

Abstract
Calpain contributes to infection-induced diaphragm dysfunction but the upstream mechanism(s) responsible for calpain activation are poorly understood. It is known, however, that cytokines activate neutral sphingomyelinase (nSMase) and nSMase has downstream effects with the potential to increase calpain activity. We tested the hypothesis that infection-induced skeletal muscle calpain activation is a consequence of nSMase activation. We administered cytomix (20 ng/ml TNF-α, 50 U/ml IL-1β, 100 U/ml IFN-γ, 10 μg/ml LPS) to C2C12 muscle cells to simulate the effects of infection in vitro and studied mice undergoing cecal ligation puncture (CLP) as an in vivo model of infection. In cell studies, we assessed sphingomyelinase activity, subcellular calcium levels, and calpain activity and determined the effects of inhibiting sphingomyelinase using chemical (GW4869) and genetic (siRNA to nSMase2 and nSMase3) techniques. We assessed diaphragm force and calpain activity and utilized GW4869 to inhibit sphingomyelinase in mice. Cytomix increased cytosolic and mitochondrial calcium levels in C2C12 cells (P < 0.001); addition of GW4869 blocked these increases (P < 0.001). Cytomix also activated calpain, increasing calpain activity (P < 0.02), and the calpain-mediated cleavage of procaspase 12 (P < 0.001). Procaspase 12 cleavage was attenuated by either GW4869 (P < 0.001), BAPTA-AM (P < 0.001), or siRNA to nSMase2 (P < 0.001) but was unaffected by siRNA to nSMase3. GW4869 prevented CLP-induced diaphragm calpain activation and diaphragm weakness in mice. These data suggest that nSMase2 activation is required for the development of infection-induced diaphragm calpain activation and muscle weakness. As a consequence, therapies that inhibit nSMase2 in patients may prevent infection-induced skeletal muscle dysfunction.
Keywords: diaphragm, sepsis, calpain, cytokines, sphingomyelinase
recent studies indicate that critically ill patients have severe reductions in diaphragm function (7, 11, 14, 30, 37). Moreover, the average diaphragm pressure-generating capacity in this population is only 22–25% of normal values (30). These studies also show that this severe diaphragm weakness results in difficulty weaning critically ill patients from mechanical ventilation. In addition, the weakest intensive care unit (ICU) patients have a markedly increased incidence of death, which may be due to withdrawal of care when liberation from mechanical ventilation proves to be difficult (7, 30).
Although several processes contribute to weakness in these patients (inactivity, use of corticosteroids, neuromuscular blocking agents), several reports indicate that infection is a major cause of skeletal muscle weakness in critically ill patients (7, 30). Animal studies have shown that infection activates calpain in respiratory skeletal muscle and administration of calpain inhibitors to infected animals attenuates muscle weakness and muscle proteolysis (1, 28, 34). However, the precise mechanisms by which infections induce calpain activation in skeletal muscle are not known. Since infections are associated with increases in systemic cytokine levels and cytokines, in turn, are known to activate skeletal muscle sphingomyelinase (5), we considered the possibility that infection-induced calpain activation is a downstream consequence of sphingomyelinase activation.
The present study was designed to study the mechanism by which cytokines activate skeletal muscle calpain and to test the specific hypothesis that cytokine-induced sphingomyelinase activation is a key mediator responsible for infection-induced calpain activation. These studies were carried out using a skeletal muscle cell line (C2C12 cells) and mice. To simulate infection, C2C12 cells were exposed to a mixture of cytokines (cytomix, a combination of TNF-α, IL-1β, IFN-γ, and LPS), and sepsis was induced in intact animals by cecal ligation puncture (CLP)-induced peritonitis. The role of sphingomyelinase in calpain activation was assessed by inhibiting neutral sphingomyelinase with chemical inhibitors (GW4869) and genetic agents (nSMase2 and nSMase3 siRNA) in cells and use of GW4869 in animals.
METHODS
Experimental protocols: cell studies.
C2C12 cells were obtained from ATCC, Manassas, VA. Myoblasts were grown to 70% confluency in DMEM (Dulbecco's modification of Eagle's medium) with 10% FBS and antibiotics. Cells were then exposed to DMEM with 2% horse serum for 7 days to induce differentiation. We first determined whether a cytokine mixture (cytomix: 20 ng/ml TNF-α, 50 U/ml IL-1β, 100 U/ml IFN-γ, and 10 μg/ml LPS) increases neutral sphingomyelinase (nSMase) activity in C2C12 cells. Since activated nSMase generates ceramide and ceramide induces calcium movement across cell membranes (36), we also determined whether an inhibitor of nSMase2 (GW4869) blocked cytokine-induced shifts in C2C12 cell calcium concentrations. To determine the effects of cytomix on cell sphingomyelinase levels, we added cytomix to well-differentiated C2C12 cells and harvested cells 24 h later. Acid and neutral sphingomyelinase activities were then assessed on cell homogenates by using the Amplex Red Assay Kit (Molecular Probes, Eugene, OR) according to the manufacturer's protocol (n = 5 plates/condition). For this assay, duplicates were prepared in which GW4869 (10 μM) was added to cell homogenates at the time of assay.
To assess the effects of cytomix and GW4869 on cellular calcium concentrations, we used two stably transfected C2C12 cell lines that contained either a cytosolic-targeted ratiometric pericam protein (first line) or a mitochondrially targeted ratiometric pericam protein (second line) (10, 19). Differentiated pericam-transfected C2C12 cells containing either the cytosolic-targeted pericam or the mitochondrially targeted pericam were then treated with culture medium containing saline, cytomix, cytomix + GW4869 (10 μM), or GW4869 alone (n = 5 plates/condition). After 24 h of exposure, we assessed calcium levels using laser fluorescent imaging (excitation 408 and 488, emission 530). The ratio of the fluorescent intensities for 408 excitation to 488 excitation images was taken as an index of the cellular calcium level. Imaging was performed with a tunable confocal laser fluorescent microscope available in the imaging center at the University of Kentucky. This microscope is equipped to permit imaging under conditions similar to those in an incubator.
We also determined the effect of cytomix exposure on C2C12 cell calpain activation. Two indexes of calpain activation were used, including direct measurement of cell calpain activity using a calpain assay (see Calpain activity assay) and assessment of calpain-specific caspase 12 cleavage (i.e., formation of the 38-kDa caspase 12 cleavage product) (15, 18) by Western blotting. For these studies, differentiated C2C12 were exposed to cytomix, with and without GW4869 (10 μM), and harvested 24 h later. We also assessed the effect of lowering cell calcium concentrations by assessing indexes of calpain activation in C2C12 cells treated with a cytosolic calcium chelator (BAPTA-AM, 40 μM). For these experiments, we studied four groups of cells, including control, cytomix, cytomix + BAPTA-AM, and BAPTA-AM groups (n = 4 plates/group).
To examine the effect of reducing protein levels of sphingomyelinase on cytokine-induced calpain activation in C2C12 cells, we used siRNA to either nSMase2 (Santa Cruz Biotechnology, Santa Cruz, CA) or nSMase3 (gift from Dr. Michael B. Reid), two muscle isoforms of nSMase. For siRNA transfections, C2C12 cells were differentiated for 2 days and then treated with siRNA to either nSMase2 or nSMase3 mixed with transfection medium and NeoFX transfection reagent (Life Technologies, Grand Island, NY). For control conditions, cells were treated with scrambled siRNA. Cells were then washed with transfection medium, and a mixture of transfection medium and siRNA was overlaid onto washed cells. After a 5-h incubation period, differentiation medium (DMEM + 2% horse serum) was added to the plates. Cells were incubated for an additional 24 h, washed, and then exposed to differentiation medium alone (n = 4 plates/condition). Following completion of differentiation, cells were exposed to cytomix, harvested, and assayed for calpain activity by measuring calpain-specific caspase 12 cleavage.
To determine whether increases in ceramide levels induce changes that mirror the effects of cytokines (i.e., activating calpain), we also examined the effects of addition of the cell permeant ceramide analogs C2-ceramide (15 μM) or C6-ceramide (20 μM) to C2C12 cells (n = 5 plates/group). For these studies, cells were exposed to either C2 ceramide or C6 ceramide, harvested 24 h later, and calpain activation was assessed on homogenates.
Finally, we examined the effect of cytomix exposure in C2C12 cells, with and without concomitant addition of GW4869, on protein levels of two important contractile proteins, myosin light chain (MLC) and myosin heavy chain (MHC). Levels of these two proteins were determined by Western blotting, as described below.
Experimental protocols: animal studies.
To provide an assessment of the effect of inhibition of sphingomyelinase activity on diaphragm force generation in an intact animal model of infection, we studied mice undergoing cecal ligation puncture (CLP; details of this procedure are described in the next section). All the animal studies were approved by the University of Kentucky Institutional Animal Care and Use Committee. We studied four groups of animals: sham-operated, saline-treated control animals; CLP-operated, saline-treated animals; sham-operated animals given GW4869 (10 μM or 5.8 mg/kg); and CLP-operated animals given GW4869 (10 μM) (n = 4–5/group). GW4869 was administered intraperitoneally immediately following surgery.
CLP-induced sepsis.
We performed either sham abdominal surgery or CLP as previously published (33). Briefly, mice were anesthetized with halothane (2–4%) and, after a steady plane of anesthesia was achieved, the abdomen was prepped with betadine and alcohol, followed by a midline incision (∼1.5 cm in length). The cecum was identified and a portion ligated (∼0.5 cm), and the ligated cecum was punctured through-and-through with a 21-gauge sterile needle. The abdominal musculature was then approximated and sutured, and the skin was closed with surgical staples. Sham surgery consisted of opening and closing the abdomen without cecal ligation or puncture. All mice were resuscitated with subcutaneous saline (60 ml/kg) following surgery, and all were treated with buprenorphine (0.05 mg/kg) subcutaneously immediately following surgery and every 12 h for analgesia. Laboratory personnel monitored animals for 2 h postoperatively, and then mice were returned to the institutional animal facility where they were allowed to eat and drink ad libitum until the time of euthanasia.
Diaphragm force-frequency curves and muscle mass.
Diaphragm force generation was assessed as previously reported (27, 31–33). In brief, diaphragms were excised, and strips were carefully dissected from the left midcostal portion and thereafter mounted vertically in water-jacketed glass organ baths containing Krebs-Henseleit solution (25°C, 50 mg/l curare, pH 7.40, 135 mM NaCl, 5 mM KCl, 11.1 mM dextrose, 2.5 mM CaCl2, 1 mM MgSO4, 14.9 mM NaHCO3, 1 mM NaHPO4, 50 units/l insulin, 95% O2-5% CO2). One end of the strip was tied to the base of the organ bath and the other to a force transducer (Scientific Instruments, Heidelberg, Germany). A biphasic constant current amplifier driven by a Grass S48 stimulator (Grass, West Warwick, RI) and platinum mesh field electrodes were employed to deliver supramaximal currents. After an equilibration period of 15 min, diaphragm muscle length was adjusted to Lo, followed by sequential stimulation of muscle strips with trains of 1, 10, 20, 50, 100, and 150 Hz stimuli (train duration 800 ms, 30 s between adjacent trains). Force was recorded with a Kipp-Zonen chart recorder (Bohemia, NY). Muscle cross-sectional area was calculated by using the weight of the muscle strip divided by muscle density (1.06) and muscle length (6). Muscle-specific force was then determined by calculating the raw force divided by the cross-sectional area. The total weight of the costal diaphragm was recorded by adding the weight of the strips used for the force-frequency curve to the weight of the remaining muscle. Diaphragm tissues not used for force-frequency determinations were frozen, stored at minus 80°C, and subsequently used to determine protein concentration by using the Bradford assay kit (Bio-Rad Laboratories, Hercules, CA). We also assessed neutral sphingomyelinase activities on diaphragm samples using the Amplex Red Assay Kit (Molecular Probes, Eugene, OR) following the manufacturer's protocol (n = 4/condition). Finally, diaphragm calpain activity (as described below) was assessed on diaphragm samples.
Calpain activity assay.
We used a commercially available kit to assay calpain activity (Abcam, Cambridge, MA) as previously reported (33). Samples were prepared in the proprietary extraction buffer according to the manufacturer's specifications. For these experiments, duplicate samples of 100 μg of protein with or without 3 μl of calpain inhibitor 3 were diluted to a final volume of 85 μl with extraction buffer. Extraction buffer alone was used as a blank. Mixtures were placed in a 96-well plate, followed by addition of 10 μl of reaction buffer and 5 μl of calpain substrate to each well. After an initial reading, the plates were incubated in the dark at 37°C for 1 h and a final reading was made (excitation 400 nm, emission 505 nm). The difference in increases in fluorescent activity over time for a given sample in the presence and absence of the specific calpain inhibitor was used as an index of calpain enzyme activity.
Protein determinations by Western blotting.
C2C12 cell and diaphragm levels of proteins of interest were examined using Western blot techniques. For these determinations, samples were diluted with an equal volume of loading buffer (126 mM Tris·HCl, 20% glycerol, 4% SDS, 1.0% 2-mercaptoethanol, 0.005% bromophenol blue, pH 6.8) and equal amounts of proteins were loaded onto Tris-glycine polyacrylamide gels, and separated by electrophoresis (Novex Minicell II, Carlsbad, CA). Proteins were transferred to polyvinylidene fluoride membranes, incubated overnight at 4°C with primary antibodies to targeted proteins, followed by incubation with horseradish peroxidase-conjugated secondary antibodies, and binding was detected by enhanced chemiluminescence (Western Lightning, Perkin Elmer, Boston, MA). A Microtek scanner (Carson, CA) and UN-SCAN-IT software (Silk Scientific, Orem, UT) were used for densitometric analysis of proteins. To verify equal loading, membranes were reprobed with antibodies to GAPDH. We chose GAPDH because this protein is not altered in skeletal muscle by sepsis. The following primary antibodies were used: anti-caspase 12 (cs2202, Cell Signaling Technology, Beverly, MA), anti-GAPDH (ab9484, Abcam, Cambridge, MA), anti-nSMase2 (sc67693, Santa Cruz Biotechnology), anti-nSMase3 (ECM Biosciences, Versailles, KY, gift from Dr. Michael B. Reid), anti-myosin heavy chain (sc20641, Santa Cruz Biotechnology), and anti-myosin light chain (M4401, Sigma-Aldrich, St. Louis, MO).
Statistical analysis.
ANOVA was used to compare variables (e.g., force) across groups of cells and animals treated with different agents, with post hoc testing (Tukey's) to determine differences between groups. A P value of less than 0.05 was taken as indicating statistical significance for experiments.
RESULTS
Sphingomyelinase activity in C2C12 cells.
We found that addition of cytomix to C2C12 cells elicited significant increases in both neutral and acid sphingomyelinase activity levels, as indicated in Fig. 1. Neutral sphingomyelinase increased ∼57% (P < 0.001), while acid sphingomyelinase activity increased ∼29% (P < 0.001). Addition of GW4869, a neutral sphingomyelinase 2 inhibitor, to the assay homogenate blocked the cytokine-induced increase in neutral sphingomyelinase activity but had no effect on acid sphingomyelinase activity levels.
Fig. 1.
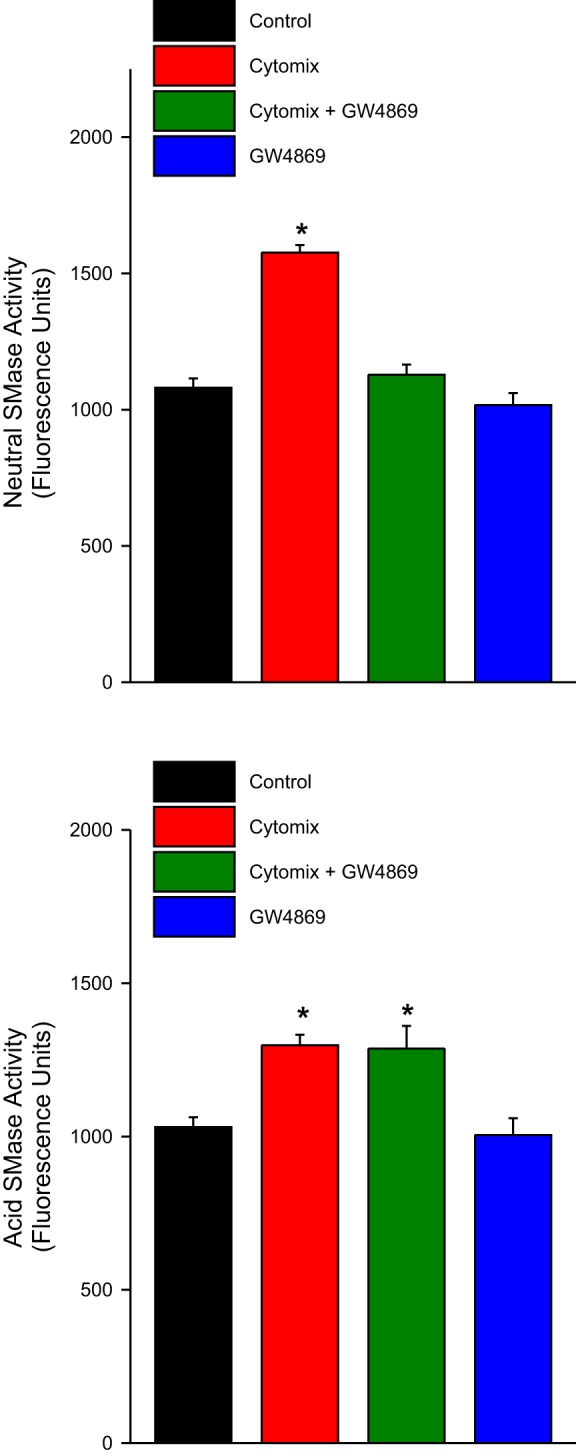
Mean neutral sphingomyelinase activity (top) and acid sphingomyelinase activity levels (bottom) for saline-treated control C2C12 cells (black bars), cytomix-treated cells (red bars), cytomix + GW4869-treated cells (green bars), and cells treated with GW4869 alone (blue bars). Cytomix elicited a significant increase in both neutral and acid sphingomyelinase activity levels (P < 0.001 for both assays; asterisks indicate statistical significance in this and all other figures). GW4869 blocked the cytokine-induced increase in neutral sphingomyelinase activity but had no effect on acid sphingomyelinase activity levels.
Cell calcium imaging studies.
We stably transfected C2C12 cells with either a cytosolic pericam protein or a mitochondrially targeted pericam protein (10, 19) and used these transfected cells to assess intracellular calcium responses to cytomix administration. Pericam is a ratiometric calcium-sensing protein, with increasing calcium concentrations eliciting an increase in the ratio of light intensity at an excitation wavelength of 488 nm to the light intensity at an excitation wavelength of 408 nm. As shown in Fig. 2, the 488-nm light intensity (green) relative to the 408-nm light intensity (blue) increased in response to cytomix administration for both cytosolic pericam-transfected C2C12 cells and mitochondrial pericam-transfected C2C12 cells. These findings indicate that cytomix increases both cytosolic and mitochondrial calcium concentrations (P < 0.001 and P < 0.002, respectively). We also found that administration of GW4869, an nSMase2 inhibitor, blocked these cytomix-induced responses, with 488 nm/408 nm intensity ratios in C2C12 cell lines treated with both cytomix and GW4869 equal to the 488 nm/408 nm intensity ratios of control C2C12 cells [not significant (NS) for comparison of the 488 nm/408 nm ratios of the cytomix + GW4869 groups to the control group].
Fig. 2.

Cell calcium imaging studies. We stably transfected C2C12 cells with either a cytosolic pericam protein (left) or a mitochondrially targeted pericam protein (right); increases in calcium levels in these compartments cause an increase in the ratio of light emission intensity at 488 nm to the light emission intensity at 408 nm for pericam. Data from representative cell studies are shown at top and group mean data for the light emission 488 nm/408 nm intensity ratios are presented at bottom. The color coding of bars for group mean data is the same as in Fig. 1. The 488-nm emission is pseudo-colored green and the 408-nm emission is pseudo-colored blue. Cytomix increased both cytosolic and mitochondrial calcium concentrations (P < 0.001 and P < 0.002, respectively). Administration of GW4869 blocked these cytomix-induced responses.
Indexes of cell calpain activity.
We used two different indexes to assess calpain activity, including a direct determination of the calpain activity of muscle homogenates and measurement of a caspase 12 fragment that previously was shown to be generated in response to calpain-mediated cleavage of intact caspase 12 (17, 20). These previous studies found that NH2-terminal prodomain processing of procaspase-12 by calpain generates a 38-kDa fragment (17). In addition, this previous work found that treatment with calpain inhibitors (calpain inhibitor I and II) inhibits caspase-12 cleavage in response to pathophysiological stress but pan-caspase inhibitors and proteasome inhibitors do not block caspase-12 cleavage (20).
As shown in Fig. 3, we found that cytomix administration to C2C12 cells elicited a dramatic increase in the formation of this calpain-dependent caspase 12 degradation product (i.e., CSCDP) (P < 0.001). We also found that coadministration of calpain inhibitor 3 blocked generation of this fragment. As shown in Fig. 4, cytomix-induced increases in CSCDP were markedly attenuated by administration of either an nSMase inhibitor (GW4869) or an intracellular calcium chelator (BAPTA-AM). These findings suggest that both nSMase activation and increases in cellular calcium levels are required for cytomix-induced increases in calpain-mediated caspase 12 cleavage. In keeping with this observation, we also found that directly determined calpain activity increased in C2C12 cells in response to cytomix administration (Fig. 3, bottom) and administration of either GW4869 or BAPTA-AM also prevented the cytomix-induced increase in calpain activity assessed by this assay.
Fig. 3.
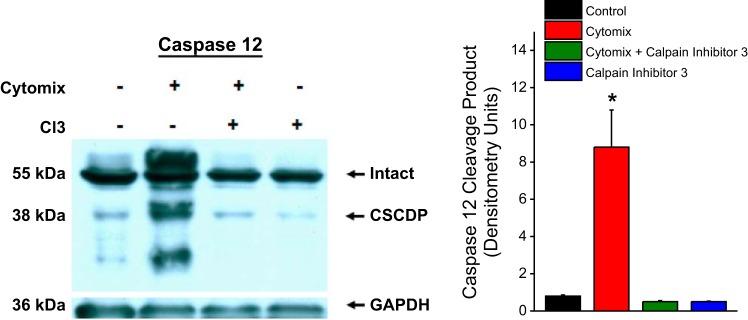
Western blots for caspase 12, with a representative caspase 12 blot at left and group mean data for the densitometry of the calpain-specific caspase 12 degradation product (CSCDP) is shown at right. Color coding of bars is the same as used in Fig. 1. Western blotting for GAPDH was used as a loading control. Cytomix administration to C2C12 cells elicited an increase in the formation of CSCDP (P < 0.001) and administration of calpain inhibitor 3 to cells blocked this cytomix-mediated increase in CSCDP.
Fig. 4.

Top: data for assessment of cell caspase 12 protein cleavage in response to cytomix and GW4869, with a representative blot shown on the left and group mean data for densitometry of the CSCDP shown on the right. Middle: data for assessment of caspase 12 cleavage in response to cytomix and BAPTA-AM, with a representative blot on the left and group mean data for CSCDP levels on the right. Bottom: data for cell calpain activity assay determinations for the various experimental groups. Bars indicate mean data for saline-treated control cells (black), cytomix-treated cells (red), GW4869 plus cytomix-treated cells (green), GW4869-treated cells (blue), BAPTA-AM plus cytomix-treated cells (yellow), and cells treated BAPTA-AM alone (gray). Cytomix induced an increase in CSCDP (P < 0.001) and this increase was blocked by administration of either GW4869 or BAPTA-AM. Calpain assay activity increased in response to cytomix (P < 0.001) and administration of either GW4869 or BAPTA-AM prevented this cytomix-induced increase.
Because chemical inhibitors, such as BAPTA-AM or GW4869, can sometimes have nonspecific actions on other cellular pathways that may artifactually influence results, we also used a genetically specific technique to reduce the levels of the two nSMase isoforms, namely nSMase2 and nSMase3 in C2C12 cells. As shown in Fig. 5, silencing nSMase2 with siRNA successfully reduced nSMase2 protein levels and also diminished generation of the CSCDP in response to cytomix. In contrast, knockdown of nSMase3 protein with siRNA to nSMase3 did not alter generation of CSCDP in C2C12 cells in response to cytomix. These data indicate that nSMase2 is necessary for cytomix-mediated calpain activation in C2C12 cells and that nSMase3 does not appear to mediate this effect of cytomix.
Fig. 5.

Top: data for assessment of cell caspase 12 protein cleavage in response to cytomix and siRNA to nSMase2, with a representative blot shown on the left and group mean data for densitometry of the CSCDP shown on the right. Bottom: data for assessment of cell caspase 12 protein cleavage in response to cytomix and siRNA to nSMase3. siRNA to nSMase2 successfully reduced nSMase2 protein levels and also diminished generation of CSCDP in response to cytomix (P < 0.001). siRNA to nSMase3 reduced nSMase3 protein levels but did not prevent cytomix-induced increases in CSCDP.
If generation of ceramide by nSMase2 is a mediator of cytomix-induced calpain activation, then administration of cell permeant analogs of ceramide (e.g., C2 ceramide and C6 ceramide) should replicate the effect of cytomix to induce calpain activation. As shown in Fig. 6, we found that administration of either of these analogs elicited in increases in both of our indexes of cell calpain activity, with both analogs increasing formation of CSCDP and directly measured calpain activity in C2C12 cells.
Fig. 6.

Effect of administration of C2 ceramide and C6 ceramide on CSCDP formation is shown at top, with a representative blot shown on the left and group mean densitometry data for CSCDP presented on the right. Bars represent mean data for control (black), C2 ceramide (yellow), and C6 ceramide (blue) groups. Bottom compares the effect of cytomix, C2 ceramide, and C6 ceramide on cell calpain activity assay levels. Bars represent mean data for control (black), cytomix (red), C2 ceramide (yellow), and C6 ceramide (blue) groups. Both C2 ceramide and C6 ceramide increased CSCDP formation relative to the control group (P < 0.001) and also directly measured calpain activity (P < 0.001).
MLC and MHC levels.
Activated calpain is capable of cleaving a number of muscle cytoskeletal and contractile proteins. Two contractile proteins that are targets of calpain-mediated cleavage are MLC and MHC (23, 26). As shown in Fig. 7, cytomix administration elicited reductions in both C2C12 MLC and MHC protein levels. Administration of an nSMase2 inhibitor, GW4869, prevented cytomix-induced reductions in both MLC and MHC. These findings are indicative that depletion of both MLC and MHC proteins are dependent on cytomix-induced nSMase2 activation.
Fig. 7.

Top: effects of cytomix and GW4869 on C2C12 myosin light chain (MLC) levels, with a representative blot shown on the left and group mean densitometry data on the right. Bottom: the effects of cytomix and GW4869 on C2C12 myosin heavy chain (MHC) levels with a representative blot shown on the left and group mean densitometry data on the right. Bars represent data for saline-treated control C2C12 cells (black), cytomix-treated cells (red), GW4869 plus cytomix-treated cells (green), and cells treated with GW4869 alone (blue). Cytomix-induced reductions in both MLC and MHC protein levels (P < 0.001 for both proteins) and concomitant administration of GW4869 blocked cytomix-induced reductions in the levels of both proteins.
Diaphragm-specific force generation, nSMase activity, and calpain activation.
In previous studies we found that CLP-induced sepsis elicits activation of calpain in the diaphragm (28, 34). We therefore determined the effect of systemic administration of GW4869 to mice with CLP to determine the effects of this nSMase inhibitor in an animal model of sepsis. We found that CLP induced significant diaphragm weakness, as assessed by examining the contractility of diaphragm strips excised from saline-treated sham-operated control and saline-treated CLP-induced septic animals. Specifically, diaphragm strips from saline-treated control animals generated a maximum specific force of 21.5 ± 4.5 N/cm2 in response to 150 Hz supramaximal electrical stimulation, whereas diaphragm strips from saline-treated CLP animals produced a maximum specific force of only 6.9 ± 3.3 N/cm2 (P < 0.001). We also found that administration of GW4869 to animals with CLP resulted in preservation of diaphragm strength, with the maximum specific force of strips from this group of animals, 20.3 ± 3.4 N/cm2, significantly greater than the force generated by saline-treated CLP animals (P < 0.001). Sham-operated GW4869-treated animals had a level of force generation similar to that of saline-treated sham-operated controls (NS). Forces generated in response to other frequencies of electrical stimulation had this same pattern, with forces generated in response to 1- to 150-Hz stimulation reduced in the saline-treated CLP group compared with the saline-treated sham-operated group, and with GW4869 treatment preventing the sepsis-induced development of diaphragm weakness (see Fig. 8, top).
Fig. 8.
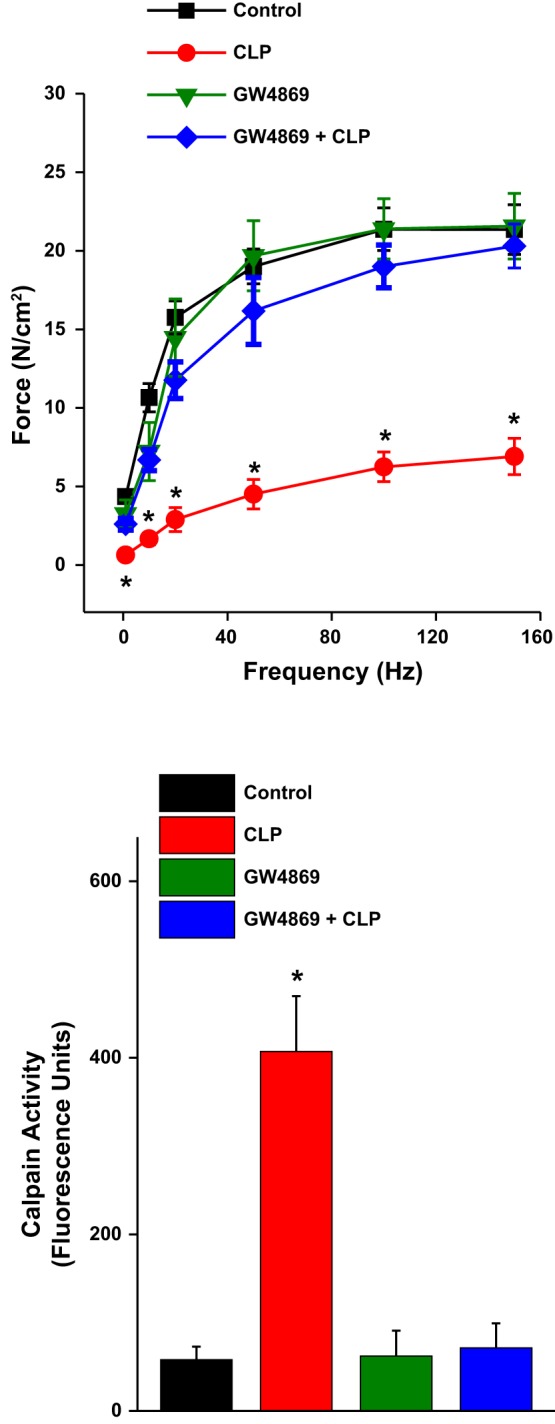
Top: mean specific force-frequency curves for diaphragm strips taken from the 4 experimental groups: saline-treated sham-operated controls (black), saline-treated cecal ligation puncture (CLP) animals (red), GW4869-treated sham-operated mice (green), and GW4869-treated CLP animals (blue). CLP induced significant diaphragm weakness, reducing force generation in response to all stimulation frequencies tested. GW4869 treatment prevented CLP-induced weakness, preserving force at all stimulation frequencies. Bottom: calpain activity levels for the 4 experimental groups. CLP induced an increase in diaphragm calpain activity (P < 0.001) and concomitant GW4869 administration prevented this CLP-induced increase.
We also found that nSMase activity (in fluorescent units per 1 h) was greatly increased in CLP, vehicle-treated animals (3,895 ± 139) compared with sham-operated, vehicle-treated controls (2,412 ± 142, P < 0.001 for comparison of the two groups). GW4869 administration attenuated the CLP-induced increase in diaphragm nSMase activity, with nSMase activity levels of 2,510 ± 199 for the GW4869+CLP group and 1,874 ± 46 for the GW4869-treated, sham-operated group (P < 0.001 for comparison of each to the vehicle-treated, CLP group). Calpain activity increased in diaphragms of CLP animals compared with controls (P < 0.001), and GW4869 prevented this CLP-induced increase in diaphragm calpain activation (see Fig. 8, bottom).
DISCUSSION
This work found that a mixture of cytokines increased cytosolic and mitochondrial calcium levels in skeletal muscle-derived C2C12 cells and that addition of an nSMase inhibitor, GW4869, blocked these increases. Cytokines also activated C2C12 cell calpain, increasing calpain enzyme activity and the calpain-mediated cleavage of procaspase 12. Cytokine-induced procaspase 12 cleavage in C2C12 cells was attenuated by either GW4869, a cell-permeant calcium chelator (BAPTA-AM), or siRNA to nSMase2 but was unaffected by siRNA to nSMase3. We also found that administration of an nMSase inhibitor prevented sepsis-induced diaphragm calpain activation and diaphragm weakness in mice.
Weakness in critically ill patients.
Critically ill mechanically ventilated patients are profoundly weak, with this patient population having an average diaphragm force-generating capacity that is only 20–25% of normal (7, 11, 14, 30, 37). Recent studies indicate, moreover, that the subgroup of critically ill patients with the weakest diaphragms has the worst outcomes (7, 30). In particular, diaphragm weakness results in difficulty weaning from mechanical ventilation, with patients having a diaphragm Pdi twitch of less than 10 cmH2O (Pdi twitch is the transdiaphragmatic pressure generated in response to bilateral magnetic stimulation of the phrenic nerves) requiring an average of 16 days to wean from mechanical ventilation, whereas patients with Pdi twitch greater than 10 cmH2O require only 4.5 days to wean from mechanical ventilation (30). The weakest patients also have an increased incidence of death, even after correction for severity of illness (7, 30).
Several factors are thought to contribute to weakness in ICU patients, including inactivity, use of neuromuscular blocking agents, and corticosteroids. Another important factor is the presence of infection, with two recent studies indicating that infected medical ICU patients have roughly half the diaphragm pressure-generating capacity of noninfected patients (7, 30). Several processes are thought to contribute to infection-induced skeletal muscle dysfunction, including reductions in protein synthesis due to inhibition of signaling pathways regulating enzymes required for protein synthesis (13, 15), enhanced free radical generation with resultant free radical-induced modification of proteins required for contractile function (3), and activation of multiple proteolytic enzyme systems (caspase, the proteasome, and the calpain proteolytic enzyme system) (8, 24, 29). Regarding this last proteolytic pathway (the calpain proteolytic system), recent work indicates that this pathway may be a major contributor to development of infection-induced diaphragm weakness (28, 33). Multiple indexes of calpain activation are present in the diaphragm for animal models of infection, including talin cleavage, spectrin cleavage, and autocatalytic cleavage of calpain 1 and calpain 2 (1, 28, 33, 34). In addition, administration of calpain inhibitors reduces the level of diaphragm weakness present in animal models of infection (endotoxin and cecal ligation perforation models of infection) (28, 34). Diaphragm weakness due to cecal ligation puncture is also markedly attenuated in animals with muscle-specific overexpression of calpastatin, the endogenous calpain inhibitor (33).
Theoretical mechanisms of calpain activation.
Although the above studies make an argument that calpain activation contributes to the development of diaphragm weakness during infections, the precise mechanism by which infections induce calpain activation in skeletal muscle are not known. Infections are known to elicit increases in circulating levels of several cytokines (18) and cytokines have been shown to directly activate calpain in isolated muscle cell lines (22). Cytokines are also known to activate sphingomyelinase in multiple cell lines (25), so it seems reasonable to postulate that cytokine-induced sphingomyelinase may be the upstream signaling process responsible for mediating cytokine-induced calpain activation. In keeping with this possibility, the present study found that incubation of muscle cells with a cytokine mixture induced large increases in neutral sphingomyelinase activity and a smaller increase in acid sphingomyelinase activity. Increases in nSMase activity were blocked by addition of GW4869, a known inhibitor of the nSMase 2 isoform, to cell homogenates.
Sphingomyelinase activation leads to release of ceramide (21), and ceramide and its metabolites are known to induce alterations in cell calcium compartmentalization (16).
Specifically, ceramide metabolites, including ceramide-1-phosphate, are known to activate channels that modulate calcium flux, including several large transient receptor protein channels, increasing calcium levels in the cytosol and other intracellular compartments (12). Since calpain is a calcium-dependent enzyme, we thought it was important to determine whether cytokines increase muscle cell cytosolic and mitochondrial calcium levels. In the present study, we found that cytokines do elicit increases in both cytosolic and mitochondrial calcium concentrations and these cytokine-mediated calcium increases are blocked by administration of GW 4869.
We also explored the relationship between cytokine-induced sphingomyelinase activation and muscle cell calpain activation. For these determinations, we assessed two indexes of calpain activity, including an assay that directly measures cell calpain activity levels and determination of the calpain-specific cleavage products of caspase 12 (17, 20). It is known that caspase 12 is an endoplasmic reticulum (ER) protein that is activated by physiological stresses and, in turn, is linked to downstream activation of three ER stress pathways (i.e., IRE1α, PERK, and ATF6) (35). As a component of this activation process, calpain is known to cleave intact procaspase 12 to a smaller, 38-kDa caspase 12 fragment. This cleavage product is thought to be a relatively specific molecular indicator of calpain activation, since its generation in response to various stresses is specifically and quantitatively prevented by use of either chemical or genetic inhibitors of calpain activation (2, 17, 20).
In keeping with this earlier work, we also found that caspase 12 cleavage in C2C12 muscle cells in response to cytokines was blocked by a calpain inhibitor (calpain inhibitor 3). In addition, cytokine-induced caspase 12 cleavage was also prevented by administration of the nSMase2 inhibitor GW4869. We also found that cytokine-mediated caspase 12 activation was inhibited by siRNA mediated nSMase2 knockdown, arguing that nSMase2 activation is necessary for cytokine-dependent muscle calpain activation. In contrast, siRNA-induced nSMase3 knockdown did not prevent cytokine-induced caspase 12 cleavage, indicating that nSMase3 activation is not required for muscle calpain activation.
In addition, we found that caspase 12 cleavage was blocked by administration of BAPTA-AM, an intracellular calcium chelator, consistent with the fact that calpain is a calcium-dependent enzyme. We also found that both the nSMase2 inhibitor GW4869 and BAPTA-AM also prevented cytokine-induced muscle calpain activation, as assessed by a standard calpain activity assay. In addition, we found that direct addition of cell-permeant ceramide analogs, C2 ceramide and C6 ceramide, to C2C12 cells resulted in increased calpain activation as demonstrated by an effect of both these agents to increase caspase 12 cleavage and increase calpain activity assay levels. The present findings, as a result, indicate that increases in cytosolic calcium levels are necessary for nSMase-dependent calpain activation in muscle cells.
Contractile proteins and diaphragm force.
We also found that addition of cytokines to C2C12 cells elicited a reduction in both myosin light chain (MLC) and myosin heavy chain (MHC) levels. Moreover, these reductions were prevented by treating cells with the nSMase2 inhibitor GW4869, arguing that MLC and MHC protein loss is the result of downstream effects of sphingomyelinase activation. Of interest, several previous reports indicate that calpain is capable of cleaving MLC and MHC (23, 26). Our observation that MHC and MLC loss are prevented by nSMase2 inhibition is therefore consistent with an effect of sphingomyelinase to induce protein degradation by activation of the calpain proteolytic pathway.
Finally, we also found that inhibition of nSMase in the diaphragms of septic animals (i.e., CLP-induced sepsis) prevented the development of sepsis-induced diaphragm weakness. This finding is consistent with recent work by Ferreira et al. (9) demonstrating that addition of active sphingomyelinase to the diaphragm in vitro can induce reductions in diaphragm force generation. Of note, another study, by Wong et al. (38) recently reported that serum sphingomyelinase activity (acid) is increased following LPS injection into intact animals. It seems unlikely, however, that circulating acid sphingomyelinase was responsible for the sepsis-induced reductions in diaphragm force observed in the present report, since GW4869 did not reduce cytokine-induced acid sphingomyelinase activity in muscle cells in the present study and would therefore not be expected to improve diaphragm force following CLP if the effects of CLP were mediated by increasing levels of acid sphingomyelinase. Our data are more consistent with the possibility that increases in diaphragm neutral sphingomyelinase activity following CLP-induced sepsis are a consequence of cytokine-mediated increases in the activity of endogenous diaphragm nSMase.
Study limitations.
It should be noted, however, that there are some important limitations to our work. In our cell based studies, we were used a variety of methods to both chemically and genetically inhibit sphingomyelinase, employing these agents as mechanistic tools to probe the relationship of muscle sphingomyelinase activation to the downstream responses to cytokine administration (i.e., calcium shifts, calpain activation, MHC and MLC protein loss). By using cell-based studies, we obviated the potential effects of our inhibitors on other cell types that might have artifactually influenced our findings if we had studied only responses in intact animals. Our cell line experiments did not, however, allow us to determine the effects of cytokines or the inhibitors tested on muscle force development. In addition, infection is a complex phenomenon and its effects on muscle function may be at least partially mediated by processes that are not dependent on cytokine production (4). Our in vivo experiments examining the effects of a sphingomyelinase inhibitor on diaphragm force-generating capacity in an animal model of infection (CLP) circumvented these latter problems, allowing us to directly assess diaphragm contractile function in an experimental model in which both the cytokine- and non-cytokine-mediated responses to infection were present. Our observations that administration of GW4869 to intact animals with CLP-induced sepsis prevented both diaphragm weakness and diaphragm calpain activation are consistent with a potential role for sphingomyelinase in the induction of infection-mediated diaphragm dysfunction. In this fashion, our in vivo experiments complement our in vitro cell findings, with each set of studies allowing forms of assessment not possible in the other.
Summary.
In summary, these data are consistent with the possibility that sequential activation of nSMase2, generation of ceramide, and increases in cytosolic and mitochondrial calcium concentrations are required for cytokine-induced skeletal muscle calpain activation. As a consequence, therapies that inhibit nSMase activation or prevent increases in resting cytosolic calcium concentrations should block cytokine-induced calpain activation and attenuate infection-induced skeletal muscle dysfunction.
GRANTS
This work was supported by Veterans Administration Merit Award 1I01BX002132 (G. S. Supinski) and National Heart, Lung, and Blood Institute Grants R01HL113494 (G. S. Supinski), R01HL080429 (G. S. Supinski), R01HL081525 (G. S. Supinski), and R01HL112085 (L. A. Callahan).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
G.S.S. conception and design of research; G.S.S., A.P.A., L.W., and X.-H.S. performed experiments; G.S.S. analyzed data; G.S.S. and L.A.C. interpreted results of experiments; G.S.S. drafted manuscript; G.S.S. and L.A.C. edited and revised manuscript; G.S.S., A.P.A., L.W., X.-H.S., and L.A.C. approved final version of manuscript; L.A.C. prepared figures.
ACKNOWLEDGMENTS
We thank Dr. Michael Reid (Department of Physiology, Lexington, KY) for kindly providing us with the nSMase3 siRNA and the anti-nSMase 3 antibody. We are also grateful to Dr. Atsushi Miyawaki (RIKEN Brain Science Institute, Wako, Japan) for providing us with the cytosolic and mitochondrial pericam constructs. These studies would not have been possible without their generosity.
REFERENCES
- 1.Ahmad S, Choudhry MA, Sayeed MM. Ca2+-dependent and Ca2+-independent proteinase contents in the skeletal muscle in septic rats. Shock 5: 247–250, 1996. [DOI] [PubMed] [Google Scholar]
- 2.Badiola N, Penas C, Minano-Molina A, Barneda-Zahonero B, Fado R, Sanchez-Opazo G, Comella JX, Sabria J, Zhu C, Blomgren K, Casas C, Rodriguez-Alvarez J. Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis 2: e149, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Callahan LA, Nethery D, Stofan D, DiMarco A, Supinski G. Free radical-induced contractile protein dysfunction in endotoxin-induced sepsis. Am J Respir Cell Mol Biol 24: 210–217, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Chang HR, Bistrian B. The role of cytokines in the catabolic consequences of infection and injury. JPEN J Parenter Enteral Nutr 22: 156–166, 1998. [DOI] [PubMed] [Google Scholar]
- 5.Clarke CJ, Cloessner EA, Roddy PL, Hannun YA. Neutral sphingomyelinase 2 (nSMase2) is the primary neutral sphingomyelinase isoform activated by tumour necrosis factor-alpha in MCF-7 cells. Biochem J 435: 381–390, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Close RI. Dynamic properties of mammalian skeletal muscles. Physiol Rev 52: 129–197, 1972. [DOI] [PubMed] [Google Scholar]
- 7.Demoule A, Jung B, Prodanovic H, Molinari N, Chanques G, Coirault C, Matecki S, Duguet A, Similowski T, Jaber S. Diaphragm dysfunction on admission to the intensive care unit. Prevalence, risk factors, and prognostic impact—a prospective study. Am J Respir Crit Care Med 188: 213–219, 2013. [DOI] [PubMed] [Google Scholar]
- 8.Fareed MU, Evenson AR, Wei W, Menconi M, Poylin V, Petkova V, Pignol B, Hasselgren PO. Treatment of rats with calpain inhibitors prevents sepsis-induced muscle proteolysis independent of atrogin-1/MAFbx and MuRF1 expression. Am J Physiol Regul Integr Comp Physiol 290: R1589–R1597, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Ferreira LF, Moylan JS, Gilliam LA, Smith JD, Nikolova-Karakashian M, Reid MB. Sphingomyelinase stimulates oxidant signaling to weaken skeletal muscle and promote fatigue. Am J Physiol Cell Physiol 299: C552–C560, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fonteriz RI, de la Fuente S, Moreno A, Lobaton CD, Montero M, Alvarez J. Monitoring mitochondrial [Ca2+] dynamics with rhod-2, ratiometric pericam and aequorin. Cell Calcium 48: 61–69, 2010. [DOI] [PubMed] [Google Scholar]
- 11.Hermans G, Agten A, Testelmans D, Decramer M, Gayan-Ramirez G. Increased duration of mechanical ventilation is associated with decreased diaphragmatic force: a prospective observational study. Crit Care 14: R127, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hinkovska-Galcheva V, Shayman JA. Ceramide-1-phosphate in phagocytosis and calcium homeostasis. Adv Exp Med Biol 688: 131–140, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klaude M, Mori M, Tjader I, Gustafsson T, Wernerman J, Rooyackers O. Protein metabolism and gene expression in skeletal muscle of critically ill patients with sepsis. Clin Sci (Lond) 122: 133–142, 2012. [DOI] [PubMed] [Google Scholar]
- 14.Laghi F, Cattapan SE, Jubran A, Parthasarathy S, Warshawsky P, Choi YS, Tobin MJ. Is weaning failure caused by low-frequency fatigue of the diaphragm? Am J Respir Crit Care Med 167: 120–127, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Laufenberg LJ, Kazi AA, Lang CH. Salutary effect of aurintricarboxylic acid on endotoxin- and sepsis-induced changes in muscle protein synthesis and inflammation. Shock 41: 420–428, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu G, Kleine L, Hebert RL. Advances in the signal transduction of ceramide and related sphingolipids. Crit Rev Clin Lab Sci 36: 511–573, 1999. [DOI] [PubMed] [Google Scholar]
- 17.Martinez JA, Zhang Z, Svetlov SI, Hayes RL, Wang KK, Larner SF. Calpain and caspase processing of caspase-12 contribute to the ER stress-induced cell death pathway in differentiated PC12 cells. Apoptosis 15: 1480–1493, 2010. [DOI] [PubMed] [Google Scholar]
- 18.Michie HR. Metabolism of sepsis and multiple organ failure. World J Surg 20: 460–464, 1996. [DOI] [PubMed] [Google Scholar]
- 19.Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc Natl Acad Sci USA 98: 3197–3202, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol 150: 887–894, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nikolova-Karakashian M, Karakashian A, Rutkute K. Role of neutral sphingomyelinases in aging and inflammation. Subcell Biochem 49: 469–486, 2008. [DOI] [PubMed] [Google Scholar]
- 22.Nozaki K, Das A, Ray SK, Banik NL. Calpeptin attenuated apoptosis and intracellular inflammatory changes in muscle cells. J Neurosci Res 89: 536–543, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pemrick SM, Grebenau RC. Qualitative analysis of skeletal myosin as substrate of Ca2+-activated neutral protease: comparison of filamentous and soluble, native, and L2-deficient myosin. J Cell Biol 99: 2297–2308, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rabuel C, Samuel JL, Lortat-Jacob B, Marotte F, Lanone S, Keyser C, Lessana A, Payen D, Mebazaa A. Activation of the ubiquitin proteolytic pathway in human septic heart and diaphragm. Cardiovasc Pathol 19: 158–164, 2010. [DOI] [PubMed] [Google Scholar]
- 25.Rybakina EG, Nalivaeva NN, Pivanovich YU, Shanin SN, Kozinets A, Korneva EA. The role of neutral sphingomyelinase in interleukin-1beta signal transduction in mouse cerebral cortex cells. Neurosci Behav Physiol 31: 439–444, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Showalter CJ, Engel AG. Acute quadriplegic myopathy: analysis of myosin isoforms and evidence for calpain-mediated proteolysis. Muscle Nerve 20: 316–322, 1997. [DOI] [PubMed] [Google Scholar]
- 27.Supinski GS, Callahan LA. β-Hydroxy-β-methylbutyrate (HMB) prevents sepsis-induced diaphragm dysfunction in mice. Respir Physiol Neurobiol 196: 63–68, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Supinski GS, Callahan LA. Calpain activation contributes to endotoxin-induced diaphragmatic dysfunction. Am J Respir Cell Mol Biol 42: 80–87, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Supinski GS, Callahan LA. Caspase activation contributes to endotoxin-induced diaphragm weakness. J Appl Physiol 100: 1770–1777, 2006. [DOI] [PubMed] [Google Scholar]
- 30.Supinski GS, Callahan LA. Diaphragm weakness in mechanically ventilated critically ill patients. Crit Care 17: R120, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Supinski GS, Callahan LA. Double-stranded RNA-dependent protein kinase activation modulates endotoxin-induced diaphragm weakness. J Appl Physiol 110: 199–205, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Supinski GS, Ji XY, Callahan LA. p38 Mitogen-activated protein kinase modulates endotoxin-induced diaphragm caspase activation. Am J Respir Cell Mol Biol 43: 121–127, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Supinski GS, Wang L, Song XH, Moylan JS, Callahan LA. Muscle-specific calpastatin overexpression prevents diaphragm weakness in cecal ligation puncture-induced sepsis. J Appl Physiol 117: 921–929, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Supinski GS, Wang W, Callahan LA. Caspase and calpain activation both contribute to sepsis-induced diaphragmatic weakness. J Appl Physiol 107: 1389–1396, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szegezdi E, Fitzgerald U, Samali A. Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann NY Acad Sci 1010: 186–194, 2003. [DOI] [PubMed] [Google Scholar]
- 36.Tornquist K, Blom T, Shariatmadari R, Pasternack M. Ceramide 1-phosphate enhances calcium entry through voltage-operated calcium channels by a protein kinase C-dependent mechanism in GH4C1 rat pituitary cells. Biochem J 380: 661–668, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watson AC, Hughes PD, Louise Harris M, Hart N, Ware RJ, Wendon J, Green M, Moxham J. Measurement of twitch transdiaphragmatic, esophageal, and endotracheal tube pressure with bilateral anterolateral magnetic phrenic nerve stimulation in patients in the intensive care unit. Crit Care Med 29: 1325–1331, 2001. [DOI] [PubMed] [Google Scholar]
- 38.Wong ML, Xie B, Beatini N, Phu P, Marathe S, Johns A, Gold PW, Hirsch E, Williams KJ, Licinio J, Tabas I. Acute systemic inflammation up-regulates secretory sphingomyelinase in vivo: a possible link between inflammatory cytokines and atherogenesis. Proc Natl Acad Sci USA 97: 8681–8686, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]