

Abstract
AIM: To study the effect of cholecystokinin-octapeptide (CCK-8) on lipopolysaccharide (LPS) -induced pulmonary artery smooth muscle cell (PASMCs) injury and the role of heme oxygenase-1 (HO-1), and to explore the regulation mechanism of c-Jun N-terminal kinase (JNK) and activator protein-1 (AP-1) signal transduction pathway in inducing HO-1 expression further.
METHODS: Cultured PASMCs were randomly divided into 4 or 6 groups: normal culture group, LPS (10 mg/L), CCK-8 (10-6 mol/L) plus LPS (10 mg/L) group, CCK-8 (10-6 mol/L) group, zinc protoporphyrin 9 (ZnPPIX) (10- 6 mol/L) plus LPS (10 mg/L) group, CCK-8 (10-6 mol/L) plus ZnPPIX and LPS (10 mg/L) group. Seven hours after LPS administration, ulterstructrual changes and content of malondialdehyde (MDA) of PASMCs in each group were investigated by electron microscopy and biochemical assay respectively. HO-1 mRNA and protein of PASMCs in the former4 groups were examined by reverse transcriptase polymerase chain reaction (RT-PCR) and immunocytochemistry staining. Changes of c-fos expression and activation of JNK of PASMCs in the former 4 groups were detected with immunocytochemistry staining and Western blot 30 min after LPS administration.
RESULTS: The injuries of PASMCs and the increases of MDA content induced by LPS were alleviated and significantly reduced by CCK-8 (P < 0.05). The specific HO-1 inhibitor-ZnPPIX could worsen LPS-induced injuries and weaken the protective effect of CCK-8. The expressions of c-fos, p-JNK protein and HO-1 mRNA and protein were all slightly increased in LPS group, and significantly enhanced by CCK-8 further (P < 0.05).
CONCLUSION: HO-1 may be a key factor in CCK-8 attenuated injuries of PASMCs induced by LPS, and HO-1 expression may be related to the activation of JNK and activator protein (AP-1).
INTRODUCTION
Lipopolysaccharide (LPS), a main component of Gram-negative bacterial endotoxin, is the main factor to induce endotoxic shock (ES). ES is a common and serious syndrome in clinic and its mortality is very high. The lung is one of the target organs easily insulted in ES. It was demonstrated that lung injury in ES was associated with oxygen free radicals (OFR)[1,2] and the content of cholecystokinin (CCK) in serum was increased when ES occurred. Our previous in vivo and in vitro studies demonstrated that CCK-8 could protect animals from LPS-induced ES and lung injury, which may be related to its effect on reducing the production of OFR[3-7].
There is a rapid increase in those substances that provide protection against oxidative stress. Among them one is heme oxygenase (HO)-1, which has generated much interest as a novel stress protein that is highly induced by many factors which induce oxidative injury and protect against oxidative stress[8-12]. However, the regulation mechanism of HO-1 expression was still unclear and there were no reports about the relationship between HO-1 and the protection of CCK-8 in LPS-induced ES and lung injury. One of the earliest responses to LPS and CCK-8 is the activation of mitogen-activated protein kinases (MAPKs), including p38, p42/p44 extracellular signal-regulated kinase (ERK) and c-Jun NH2-terminal kinase (JNK)[13,14]. A slightly later cellular response is the activation of activator protein (AP)-1. AP-1 is a dimeric protein complex containing 2 members of the real family of transcription factors, c-fos and c-Jun. Recent studies showed that, one or more members of AP-1 were likely involved in HO-1 gene transcription[15-18]. The relationship between the activation of these signaling molecules and downstream HO-1 expression represents an active line of investigation. This can provide experimental evidences to elucidate whether the protective mechanism of CCK-8 is associated with HO-1.
Pulmonary artery hypertension (PAH) is the typical pathological change in the early phase of ES. It was reported that the degree and duration of PAH were the important factors of ES accompanied by acute lung injury, and pulmonary artery smooth muscle cells (PASMCs) played an important role in maintaining the tone of pulmonary artery. In the present study, the effect of CCK-8 pretreatment on the injuries of PASMCs induced by LPS was observed, and further investigated the role of HO-1 and the regulation mechanism of c-Jun N-terminal kinase (JNK) and AP-1 signal transduction pathway in inducing HO-1 expression were further investigated.
MATERIALS AND METHODS
Materials
CCK-8 (sulfated), LPS (E.coli LPS,serotype 0111:B4), ZnPPIX and Triton X-100 were all purchased from Sigma. Mouse anti-rat phosphorylate JNK (p-JNK) monoclonal antibody, rabbit anti- rat c-fos and HO-1 polyclonal antibody were all purchased from Santa Cruz. DMEM and fetal calf serum (FCS) were purchased from GibcoBRL. Total RNA isolation system and access RT-PCR system were purchased from Promega (USA). SABC kit was purchased from Boshide (China). All other reagents used were of analytic grade.
Methods
Cell isolation and culture Rat PASMCs were prepared as previously described[19]. Briefly, male healthy Sprague-Dawley rats (100-150 g BM, Experimental Animal Center of Hebei Province) were anesthetized with intraperitoneal administration of pentobarbital sodium (35 mg/kg), and pulmonary arteries were obtained. The isolated pulmonary arteries were cleaned of connective tissues, and aseptically opened longitudinally. The adventitia was carefully removed and the luminal surface was scraped with forceps to remove endothelial cells and then minced into 1 mm2 pieces and plated on culture flasks at 37 °C in humidified air containing 50 mL/L CO2 for 2-3 h , and then cultured in Dulbecco modified Eagle medium (DMEM) supplemented with heat-inactivated 10 mL/L FCS and antibiotics (100 U penicillin, 100 μg/mL streptomycin) and grew until confluence. The medium was changed every 3-4 d, and confluent cells were passaged with 1.25 g/L trypsin solution every 5-7 d, and experiments were performed in an 80% confluent state on the sixth- to eighth-passages from primary culture. Cells were made quiescent by incubation in each medium without FCS for 24 h before LPS or CCK-8 addition. PASMCs in culture were elongated and spindle shaped, grown with the typical hill-and-valley appearance, and characterized by immunocytochemical assay with anti-α-actin monoclonal antibody, demonstrating positive staining in > 95% of cells. The cultured PASMCs were randomly divided into 4 or 6 groups: normal culture (control) group, LPS (10 mg/L) group, CCK-8 (10-6 mol/L) group, CCK-8 (10-6 mol/L) plus LPS (10 mg/L) group, ZnPPIX (10 -6 mol/L) plus LPS (10 mg/L) group, and CCK-8 (10-6 mol/L) plus ZnPPIX (10-6 mol/L) and LPS (10 mg/L) group.
Observation of ultrastructrual changes with transmission electron microscopy After treated with LPS, CCK-8 or ZnPPIX, the cells were washed rapidly with PBS and harvested, then fixed with 40 g/L paraformaldehyde/5 g/L glutaraldehyde and postfixed with 40 g/L OsO4/potassium hexacyanoferrate. After embedded in Epon, thin sections were cut, contrasted with uranyl acetate (20 g/L)/lead citrate (27 g/L) and examined with an EM10 electron microscope as described previously[20].
Assessments of PASMCs malondialdehyde (MDA) content The cells were harvested after treated with LPS or CCK-8 or ZnPPIX and washed rapidly with PBS, then immediately homogenized on ice in 9 volumes of saline. The homogenates were centrifuged at 4 000 r/min at 4 °C for 10 min. The MDA content in the supernatants was measured using a MDA assay kit (Nanjing Jiancheng Corp. China).
Analysis of c-fos and HO-1 protein expression by immunocytochemistry Confluent cells were passaged with 2.5 g/L trypsin solution onto 20 mm×20 mm glass sheets in a 6-well plate. After treatment with LPS, CCK-8 or both LPS and CCK-8 for 30 min or 7 h, the cells were treated with 3 mL/L H2O2 in methanol to block endogenous peroxide activity. Immunocytochemical staining was performed using rabbit polyclonal antibody against c-fos and HO-1 by an indirect streptavidin/peroxidase technique. Experiments were performed following the manufacturer’s recommendations. The cells were incubated with polyclonal anti-rat c-fos and HO-1 antibody for 12 h at 4 °C after antigen repair. Biotinylated IgG was added as the second antibody. Horseradish peroxidase labeled streptomycin-avidin complex was used to detect the second antibody. Slides were stained with DAB and examined under a light microscope. The brown or dark brown stained cytoplasm or cell nucleus was considered as positive. Phosphate-buffered saline (PBS) solution was used as negative control. The result of immunocytochemistry was analysed by using the CMIAS-8 image analysis system.
Analysis of HO-1 mRNA by RT-PCR After treatment with LPS, CCK-8 or both LPS and CCK-8 for 7 h, total RNA was extracted from PASMCs. The concentration of RNA was determined from absorbent at 260 nm. The primers for HO-1 and β-actin were as follows: HO-1 (309 bp), 5’-CTT TCA GAA GGG TCA GGT GTC CA-3’ , 5’-CTG AGA GGT CAC CCA GGT AGC GG-3’; β-actin (224 bp), 5’-CGT GGG CCG CCC TAG GCA CCA-3’ , 5’-CGG TTG CCT TAG GGT TCA GAG GGG-3’. Polymerase chain reactions(PCR) were performed in a 50 μL reaction volume. Reverse transcriptase polymerase chain reaction (RT-PCR) was run in the following procedures: at 42 °C for 45 min, 1 circle; at 95 °C for 3 min, at 60 °C for 30 s, at 72 °C for 30 s, 1 circle; at 94 °C for 30 s , at 60 °C for 30 s, at 72 °C for 30 s, 30 circles; at 94 °C for 30 s, at 60 °C for 30 s, at 72 °C for 6 min, 1 circle. A 10 μL PCR product was placed on to 15 g /L agarose gel and observed by EB staining using a Gel-Pro analyzer.
Western blot analysis of p-JNK protein in PASMCs After treatment with LPS or CCK-8 or both LPS and CCK-8 for 30 min, the cells were harvested and then lysed with ice-cold lysis buffer [50 mmol/LTris (pH7.5), 150 mmol/LNaCl, 10 g/L Triton X-100, 5 g/L deoxycholic acid, 1 g/L sodium dodecyl sulfate, 1 mmol/L phenylmethysulfonyl (PMSF), 10 mmol/L NaF, 1 mmol/L sodium vanadate, 5 mmol/L EDTA (pH8.0) and a 40 μg/mL protease inhibitor cocktail] as described previously for 1 h and then centrifuged at 12 000 g at 4 °C for 10 min. After precipitated unsolubilized fraction was discarded, protein concentration in the supernatant was determined by Coomassie blue dye-binding assay (Nanjing Jiancheng Corp. China). The supernatant containing 30 μg protein was treated with 2×Laemmli buffer, then subjected to SDS-PAGE using 100 g/L running gel for 3 h at 100 V. Protein was transferred to nitrocellulose membrane, and immunoblot analysis was performed as described previously. Briefly, the membrane was incubated successively with 3 mL/L milk in TPBS at room temperature for 1 h, with 1:1 000 diluted mouse anti-rat specific antibody of p-JNK at 37 °C for 2 h, and then with horseradish peroxidase-labeled secondary antibody at 37 °C for 30 min. After each incubation, the membrane was washed extensively with TPBS and the immunoreactive band was stained with diaminobenzidine (DAB). The brown or dark brown stained strap was considered as positive.
Statistical analysis
Data were reported as mean ± SD. Statistical differences between values from different groups were determined by one way ANOVA and Newman-Keuls q test. Significance was set at P < 0.05.
RESULTS
CCK-8 alleviated PASMC injury induced by LPS
PASMCs were harvested at 7 h. There were significantly ultrastructural injuries in LPS group such as seriously swollen mitochondria and mitochondria without or partly without cristallins. CCK-8 could significantly alleviate the above-mentioned changes. While the ultrastructrual injuries induced by LPS were aggravated and the protective effect of CCK-8 on cells was weakened by ZnPPIX. There was no significant difference between CCK-8 group and control group (Figure 1).
Figure 1.
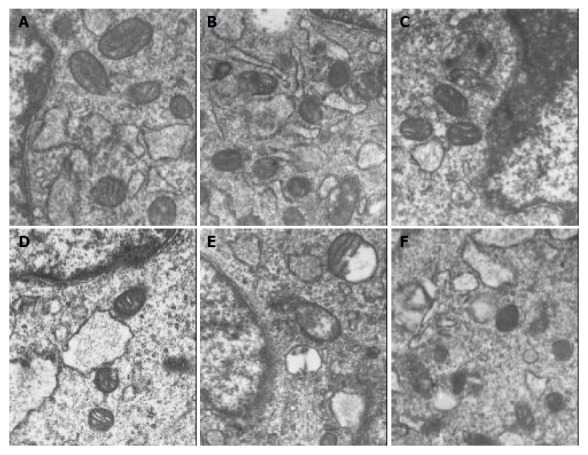
Mitochondria ultrastructural changes shown in EM photograph of PASMCs (×25 000). A: Control group, B: LPS group, C: CCK-8+LPS group, D: CCK-8 group, E: LPS+ZnPP group, F: CCK-8+LPS+ZnPP group.
CCK-8 inhibited MDA production
The MDA content of PASMCs was significantly increased in LPS group when compared with control group (P < 0.05). Compared with the LPS group, the MDA content was further increased in LPS+ZnPPIX group but markedly decreased in CCK-8+L PS group, however the protective effect of CCK-8 was weakened by ZnPPIX. There was no significant difference between CCK-8 group and control group (P < 0.05) (Table 1).
Table 1.
Change of MDA content in PASMCs (mean ± SD, n = 8)
| Group | MDA content (nmol/mL ) |
| Control | 4.02 ± 2.18 |
| LPS | 23.44 ± 1.25a |
| LPS+CCK-8 | 14.66 ± 1.66e |
| CCK-8 | 3.00 ± 2.18 |
| LPS+ZnPP | 34.97 ± 1.54ae |
| LPS+CCK-8+ZnPP | 29.68 ± 2.18ec |
P < 0.05 vs Control group,
P < 0.05 vs LPS+CCK-8 group,
P < 0.05 vs LPS group.
Change of c-fos and HO-1 protein expression
The expressions of c-fos and HO-1 protein in PASMCs were demonstrated by immunocytochemical staining. The results showed that in control group, no brown deposits were present in PASMCs. In contrast, LPS slightly increased the expressions of c-fos and HO-1 protein, some strong positive signals were observed in PASMCs from LPS group. CCK-8 could further enhance the increased expressions of c-fos and HO-1. The dimension and intensity of positive signals were increased also in CCK-8 group (Figure 2),(Table 2).
Figure 2.
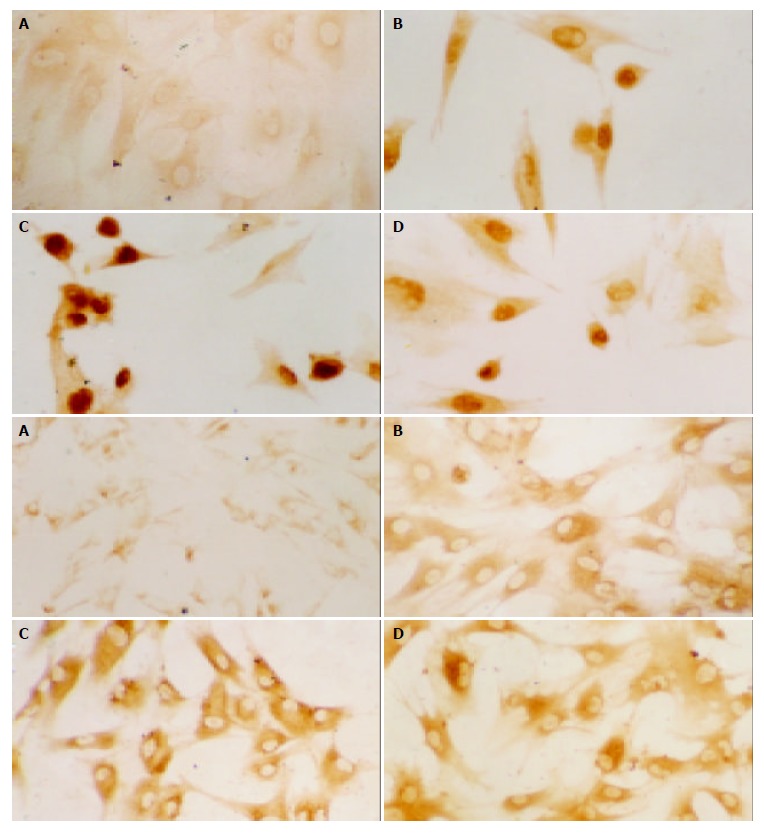
Effect of CCK-8 on LPS-induced c-fos (upper, ×400) and HO-1 (lower, ×200) protein expression in PASMCs. A: Control group, B: LPS group, C: CCK-8+LPS group, D: CCK-8 group.
Table 2.
Changes of HO-1 and c-fos protein expression by immumocytochemistry in PASMCs (mean ± SD, n = 6)
| Group |
Area (%) |
(A) |
||
| HO-1 | c-fos | HO-1 | c-fos | |
| Control | 2.10 ± 0.01 | 1.18 ± 0.02 | 0.02 ± 0.009 | 0.03 ± 0.021 |
| LPS | 20.18 ± 0.08a | 18.95 ± 0.07a | 0.19 ± 0.07a | 0.20 ± 0.01a |
| LPS+CCK-8 | 28.91 ± 0.36c | 29.87 ± 0.33c | 0.47 ± 0.04c | 0.45 ± 0.02c |
| CCK-8 | 21.27 ± 1.05 | 20.16 ± 0.98 | 0.31 ± 0.02 | 0.33 ± 0.01 |
P < 0.05 vs Control group,
P < 0.05 vs LPS group.
RT-PCR detection of HO-1 mRNA
HO-1 mRNA in PASMCs was detected by PT-PCR analysis. The results showed that the PASMCs of rats 7 h after LPS administration expressed the gene coding for HO-1, because RT-PCR generated a DNA fragment corresponding to the predicted length, 309 bp, of the HO-1 amplification product. The expression of HO-1 increased significantly in LPS and CCK-8 groups compared with control group. The rate of β-actin was (11 ± 2) % in control group, while it increased to (30 ± 6) % and (47 ± 7) % respectively in LPS and CCK-8 groups. The expression of HO-1 increased further in LPS+CCK-8 group compared with LPS group, it increased to (80 ± 8) % in LPS+CCK-8 group. In each cell sample, all β-actin amplification products were 224 bp in length and there was no significant difference in β-actin expression (Figure 3).
Figure 3.
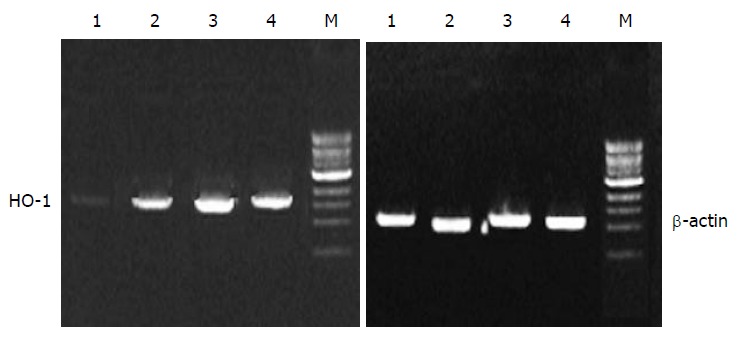
RT-PCR product gel electrophoresis. M: DNA marker, 1: Control, 2: LPS, 3: CCK-8 + LPS, 4: CCK-8.
Western blot analysis of p-JNK protein
There were a few of activated JNK proteins in control group. JNK could be activated by LPS, significant phosphorylation of JNK was observed in PASMCs 30 min after LPS administration. CCK-8 could significantly enhance LPS-induced phosphorylation of JNK. Phosphorylation of JNK was also observed in cells receiving CCK-8 (Figure 4).
Figure 4.
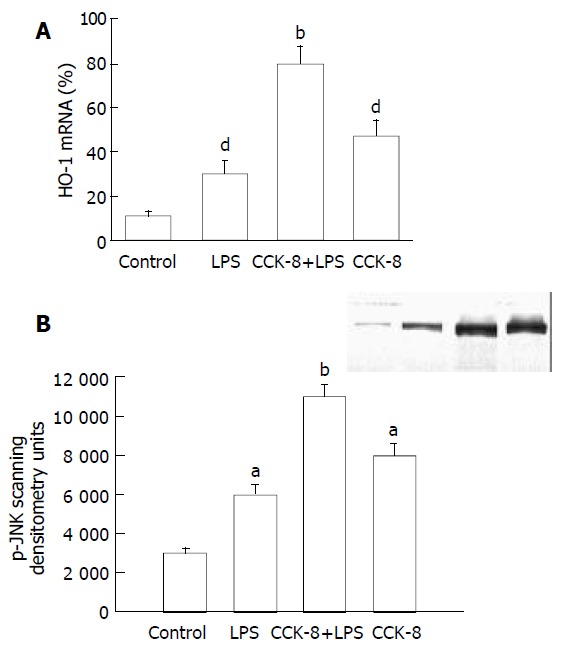
Effect of CCK-8 on LPS-induced HO-1 mRNA ex-pression and JNK activation in PASMCs. A: Effect of CCK-8 on LPS-induced HO-1 mRNA expression in PASMCs. n = 3. dP < 0.01 vs Control; bP < 0.01 vs LPS. B: CCK-8 increases JNK activation induced by LPS in PASMCs. n = 3. aP < 0.05 vs Control; bP < 0.01 vs LPS.
DISCUSSION
CCK is a member of the gastrin-CCK family, first isolated from hog intestine, and shows a widespread distribution in different organs and tissues. The sulfated carboxy-terminal octapeptide (CCK-8), isolated from the central nervous system and digestive tract, is the predominant active form. Our previous in vivo and in vitro studies demonstrated that CCK-8 could protect animals from LPS-induced ES. Treatment of ES rats and rabbits with CCK-8 could lead to an increase in decreasing mean arterial pressure and a reduction in increasing pulmonary artery pressure. Pathological changes of lung could be ameliorated when CCK-8 was via vessel in advance. CCK-8 could protect pulmonary artery endothelia against detrimental effects and then reverse the inhibition of endothelia-dependent relaxation of pulmonary artery induced by LPS, which might be associated with the above-mentioned protective effect of CCK-8[5-10]. Despite convincing data indicating the protective function of CCK-8 to the organisms in endotoxic shock or to the cells exposed to LPS, the precise mechanism remains unclear.
During stress state induced by administration of LPS, inflammation or therapeutic doses of inhaled oxygen, the bodies would evolve a complex, and redundant network of antioxidants. It has been found that an important arm of the antioxidant response consists of antioxidant enzymes and stress-response proteins[21-23]. One such stress-response protein is HO-1, the rate-limiting enzyme in the oxidative degradation of heme into bilirubin, carbon monoxide (CO) and free iron. HO exists in three isoforms, whereas HO-2 and HO-3 are primarily constitutive. HO-1 also known as heat shock protein 32, has been found to be the only inducible HO isoform[24-27]. There is a strong evidence to support the emerging paradigm that HO-1 is essential in maintaining cellular and tissue homeostasis in various in vitro and in vivo models of oxidant-induced injury. Recent analyses of HO-1 null mice as well as the first reported HO-1-deficient human have strengthened the emerging paradigm that HO-1 is indeed an important molecule in the host’s defense against oxidant stresses such as hypoxia, hyperthermia, and inflammation. It has been considered as one of the most sensitive indicators of cellular injury[28-31]. Recent studies showed that HO-1 was an important regulator of the vascular response to injury. In this experiment we used the specific HO-1 inhibitor-ZnPPIX to study the relationship between induced HO-1 and injuries of PASMCs. The results showed that HO-1 mRNA and protein expression increased after LPS administration and ZnPPIX could worsen LPS-induced injuries, indicating that overexpression of HO-1 in PASMCs can attenuate their injuries induced by LPS. It is undoubtedly one of the adaptive and self-protective responses to the injury.
Given the overall consistency of data that show HO-1 expression is generally a protective response, it is necessary to study the relationship between HO-1 expression and the protection of CCK-8. We found that CCK-8 could ameliorate the ultrastructural injuries of PASMCs induced by LPS. However, the protective effect of CCK-8 was impaired by ZnPPIX. To further demonstrate the role of HO-1 in CCK-8 attenuated injury of PASMCs induced by LPS, the HO-1 expression and signal pathway of HO-1 induction in PASMCs were studied. The candidate upstream signaling pathways for HO-1 regulation were MAPKs, a group of protein kinases that could mediate the nuclear responses of cells to a wide variety of extracellular stresses such as inflammatory cytokines, growth factors, ultraviolet light, and osmotic stress[32-34]. The “classical” MAPKs are the p44 and p42 isoforms. Recently, two novel MAPK-related enzymes have been identified, one is JNK and the other is p38[35]. Although three distinct subfamilies have been described, there is a significant cross talk between the pathway and common downstream target[36]. In our experiment, we found that CCK-8 significantly enhanced LPS-induced overexpression of HO-1 mRNA and protein. We also observed that LPS could also activate c-fos (one of the two members of the real family of AP-1) and JNK in PASMCs, while CCK-8 could further enhance the activation of c-fos and JNK induced by LPS. From all above, the role of MAPKs was potential in HO-1 signal pathway after LPS and CCK-8 administration to PASMCs, HO-1 might be the key for CCK-8 to exert its protective effect.
In summary, administration of CCK-8 can prevent injuries of PASMCs induced by LPS through overexpression of HO-1 mRNA and protein, which may involve c-fos and JNK.
Footnotes
Edited by Wang XL and Xu FM
References
- 1.Liaudet L, Szabó C, Evgenov OV, Murthy KG, Pacher P, Virág L, Mabley JG, Marton A, Soriano FG, Kirov MY, et al. Flagellin from gram-negative bacteria is a potent mediator of acute pulmonary inflammation in sepsis. Shock. 2003;19:131–137. doi: 10.1097/00024382-200302000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Molina PE, Abumrad NN. Differential effects of hemorrhage and LPS on tissue TNF-α, IL-1 and associate neuro-hormonal and opioid alterations. Life Sci. 2000;66:399–409. doi: 10.1016/s0024-3205(99)00606-2. [DOI] [PubMed] [Google Scholar]
- 3.Bertolini A, Guarini S, Ferrari W, Rompianesi E. Caerulein and cholecystokinin reverse experimental hemorrhagic shock. Neuropeptides. 1986;8:25–31. doi: 10.1016/0143-4179(86)90061-2. [DOI] [PubMed] [Google Scholar]
- 4.Guarini S, Bazzani C, Leo L, Bertolini A. Haematological changes induced by the intravenous injection of CCK-8 in rats subjected to haemorrhagic shock. Neuropeptides. 1988;11:69–72. doi: 10.1016/0143-4179(88)90012-1. [DOI] [PubMed] [Google Scholar]
- 5.Ling YL, Huang SS, Wang LF, Zhang JL, Wan M, Hao RL. [Cholecystokinin-octapeptide (CCK-8) reverses experimental endotoxin shock] Shengli Xuebao. 1996;48:390–394. [PubMed] [Google Scholar]
- 6.Meng AH, Ling YL, Wang DH, Gu ZY, Li SJ, Zhu TN. [Cholecystokinin-octapeptide alleviates tumor necrosis factor-α induced changes in rabbit pulmonary arterial reactivity and injuries of endothelium in vitro] Shengli Xuebao. 2000;52:502–506. [PubMed] [Google Scholar]
- 7.Ling YL, Meng AH, Zhao XY, Shan BE, Zhang JL, Zhang XP. Effect of cholecystokinin on cytokines during endotoxic shock in rats. World J Gastroenterol. 2001;7:667–671. doi: 10.3748/wjg.v7.i5.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Otani K, Shimizu S, Chijiiwa K, Morisaki T, Yamaguchi T, Yamaguchi K, Kuroki S, Tanaka M. Administration of bacterial lipopolysaccharide to rats induces heme oxygenase-1 and formation of antioxidant bilirubin in the intestinal mucosa. Dig Dis Sci. 2000;45:2313–2319. doi: 10.1023/a:1005626622203. [DOI] [PubMed] [Google Scholar]
- 9.Zampetaki A, Minamino T, Mitsialis SA, Kourembanas S. Effect of heme oxygenase-1 overexpression in two models of lung inflammation. Exp Biol Med (Maywood) 2003;228:442–446. doi: 10.1177/15353702-0322805-02. [DOI] [PubMed] [Google Scholar]
- 10.Henningsson R, Alm P, Lundquist I. Evaluation of islet heme oxygenase-CO and nitric oxide synthase-NO pathways during acute endotoxemia. Am J Physiol Cell Physiol. 2001;280:C1242–C1254. doi: 10.1152/ajpcell.2001.280.5.C1242. [DOI] [PubMed] [Google Scholar]
- 11.Otterbein LE, Choi AM. Heme oxygenase: colors of defense against cellular stress. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1029–L1037. doi: 10.1152/ajplung.2000.279.6.L1029. [DOI] [PubMed] [Google Scholar]
- 12.González A, Schmid A, Salido GM, Camello PJ, Pariente JA. XOD-catalyzed ROS generation mobilizes calcium from intracellular stores in mouse pancreatic acinar cells. Cell Signal. 2002;14:153–159. doi: 10.1016/s0898-6568(01)00247-9. [DOI] [PubMed] [Google Scholar]
- 13.Jarvis BW, Harris TH, Qureshi N, Splitter GA. Rough lipopolysaccharide from Brucella abortus and Escherichia coli differentially activates the same mitogen-activated protein kinase signaling pathways for tumor necrosis factor alpha in RAW 264.7 macrophage-like cells. Infect Immun. 2002;70:7165–7168. doi: 10.1128/IAI.70.12.7165-7168.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zieger M, Oehrl W, Wetzker R, Henklein P, Nowak G, Kaufmann R. Different signaling pathways are involved in CCK(B) receptor-mediated MAP kinase activation in COS-7 cells. Biol Chem. 2000;381:763–768. doi: 10.1515/BC.2000.097. [DOI] [PubMed] [Google Scholar]
- 15.Yuksel M, Okajima K, Uchiba M, Okabe H. Gabexate mesilate, a synthetic protease inhibitor, inhibits lipopolysaccharide-induced tumor necrosis factor-alpha production by inhibiting activation of both nuclear factor-kappaB and activator protein-1 in human monocytes. J Pharmacol Exp Ther. 2003;305:298–305. doi: 10.1124/jpet.102.041988. [DOI] [PubMed] [Google Scholar]
- 16.Camhi SL, Alam J, Wiegand GW, Chin BY, Choi AM. Transcriptional activation of the HO-1 gene by lipopolysaccharide is mediated by 5' distal enhancers: role of reactive oxygen intermediates and AP-1. Am J Respir Cell Mol Biol. 1998;18:226–234. doi: 10.1165/ajrcmb.18.2.2910. [DOI] [PubMed] [Google Scholar]
- 17.Lee PJ, Camhi SL, Chin BY, Alam J, Choi AM. AP-1 and STAT mediate hyperoxia-induced gene transcription of heme oxygenase-1. Am J Physiol Lung Cell Mol Physiol. 2000;279:L175–L182. doi: 10.1152/ajplung.2000.279.1.L175. [DOI] [PubMed] [Google Scholar]
- 18.Ryter SW, Xi S, Hartsfield CL, Choi AM. Mitogen activated protein kinase (MAPK) pathway regulates heme oxygenase-1 gene expression by hypoxia in vascular cells. Antioxid Redox Signal. 2002;4:587–592. doi: 10.1089/15230860260220085. [DOI] [PubMed] [Google Scholar]
- 19.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268(4 Pt 1):C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- 20.Wang YX, Zheng YM, Abdullaev I, Kotlikoff MI. Metabolic inhibition with cyanide induces calcium release in pulmonary artery myocytes and Xenopus oocytes. Am J Physiol Cell Physiol. 2003;284:C378–C388. doi: 10.1152/ajpcell.00260.2002. [DOI] [PubMed] [Google Scholar]
- 21.Haddad JJ. Glutathione depletion is associated with augmenting a proinflammatory signal: evidence for an antioxidant/pro-oxidant mechanism regulating cytokines in the alveolar epithelium. Cytokines Cell Mol Ther. 2000;6:177–187. doi: 10.1080/mccm.6.4.177.187. [DOI] [PubMed] [Google Scholar]
- 22.Haddad JJ, Land SC. Redox signaling-mediated regulation of lipopolysaccharide-induced proinflammatory cytokine biosynthesis in alveolar epithelial cells. Antioxid Redox Signal. 2002;4:179–193. doi: 10.1089/152308602753625942. [DOI] [PubMed] [Google Scholar]
- 23.Wiesel P, Foster LC, Pellacani A, Layne MD, Hsieh CM, Huggins GS, Strauss P, Yet SF, Perrella MA. Thioredoxin facilitates the induction of heme oxygenase-1 in response to inflammatory mediators. J Biol Chem. 2000;275:24840–24846. doi: 10.1074/jbc.M000835200. [DOI] [PubMed] [Google Scholar]
- 24.Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci U S A. 1968;61:748–755. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCoubrey WK, Huang TJ, Maines MD. Isolation and characterization of a cDNA from the rat brain that encodes hemoprotein heme oxygenase-3. Eur J Biochem. 1997;247:725–732. doi: 10.1111/j.1432-1033.1997.00725.x. [DOI] [PubMed] [Google Scholar]
- 26.Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557–2568. [PubMed] [Google Scholar]
- 27.Fujiwara T, Takahashi T, Suzuki T, Yamasaki A, Hirakawa M, Akagi R. Differential induction of brain heme oxygenase-1 and heat shock protein 70 mRNA in sepsis. Res Commun Mol Pathol Pharmacol. 1999;105:55–66. [PubMed] [Google Scholar]
- 28.Agarwal A, Nick HS. Renal response to tissue injury: lessons from heme oxygenase-1 GeneAblation and expression. J Am Soc Nephrol. 2000;11:965–973. doi: 10.1681/ASN.V115965. [DOI] [PubMed] [Google Scholar]
- 29.Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, Ohta K, Kasahara Y, Koizumi S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest. 1999;103:129–135. doi: 10.1172/JCI4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. 2002;8:240–246. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 31.Inoue S, Suzuki M, Nagashima Y, Suzuki S, Hashiba T, Tsuburai T, Ikehara K, Matsuse T, Ishigatsubo Y. Transfer of heme oxygenase 1 cDNA by a replication-deficient adenovirus enhances interleukin 10 production from alveolar macrophages that attenuates lipopolysaccharide-induced acute lung injury in mice. Hum Gene Ther. 2001;12:967–979. doi: 10.1089/104303401750195926. [DOI] [PubMed] [Google Scholar]
- 32.Chan ED, Riches DW, White CW. Redox paradox: effect of N-acetylcysteine and serum on oxidation reduction-sensitive mitogen-activated protein kinase signaling pathways. Am J Respir Cell Mol Biol. 2001;24:627–632. doi: 10.1165/ajrcmb.24.5.4280. [DOI] [PubMed] [Google Scholar]
- 33.Haddad JJ, Land SC. Redox/ROS regulation of lipopolysaccharide-induced mitogen-activated protein kinase (MAPK) activation and MAPK-mediated TNF-alpha biosynthesis. Br J Pharmacol. 2002;135:520–536. doi: 10.1038/sj.bjp.0704467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 35.Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 36.Whitmarsh AJ, Shore P, Sharrocks AD, Davis RJ. Integration of MAP kinase signal transduction pathways at the serum response element. Science. 1995;269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]