

Abstract
Arterial smooth muscle cells (myocytes) express large-conductance Ca2+-activated K+ (BK) channel α and auxiliary β1 subunits that modulate arterial contractility. In arterial myocytes, β1 subunits are stored within highly mobile rab11A-positive recycling endosomes. In contrast, BKα subunits are primarily plasma membrane-localized. Trafficking pathways for BKα and whether physiological stimuli that regulate arterial contractility alter BKα localization in arterial myocytes are unclear. Here, using biotinylation, immunofluorescence resonance energy transfer (immunoFRET) microscopy, and RNAi-mediated knockdown, we demonstrate that rab4A-positive early endosomes traffic BKα to the plasma membrane in myocytes of resistance-size cerebral arteries. Angiotensin II (ANG II), a vasoconstrictor, reduced both surface and total BKα, an effect blocked by bisindolylmaleimide-II, concanavalin A, and dynasore, protein kinase C (PKC), internalization, and endocytosis inhibitors, respectively. In contrast, ANG II did not reduce BKα mRNA, and sodium nitroprusside, a nitric oxide donor, did not alter surface BKα protein over the same time course. MG132 and bafilomycin A, proteasomal and lysosomal inhibitors, respectively, also inhibited the ANG II-induced reduction in surface and total BKα, resulting in intracellular BKα accumulation. ANG II-mediated BK channel degradation reduced BK currents in isolated myocytes and functional responses to iberiotoxin, a BK channel blocker, and NS1619, a BK activator, in pressurized (60 mmHg) cerebral arteries. These data indicate that rab4A-positive early endosomes traffic BKα to the plasma membrane in arterial myocytes. We also show that ANG II stimulates PKC-dependent BKα internalization and degradation. These data describe a unique mechanism by which ANG II inhibits arterial myocyte BK currents, by reducing surface channel number, to induce vasoconstriction.
Keywords: trafficking, BK channel, angiotensin II
large-conductance Ca2+-activated K+ (BKα, MaxiK, Slo1) channels are seven-transmembrane proteins with a short extracellular amino (NH2) and long intracellular carboxy (COOH) terminus (14, 16). Functional BK channels contain four α subunits that assemble to form a K+-selective pore (16, 39). The BKα COOH terminus contains RCK (regulator of conductance for K+) domains and a “Ca2+ bowl,” which endow Ca2+ sensitivity (23). BK accessory β and γ subunits modify channel properties and can exhibit tissue-specific expression to elicit these effects in a cell type-dependent manner (3, 39, 43).
Arterial smooth muscle cell (myocyte) BK channels serve as a negative regulator of pressure-induced vasoconstriction (the myogenic response) and are modulated by multiple vasoconstrictor and vasodilator agonists and stimuli (11, 24, 31–33). Arterial myocytes express BKα, β1, and γ1 subunits (12, 39). β1 Subunits elevate channel apparent Ca2+ sensitivity into a micromolar concentration range sufficient to permit sensing of local intracellular Ca2+ transients termed Ca2+ sparks, which control BK channel activity in arterial myocytes (18–20). Arterial myocytes also express LRRC26, an auxiliary γ subunit that elevates channel voltage sensitivity to induce vasodilation (12).
Current (I) generated by an ion channel population, including BK channels, is determined by N, the number of channels, Po, the open probability, and i, the single channel current, such that I = N·Po·i. Previous studies have focused primarily on identifying mechanisms that control plasma membrane (surface) BK channel activity in cells. In contrast, mechanisms that regulate channel surface expression in native cell types, including arterial myocytes, are unclear. Similarly uncertain is whether vasoregulatory stimuli can modulate the number of functional channels (N) in the plasma membrane to control arterial myocyte contractility.
Rab-GTPases are a family of more than 60 proteins that control protein trafficking (36, 45). Rab proteins regulate vesicular transport and are implicated in vesicle docking and fusion (45). Recently, we demonstrated that β1 subunits are primarily intracellular, whereas BKα channel are located at the surface in arterial myocytes (25). Nitric oxide (NO) stimulates rapid surface trafficking of β1 subunits within rab11A-positive recycling endosomes, leading to an increase in plasma membrane BK channel apparent Ca2+ sensitivity and vasodilation (25). These data also suggested that BKα and β1 subunits are independently trafficked to the plasma membrane.
Here, we investigated mechanisms that control BKα surface trafficking and regulation by angiotensin II (ANG II), a vasoconstrictor, in cerebral artery myocytes. We show that rab4A regulates BKα anterograde trafficking to control surface BKα levels. Our data show that ANG II stimulates protein kinase C (PKC)-dependent internalization of BKα subunits, which are subsequently targeted for degradation. This mechanism reduces myocyte BK currents, leading to contraction. In contrast, nitric oxide does not regulate BKα surface expression or localization. These data indicate that ANG II stimulates internalization and degradation of plasma membrane BK channels to reduce surface number (N) and induce vasoconstriction.
MATERIALS AND METHODS
Tissue preparation and smooth muscle cell isolation.
All animal protocols were reviewed and approved by the Animal Care and Use Committee at the University of Tennessee Health Science Center. Male Sprague-Dawley rats (6–8 wk) were euthanized by intraperitoneal injection of pentobarbital sodium (150 mg/kg). The brain was removed and cerebral arteries were harvested and maintained in ice-cold (4°C), oxygenated (21% O2-5% CO2-74% N2) physiological saline solution (PSS) containing (in mM) 119 NaCl, 4.7 KCl, 24 NaHCO3, 1.2 KH2PO4, 1.6 CaCl2, 1.2 MgSO4, and 11 glucose (pH 7.4). Smooth muscle cells (myocytes) were dissociated from cerebral arteries as previously described (47). Experiments involving myocytes were completed on the same day of isolation.
Transfection of intact cerebral arteries.
Rab11AshV, rab4AshV, and scrambled short hairpin vector (shV) were purchased from OriGene Technologies (Rockville, MD). Rab11B and scrambled siRNA were purchased from Life Technologies (Grand Island, NY). Cerebral arteries were transfected using a CUY21Vivo-SQ electroporator (Bex, Japan), as previously described (29). Briefly, cerebral arteries were placed in an electroporation chamber containing Ca2+-free phosphate-buffered saline (PBS) containing the vector. Tandem pulse electroporation was then applied. Arteries were removed from the chamber and kept in Ca2+-free PBS for 10 min before being placed in serum-free DMEM-F12 media supplemented with 1% penicillin-streptomycin (Sigma-Aldrich, St. Louis, MO) for 3 days under standard conditions (21% O2-5% CO2-74% N2, 37°C). Transfected arteries were then washed once in PSS and used for surface biotinylation or immuno-FRET experiments.
Surface biotinylation.
Cellular distribution of BKα and β1 subunit proteins in cerebral arteries was determined using surface biotinylation (1). Arteries were incubated for 1 h in a 1 mg/ml mixture each of EZ-Link Sulfo-NHS-LC-LC-Biotin and EZ-Link Maleimide-PEG2-Biotin reagents (Pierce) in PSS. Unbound biotin was removed by quenching using 100 mM glycine in PBS followed by a wash in PBS. Biotinylated arteries were homogenized in lysis buffer consisting of 50 mM Tris·HCl, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, and 0.1% SDS. Total protein was quantified as previously described (15) to allow normalization for pulldown. Biotinylated proteins were pulled down using monomeric avidin beads (Pierce). The supernatant contained the nonbiotinylated, intracellular protein fraction. Avidin-bead bound biotinylated plasma membrane proteins were eluted by boiling in Laemmli buffer (with 5% β-mercaptoethanol). Western blot analysis was used to identify surface and intracellular proteins and was calculated as a percentage of total protein.
Western blot analysis.
Proteins were separated on a 7.5% SDS-polyacrylamide gel and transferred onto nitrocellulose membranes. Blots were physically cut at ∼60 kDa to produce two membrane sections from the same protein sample that were probed for BKα or β1, rab11A, rab11B, or rab4. Membrane sections were incubated with mouse monoclonal anti-BKα (1:500 dilution, Neuromab, UC Davis, http://neuromab.ucdavis.edu/datasheet/L6_60.pdf), rabbit polyclonal anti-BKβ1 (1:500, Abcam), rabbit polyclonal anti-rab4 (1:300, Sigma-Aldrich), mouse monoclonal anti-rab11A (1:500, Abcam), or rabbit polyclonal anti-rab11B (1:500, Cell Signaling) antibodies overnight at 4°C in Tris-buffered saline (TBS) with 0.1% Tween 20 (TBS-T) and 5% nonfat dry milk. Proteins were visualized using appropriate horseradish peroxidase-conjugated secondary antibody (Pierce) and a chemiluminescent detection kit (Pierce). Band intensity was quantified by digital densitometry using Quantity One software (Bio-Rad).
Immunofluorescence resonance energy transfer (ImmunoFRET) and confocal imaging.
ImmunoFRET was performed following a similar protocol as previously described (48). Briefly, freshly isolated myocytes were fixed with paraformaldehyde and permeabilized with 0.1% Triton X-100 for 2 min at room temperature. After a 1-h incubation in PBS containing 5% bovine serum albumin (BSA), the myocytes were treated overnight at 4°C with mouse monoclonal anti-BKα (Neuromab, UC Davis), rabbit polyclonal anti-rab4 (Sigma-Aldrich), or rabbit polyclonal rab11A (Abcam). In a separate set of experiments mouse monoclonal anti-BKβ1 (Santa Cruz) was incubated with rabbit polyclonal anti-rab4 (Sigma-Aldrich). Primary antibodies were used at a dilution of 1:100 each in PBS containing 5% BSA. After a wash, cells were incubated for 1 h at 37°C with secondary antibodies: Alexa 546- or Alexa 488-conjugated (1:100 dilution; Life Technologies). Cover slips were then washed and mounted on glass slides. Fluorescence images were acquired using a Zeiss LSM Pascal laser-scanning confocal microscope. Alexa 488 and Alexa 546 were excited at 488 and 543 nm and emission collected at 505–530 and >560 nm, respectively. Images were acquired using a z-resolution of ∼1 μm. Images were background-subtracted, and normalized FRET (N-FRET) was calculated on a pixel-by-pixel basis for the entire image and in regions of interest (within the boundaries of the cell) using the Xia method (41) and Zeiss LSM FRET Macro tool version 2.5 as previously described (48).
Quantitative real-time PCR.
Quantitative Taqman real-time PCR reactions were performed using an LC480 light cycler (Roche Applied Science, Indianapolis, IN). Reaction conditions were an initial denaturation step at 95°C for 5 min followed by 40 cycles of denaturation at 95°C for 10 s, annealing at 60°C for 30 s, and extension at 72°C for 10 s. Negative controls without cDNA were run for each reaction. Differences between fluorescence (Ct) values (ΔCt) of BKα or β1 and Rps5 were calculated. ΔΔCt was calculated from the difference between the ΔCt values for BKα and β1. Relative transcript expression was calculated using the formula 100 × 2(−ΔΔCt) (26). Each PCR reaction, including standard curves, was performed in triplicate. Gene-specific primers and probes were designed using the Universal Probe Library (UPL; http://www.roche-applied-science.com/sis/rtpcr/upl/index.jsp). Standard curves using four 10-fold dilutions of cDNA were run for all probe and primer pairs to determine PCR efficiency.
Patch-clamp electrophysiology.
Cerebral arteries were divided into three groups, control (no treatment), ANG II (100 nM), or ANG II (100 nM) + dynasore (100 μM), and placed in culture for 8 h in serum-free DMEM F-12 media. Arteries were then removed and processed for cell isolation. Potassium (K+) currents were measured using the conventional whole cell configuration with an Axopatch 200B amplifier equipped with a CV203BU headstage, Digidata 1332A, and Clampex 10 (Molecular Devices). The bath solution contained (in mM) 134 NaCl, 6 KCl, 10 HEPES, 10 glucose, 2 CaCl2, 1 MgCl2 with the pH adjusted to 7.4 with NaOH. The pipette solution contained (in mM) 140 KCl, 1.9 MgCl2, 0.075 CaCl2, 10 HEPES, 0.1 EGTA, 2 Na2ATP with the pH adjusted to 7.2 with KOH. The calculated free Ca2+ and Mg2+ concentrations for this solution are 500 nM and 1 mM, respectively (http://www.stanford.edu/∼cpatton/webmaxcS.htm). Whole cell K+ currents were measured by applying 1-s voltage steps between −80 mV and +80 mV in 20-mV increments using an interpulse holding potential of −80 mV. Iberiotoxin (100 nM) was used to block BK currents. Recordings were filtered at 1 kHz using a low-pass Bessel filter and digitized at 4 kHz. Currents were calculated by measuring the average across the final 20 ms of a single sweep.
Pressurized artery myography.
Endothelium was denuded by introducing an air bubble into the lumen for ∼1 min. Artery segments 1–2 mm in length were cannulated in a perfusion chamber (Living Systems Instrumentation, St. Albans, VT) maintained at 37°C and continuously perfused with PSS (pH 7.4) gassed with 21% O2-5% CO2-74% N2. Arterial diameter was measured at 1 Hz using a CCD camera attached to a Nikon TS100-F microscope and the automatic edge-detection function of IonWizard software (Ionoptix, Milton, MA). There was no luminal flow during experiments. Myogenic tone (%) was calculated as: 100 × (1 − Dactive/Dpassive), where Dactive is active arterial diameter and Dpassive is the diameter determined in the presence of Ca2+-free PSS supplemented with 5 mmol/l EGTA.
Statistical analysis.
Data are presented as means ± SE. GraphPad Prism v4.0 was used for statistical analysis. Student's t-test was used for comparing paired and unpaired data from two populations, and ANOVA with Student-Newman-Keuls post hoc test was used for multiple group comparisons. P < 0.05 was considered significant. Where P > 0.05, power analysis was performed to verify that sample size gave a value of >0.8.
RESULTS
Surface trafficking of BKα occurs via a rab4-dependent pathway in arterial myocytes.
We have previously shown that BK β1 subunits are primarily intracellular in resting arterial myocytes and can be stimulated by nitric oxide (NO) to rapidly traffic to the plasma membrane via rab11A-dependent recycling endosomes (25). To study involvement of Rab11 proteins in BKα surface expression, we used RNAi as an approach. Vectors encoding Rab11A short hairpin RNA (Rab11AshV) or rab11B siRNA reduced rab11A and rab11B total proteins to ∼53% and 67% of scrambled controls, respectively, in cerebral arteries (Fig. 1, A and B). In contrast, rab11AshV or rab11B siRNA did not change BKα total protein (Fig. 1, A and B). Surface biotinylation experiments demonstrated that rab11A and rab11B knockdown also did not alter plasma membrane BKα protein levels (Fig. 1, C and D). These data suggest that BKα and β1 are trafficked via independent pathways.
Fig. 1.

Rab11A and rab11B do not regulate large-conductance Ca2+-activated K+ channel (BK)α protein trafficking in cerebral artery myocytes. A: representative Western blots illustrating BKα, rab11A, rab11B, and actin total protein in cerebral arteries transfected with scrambled, rab11A short hairpin vector (shV) or rab11B siRNA. B: mean data (n = 6 for each). *P < 0.05 vs. control shV. C: representative Western blots illustrating plasma membrane (M) and intracellular (I) BKα protein obtained from surface biotinylation experiments in cerebral arteries transfected with scrambled, rab11A shV or rab11B siRNA. Lanes are contiguous in each blot. D: mean data (n = 6 for each). *P < 0.05 vs. respective controls.
Early endosomes are characterized by expression of rab4, a regulatory protein required for vesicle function (7). Immuno-FRET experiments indicated close spatial proximity between BKα- and rab4-bound antibodies in isolated cerebral artery myocytes (Fig. 2A). In contrast, N-FRET between BKα- and rab11A-bound antibodies was similar to background, indicating that these proteins are spatially distant (Fig. 2, A and B). The FRET signal between β1 and rab4-bound antibodies was low, but slightly above background. (Fig. 2, A and B) The involvement of rab4 in BKα surface trafficking was studied using RNAi in cerebral arteries. Rab4A-specific shRNA reduced rab4 total protein to ∼52% of scrambled shRNA controls (Fig. 2, C and D). Rab4A knockdown also reduced BKα total protein to ∼60% of control (Fig. 2, C and D). Surface biotinylation of cerebral arteries revealed that rab4A knockdown reduced plasma membrane BKα to ∼57% of scrambled controls (Fig. 2, E and F). Rab4A knockdown also slightly reduced (∼7%) total β1 protein (Fig. 2, C and D). These data suggest that a rab4A-dependent pathway traffics BKα to the plasma membrane and that inhibition of this anterograde trafficking pathway reduces surface and total BKα protein. These data also indicate that BKα and β1 are surface-trafficked by different rab proteins.
Fig. 2.

Rab4A controls BKα surface expression in arterial myocytes. A: immunofluorescence and immunoFRET images for BKα-Rab4, β1-Rab4 and BKα-Rab11A in isolated arterial myocytes. B: mean data for immuno-FRET experiments (n = 7–10). *P < 0.05 compared with background. Scale bars, 10 μm. Rab4A knockdown decreases BKα total and membrane protein. C: representative Western blots illustrating BKα, β1 and Rab4 total protein in cerebral arteries transfected with control or Rab4A shV. D: mean data (n = 6 for each). *P < 0.05 vs. control shV. E: representative Western blots illustrating plasma membrane (M) and intracellular (I) BKα protein obtained from surface biotinylation experiments in cerebral arteries transfected with scrambled or rab4A shV. Lanes are contiguous in the blot. F: mean data (n = 6 for each). *P < 0.05 vs. scrambled control.
ANG II-induced PKC activation reduces BKα surface and total protein.
We next studied whether BKα is mobile and surface expression can be modulated by physiological stimuli. Biotinylation experiments indicated that a 1-h exposure to sodium nitroprusside (SNP), a NO donor, or ANG II did not affect BKα surface protein in cerebral arteries (Fig. 3, A and B). ANG II (1 h) also did not alter surface β1, although SNP elevated surface β1, as we have previously shown (Fig. 3, A and B) (25). In contrast, a longer (8 h) exposure to ANG II decreased BKα total and surface protein to ∼66 and 63% of untreated controls, respectively (Fig. 3, C–F). ANG II (8 h) did not change the plasma membrane to intracellular (M:I) distribution of BKα protein (% of total protein at surface: control 95.3 ± 1.1, ANG II 93.8 ± 1.6, n = 8 for each, P > 0.05) (Fig. 3, C and D). These data indicate that ANG II reduces BKα total and surface levels in arterial myocytes.
Fig. 3.

ANG II decreases surface BKα in cerebral arteries. Representative Western blots illustrating plasma membrane (M) and intracellular (I) BKα and β1 proteins in rat cerebral arteries obtained using arterial surface biotinylation and regulation by 1 h treatment with sodium nitroprusside (SNP) (10 μM) or ANG II (100 nM) (A). B: mean data for regulation of surface BKα and β1 by 1 h ANG II (n = 6 for each). *P < 0.05 vs. control. C: representative Western blots illustrating plasma membrane (M) and intracellular (I) BKα and β1 protein and modulation by ANG II (100 nM, 8 h). D: mean data (n = 6 for each). *P < 0.05 vs. control. E: representative Western blots illustrating ANG II (100 nM, 8 h) regulation of BKα and β1 total protein. F: mean total protein data (n = 6 for each). *P < 0.05 vs. control. G: real-time PCR data indicating fold change in mRNA for BKα and β1 in myocytes isolated from arteries treated with ANG II (100 nM, 8 h, n = 4). *P < 0.05 vs. control.
Conceivably, the ANG II-induced decrease in BKα protein may be due to inhibition of transcription. Real-time PCR was performed using primers designed to amplify BKα and β1 transcripts in pure populations of arterial myocytes that were harvested as previously described (29). ANG II (8 h) increased BKα mRNA message ∼1.4 fold, whereas β1 message was unchanged, compared with 0 h controls (Fig. 3G). These data indicate that ANG II (8 h) does not reduce BKα protein by inhibiting transcription.
ANG II activates PKC, which inhibits BK channels (40). We tested the hypothesis that ANG II-induced PKC activation reduces BKα plasma membrane protein in cerebral arteries. Bisindolylmaleimide II (BIM-II, 10 μM), a PKC blocker, prevented the ANG II-induced reduction in BKα surface protein (Fig. 4, A and B). In contrast, BIM-II alone did not alter surface BKα (Fig. 4B). These experiments indicate that ANG II-induced PKC activation decreases BKα surface expression.
Fig. 4.

Protein kinase C (PKC) inhibition inhibited ANG II-induced decrease in BKα total and membrane protein. A: representative Western blots illustrating plasma membrane (M) and intracellular (I) BKα and β1 protein and regulation by ANG II (100 nM, 8 h), bisindolylmaleimide II (BIM II, 10 μM), or no treatment. B: mean data (n = 6 for each). *P < 0.05 vs. control, #P < 0.05 vs. ANG II. Lanes are contiguous.
ANG II stimulates internalization and degradation of BKα protein by proteasomal and lysosomal pathways.
ANG II reduced surface BKα, but this protein did not appear in the intracellular fraction. Therefore, we tested the hypothesis that ANG II stimulates internalization and degradation of plasma membrane BKα protein. Consistent with this hypothesis, concanavalin A, an internalization inhibitor, and dynasore, an endocytosis inhibitor, each attenuated the ANG II-induced reduction in surface BKα protein (Fig. 5, A, B, and D). Concanavalin A and dynasore alone did not alter surface BKα protein (Fig. 5D).
Fig. 5.

ANG II-induced decrease in surface BKα is reversed by inhibitors of endocytosis and proteasomal and lysosomal degradation. A: representative Western blots illustrating plasma membrane (M) and intracellular (I) BKα protein in rat cerebral arteries obtained using arterial surface biotinylation. Regulation by ANG II and ANG II + concanavalin A (ConA, 250 μg/ml) is shown. B: regulation of plasma membrane and intracellular BKα by ANG II (100 nM, 8 h) and ANG II + dynasore (100 μM). C: representative Western blots using arterial surface biotinylation and modulation by ANG II (100 nM, 8 h), ANG II + bafilomycin A (BafA, 50 nM), or ANG II + MG132 (15 μM). D: mean data for surface BKα (n = 6 for each dataset). *P < 0.05 vs. control, #P < 0.05 vs. ANG II. E: representative Western blots illustrating BKα and β1 total protein in control, ANG II (100 nM, 8 h), ANG II + bafilomycin A (BafA, 50 nM) or ANG II + MG132 (15 μM). F: mean data normalized to control (n = 6 for each). *P < 0.05 vs. control. Lanes are contiguous in each blot.
Membrane proteins, once internalized, can undergo degradation by proteasomal or lysosomal mechanisms (4, 30, 45). To determine pathways involved in BKα degradation, MG132, an inhibitor of the 26S proteasome, and bafilomycin A, a lysosomal v-ATPase blocker, were used. MG132 and bafilomycin A each attenuated the ANG II-induced reduction in total and surface BKα protein. MG132 and bafilomycin A also increased intracellular BKα protein 2.5 ± 0.3- and 2.4 ± 0.5-fold, respectively, compared with ANG II-treated arteries (n = 6 for each, P < 0.05, Fig. 5C). In contrast, MG132 or bafilomycin A did not alter surface BKα when applied alone (Fig. 5D). MG132 and bafilomycin also blocked the ANG II-induced decrease in total BKα protein (Fig. 5, E and F). These results indicate that ANG II stimulates BKα protein internalization and degradation by proteasomal and lysosomal pathways and that inhibition of degradation leads to BKα recycling to the plasma membrane.
Sustained ANG II treatment inhibits BK channel whole cell currents.
Cerebral arteries were placed in DMEM with or without (control) ANG II for 8 h, after which myocytes were isolated and whole cell K+ currents measured in the absence of ANG II (Fig. 6, A–D). In control myocytes, mean peak whole cell current density was 157.3 ± 14.3 pA/pF (at +80 mV, n = 10). Iberiotoxin (100 nM), a BK channel-specific inhibitor, reduced mean peak current density to 36.9 ± 4.64 pA/pF, indicating that the mean iberiotoxin-sensitive current (or BK current) was 120.6 ± 17.6 pA/pF (at +80 mV) in control myocytes (Fig. 6, A and D). ANG II reduced mean peak whole cell current density in myocytes to 116.2 ± 9.0 pA/pF or to ∼73.9% of that in control myocytes (P < 0.05, at +80 mV, Fig. 6B, n = 8). Mean iberiotoxin-sensitive current density in ANG II-treated myocytes was 67.5 ± 13.7 pA/pF, or 56% of that in untreated controls (P < 0.05, Fig. 6, B and D). In myocytes treated with both ANG II and dynasore, mean whole cell current density was 173.5 ± 14.7 pA (at +80 mV, n = 7), indicating that dynasore blocked the ANG II-induced reduction in currents. Dynasore also prevented the decrease in iberiotoxin-sensitive currents, which were 118.3 ± 13.9 pA/pF, or ∼98.1% of untreated control myocytes (at +80 mV, P > 0.05, Fig. 6, C and D). These data indicate that ANG II-induced BK channel internalization reduces whole cell BK currents in arterial myocytes.
Fig. 6.
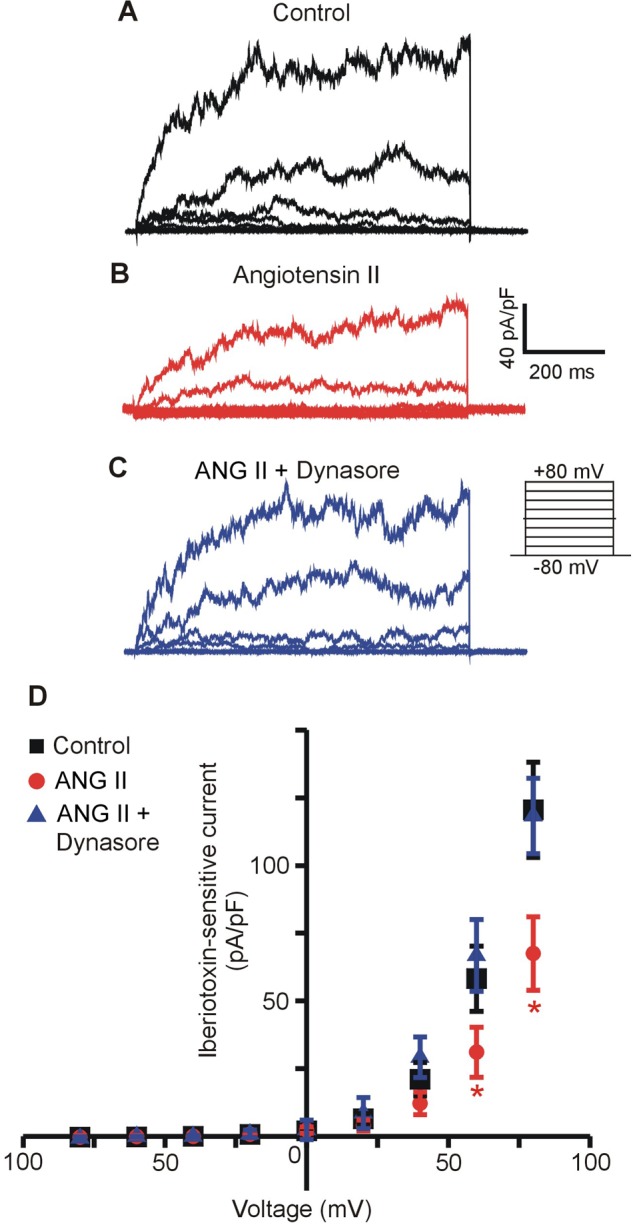
ANG II (8 h) decreases iberiotoxin-sensitive whole cell currents in arterial myocytes. Representative traces illustrating whole cell iberiotoxin-sensitive BK currents in isolated arterial myocytes from control (black trace) (A), ANG II (100 nM, 8 h, red trace) (B), and ANG II + dynasore (100 μM, blue trace) (C)- treated arteries. D: mean data for iberiotoxin-sensitive currents in freshly isolated arterial myocytes from control (n = 10), ANG II (n = 8), and ANG II + dynasore (n = 7)-treated arteries. *P < 0.05 vs. control untreated arteries.
ANG II inhibits functional BK channel activity in pressurized arteries.
The functional significance of an ANG II-induced decrease in BKα protein was studied by measuring arterial contractility. Cerebral arteries were incubated with or without ANG II for 8 h. ANG II was then removed after which the endothelium was denuded and arteries pressurized to 60 mmHg. Pressure-induced vasoconstriction (myogenic tone), responses to iberiotoxin and to NS1619, a BK channel opener, and depolarization-induced vasoconstriction (60 mM K+), were measured. Control arteries developed ∼21% myogenic tone, whereas ANG II-treated arteries developed ∼30% myogenic tone, or 1.42-fold more (Fig. 7A). Passive diameter of control (301 ± 18 μm) and ANG II-treated (287 ± 31 μm) arteries were similar (P > 0.05, n = 6 for each). Iberiotoxin (100 nM) constricted control arteries by ∼31 μm and ANG II-treated arteries by ∼16.5 μm, or ∼47% less (Fig. 7, B and D). Similarly, NS1619 dilated control arteries by ∼37 μm and ANG II-treated arteries by ∼24.4 μm, or ∼34% less (Fig. 7, C and D). In contrast, ANG II treatment did not alter constriction to 60 mM K+ PSS (Fig. 7A). These data indicate that ANG II stimulates BK channel internalization and degradation, leading to an elevation in myogenic tone.
Fig. 7.

ANG II (8 h) constricts pressurized cerebral arteries due to loss of BK channel activity. A: mean data for myogenic tone development at an intravascular pressure of 60 mmHg and responses to 60 mM K+ in control and ANG II (100 nM, 8 h)-treated arteries. *P < 0.05 vs. control untreated arteries. Original recordings from control (black trace) and ANG II-treated (red trace) arteries illustrating constriction to iberiotoxin (10 nM, IbTX) (B) and dilation to NS1619 (10 μM) (C). D: mean data. n = 6 for each. *P < 0.05 vs. control untreated arteries.
DISCUSSION
Intracellular trafficking pathways that control surface expression of vascular myocyte ion channels are poorly understood. The aim of this study was to identify mechanism(s) and functional significance of BKα trafficking and to investigate regulation by vasoactive agents in arterial myocytes. Our data indicate that 1) BKα is primarily plasma membrane-localized and is trafficked to the plasma membrane via a rab4A-dependent, but not rab11A or rab11B-dependent, pathway; 2) ANG II stimulates a PKC-dependent reduction in surface and total BKα, whereas nitric oxide (NO) does not alter BKα surface expression; 3) ANG II did not alter surface or total β1 protein; 4) ANG II-induced reduction in surface BKα is blocked by inhibitors of endocytosis and lysosomal and proteasomal degradation; and 5) ANG II-induced BKα internalization and degradation reduces BK currents in arterial myocytes, elevates myogenic tone, and decreases vasoactive responses to iberiotoxin and NS1619. These data indicate that ANG II stimulates BKα internalization and degradation via a PKC-dependent pathway to inhibit channel activity and induce vasoconstriction. This action of ANG II is specific for BKα subunits and does not alter surface or total β1 protein.
Rab proteins are small GTPases that can control both anterograde and retrograde trafficking of cellular proteins (36). Rab proteins have been shown to traffic several different ion channels or their auxiliary subunits, with many of these studies performed by investigating recombinant proteins (5, 9, 27, 38). BK channel activity controls vascular tone and is regulated by a wide variety of vasoactive agonists and stimuli (21). Recently, we demonstrated that BKα subunits are primarily plasma membrane-localized, whereas β1 subunits are intracellular, in rat and human cerebral and rat mesenteric arteries. NO stimulated rapid rab11A-dependent β1 surface trafficking, but did not alter BKα subunit localization (25). This led us to suggest that BKα was trafficked to the plasma membrane independently of β1 subunits, likely via a different pathway. Data here and in our previous study strongly indicate that BKα and β1 undergo anterograde trafficking via distinct pathways.
Here, immuno-FRET analysis in isolated myocytes revealed significant N-FRET between BKα and rab4-bound secondary antibodies, indicating close spatial proximity of these two proteins. In contrast, BKα and rab11A immunoFRET was not significant (25). Recombinant BK channels are associated with rab11B in CHO cells (34). Together, data here and in our earlier study indicate that BKα localization was not affected by rab11A or rab11B knockdown in arterial myocytes (25). These data indicate that BKα is in an intracellular compartment located in close spatial proximity to rab4. Immuno-FRET images reveal the FRET signal to be primarily at the surface, suggesting that BKα and rab4A remain colocalized after early endosomes fuse with the plasma membrane. Early endosomes (EE) are intracellular protein storage and trafficking vesicles characterized by their expression of rab4 (36). EE can recycle proteins to and from the cell surface (6, 36, 45). Here, rab4A knockdown decreased rab4 total protein and BKα total and membrane protein in cerebral arteries. Rab4A knockdown did not result in BKα intracellular accumulation, suggesting that internalized protein was targeted for degradation. A lack of intracellular protein accumulation was also observed with GLUT4 following rab4 knockdown (22). Data indicate that β1- and rab4-bound fluorescent antibodies generate a low FRET signal and that rab4A knockdown slightly decreased (∼7%) β1 total protein. Rab4 and rab11 can exist together in the same endosomal compartment but within distinct domains (35). Early endosomes and recycling endosomes can also fuse with each other to transfer protein cargo (8, 35). Conceivably, β1 may be associated with a similar protein sorting mechanism, but the relatively small decrease in β1 protein after rab4 knockdown indicates that this pathway is relatively minor for β1 recycling in arterial myocytes. In summary, our data indicate that BKα and β1 undergo anterograde trafficking via distinct pathways: BKα via rab4A-dependent early endosomes and β1 via rab11A-positive recycling endosomes.
Several vasoregulatory stimuli modify BK currents (I), including vasoconstrictors such as ANG II, and vasodilators, including NO (28, 37, 42). Studies have shown that PKG, PKA, and PKC modify BK channel open probability (Po) (13, 32, 33, 49). Our data here show that ANG II decreases BK channel currents, in part by reducing surface channel number (N). To our knowledge this is the first time it has been shown that a vasoconstrictor inhibits BK channel currents by reducing BKα N in arterial myocytes. Concanavalin and dynasore each inhibited the ANG II-induced decrease in surface BKα protein. ANG II altered BKα surface expression over a longer time scale than the rapid regulation of surface β1 expression by NO, which took seconds (25). ANG II-induced BKα internalization may occur faster than 8 h and the signal for degradation may be initiated before 8 h. It was not a focus of this study to identify the precise time period by which BK channel activity was reduced by internalization. Regardless, brain blood flow can be altered by both rapid and slower regulatory mechanisms. Collectively, our data indicate that NO rapidly elevates β1 to induce immediate vasodilation, whereas ANG II reduces BKα surface levels to promote long-term vasoconstriction.
Many vasoconstrictors activate PKC, including ANG II (40). PKC can directly inhibit BK channels in arterial myocytes, but involvement of channel internalization was unclear (33). PKC stimulates degradation of ENaC in adrenal chromaffin cells (44) and recombinant KATP channels expressed in COS-7 cells (17). Our data indicate that PKC also stimulates internalization and degradation of functional surface BK channels, thereby reducing channel number (N). Taken together, these results suggest that protein internalization and degradation is a long-term regulatory mechanism that controls BK channel activity.
Involvement of PKC indicates G protein activation by ANG II. Recently, angiotensin type-1 receptors (AT1R) were shown to be physically associated with BKα in rat renal artery myocytes and when overexpressed in HEK293 cells (46). ANG II activation of AT1 receptors inhibited BK channels by a G protein-independent mechanism (46). Conceivably, BK channel inhibition by ANG II may be mediated in the short term by a G protein-independent mechanism and in the long term by the G protein-dependent pathway we describe here.
Cargo proteins contained within early endosomes that are not trafficked to the plasma membrane can be shuttled for degradation (10). Our data indicate that internalized BKα is degraded via both lysosomal and proteasomal pathways. Ion channels, including recombinant KCa3.1 and ENaC also undergo degradation via both pathways (2, 10). Here, degradation inhibitors slightly increased intracellular BKα, but in the continued presence of ANG II most of the rescued protein was detected at the plasma membrane. These data suggest that PKC primarily stimulates BKα degradation, but also activates internalization and/or inhibits anterograde trafficking.
Whole cell patch-clamp studies confirmed that ANG II pretreatment inhibited BK currents via a mechanism that was inhibited by dynasore, an endocytosis blocker. Functional studies also indicated that ANG II pretreatment leads to vasoconstriction in the absence of ANG II that was observed as an increase in myogenic tone and smaller responses to BK channel modulators. Taken together these results indicate that ANG II reduces the number of functional channels in the myocyte plasma membrane, leading to vasoconstriction.
In summary, we show that in cerebral artery myocytes, surface trafficking of BKα occurs via a rab4A-dependent pathway. ANG II-stimulated PKC promotes internalization of BKα. Internalized BKα is then degraded by both lysosomal and proteasomal pathways, inhibition of which recycles BKα back to the plasma membrane. This study indicates that ANG II stimulates endocytosis and degradation of BK channels to reduce activity (NPo) and induce vasoconstriction.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants RO1-HL-67661, HL-110347, and HL-094378 to J. H. Jaggar and American Heart Association (AHA)-SDG to M. D. Leo.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.D.L. and J.H.J. conception and design of research; M.D.L., S.B., J.P.B., K.P.K., and D.N. performed experiments; M.D.L., S.B., J.P.B., K.P.K., and D.N. analyzed data; M.D.L. and J.H.J. interpreted results of experiments; M.D.L. prepared figures; M.D.L. and J.H.J. drafted manuscript; M.D.L. and J.H.J. edited and revised manuscript; J.H.J. approved final version of manuscript.
REFERENCES
- 1.Bannister JP, Adebiyi A, Zhao G, Narayanan D, Thomas CM, Feng JY, Jaggar JH. Smooth muscle cell a2d-1 subunits are essential for vasoregulation by CaV1.2 channels. Circ Res 105: 948–955, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertuccio CA, Lee SL, Wu G, Butterworth MB, Hamilton KL, Devor DC. Anterograde trafficking of KCa3.1 in polarized epithelia is Rab1- and Rab8-dependent and recycling endosome-independent. PLoS ONE 9: e92013, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature 407: 870–876, 2000. [DOI] [PubMed] [Google Scholar]
- 4.Burd CG. Physiology and pathology of endosome-to-Golgi retrograde sorting. Traffic 12: 948–955, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiu YH, Alvarez-Baron C, Kim EY, Dryer SE. Dominant-negative regulation of cell surface expression by a pentapeptide motif at the extreme COOH terminus of an Slo1 calcium-activated potassium channel splice variant. Mol Pharmacol 77: 497–507, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daro E, van der Sluijs P, Galli T, Mellman I. Rab4 and cellubrevin define different early endosome populations on the pathway of transferrin receptor recycling. Proc Natl Acad Sci USA 93: 9559–9564, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De RS, Sonnichsen B, Zerial M. Divalent Rab effectors regulate the sub-compartmental organization and sorting of early endosomes. Nat Cell Biol 4: 124–133, 2002. [DOI] [PubMed] [Google Scholar]
- 8.De RS, Sonnichsen B, Zerial M. Divalent Rab effectors regulate the sub-compartmental organization and sorting of early endosomes. Nat Cell Biol 4: 124–133, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Deutsch E, Weigel AV, Akin EJ, Fox P, Hansen G, Haberkorn CJ, Loftus R, Krapf D, Tamkun MM. Kv2.1 cell surface clusters are insertion platforms for ion channel delivery to the plasma membrane. Mol Biol Cell 23: 2917–2929, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eaton DC, Malik B, Bao HF, Yu L, Jain L. Regulation of epithelial sodium channel trafficking by ubiquitination. Proc Am Thorac Soc 7: 54–64, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edwards G, Feletou M, Weston AH. Endothelium-derived hyperpolarising factors and associated pathways: a synopsis. Pflügers Arch 459: 863–879, 2010. [DOI] [PubMed] [Google Scholar]
- 12.Evanson KW, Bannister JP, Leo MD, Jaggar JH. LRRC26 is a functional BK channel auxiliary gamma subunit in arterial smooth muscle cells. Circ Res 115: 423–431, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukao M, Mason HS, Britton FC, Kenyon JL, Horowitz B, Keef KD. Cyclic GMP-dependent protein kinase activates cloned BKCa channels expressed in mammalian cells by direct phosphorylation at serine 1072. J Biol Chem 274: 10927–10935, 1999. [DOI] [PubMed] [Google Scholar]
- 14.Ghatta S, Nimmagadda D, Xu X, O'Rourke ST. Large-conductance, calcium-activated potassium channels: structural and functional implications. Pharmacol Ther 110: 103–116, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Henkel AW, Bieger SC. Quantification of proteins dissolved in an electrophoresis sample buffer. Anal Biochem 223: 329–331, 1994. [DOI] [PubMed] [Google Scholar]
- 16.Hill MA, Yang Y, Ella SR, Davis MJ, Braun AP. Large conductance, Ca2+-activated K+ channels (BKCa) and arteriolar myogenic signaling. FEBS Lett 584: 2033–2042, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu K, Huang CS, Jan YN, Jan LY. ATP-sensitive potassium channel traffic regulation by adenosine and protein kinase C. Neuron 38: 417–432, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol Cell Physiol 278: C235–C256, 2000. [DOI] [PubMed] [Google Scholar]
- 19.Jaggar JH, Wellman GC, Heppner TJ, Porter VA, Perez GJ, Gollasch M, Kleppisch T, Rubart M, Stevenson AS, Lederer WJ, Knot HJ, Bonev AD, Nelson MT. Ca2+ channels, ryanodine receptors and Ca2+-activated K+ channels: a functional unit for regulating arterial tone. Acta Physiol Scand 164: 577–587, 1998. [DOI] [PubMed] [Google Scholar]
- 20.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508: 199–209, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kyle BD, Braun AP. The regulation of BK channel activity by pre- and post-translational modifications. Front Physiol 5: 316, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JO, Lee SK, Jung JH, Kim JH, You GY, Kim SJ, Park SH, Uhm KO, Kim HS. Metformin induces Rab4 through AMPK and modulates GLUT4 translocation in skeletal muscle cells. J Cell Physiol 226: 974–981, 2011. [DOI] [PubMed] [Google Scholar]
- 23.Lee US, Cui J. BK channel activation: structural and functional insights. Trends Neurosci 33: 415–423, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leffler CW, Parfenova H, Jaggar JH. Carbon monoxide as an endogenous vascular modulator. Am J Physiol Heart Circ Physiol 301: H1–H11, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leo MD, Bannister JP, Narayanan D, Nair A, Grubbs JE, Gabrick KS, Boop FA, Jaggar JH. Dynamic regulation of beta1 subunit trafficking controls vascular contractility. Proc Natl Acad Sci USA 111: 2361–2366, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCt) Method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 27.Makhina EN, Nichols CG. Independent trafficking of KATP channel subunits to the plasma membrane. J Biol Chem 273: 3369–3374, 1998. [DOI] [PubMed] [Google Scholar]
- 28.Minami K, Fukuzawa K, Nakaya Y. Protein kinase C inhibits the Ca2+-activated K+ channel of cultured porcine coronary artery smooth muscle cells. Biochem Biophys Res Commun 190: 263–269, 1993. [DOI] [PubMed] [Google Scholar]
- 29.Narayanan D, Bulley S, Leo MD, Burris SK, Gabrick KS, Boop FA, Jaggar JH. Smooth muscle cell transient receptor potential polycystin-2 (TRPP2) channels contribute to the myogenic response in cerebral arteries. J Physiol 591: 5031–5046, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 122: 735–749, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Robertson BE, Schubert R, Hescheler J, Nelson MT. cGMP-dependent protein kinase activates Ca2+-activated K+ channels in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol 265: C299–C303, 1993. [DOI] [PubMed] [Google Scholar]
- 32.Schubert R, Nelson MT. Protein kinases: tuners of the BKCa channel in smooth muscle. Trends Pharmacol Sci 22: 505–512, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Schubert R, Noack T, Serebryakov VN. Protein kinase C reduces the KCa current of rat tail artery smooth muscle cells. Am J Physiol Cell Physiol 276: C648–C658, 1999. [DOI] [PubMed] [Google Scholar]
- 34.Sokolowski S, Harvey M, Sakai Y, Jordan A, Sokolowski B. The large conductance calcium-activated K+ channel interacts with the small GTPase Rab11b. Biochem Biophys Res Commun 426: 221–225, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sonnichsen B, De RS, Nielsen E, Rietdorf J, Zerial M. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J Cell Biol 149: 901–914, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 10: 513–525, 2009. [DOI] [PubMed] [Google Scholar]
- 37.Toro L, Amador M, Stefani E. ANG II inhibits calcium-activated potassium channels from coronary smooth muscle in lipid bilayers. Am J Physiol Heart Circ Physiol 258: H912–H915, 1990. [DOI] [PubMed] [Google Scholar]
- 38.Vithlani M, Terunuma M, Moss SJ. The dynamic modulation of GABA(A) receptor trafficking and its role in regulating the plasticity of inhibitory synapses. Physiol Rev 91: 1009–1022, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu RS, Marx SO. The BK potassium channel in the vascular smooth muscle and kidney: alpha- and beta-subunits. Kidney Int 78: 963–974, 2010. [DOI] [PubMed] [Google Scholar]
- 40.Wynne BM, Chiao CW, Webb RC. Vascular smooth muscle cell signaling mechanisms for contraction to angiotensin II and endothelin-1. J Am Soc Hypertens 3: 84–95, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xia Z, Liu Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys J 81: 2395–2402, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamakage M, Hirshman CA, Croxton TL. Sodium nitroprusside stimulates Ca2+-activated K+ channels in porcine tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 270: L338–L345, 1996. [DOI] [PubMed] [Google Scholar]
- 43.Yan J, Aldrich RW. LRRC26 auxiliary protein allows BK channel activation at resting voltage without calcium. Nature 466: 513–516, 2010. [DOI] [PubMed] [Google Scholar]
- 44.Yanagita T, Kobayashi H, Yamamoto R, Kataoka H, Yokoo H, Shiraishi S, Minami S, Koono M, Wada A. Protein kinase C-alpha and -epsilon down-regulate cell surface sodium channels via differential mechanisms in adrenal chromaffin cells. J Neurochem 74: 1674–1684, 2000. [DOI] [PubMed] [Google Scholar]
- 45.Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2: 107–117, 2001. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Z, Li M, Lu R, Alioua A, Stefani E, Toro L. The angiotensin II type 1 receptor (AT1R) closely interacts with large conductance voltage- and Ca2+-activated K+ (BK) channels and inhibits their activity independent of G-protein activation. J Biol Chem 289: 25678–25689, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao G, Adebiyi A, Blaskova E, Xi Q, Jaggar JH. Type 1 inositol 1,4,5-trisphosphate receptors mediate UTP-induced cation currents, Ca2+ signals, and vasoconstriction in cerebral arteries. Am J Physiol Cell Physiol 295: C1376–C1384, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao G, Neeb ZP, Leo MD, Pachuau J, Adebiyi A, Ouyang K, Chen J, Jaggar JH. Type 1 IP3 receptors activate BKCa channels via local molecular coupling in arterial smooth muscle cells. J Gen Physiol 136: 283–291, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou XB, Arntz C, Kamm S, Motejlek K, Sausbier U, Wang GX, Ruth P, Korth M. A molecular switch for specific stimulation of the BKCa channel by cGMP and cAMP kinase. J Biol Chem 276: 43239–43245, 2001. [DOI] [PubMed] [Google Scholar]