

Abstract
Despite greater understanding of acute kidney injury (AKI) in animal models, many of the preclinical studies are not translatable. Most of the data were derived from a bilateral renal pedicle clamping model with warm ischemia. However, ischemic injury of the kidney in humans is distinctly different and does not involve clamping of renal vessel. Permanent ligation of the left anterior descending coronary artery model was used to test the role of microRNA (miR)-150 in AKI. Myocardial infarction in this model causes AKI which is similar to human cardiac bypass surgery. Moreover, the time course of serum creatinine and biomarker elevation were also similar to human ischemic injury. Deletion of miR-150 suppressed AKI which was associated with suppression of inflammation and interstitial cell apoptosis. Immunofluorescence staining with endothelial marker and marker of apoptosis suggested that dying cells are mostly endothelial cells with minimal epithelial cell apoptosis in this model. Interestingly, deletion of miR-150 also suppressed interstitial fibrosis. Consistent with protection, miR-150 deletion causes induction of its target gene insulin-like growth factor-1 receptor (IGF-1R) and overexpression of miR-150 in endothelial cells downregulated IGF-1R, suggesting miR-150 may mediate its detrimental effects through suppression of IGF-1R pathways.
Keywords: myocardial infarction, acute kidney injury, bilateral renal pedicle clamping model, miR-150
acute kidney injury (AKI) due to ischemia is a serious and frequent problem in hospitalized patients and patients who undergo surgery (7, 16, 30). Currently, there are no therapies available to treat or prevent the development of AKI. Moreover, therapies developed based on the currently existing renal ischemia-reperfusion injury were not translatable. This is mainly due to the inadequacy of currently used animal models of bilateral renal pedicle clamping in mimicking hypoxic renal injury in humans during bypass surgery or artificial ventilation. Except during partial nephrectomy, all other forms of AKI in humans develop without complete stoppage of blood supply as opposed to the mouse model of bilateral clamping. Therefore, underlying mechanisms and cell types that may be affected could be quite different. In addition, the definition of 50% increase of creatinine over baseline (2-fold increase) for diagnosis of AKI (26, 31) as opposed to 10-fold increase in the mouse model with significant necrosis in the tubules suggests extreme difference in the severity. Induction of myocardial infarction (MI) through ligation of left coronary artery with artificial ventilation of lung in the mouse is a much closer and ideal model that is closely related to human cardiopulmonary bypass surgery-induced AKI in humans. Due to its time-consuming nature and complicated surgical protocol requiring much needed surgical skills, it is not practiced as a routine model for inducing AKI in the mouse. With the help of a cardiac surgeon, we performed this model to determine the underlying mechanism of MI-induced AKI and role of miRNA-150 in AKI.
MicroRNAs constitute a class of noncoding RNAs that play key roles in the regulation of gene expression. Acting at the posttranscriptional level, these fascinating molecules may fine-tune the expression of as much as 60% of all mammalian protein-encoding genes. Recent studies have shown that microRNAs (miRs) are involved in the pathogenesis of chronic kidney disease and renal fibrosis (6, 17, 21). MiR-150 is shown to mediate its profibrotic effects through suppression of SOCS1 in tubular epithelium and mesangial cells (37). However, its role in AKI is unknown. Our results demonstrate for the first time that miR-150 deletion in mice protects the kidney against MI-induced AKI as well as bilateral renal ischemia-reperfusion injury.
MATERIALS AND METHODS
Mouse strains.
MiR-150 knockout mice and their wild-type (WT; C57BL/6J) counterparts were purchased from Jackson Laboratories. The Institutional Animal Care and Use Committee of the Georgia Regents University approved all of the protocols and procedures for using animals (approval number BR10-10-369).
Mouse model of MI-induced AKI.
Neck area and the left side of the ribcage were shaved and disinfected using 80% ethanol. The mouse was placed on its back and a facemask was placed over its nose and mouth to keep up the anesthesia, isoflurane. Unconsciousness of the mouse was confirmed by pinching the toe. Midline cervical incision was performed by separating the skin, muscle, and tissue covering the trachea under the dissection microscope. A small hole was cut in the exposed trachea between two cartridge rings below the glottis to insert the endotracheal tube and then a tube was inserted. The thoracic movement was checked to make sure that both lungs were well-ventilated. The respiration rate was ∼110 breaths/min, with an inspiratory pressure of 17 to 18 cmH2O. The mouse was turned carefully to its right side, facing its left side. Left-sided thoracotomy was performed between the third and the fourth rib, and the tissue and muscle were dissected carefully using a cauter to prevent bleeding. The thorax was opened carefully and the heart was located without touching the lung with any sharp object. After that, part of the pericardial sac that is covering the heart was removed. The left anterior descending artery (LAD) was identified which is located between the pulmonary artery and the left auricle. With the use of an 8-0 Prolene suture (Ethicon, Norderstedt, Germany), the LAD was ligated in the proximal region with one single suture. A chest tube (28-gauge, venal catheter) was placed between the fourth and the fifth rib and then a thoracic incision was closed in layers, using 6-0 Prolene running sutures to adapt the ribs and 4-0 Prolene running sutures to close the skin. The thorax was carefully drained with the help of a 2-ml syringe. The mouse was placed on its back and we removed the endotracheal tube out and adopted the tracheal cartridge rings with one single stitch using 7-0 Prolene sutures. We then closed the skin using 4-0 Prolene running sutures. Animals were monitored for heart function by echocardiography (29) and the echocardiography data are provided for these mice in the same manuscript (29). Kidney function was monitored by measuring serum creatinine. Animals were killed at 24 h or 8 wk after surgery.
Renal ischemia reperfusion.
Eight- to nine-week-old miR-150 knockout and their WT counterparts were anesthetized with pentobarbital sodium (50 mg/kg body wt ip) and were placed on a heating pad to maintain body temperature at 37°C. Both renal pedicles were identified through dorsal incisions and clamped for a period of 26 min. Reperfusion was confirmed visually upon release of the clamps. As a control, sham-operated animals were subjected to the same surgical procedure except the renal pedicles were not clamped. Surgical wounds were closed and mice were given 1 ml of warm saline intraperitoneally. The mice were kept in a warm incubator until they regained consciousness.
Renal function.
Renal function was assessed by measurements of serum creatinine (DZ072B, Diazyme Laboratories, Poway, CA) (28, 32).
TACS TdT in situ apoptosis detection.
To identify apoptotic cells, tissue sections were stained using TACS TdT in situ Apoptosis Detection kit (R&D Systems) according to the manufacturer's instructions. Briefly, tissue sections were deparaffinized, hydrated, and washed with PBS. Sections were digested with proteinase K for 15 min at 24°C. Slides were then washed and endogenous peroxidase activity was quenched with 3% H2O2 in methanol. Slides were washed and incubated with TdT labeling reaction mix at 37°C for 1 h and then with streptavidin-horseradish peroxidase. Color was developed using TACS blue label substrate solution. Slides were washed, counterstained, and mounted with Permount. Sections were photographed and labeled cells were counted and quantified.
Quantitation of mRNA by real-time RT-PCR.
Real-time RT-PCR was performed in an Applied Biosystems 7700 Sequence Detection System (Foster City, CA). Total RNA (1.5 μg) was reverse transcribed in a reaction volume of 20 μl using Omniscript RT kit and random primers. The product was diluted to a volume of 150 μl, and 6-μl aliquots were used as templates for amplification using the SYBR Green PCR amplification reagent (Qiagen) and gene-specific primers. The primer sets used were as follows: mouse TNF-α (forward: GCATGATCCGCGACGTGGAA; reverse: AGATCCATGCCGTTG GCCAG), MCP-1 (forward: ATGCAGGTCCCTGTCATG; reverse: GCTTGAGGTGGTTGTGGA), ICAM-1 (forward: AGATCACATTCACGGTGCTG; reverse: CTTCAGAGGCAGGAAACAGG), KIM-1 (forward: ATGAATCAGATTCAAGTCTTC; reverse: TCTGGTTTGTGAGTCCATGTG), and NGAL (forward: CCCCATCTCTGCTCACTGTC; reverse: TTTTTCTGGACCGCATTG). The amount of cDNA was normalized to the β-actin signal amplified in a separate reaction (forward primer: AGAGGGAAATCGTGCGTGAC; reverse: CAATAGTGATGACCTGGCCGT).
Histology and immunostaining.
Kidney tissue was fixed in buffered 10% formalin for 12 h and then embedded in paraffin wax. For assessment of injury, 5-μm sections were stained with periodic acid Schiff (PAS) followed by hematoxylin. To quantify leukocyte infiltration, sections were stained with rat anti-mouse neutrophil antibody (Abcam, Cambridge, MA; 1:200 dilution) followed by goat anti-rat biotin conjugate. Color was developed after incubation with ABC reagent (Vector Lab). Stained sections were photographed and five ×40 fields of neutrophils were examined for quantification of leukocytes. To determine whether TdT-mediated dUTP nick end labeling (TUNEL)-positive cells are endothelial cells, after staining the section for FITC-based TUNEL assay (R&D Systems), sections were counterstained with CD31 antibody (Abcam) followed by Cy5-conjugated secondary antibody. Stained sections were photographed using an Olympus inverted microscope with color CCD camera.
Cell culture and transfection.
Immortalized mouse cardiac endothelial cell line was purchased from Cedarlane and maintained according to the company's recommendation. For gain-of-function studies, we transfected cytomegalovirus expression plasmids for miR-150 (Origene, SC400788) or miR-150 mimics (Life Technologies, MC10070). All in vitro assays were performed 60–72 h after transfection when overexpression is maximum. Overexpression was confirmed by RT-PCR and showed over 20,000-fold more miR-150 than control transfected cells.
Statistical methods.
All assays were performed in duplicate or triplicate. The data are reported as means ± SE. Statistical significance was assessed by an unpaired, two-tailed Student's t-test for single comparison or ANOVA for multiple comparisons. P < 0.05 is considered significant.
RESULTS
MI-induced AKI in mice follows a similar time course of human cardiopulmonary bypass surgery.
Immediately after the MI surgical procedure (Fig. 1A), kidney function was monitored by measuring serum creatinine. Sham-operated WT mice with just ventilation showed a small but insignificant increase in serum creatinine. However, WT mice subjected to left coronary artery ligation with ventilation developed a significant rise in serum creatinine on day 1 and peaked at day 2 (Fig. 1B). In the subsequent days, serum creatinine started falling to reach sham-operated animal levels. The time course and serum creatinine rise were very similar to previously reported pediatric human cardiopulmonary bypass surgery (20, 23).
Fig. 1.

microRNA (miR)-150 deletion protects the kidney against myocardial infarction (MI)-induced acute kidney injury (AKI). A: description of MI surgery protocol. B: time course of serum creatinine rise after sham and MI surgery in wild-type (WT) mice. *P < 0.001 vs. corresponding time point in sham; n = 4–6. C: serum creatinine at 48 h after sham and MI surgery in WT and miR-150 knockout (KO) animals. *P < 0.05 vs. other groups; n = 4–6. D: RT-PCR analysis of inflammatory cytokine and kidney injury markers in WT and miR-150 KO animals. *P < 0.05 vs. other groups; n = 4. E: periodic acid Schiff (PAS)-stained section from sham and MI-operated WT and miR-150 KO animals showing no visible structural changes in the kidney with MI.
Interestingly, we carried a similar procedure in miR-150 knockout mice and they are protected against MI-induced kidney dysfunction as seen by reduced serum creatinine rise at 48 h (Fig. 1C). Kidney dysfunction in WT mice was associated with a significant increase in inflammatory cytokines such as TNF-α, IL-6, and MCP-1 as well as kidney injury markers such as NGAL and KIM-1 (Fig. 1D). These changes were minimal in miR-150 knockout mice. Interestingly, PAS-stained sections did not show any visible structural alteration such as cast formation and brush-border damage in both WT and miR-150 knockout mice (Fig. 1E).
Despite increased inflammatory cytokine expression, there was no neutrophil infiltration seen in the kidney after MI (not shown). In addition, macrophage/monocyte infiltration was not increased after MI in the WT and miR-150 knockout animal kidney (Fig. 2) in both the cortex and medulla.
Fig. 2.

Immunolocalization of monocyte/macrophage in the kidney. No change in macrophage infiltration was seen with MI surgery in WT and miR-150 KO.
MI with ventilation induced a prominent peritubular interstitial cell apoptosis.
To determine whether kidney dysfunction was associated with tubular epithelial cell apoptosis similar to the bilateral renal pedicle clamping model, a kidney section was stained with TUNEL staining to determine apoptosis. TUNEL staining showed a large increase in apoptotic positive cells in WT. Surprisingly, very little apoptosis was found in the tubular epithelium of the cortex. A large number of interstitial cells were positive for TUNEL in WT mice subjected to MI and maximum apoptosis was found in the inner medulla (Fig. 3, A–C). Moreover, many cells were positive for TUNEL within the vessels. miR-150 knockout mice showed a significant reduction in MI-induced apoptosis of interstitial cells (Fig. 3, A–C).
Fig. 3.

Quantification of apoptosis after sham and MI surgery in WT and miR-150 KO animals. A: quantification of TdT-mediated dUTP nick end labeling (TUNEL)-positive cells in the kidney cortex. B: quantification of TUNEL-positive cells in the kidney medulla. *P < 0.001 vs. sham. #P < 0.001 vs. WT MI; n = 4–6. C: representative images showing TUNEL staining in WT and miR-150 KO mouse kidney.
To determine whether apoptotic interstitial cells are endothelial cells, immunofluorescence double staining was done for TUNEL and endothelial cell marker CD31. As shown in Fig. 4, apoptotic cells are indeed endothelial cells. However, we also saw few cells that were negative for endothelial cell marker. The identity of these CD31-negative cells is unknown.
Fig. 4.

Colocalization of CD31 (endothelial marker) with TUNEL-positive cells in the WT kidney. A: CD31 staining shown in red. B: TUNEL-positive cells shown in green. C: nuclear staining shown in blue. D: overlay of 3 images. Yellow arrow indicates colocalization of CD31 cells and TUNEL-positive nuclei.
Mild interstitial fibrosis seen at 8 wk was suppressed in mir-150 knockout mice.
Previous studies have shown that miR-150 blockade suppressed interstitial fibrosis in a model of lupus nephritis (37). However, it was not clear whether such phenomenon is also seen in other models of interstitial fibrosis. To determine the effect of miR-150 deletion on AKI-induced fibrosis in vivo, kidney tissue sections were stained with Trichrome. As shown in Fig. 5, mild interstitial fibrosis was seen in WT mice that were subjected to MI which was completely suppressed in miR-150 knockout mice. Consistent with trichrome staining, a modest but significant increase in collagen I and connective tissue growth factor mRNA was seen in WT kidney after MI that was suppressed in the miR-150 knockout animal kidney with MI.
Fig. 5.
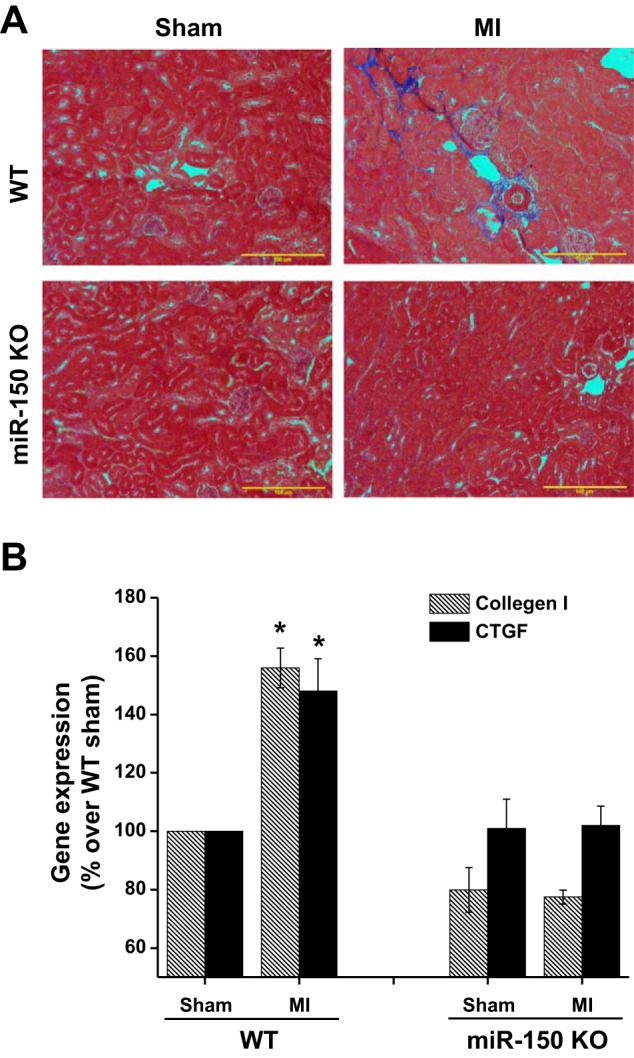
Kidney fibrosis was assessed with Trichrome staining and RT-PCR. A: mild interstitial fibrosis seen at 8 wk after MI surgery that was suppressed in miR-150 KO animals. B: collagen and connective tissue growth factor (CTGF) expression was significantly increased in WT mice kidney which was suppressed in miR-150 KO animal kidney. *P < 0.05 vs. other groups; n = 4–6.
MiR-150 knockout mice are resistant to bilateral renal ischemia-reperfusion injury model as well.
To determine whether miR-150 knockout mice are also resistant to bilateral renal clamping model of ischemia-reperfusion injury, the renal pedicle was clamped for 26 min and then the kidney function and inflammatory responses were measured at 24 h. As shown in Fig. 6, WT mice developed severe renal injury as shown by increased serum creatinine over sham-operated animals. However, miR-150 knockout mice showed significantly lower levels of serum creatinine compared with WT mice that were subjected to ischemia-reperfusion injury. Consistent with improved renal function, the biomarker expressions of KIM-1 and NGAL were significantly reduced in miR-150 knockout mice compared with WT mice after ischemia-reperfusion injury. In addition, the expressions of inflammatory cytokines were also reduced in the kidney of miR-150 knockout compared with WT mice.
Fig. 6.

miR-150 deletion renders mice resistant to bilateral renal pedicle clamping model of ischemia-reperfusion (IR) injury. A: serum creatinine at 24 h after ischemia reperfusion. *P < 0.001 vs. sham. #P < 0.05 vs. WT IR. B: RT-PCR analysis of kidney injury molecule-1 (KIM-1) expression in the kidney. *P < 0.01 vs. sham. #P < 0.01 vs. WT IR. C: RT-PCR analysis of neutrophil gelatinase-associated lipocalin (NGAL) expression in the kidney. *P < 0.01 vs. sham. #P < 0.01 vs. WT IR. D: RT-PCR analysis of inflammatory cytokine expression in the kidney. *P < 0.01 vs. other groups; n = 6.
miR-150 represses IGF-1R expression in the kidney.
IGF-1 pathways play a critical protective role in vascular damage and kidney reperfusion injury (8, 13, 14, 22). Recent studies showed that miR-150 targets insulin-like growth factor-1 receptor (IGF-1R) expression in pancreatic cancer cells through c-myb transcription factor thereby inducing apoptosis (11). Therefore, we determined whether miR-150 induces apoptosis through downregulation of IGF-1R expression. As shown in Fig. 7A, in response to reperfusion injury the expression of miR-150 was downregulated but the downregulation was not complete. However, complete deletion of miR-150 in mice causes upregulation of IGF-1R expression in the kidney (Fig. 7, B and C). Interestingly, when we overexpressed miRNA in endothelial cells, IGF-1R and c-myb expression were downregulated, suggesting that IGF-1R is a target for miR-150 that may be regulated indirectly through c-myb downregulation.
Fig. 7.

Effect of miR-150 deletion and overexpression in endothelial cells on insulin-like growth factor-1 receptor (IGF-1R) expression. A: miR-150 expression is downregulated in the kidney after ischemia reperfusion. *P < 0.05 vs. sham; n = 3. B: IGF-1R mRNA expression in WT and miR-150 KO mice after sham and MI surgery. *P < 0.05 vs. other groups; n = 4. C: Western blot analysis of IGF-1R protein expression in WT and miR-150 KO mice after sham and MI surgery. D: densitometric quantification of Western blot in C. #P < 0.05 vs. sham. *P < 0.05 vs. WT; n = 3. E: miR-150 overexpression in endothelial cells causes downregulation of IGF-1R expression. *P < 0.001 vs. control. F: miR-150 overexpression in endothelial cells causes downregulation of c-myb expression. *P < 0.001 vs. control; n = 3.
DISCUSSION
For more than two decades, bilateral renal pedicle clamping model (warm ischemia reperfusion) was used to study human ischemic kidney disease (24, 34). Simplicity and uniform kidney injury response lead to widespread use of this model among renal researchers. Although a large amount of knowledge was generated from this model, whether it is an appropriate reflection of human ischemic kidney disease remains a big question. Except during partial nephrectomy, renal pedicle clamping was not employed in any of the ischemic kidney diseases in humans. Rather, reduced renal blood flow or reduced oxygenation of the kidney is the common underlying cause of ischemic injury in humans. This is mainly due to ventilation and/or in combination with surgery. Difficulties of translating preclinical mouse studies for therapies in humans suggest that the model system used may not be appropriate. To find an optimal model that has a similar course of kidney pathogenesis, we employed the MI model in combination with ventilation similar to cardiopulmonary bypass surgery in humans.
The creatinine rise and time course are very similar to that as seen after cardiopulmonary bypass surgery (20). Moreover, the severity of AKI is also similar to human AKI. Consistent with human studies (12, 20), both NGAL and KIM-1 expression increased significantly in the kidney. These data suggest that the model system we chose is close to human ischemic kidney disease.
Tubular necrosis and epithelial cell apoptosis were shown to be prominent features of ischemia-reperfusion injury of the kidney using the bilateral renal pedicle clamping model (4, 5, 25, 33). This was attributed to the sensitivity of S3 segment of the proximal tubular epithelium unable to adopt anaerobic metabolism. Although in vitro studies did not support this notion (27), it is still widely believed in the field. In contrast to this, the currently employed model does not induce a large amount of apoptosis in S3 segment of the nephron, rather it induced apoptosis in the circulating and interstitial cells. Our double staining with endothelial cell marker CD31 suggests that these apoptotic cells are indeed endothelial cells. In addition, we also see CD31-negative interstitial cells that are positive for TUNEL. The identities of these interstitial cells are not clear.
MiR-150 deletion suppressed both MI-induced AKI as well as renal clamping-induced AKI despite differences in their primary injury mechanism. The best understood role of miR-150 in the hematopoietic system is its regulation of the transcription factor Myb during B cell development. It is becoming clear, however, that miR-150 has additional relevant targets that affect growth, maturation, and the immune response in both B and T lymphocytes and that downregulation of miR-150 in lymphatic tissue results in unregulated proliferation that contributes to tumorigenesis (1, 3, 36). Overexpression of miR-150 induced apoptosis of pancreatic cancer by targeting IGF-1R (11), suggesting the proapoptotic role of this miR-150. This downregulation of IGF-1R is an indirect mechanism. MiR-150 targets the c-myb expression and, therefore, miR-150 overexpression causes suppression of c-myb expression. c-myb has a binding site in IGF-1R promoter as well as Bcl-2 promoter and promotes IGF-1R and Bcl-2 expression and cell survival. Therefore, in the absence of miR-150, IGF-1R is induced (11, 35). Consistent with these data, our studies also demonstrate that miR-150 overexpression downregulated both c-myb and IGF-1R, suggesting that a similar mechanism may exist in endothelial cells as well. IGF-1 and IGF-1R play a critical role in endothelial cell survival, proliferation, and regeneration (8, 14). Consistent with this view, our data show that IGF-1R is upregulated in the kidney of miR-150 knockout mice, whereas overexpression of miR-150 downregulated IGF-1R in endothelial cells. Moreover, administration of recombinant IGF-1 suppressed AKI and accelerated recovery of the kidney from AKI in the animal model (10, 19), suggesting IGF-1 pathways are protective in the kidney. Therefore, it is possible that deletion of miR-150 could abolish ischemia-induced changes in IGF-1R expression and enhance endothelial cell survival. Recent studies also show that miR-150 plays a critical role in kidney fibrosis through suppression of SOCS1 (37). Consistent with this report, our results also show that miR-150 deletion suppressed MI-induced interstitial fibrosis at 8 wk after surgery. It is interesting to note that miR-150 exhibits tissue specificity. Recent studies showed that miR-150 knockout exacerbated MI-induced cardiac dysfunction and fibrosis (29), whereas the same mice show protection in the kidney against MI-induced AKI as well as kidney fibrosis (37). A similar tissue-specific difference in microRNA activity was seen for miR-214 as well (2, 9, 18). To determine whether hypoperfusion due to decreased fractional shortening may contribute to observed sensitivity of the kidney in different groups, we could not find any relation with decrease in fractional shortening and development of AKI (data not shown), suggesting that an additional mechanism may exist for protective effects in miR-150 knockout mice.
In conclusion, our studies demonstrate that MI-induced AKI shows a distinct pattern of interstitial cell apoptosis and serum creatinine rise similar to human cardiac bypass surgery. Deletion of miR-150 suppressed interstitial cell apoptosis and these interstitial cells are mostly endothelial cells and miR-150 represses IGF-1R expression in endothelial cells. Our studies suggest that inhibition of miR-150 may be useful for treatment of MI-induced AKI in humans.
GRANTS
This work was supported by American Heart Association (AHA) Greater Southeast Affiliate Postdoctoral Fellowship 13POST16840074 to P. Ranganathan, National Institutes of Health (NIH) R01 DK083379 to G. Ramesh, AHA National Scientist Development Grant 11SDG6960011 and NIH R01HL124248 to H. Su, and AHA Grant-in-Aid 12GRNT12100048, AHA National Scientist Development Grant 14SDG18970040, and NIH R01 HL124251 to I. Kim.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: P.R., C.J., Y.T., K.-m.P., J.-p.T., and J.L. performed experiments; P.R., Y.T., K.-m.P., J.-p.T., H.S., J.L., I.-m.K., and G.R. analyzed data; P.R., K.-m.P., J.-p.T., H.S., I.-m.K., and G.R. interpreted results of experiments; P.R. prepared figures; P.R. and C.J. drafted manuscript; P.R., C.J., Y.T., K.-m.P., J.-p.T., H.S., J.L., I.-m.K., and G.R. edited and revised manuscript; P.R., C.J., Y.T., K.-m.P., J.-p.T., H.S., J.L., I.-m.K., and G.R. approved final version of manuscript; I.-m.K. and G.R. conception and design of research.
REFERENCES
- 1.Adams B, Guo S, Bai H, Guo Y, Megyola C, Cheng J, Heydari K, Xiao C, Reddy E, Lu J. An in-vivo functional screen uncovers miR-150-mediated regulation of hematopoietic injury response. Cell Rep 2: 1048–1060, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aurora AB, Mahmoud AI, Luo X, Johnson BA, van Rooij E, Matsuzaki S, Humphries KM, Hill JA, Bassel-Duby R, Sadek HA, Olson EN. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca2+ overload and cell death. J Clin Invest 122: 1222–1232, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bezman NA, Chakraborty T, Bender T, Lanier LL. miR-150 regulates the development of NK and iNKT cells. J Exp Med 208: 2717–2731, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonventre JV. Mechanisms of ischemic acute renal failure. Kidney Int 43: 1160–1178, 1993. [DOI] [PubMed] [Google Scholar]
- 5.Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol 14: 2199–2210, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Chandrasekaran K, Karolina DS, Sepramaniam S, Armugam A, Wintour EM, Bertram JF, Jeyaseelan K. Role of microRNAs in kidney homeostasis and disease. Kidney Int 81: 617–627, 2012. [DOI] [PubMed] [Google Scholar]
- 7.Chertow GM, Levy EM, Hammermeister KE, Grover F, Daley J. Independent association between acute renal failure and mortality following cardiac surgery. Am J Med 104: 343–348, 1998. [DOI] [PubMed] [Google Scholar]
- 8.Delafontaine P, Song YH, Li Y. Expression, regulation, and function of IGF-1, IGF-1R, and IGF-1 binding proteins in blood vessels. Arterioscler Thromb Vasc Biol 24: 435–444, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Denby L, Ramdas V, Lu R, Conway BR, Grant JS, Dickinson B, Aurora AB, McClure JD, Kipgen D, Delles C, van Rooij E, Baker AH. MicroRNA-214 antagonism protects against renal fibrosis. J Am Soc Nephrol 25: 65–80, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding H, Kopple JD, Cohen A, Hirschberg R. Recombinant human insulin-like growth factor-I accelerates recovery and reduces catabolism in rats with ischemic acute renal failure. J Clin Invest 91: 2281–2287, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farhana L, Dawson MI, Murshed F, Das JK, Rishi AK, Fontana JA. Upregulation of miR-150* and miR-630 induces apoptosis in pancreatic cancer cells by targeting IGF-1R. PLos One 8: e61015, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney injury molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int 62: 237–244, 2002. [DOI] [PubMed] [Google Scholar]
- 13.Hladunewich MA, Corrigan G, Derby GC, Ramaswamy D, Kambham N, Scandling JD, Myers BD. A randomized, placebo-controlled trial of IGF-1 for delayed graft function: a human model to study postischemic ARF. Kidney Int 64: 593–602, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Imrie H, Viswambharan H, Sukumar P, Abbas A, Cubbon RM, Yuldasheva N, Gage M, Smith J, Galloway S, Skromna A, Rashid ST, Futers TS, Xuan S, Gatenby VK, Grant PJ, Channon KM, Beech DJ, Wheatcroft SB, Kearney MT. Novel role of the IGF-1 receptor in endothelial function and repair: studies in endothelium-targeted IGF-1 receptor transgenic mice. Diabetes 61: 2359–2368, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolk MV, Meyberg D, Deuse T, Tang-Quan KR, Robbins RC, Reichenspurner H, Schrepfer S. LAD-ligation: a murine model of myocardial infarction. J Vis Exp 32: 1438, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loef BG, Epema AH, Smilde TD, Henning RH, Ebels T, Navis G, Stegeman CA. Immediate postoperative renal function deterioration in cardiac surgical patients predicts in-hospital mortality and long-term survival. J Am Soc Nephrol 16: 195–200, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Lorenzen JM, Haller H, Thum T. MicroRNAs as mediators and therapeutic targets in chronic kidney disease. Nat Rev Nephrol 7: 286–294, 2011. [DOI] [PubMed] [Google Scholar]
- 18.Lv G, Shao S, Dong H, Bian X, Yang X, Dong S. MicroRNA-214 protects cardiac myocytes against H2O2-induced injury. J Cell Biochem 115: 93–101, 2014. [DOI] [PubMed] [Google Scholar]
- 19.Miller SB, Martin DR, Kissane J, Hammerman MR. Insulin-like growth factor I accelerates recovery from ischemic acute tubular necrosis in the rat. Proc Natl Acad Sci USA 89: 11876–11880, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mishra J, Dent C, Tarabishi R, Mitsnefes MM, Ma Q, Kelly C, Ruff SM, Zahedi K, Shao M, Bean J, Mori K, Barasch J, Devarajan P. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet 365: 1231–1238, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Patel V, Noureddine L. MicroRNAs and fibrosis. Curr Opin Nephrol Hypertens 21: 410–416, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rae FK, Suhaimi N, Li J, Nastasi T, Slonimsky E, Rosenthal N, Little MH. Proximal tubule overexpression of a locally acting IGF isoform, Igf-1Ea, increases inflammation after ischemic injury. Growth Horm IGF Res 22: 6–16, 2012. [DOI] [PubMed] [Google Scholar]
- 23.Ramesh G, Krawczeski CD, Woo JG, Wang Y, Devarajan P. Urinary netrin-1 is an early predictive biomarker of acute kidney injury after cardiac surgery. Clin J Am Soc Nephrol 5: 395–401, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramesh G, Ranganathan P. Mouse models and methods for studying human disease, acute kidney injury (AKI). In: Mouse Genetics, edited by Singh SR and Coppola V. New York: Springer, 2014, p. 421–436. [DOI] [PubMed] [Google Scholar]
- 25.Ranganathan P, Jayakumar C, Navankasattusas S, Li DY, Kim Im, Ramesh G. UNC5B receptor deletion exacerbates tissue injury in response to AKI. J Am Soc Nephrol 25: 239–249, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ricci Z, Cruz DN, Ronco C. Classification and staging of acute kidney injury: beyond the RIFLE and AKIN criteria. Nat Rev Nephrol 7: 201–208, 2011. [DOI] [PubMed] [Google Scholar]
- 27.Shanley PF, Brezis M, Spokes K, Silva P, Epstein FH, Rosen S. Differential responsiveness of proximal tubule segments to metabolic inhibitors in the isolated perfused rat kidney. Am J Kidney Dis 7: 76–83, 1986. [DOI] [PubMed] [Google Scholar]
- 28.Tadagavadi RK, Wang W, Ramesh G. Netrin-1 regulates Th1/Th2/Th17 cytokine production and inflammation through UNC5B receptor and protects kidney against ischemia-reperfusion injury. J Immunol 185: 3750–3758, 2010. [DOI] [PubMed] [Google Scholar]
- 29.Tang Y, Wang Y, Park Km, Hu Q, Teoh Jp, Broskova Z, Ranganathan P, Jayakumar C, Li J, Su H, Tang Y, Ramesh G, Kim Im. MicroRNA-150 protects the mouse heart from ischaemic injury by regulating cell death. Cardiovasc Res 106: 387–397, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tuttle KR, Worrall NK, Dahlstrom LR, Nandagopal R, Kausz AT, Davis CL. Predictors of ARF after cardiac surgical procedures. Am J Kidney Dis 41: 76–83, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Utsumi M, Umeda Y, Sadamori H, Nagasaka T, Takaki A, Matsuda H, Shinoura S, Yoshida R, Nobuoka D, Satoh D, Fuji T, Yagi T, Fujiwara T. Risk factors for acute renal injury in living donor liver transplantation: evaluation of the RIFLE criteria. Transpl Int 26: 842–852, 2013. [DOI] [PubMed] [Google Scholar]
- 32.Wang W, Brian RW, Ramesh G. Netrin-1 and kidney injury. I. Netrin-1 protects against ischemia-reperfusion injury of the kidney. Am J Physiol Renal Physiol 294: F739–F747, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang W, Reeves WB, Pays L, Mehlen P, Ramesh G. Netrin-1 overexpression protects kidney from ischemia reperfusion injury by suppressing apoptosis. Am J Pathol 175: 1010–1018, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei Q, Dong Z. Mouse model of ischemic acute kidney injury: technical notes and tricks. Am J Physiol Renal Physiol 303: F1487–F1494, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, Rajewsky N, Bender TP, Rajewsky K. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell 131: 146–159, 2007. [DOI] [PubMed] [Google Scholar]
- 36.Zhou B, Wang S, Mayr C, Bartel DP, Lodish HF. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc Natl Acad Sci USA 104: 7080–7085, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou H, Hasni SA, Perez P, Tandon M, Jang SI, Zheng C, Kopp JB, Austin H, Balow JE, Alevizos I, Illei GG. miR-150 promotes renal fibrosis in lupus nephritis by downregulating SOCS1. J Am Soc Nephrol 24: 1073–1087, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]