effects of homocysteine. Biochem J. 1998;332(Pt 1):213–221. doi: 10.1042/bj3320213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dayal S, Brown KL, Weydert CJ, Oberley LW, Arning E, Bottiglieri T, Faraci FM, Lentz SR. Deficiency of glutathione peroxidase-1 sensitizes hyperhomocysteinemic mice to endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2002;22:1996–2002. doi: 10.1161/01.atv.0000041629.92741.dc. [DOI] [PubMed] [Google Scholar]
- 60.Ylä-Herttuala S, Palinski W, Rosenfeld ME, Parthasarathy S, Carew TE, Butler S, Witztum JL, Steinberg D. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J Clin Invest. 1989;84:1086–1095. doi: 10.1172/JCI114271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jakubowski H. Protein homocysteinylation: possible mechanism underlying pathological consequences of elevated homocysteine levels. FASEB J. 1999;13:2277–2283. [PubMed] [Google Scholar]
- 62.Ferretti G, Bacchetti T, Marotti E, Curatola G. Effect of homocysteinylation on human high-density lipoproteins: a correlation with paraoxonase activity. Metabolism. 2003;52:146–151. doi: 10.1053/meta.2003.50033. [DOI] [PubMed] [Google Scholar]
- 63.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 64.Welihinda AA, Tirasophon W, Kaufman RJ. The cellular response to protein misfolding in the endoplasmic reticulum. Gene Expr. 1999;7:293–300. [PMC free article] [PubMed] [Google Scholar]
- 65.Kaufman RJ, Scheuner D, Schröder M, Shen X, Lee K, Liu CY, Arnold SM. The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol Cell Biol. 2002;3:411–421. doi: 10.1038/nrm829. [DOI] [PubMed] [Google Scholar]
- 66.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ma Y, Hendershot LM. The mammalian endoplasmic reticulum as a sensor for cellular stress. Cell Stress Chaperones. 2002;7:222–229. doi: 10.1379/1466-1268(2002)007<0222:tmeraa>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 69.Liu CY, Schröder M, Kaufman RJ. Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J Biol Chem. 2000;275:24881–24885. doi: 10.1074/jbc.M004454200. [DOI] [PubMed] [Google Scholar]
- 70.Liu CY, Wong HN, Schauerte JA, Kaufman RJ. The protein kinase/endoribonuclease IRE1a that signals the unfolded protein response has a luminal N-terminal ligand-independent dimerization domain. J Biol Chem. 2002;277:18346–18356. doi: 10.1074/jbc.M112454200. [DOI] [PubMed] [Google Scholar]
- 71.Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–380. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- 72.Kaufman RJ. Regulation of mRNA translation by protein folding in the endoplasmic reticulum. Trends Biochem Sci. 2004;29:152–158. doi: 10.1016/j.tibs.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 73.Pahl HL, Baeuerle PA. The ER-overload response: activation of NF-kappaB. Trends Biochem Sci. 1997;22:63–67. doi: 10.1016/s0968-0004(96)10073-6. [DOI] [PubMed] [Google Scholar]
- 74.Outinen PA, Sood SK, Pfeifer SI, Pamidi S, Podor TJ, Li J, Weitz JI, Austin RC. Homocysteine-induced endoplasmic reticulum stress and growth arrest leads to specific changes in gene expression in human vascular endothelial cells. Blood. 1999;94:959–967. [PubMed] [Google Scholar]
- 75.Althausen S, Paschen W. Homocysteine-induced changes in mRNA levels of genes coding for cytoplasmic- and endoplasmic reticulum-resident stress proteins in neuronal cell cultures. Brain Res Mol Brain Res. 2000;84:32–40. doi: 10.1016/s0169-328x(00)00208-4. [DOI] [PubMed] [Google Scholar]
- 76.Roybal CN, Yang S, Sun CW, Hurtado D, Vander Jagt DL, Townes TM, Abcouwer SF. Homocysteine increases the expression of vascular endothelial growth factor by a mechanism involving endoplasmic reticulum stress and transcription factor ATF4. J Biol Chem. 2004;279:14844–14852. doi: 10.1074/jbc.M312948200. [DOI] [PubMed] [Google Scholar]
- 77.Zhang C, Cai Y, Adachi MT, Oshiro S, Aso T, Kaufman RJ, Kitajima S. Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. J Biol Chem. 2001;276:35867–35874. doi: 10.1074/jbc.M100747200. [DOI] [PubMed] [Google Scholar]
- 78.Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- 79.Lluis JM, Colell A, García-Ruiz C, Kaplowitz N, Fernández-Checa JC. Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology. 2003;124:708–724. doi: 10.1053/gast.2003.50089. [DOI] [PubMed] [Google Scholar]
- 80.Agarwala KL, Kokame K, Kato H, Miyata T. Phosphorylation of RTP, an ER stress-responsive cytoplasmic protein. Biochem Biophys Res Commun. 2000;272:641–647. doi: 10.1006/bbrc.2000.2833. [DOI] [PubMed] [Google Scholar]
- 81.Kokame K, Agarwala KL, Kato H, Miyata T. Herp, a new ubiquitin-like membrane protein induced by endoplasmic reticulum stress. J Biol Chem. 2000;275:32846–32853. doi: 10.1074/jbc.M002063200. [DOI] [PubMed] [Google Scholar]
- 82.Dimitrova KR, DeGroot K, Myers AK, Kim YD. Estrogen and homocysteine. Cardiovasc Res. 2002;53:577–588. doi: 10.1016/s0008-6363(01)00462-x. [DOI] [PubMed] [Google Scholar]
- 83.Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, Boorstein R, Kreibich G, Hendershot LM, Ron D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153) Mol Cell Biol. 1996;16:4273–4280. doi: 10.1128/mcb.16.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cai Y, Zhang C, Nawa T, Aso T, Tanaka M, Oshiro S, Ichijo H, Kitajima S. Homocysteine-responsive ATF3 gene expression in human vascular endothelial cells: activation of c-Jun NH(2)-terminal kinase and promoter response element. Blood. 2000;96:2140–2148. [PubMed] [Google Scholar]
- 85.Zhang C, Kawauchi J, Adachi MT, Hashimoto Y, Oshiro S, Aso T, Kitajima S. Activation of JNK and transcriptional repressor ATF3/LRF1 through the IRE1/TRAF2 pathway is implicated in human vascular endothelial cell death by homocysteine. Biochem Biophys Res Commun. 2001;289:718–724. doi: 10.1006/bbrc.2001.6044. [DOI] [PubMed] [Google Scholar]
- 86.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 87.Chen YR, Meyer CF, Tan TH. Persistent activation of c-Jun N-terminal kinase 1 (JNK1) in gamma radiation-induced apoptosis. J Biol Chem. 1996;271:631–634. doi: 10.1074/jbc.271.2.631. [DOI] [PubMed] [Google Scholar]
- 88.Watanabe M, Osada J, Aratani Y, Kluckman K, Reddick R, Malinow MR, Maeda N. Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci USA. 1995;92:1585–1589. doi: 10.1073/pnas.92.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lentz SR, Erger RA, Dayal S, Maeda N, Malinow MR, Heistad DD, Faraci FM. Folate dependence of hyperhomocysteinemia and vascular dysfunction in cystathionine beta-synthase-deficient mice. Am J Physiol Heart Circ Physiol. 2000;279:H970–H975. doi: 10.1152/ajpheart.2000.279.3.H970. [DOI] [PubMed] [Google Scholar]
- 90.Lentz SR, Piegors DJ, Malinow MR, Heistad DD. Supplementation of atherogenic diet with B vitamins does not prevent atherosclerosis or vascular dysfunction in monkeys. Circulation. 2001;103:1006–1011. doi: 10.1161/01.cir.103.7.1006. [DOI] [PubMed] [Google Scholar]
- 91.Dayal S, Bottiglieri T, Arning E, Maeda N, Malinow MR, Sigmund CD, Heistad DD, Faraci FM, Lentz SR. Endothelial dysfunction and elevation of S-adenosylhomocysteine in cystathionine beta-synthase-deficient mice. Circ Res. 2001;88:1203–1209. doi: 10.1161/hh1101.092180. [DOI] [PubMed] [Google Scholar]
- 92.Eberhardt RT, Forgione MA, Cap A, Leopold JA, Rudd MA, Trolliet M, Heydrick S, Stark R, Klings ES, Moldovan NI, et al. Endothelial dysfunction in a murine model of mild hyperhomocyst(e)inemia. J Clin Invest. 2000;106:483–491. doi: 10.1172/JCI8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Robert K, Chassé JF, Santiard-Baron D, Vayssettes C, Chabli A, Aupetit J, Maeda N, Kamoun P, London J, Janel N. Altered gene expression in liver from a murine model of hyperhomocysteinemia. J Biol Chem. 2003;278:31504–31511. doi: 10.1074/jbc.M213036200. [DOI] [PubMed] [Google Scholar]
- 94.Chen Z, Karaplis AC, Ackerman SL, Pogribny IP, Melnyk S, Lussier-Cacan S, Chen MF, Pai A, John SW, Smith RS, et al. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum Mol Genet. 2001;10:433–443. doi: 10.1093/hmg/10.5.433. [DOI] [PubMed] [Google Scholar]
- 95.Schwahn BC, Chen Z, Laryea MD, Wendel U, Lussier-Cacan S, Genest J, Mar MH, Zeisel SH, Castro C, Garrow T, et al. Homocysteine-betaine interactions in a murine model of 5,10-methylenetetrahydrofolate reductase deficiency. FASEB J. 2003;17:512–514. doi: 10.1096/fj.02-0456fje. [DOI] [PubMed] [Google Scholar]
- 96.Swanson DA, Liu ML, Baker PJ, Garrett L, Stitzel M, Wu J, Harris M, Banerjee R, Shane B, Brody LC. Targeted disruption of the methionine synthase gene in mice. Mol Cell Biol. 2001;21:1058–1065. doi: 10.1128/MCB.21.4.1058-1065.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tsukamoto H, Lu SC. Current concepts in the pathogenesis of alcoholic liver injury. FASEB J. 2001;15:1335–1349. doi: 10.1096/fj.00-0650rev. [DOI] [PubMed] [Google Scholar]
- 98.Bleich S, Bleich K, Kropp S, Bittermann HJ, Degner D, Sperling W, Rüther E, Kornhuber J. Moderate alcohol consumption in social drinkers raises plasma homocysteine levels: a contradiction to the 'French Paradox'. Alcohol Alcohol. 2001;36:189–192. doi: 10.1093/alcalc/36.3.189. [DOI] [PubMed] [Google Scholar]
- 99.Stickel F, Choi SW, Kim YI, Bagley PJ, Seitz HK, Russell RM, Selhub J, Mason JB. Effect of chronic alcohol consumption on total plasma homocysteine level in rats. Alcohol Clin Exp Res. 2000;24:259–264. [PubMed] [Google Scholar]
- 100.Carmel R, James SJ. Alcohol abuse: an important cause of severe hyperhomocysteinemia. Nutr Rev. 2002;60:215–221. doi: 10.1301/00296640260184309. [DOI] [PubMed] [Google Scholar]
- 101.Bleich S, Bandelow B, Javaheripour K, Müller A, Degner D, Wilhelm J, Havemann-Reinecke U, Sperling W, Rüther E, Kornhuber J. Hyperhomocysteinemia as a new risk factor for brain shrinkage in patients with alcoholism. Neurosci Lett. 2003;335:179–182. doi: 10.1016/s0304-3940(02)01194-1. [DOI] [PubMed] [Google Scholar]
- 102.Reynolds K, Lewis B, Nolen JD, Kinney GL, Sathya B, He J. Alcohol consumption and risk of stroke: a meta-analysis. JAMA. 2003;289:579–588. doi: 10.1001/jama.289.5.579. [DOI] [PubMed] [Google Scholar]
- 103.Bleich S, Kornhuber J. Relationship between plasma homocysteine levels and brain atrophy in healthy elderly individuals. Neurology. 2003;60:1220; author reply 1220. doi: 10.1212/wnl.60.7.1220. [DOI] [PubMed] [Google Scholar]
- 104.Lu SC, Huang ZZ, Yang H, Mato JM, Avila MA, Tsukamoto H. Changes in methionine adenosyltransferase and S-adenosylmethionine homeostasis in alcoholic rat liver. Am J Physiol Gastrointest Liver Physiol. 2000;279:G178–G185. doi: 10.1152/ajpgi.2000.279.1.G178. [DOI] [PubMed] [Google Scholar]
- 105.Barak AJ, Beckenhauer HC, Junnila M, Tuma DJ. Dietary betaine promotes generation of hepatic S-adenosylmethionine and protects the liver from ethanol-induced fatty infiltration. Alcohol Clin Exp Res. 1993;17:552–555. doi: 10.1111/j.1530-0277.1993.tb00798.x. [DOI] [PubMed] [Google Scholar]
- 106.Trimble KC, Molloy AM, Scott JM, Weir DG. The effect of ethanol on one-carbon metabolism: increased methionine catabolism and lipotrope methyl-group wastage. Hepatology. 1993;18:984–989. doi: 10.1002/hep.1840180433. [DOI] [PubMed] [Google Scholar]
- 107.Kenyon SH, Nicolaou A, Gibbons WA. The effect of ethanol and its metabolites upon methionine synthase activity in vitro. Alcohol. 1998;15:305–309. doi: 10.1016/s0741-8329(97)00134-1. [DOI] [PubMed] [Google Scholar]
- 108.Barak AJ, Beckenhauer HC, Tuma DJ. Betaine, ethanol, and the liver: a review. Alcohol. 1996;13:395–398. doi: 10.1016/0741-8329(96)00030-4. [DOI] [PubMed] [Google Scholar]
- 109.Barak AJ, Beckenhauer HC, Tuma DJ. Hepatic transmethylation and blood alcohol levels. Alcohol Alcohol. 1991;26:125–128. doi: 10.1093/oxfordjournals.alcalc.a045092. [DOI] [PubMed] [Google Scholar]
- 110.Tuma DJ, Keefer RC, Beckenhauer HC, Barak AJ. Effect of ethanol on uptake of choline by the isolated perfused rat liver. Biochim Biophys Acta. 1970;218:141–147. doi: 10.1016/0005-2760(70)90101-3. [DOI] [PubMed] [Google Scholar]
- 111.Thompson JA, Reitz RC. Studies of the acute and chronic effects of ethanol ingestion on choline oxidation. Ann N Y Acad Sci. 1976;273:194–204. doi: 10.1111/j.1749-6632.1976.tb52882.x. [DOI] [PubMed] [Google Scholar]
- 112.Halsted CH, Villanueva J, Chandler CJ, Stabler SP, Allen RH, Muskhelishvili L, James SJ, Poirier L. Ethanol feeding of micropigs alters methionine metabolism and increases hepatocellular apoptosis and proliferation. Hepatology. 1996;23:497–505. doi: 10.1002/hep.510230314. [DOI] [PubMed] [Google Scholar]
- 113.Halsted CH, Villanueva JA, Devlin AM, Niemelä O, Parkkila S, Garrow TA, Wallock LM, Shigenaga MK, Melnyk S, James SJ. Folate deficiency disturbs hepatic methionine metabolism and promotes liver injury in the ethanol-fed micropig. Proc Natl Acad Sci USA. 2002;99:10072–10077. doi: 10.1073/pnas.112336399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim SK, Kim YC. Attenuation of bacterial lipopolysaccharide-induced hepatotoxicity by betaine or taurine in rats. Food Chem Toxicol. 2002;40:545–549. doi: 10.1016/s0278-6915(01)00102-8. [DOI] [PubMed] [Google Scholar]
- 115.Rose ML, Rivera CA, Bradford BU, Graves LM, Cattley RC, Schoonhoven R, Swenberg JA, Thurman RG. Kupffer cell oxidant production is central to the mechanism of peroxisome proliferators. Carcinogenesis. 1999;20:27–33. doi: 10.1093/carcin/20.1.27. [DOI] [PubMed] [Google Scholar]
- 116.Rivera CA, Bradford BU, Seabra V, Thurman RG. Role of endotoxin in the hypermetabolic state after acute ethanol exposure. Am J Physiol. 1998;275:G1252–G1258. doi: 10.1152/ajpgi.1998.275.6.G1252. [DOI] [PubMed] [Google Scholar]
- 117.Rivera CA, Wheeler MD, Enomoto N, Thurman RG. A choline-rich diet improves survival in a rat model of endotoxin shock. Am J Physiol. 1998;275:G862–G867. doi: 10.1152/ajpgi.1998.275.4.G862. [DOI] [PubMed] [Google Scholar]
- 118.Zhang F, Warskulat U, Wettstein M, Häussinger D. Identification of betaine as an osmolyte in rat liver macrophages (Kupffer cells) Gastroenterology. 1996;110:1543–1552. doi: 10.1053/gast.1996.v110.pm8613062. [DOI] [PubMed] [Google Scholar]
- 119.Graf D, Kurz AK, Reinehr R, Fischer R, Kircheis G, Häussinger D. Prevention of bile acid-induced apoptosis by betaine in rat liver. Hepatology. 2002;36:829–839. doi: 10.1053/jhep.2002.35536. [DOI] [PubMed] [Google Scholar]
- 120.Lieber CS, Casini A, DeCarli LM, Kim CI, Lowe N, Sasaki R, Leo MA. S-adenosyl-L-methionine attenuates alcohol-induced liver injury in the baboon. Hepatology. 1990;11:165–172. doi: 10.1002/hep.1840110203. [DOI] [PubMed] [Google Scholar]
- 121.Lu SC, Alvarez L, Huang ZZ, Chen L, An W, Corrales FJ, Avila MA, Kanel G, Mato JM. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc Natl Acad Sci USA. 2001;98:5560–5565. doi: 10.1073/pnas.091016398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.García-Trevijano ER, Latasa MU, Carretero MV, Berasain C, Mato JM, Avila MA. S-adenosylmethionine regulates MAT1A and MAT2A gene expression in cultured rat hepatocytes: a new role for S-adenosylmethionine in the maintenance of the differentiated status of the liver. FASEB J. 2000;14:2511–2518. doi: 10.1096/fj.00-0121com. [DOI] [PubMed] [Google Scholar]
- 123.Teckman JH, Qu D, Perlmutter DH. Molecular pathogenesis of liver disease in alpha1-antitrypsin deficiency. Hepatology. 1996;24:1504–1516. doi: 10.1002/hep.510240635. [DOI] [PubMed] [Google Scholar]
- 124.Perlmutter DH. Liver injury in alpha1-antitrypsin deficiency: an aggregated protein induces mitochondrial injury. J Clin Invest. 2002;110:1579–1583. doi: 10.1172/JCI16787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Teckman JH, An JK, Loethen S, Perlmutter DH. Fasting in alpha1-antitrypsin deficient liver: constitutive [correction of consultative] activation of autophagy. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1156–G1165. doi: 10.1152/ajpgi.00041.2002. [DOI] [PubMed] [Google Scholar]
- 126.Xu Z, Jensen G, Yen TS. Activation of hepatitis B virus S promoter by the viral large surface protein via induction of stress in the endoplasmic reticulum. J Virol. 1997;71:7387–7392. doi: 10.1128/jvi.71.10.7387-7392.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tardif KD, Mori K, Siddiqui A. Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an in-tracellular signaling pathway. J Virol. 2002;76:7453–7459. doi: 10.1128/JVI.76.15.7453-7459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Waris G, Tardif KD, Siddiqui A. Endoplasmic reticulum (ER) stress: hepatitis C virus induces an ER-nucleus signal transduction pathway and activates NF-kappaB and STAT-3. Biochem Pharmacol. 2002;64:1425–1430. doi: 10.1016/s0006-2952(02)01300-x. [DOI] [PubMed] [Google Scholar]
- 129.Pavio N, Romano PR, Graczyk TM, Feinstone SM, Taylor DR. Protein synthesis and endoplasmic reticulum stress can be modu-lated by the hepatitis C virus envelope protein E2 through the eukaryotic initiation factor 2alpha kinase PERK. J Virol. 2003;77:3578–3585. doi: 10.1128/JVI.77.6.3578-3585.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tardif KD, Siddiqui A. Cell surface expression of major histocompatibility complex class I molecules is reduced in hepatitis C virus subgenomic replicon-expressing cells. J Virol. 2003;77:11644–11650. doi: 10.1128/JVI.77.21.11644-11650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tardif KD, Mori K, Kaufman RJ, Siddiqui A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J Biol Chem. 2004;279:17158–17164. doi: 10.1074/jbc.M312144200. [DOI] [PubMed] [Google Scholar]
- 132.Lieber CS. Hepatitis C and alcohol. J Clin Gastroenterol. 2003;36:100–102. doi: 10.1097/00004836-200302000-00003. [DOI] [PubMed] [Google Scholar]
- 133.Szabo G. Pathogenic interactions between alcohol and hepatitis C. Curr Gastroenterol Rep. 2003;5:86–92. doi: 10.1007/s11894-003-0014-x. [DOI] [PubMed] [Google Scholar]
- 134.Peters MG, Terrault NA. Alcohol use and hepatitis C. Hepatology. 2002;36:S220–S225. doi: 10.1053/jhep.2002.36811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bhattacharya R, Shuhart MC. Hepatitis C and alcohol: interactions, outcomes, and implications. J Clin Gastroenterol. 2003;36:242–252. doi: 10.1097/00004836-200303000-00012. [DOI] [PubMed] [Google Scholar]