

Abstract
Appropriate diagnosis and treatment of eumycetoma may vary significantly depending on the causative agent. To date, the most common fungus causing mycetoma worldwide is Madurella mycetomatis. This species fails to express any recognizable morphological characteristics, and reliable identification can therefore only be achieved with the application of molecular techniques. Recombinase polymerase amplification (RPA) and loop-mediated isothermal amplification (LAMP) are proposed as alternatives to phenotypic methods. Species-specific primers were developed to target the ribosomal DNA (rDNA) internal transcribed spacer (ITS) region of M. mycetomatis. Both isothermal amplification techniques showed high specificity and sufficient sensitivity to amplify fungal DNA and proved to be appropriate for detection of M. mycetomatis. Diagnostic performance of the techniques was assessed in comparison to conventional PCR using biopsy specimens from eumycetoma patients. RPA is reliable and easy to operate and has the potential to be implemented in areas where mycetoma is endemic. The techniques may be expanded to detect fungal DNA from environmental samples.
INTRODUCTION
Madurella mycetomatis is the most common causative agent of human eumycetoma worldwide, as evidenced in a recent review and meta-analysis on the disease (1). This highly morbid and destructive infection mainly affects individuals of low socioeconomic status in arid climate zones in the tropics (2). Although mycetoma was introduced into the medical literature in the 18th century, it still remains neglected and it has recently been recognized by the World Health Organization as a neglected tropical condition (2–4). The prevalence of the infection in villages in Sudan where the condition is endemic is estimated to be 14 per 1,000 inhabitants (5). At the Mycetoma Research Centre, Khartoum, Sudan, the main reference center in the country, the annual incidence of new cases is >300 (6). Despite this high incidence, appropriate diagnoses and therapies for this disease are still lacking (7).
Mycetoma is a subcutaneous infection that mainly affects the extremities and leads to massive disfigurement and disability (8, 9). The infection is thought to occur as a result of transcutaneous trauma, which allows insertion of the causative agent into skin and subcutaneous tissue. A distinctive and unique feature of mycetoma agents is their ability to form compact grains in infected tissue that will be discharged through the sinuses (10). Since mycetoma can be caused by both bacteria (actinomycetoma) and fungi (eumycetoma), the discharged grains are different (11). Black grains are caused by fungi, while actinomycetoma grains are white, yellow, or red (2, 10, 12). Molecular phylogeny of the eumycetoma causative agents has shown that the black-grain agents belong to entirely different fungal species, and these fungi differ in their antifungal susceptibility profiles (13, 14). Therefore, precise identification of the causative agent is necessary.
Current techniques for mycetoma diagnostics mostly comprise imaging, cytology, and histopathology, in addition to serological and cultural techniques. However, these methods are nonspecific and can only differentiate eumycetoma from actinomycetoma (6, 7). Madurella mycetomatis, which causes >70% of mycetoma worldwide, grows as sterile mycelia on routine culture media and should be identified by molecular methods (15). Sequencing of the internal transcribed spacer (ITS) of the rRNA operon has been used as a standard approach (16). In addition, amplification by specific primers combined with restriction fragment length polymorphism analysis has been developed to identify M. mycetomatis (17). Despite the specificity of these techniques, they are infrequently used in practice because of their high costs and because they require instrumentation and skilled laboratory personnel. Furthermore, application of DNA-based diagnostics for direct detection of M. mycetomatis from clinical material is challenging because of the difficulty in obtaining sufficient, purified DNA from the grains. In a previous study, we developed rolling-circle amplification (RCA) for specific identification of agents of black-grain eumycetoma (18). The technique is highly specific, rapid, and cost-effective, but culturing of the pathogen and PCR amplification are still required (18). Therefore, the purpose of the present study was to develop more simple isothermal amplification techniques that can be used for direct detection of M. mycetomatis from clinical specimens.
Since its introduction by Notomi et al. (19), LAMP has been widely applied for infectious diseases (20–22). The method was successfully commercialized for the diagnosis of tuberculosis because it suits resource-limited settings and therefore can be used in developing countries (23). The technique is based on the use of four primers that recognize six regions in the target DNA, followed by amplification through autocycling and strand displacement with a specialized DNA polymerase (19, 24).
More recently, RPA was introduced as a rapid, simple, sensitive, and highly specific exponential DNA isothermal amplification technique (25). It is based on the use of two opposing primers, similar to PCR, but a recombinase facilitates the insertion of the primers in the template DNA without requiring denaturation. With the help of single-stranded DNA binding proteins and of a strand-displacing DNA polymerase, amplification of the primer-bound complex occurs at temperatures between 25°C and 42°C. After amplification, the RPA product can be detected within as short a time as 15 to 40 min (25, 26). The method has been successfully applied for the world's major infectious diseases, including HIV, malaria, and tuberculosis (26–29).
Considering the advantages of these isothermal methods and the urgent need for simple point-of-care diagnostics for mycetoma, we thus introduce LAMP and RPA for the rapid detection of M. mycetomatis. To the best of our knowledge, no studies on the use of LAMP and RPA in mycetoma have been published to date.
MATERIALS AND METHODS
Fungal strains.
Fifty-seven fungal strains were included in this study, of which 32 belonged to the genus Madurella, 5 to other black-grain mycetoma species, 18 to the family Chaetomiaceae, and 2 to the genus Aspergillus (see Table S1 in the supplemental material). They were obtained from the Fungal Biodiversity Centre (CBS), Netherlands; the Mycetoma Research Centre (MRC), Khartoum, Sudan; the Pasteur Collection of Fungi (UMIP), France; and the National Reference Center for Invasive Mycoses and Antifungals (CNRMA), France.
Clinical samples.
Twelve surgical biopsy specimens from patients seen at the MRC were included and used for diagnostic purposes. Ten of the biopsy specimens were from patients with black-grain mycetoma, and two were from patients with other infections and were used as negative controls.
Ethics approval.
Ethical clearance was obtained from the Ethical Review Board of the Soba University Hospital Ethical Committee. Patient data were processed anonymously after obtaining written informed consent.
DNA extraction.
Strains were grown on 2% malt extract agar for 10 days at a temperature range of 21°C to 37°C. DNA was extracted using a protocol adapted from Möller et al. (30). In summary, ∼1 cm2 of fungal biomass was added to a screw-cap tube containing 6 to 10 acid-washed glass beads and homogenized in a tissue lyser (Qiagen, Hilden, Germany). Tubes were filled with 490 μl of cetyltrimethylammonium bromide (CTAB) and 10 μl of proteinase K (10 mg/ml; Sigma-Aldrich, St. Louis, MO) and were incubated for 1 h at 60°C mixed every 20 min. After incubation, 500 μl of a 24:1 chloroform-isoamylalcohol solution was added and shaken for 2 min and then centrifuged for 10 min at 14,000 rpm. The supernatant was collected, and ∼270 μl of ice-cold isopropanol was added to ∼400 μl of supernatant, followed by another centrifugation. In the final step, the pellets were washed with 70% ethanol, air-dried, and resuspended in 100 μl of TE buffer (Tris 0.12%, wt/vol, plus Na-EDTA 0.04%, wt/vol; pH 8.0).
For biopsy specimens, infected tissue containing visible grains was collected, washed several times in sterile physiological saline if necessary, and maintained at −20°C until use. DNA was extracted from the tissue and the grains using the CTAB protocol described previously (30). In addition, two alternative DNA extraction methods were used. Bead beating with a ZR Fungal/Bacterial DNA Miniprep kit (Zymo Research, Irvine, CA) was applied according to the manufacturer′s instructions. The DNeasy Plant minikit (Qiagen) was also used according to the manufacturer′s instructions but combined with bead beating using 2-mm metal beads.
Quality of DNA was verified by electrophoresis in 1% agarose gels, and concentrations were measured with a spectrophotometer at 260 nm (ND-1000 spectrophotometer, NanoDrop, Thermo Scientific, Wilmington, DE).
Loop-mediated isothermal amplification (LAMP).
For the design of LAMP primers, the rDNA ITS regions and partial β-tubulin were selected. Sequences of reference strains of the Madurella species and of closely related taxa in the Chaetomiaceae were aligned with Bionumerics v. 4.6 (Applied Maths, Sint-Martens-Latem, Belgium). Four primer sets were designed using PrimerExplorer v. 4 (https://primerexplorer.jp/elamp4.0.0/), i.e., three for ITS and one for β-tubulin.
LAMP was carried out in 25-μl reaction volumes containing 2 μl DNA, 8 U Bst DNA polymerase (New England BioLabs, Beverly, MA), and 1× reaction buffer provided with the enzyme [20 mM Tris-HCl, pH 8.8, 10 mM KCl, 2 mM MgSO4, 10 mM (NH4)2SO4, 0.1% Triton X-100]. Additional concentrations of 4, 6, 8, and 10 mM MgSO4 were tested. Primers were used in concentrations of 40 pmol of each forward inner primer (FIP) and backward inner primer (BIP) and 5 pmol each of forward and backward outer primers (F3 and B3) (Life Technologies, Breda, Netherlands). Alternative ratios of inner and outer primers were tested, including 6:1 and 10:1. Betaine was used in a concentration of 1 M (reaction without betaine was also tested), and deoxynucleoside triphosphates (dNTPs) at 1 mM each were added. LAMP mixtures were prepared in a laminar flow cabinet with use of DNase/RNase-free equipment; sterile water was used as the negative control. LAMP reaction mixtures were incubated, and three incubation temperatures (60°C, 63°C, and 65°C) and incubation periods (60, 90, and 120 min) were evaluated. Final products were examined in 2% agarose gels, and the results were considered positive if ladder-like band patterns were revealed. Results were also visualized by adding 1.0 μl of a 10-fold-diluted solution of SYBR green I (Cambrex BioScience, Workingham, United Kingdom) and with a UV transilluminator (Vilber Lourmat, Marne-la-Vallée, France).
Recombinase polymerase amplification (RPA).
Six primers of 29 to 34 bases were designed on the basis of ITS sequences of the M. mycetomatis type strain CBS 109801 and were checked for a secondary structure using PrimerSelect (DNAStar Lasergene, WI). The designed primers were used in seven pairs of different product sizes. Oligonucleotides were synthesized by Life Technologies (Breda, Netherlands), and the sequences are listed in Table 1.
TABLE 1.
Oligonucleotide primers
| Primer | Sequence |
|---|---|
| M. mycetomatis PCRa | |
| 26.1A | 5′-AATGAGTTGGGCTTTAACGG-3′ |
| 28.3A | 5′-TCCCGGTAGTGTAGTGTCCCT-3′ |
| RPA primers | |
| P1MAduF | 5′-TTGTGAACATACCCCCAAAACCGTTGCTTCGGC-3′ |
| P2MAduF | 5′-GTAGTGTAGTGTCCCTCGGCCCCCTCCGC-3′ |
| P3MAduF | 5′-ATTATACAACACCCTATTTGCTCTGTACGG-3′ |
| P3MAduR | 5′-CCGTACAGAGCAAATAGGGTGTTGTATAAT-3′ |
| P4MAduR | 5′-GCAGCACGCCCTGGGCGAGTTGGATATAA-3′ |
| P5MAduR | 5′-AAATGAGTTGGGCTTTAACGGCCGGAGCCCGCAG-3′ |
| Primer set for Madurella mycetomatis LAMP (MM) based on ITS | |
| MMF3 | 5′-TCCCCAAACCATTGTGAA-3′ |
| MMF3 | 5′-AGAGATCCGTTGTTGAAAGT-3′ |
| MMFIP | 5′-GACACTACACTACCGGGAGG-CATACCCCCAAAACCGTT-3′ |
| MMBIP | 5′-TCCGCCGGAGGATTATACAAC-CTTATTCAGTACAGAAGACTCAGA-3′ |
See reference 3.
Reaction conditions of RPA were optimized, and different primer sets were screened using materials and protocols of the TwistAmp basic kit (TwistDx, Cambridge, United Kingdom). For optimization, DNAs of the type strains of four Madurella species were analyzed. Reaction mixtures contained 2.4 μl of each primer (10 μM), 29.5 μl of rehydration buffer, 2 μl of template DNA, and 11.2 μl of sterile distilled H2O. The mixtures were added to 0.2-ml tubes containing freeze-dried enzyme pellets. A reaction mixture with 2 μl sterile water was used as a negative control, while a positive control was provided in the kit and was used to assay the reagent and the reaction performance.
To initiate the RPA, 2.5 μl of magnesium acetate (280 mM) was added to each reaction, and the tubes were vortexed and incubated on a heat block at 39°C to 40°C for 40 min. Tubes were vortexed every 6 min, centrifuged, and reincubated. RPA products were purified using a PCR purification kit (Qiagen) and analyzed in 2% agarose electrophoresis gels. Bands of expected size were cut out of the gels and applied for extraction using a gel extraction kit (Qiagen). Extraction products were amplified and sequenced with the same RPA primers.
Analytical sensitivity and specificity of LAMP and RPA.
In order to assay analytical sensitivities of both RPA and LAMP, 15 ng DNA of M. mycetomatis (CBS 109801) was used as the template in 2-fold serial dilutions for up to 12 × 10−2 ng. Analytical specificity of LAMP was determined using a panel of 25 M. mycetomatis and 32 isolates belonging to other species in the genus Madurella, as well as close relatives. Specificity of RPA was determined using 16 isolates of M. mycetomatis in addition to 11 isolates of which 7 belonged to agents of black-grain mycetoma (see Table S1 in the supplemental material).
Detection of M. mycetomatis DNA in clinical samples.
Twelve biopsy specimens were included to evaluate the performance of direct PCR, LAMP, and RPA on clinical specimens. The PCR was done using M. mycetomatis-specific primers (17) and performed using the Go Taq Flexi polymerase kit (Promega, WI) with 25 μl reaction mixture containing 2 μl template DNA (30 to 50 ng), 2.5 μl of 5× colorless Flexi buffer, 0.1 μl of GoTaq DNA polymerase, 1.4 μl of dimethyl sulfoxide (DMSO) (99.9%), 2 μl of MgCL2 (25 mM), 0.5 μl of each primer (10 pmol; Table 1), 1 μl of dNTP mix (2.5 mM), and 15 μl of sterile water. Reactions were performed in an ABI Prism 2720 (Applied Biosystems, CA) with the following amplification conditions: denaturation at 95°C for 5 s, 35 cycles at 95°C for 30 s, 48°C for 30 s, 72°C for 90 min, and final postelongation at 72°C for 10 min. The resulting amplicons were analyzed by agarose gel electrophoresis, and samples with target bands of ∼400 bp were subjected to sequencing. Sequencing was performed using the same PCR primers and BigDye Terminator cycle sequencing kit v. 3.1 (Applied Biosystems) and analyzed in an ABI 3730xl DNA analyzer (Applied Biosystems). The DNAs of 12 biopsy specimens were also analyzed with LAMP and RPA as described previously, except that the RPA products were further subjected to sequencing and compared to M. mycetomatis reference data.
RESULTS
Of the four primer sets designed for M. mycetomatis LAMP assay, only one showed optimal results (Table 1). The remaining three primers, including the one targeting β-tubulin, showed cross-reactivity between Madurella species (data not shown). Optimal LAMP reaction was achieved with the use of 1 M betaine, 4 to 8 mM MgSO4, and a ratio of 8:1 between inner and outer primer. The reaction was performed optimally at 65°C with an incubation time of 2 h. LAMP results were easily detected in agarose gel (Fig. 1) and by direct visualization using SYBR green.
FIG 1.
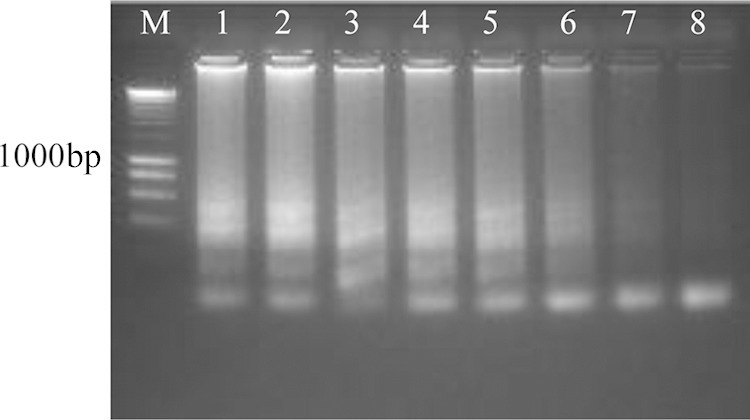
Sensitivity of Madurella mycetomatis LAMP using serial dilution of CBS 109801T DNA seen in 2% agarose gel electrophoresis. Lane M, 200-bp DNA marker; lanes 1 to 8, 15, 7.5, 3.75, 1.88, 0.94, 0.47, 0.23, and 0.12 ng.
LAMP reactions were positive only with the 25 M. mycetomatis strains, despite their divergent geographic origins, and negative with all 32 non-M. mycetomatis strains (see Table S1 in the supplemental material and Fig. 2). However, unspecific amplification was detected when high concentrations of DNA were used. Optimal concentration was 10 ng of pure culture DNA. Regarding the analytical sensitivity, LAMP was found to be sensitive and yielded positive results in all DNA concentrations tested up to 0.12 ng. However, a clear cutoff, where results were unambiguously positive both visually and in gel, was 0.47 ng, and this was considered the lowest detection limit (Fig. 1).
FIG 2.

Specificity of Madurella mycetomatis LAMP detected in 2% agarose gel electrophoresis. Lane M, DNA ladder; lane 1, M. mycetomatis CBS 109801T; lane 2 M. tropicana CBS 201.38T; lane 3, M. pseudomycetomatis CBS 129177T; lane 4, M. fahalii CBS 129176T; lane 5, Trematosphaeria grisea CBS 332.50T; lane 6, Falciformispora senegalensis CBS 196.79T; lane 7, F. tompkinsii CBS 200.70; lane 8, Medicopsis romeroi CBS 252.60T.
With RPA, of the seven primer combinations tested, six successfully amplified the target and resulted in clearly visible bands of expected size. Two pairs, P2MAduF-P5MAduR with a product size of 429 bp and P3MAduF-P4MAduR with a product size 326 bp, were selected for further analysis on the basis of band intensity (Fig. 3). RPA was performed optimally at temperatures of 39°C to 40°C with an incubation time of 40 min. RPA yielded positive results with 16 M. mycetomatis strains and negative results with other agents of black-grain mycetoma and with isolates of Chaetomium species (Fig. 4). The lowest detection limit of RPA according to the used protocol was 0.23 ng of DNA (Fig. 3).
FIG 3.
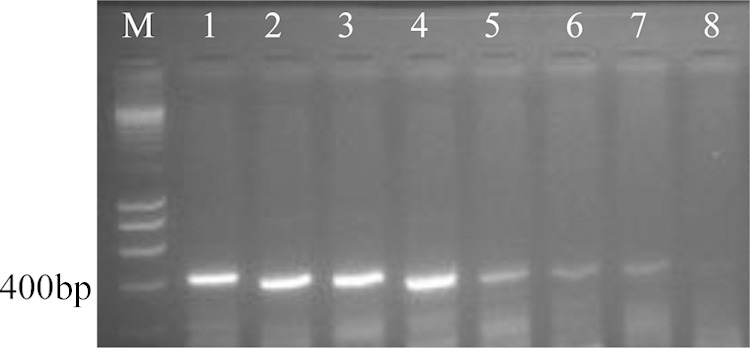
Gel representation of the sensitivity of Madurella mycetomatis-specific RPA using primer pair P2MaduF and P5MAduR. Serial dilutions of CBS 109801T DNA were used. Lane M, 200-bp DNA marker; lanes 1 to 8, 15, 7.5, 3.75, 1.88, 0.94, 0.47, 0.23, and 0.12 ng.
FIG 4.
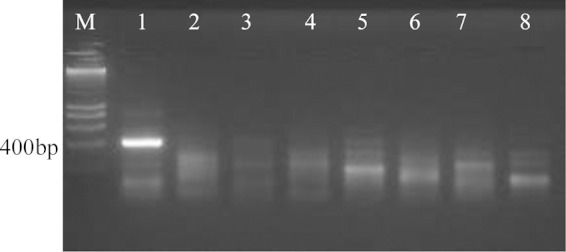
Specificity of Madurella mycetomatis RPA using primer pair P2MaduF and P5MAduR and seen in 2% agarose gel electrophoresis. Lane M, 200-bp DNA marker; lane 1, M. mycetomatis CBS 109801T; lane 2, M. tropicana CBS 201.38T lane 3, M. pseudomycetomatis CBS 129177T; lane 4, M. fahalii CBS 129176T; lane 5, Trematosphaeria grisea CBS 332.50T; lane 6, Falciformispora senegalensis CBS 196.79T; lane 7, F. tompkinsii CBS 200.70; lane 8, Medicopsis romeroi CBS 252.60T.
To evaluate the diagnostic performance of the DNA-based methods, patient biopsy specimens were analyzed. For DNA extraction from clinical samples, with CTAB protocol even when followed by several cleaning steps, the DNA pellet remained brown due to the presence of melanin and could not be used for downstream analysis. Of the extraction kits used, ZR Fungal/Bacterial DNA Miniprep with ultrahigh-density BashingBeads yielded no DNA or very low concentrations. The grains remained compacted and were not disrupted even after the increase in disruption time. Higher DNA concentrations with high purity were achieved when the grains were initially disrupted with bead beating using metal beads for 3 to 5 min followed by extraction using the Qiagen DNeasy Plant minikit.
In clinical samples, RPA and LAMP showed performances equal to that of PCR (10 positive for M. mycetomatis and 2 negative). Diagnostic sensitivity of 100% (95% confidence interval [CI], 71.33% to 100.00%) and specificity of 100% (95% CI, 19.29% to 100.00%) were obtained with RPA and LAMP, as well as with PCR. However, LAMP yielded false-negative results when older reagents were used and positive results when the procedure was repeated with freshly prepared reagents. A comparison was made among all techniques used for M. mycetomatis identification, showing that RPA and LAMP had the shortest turnaround times combined with sufficient sensitivity and specificity (Table 2). In our laboratory setting, we found that RPA is the easiest method to perform, with a lower risk of contamination than with LAMP.
TABLE 2.
Comparison among conventional and molecular-based methods for identification of mycetoma causative agents
| Assay | Culture required | DNA extraction required | PCR thermal cycler required | Product detection method | Estimated cost ($)a | Turnaround time (h) | Sensitivity | Specificity |
|---|---|---|---|---|---|---|---|---|
| Conventional (culture and phenotypic) | Yes | No | No | Eye/microscopy | 5 (41) | 168 | Low | Low |
| PCR plus sequencing | Yesb | Yes | Yes | Gel/sequencer | 7.7 (42) | 176 | High | High |
| RCA | Yes | Yes | Yes | Gel/eye | —e | 174 | Lowc | High |
| LAMP | No | Yes | No | Gel/eye | <1–5.3 (43) | 3 | High | High |
| RPA | No | Yes | No | Geld | 4.25 (26) | 3 | High | High |
Numbers in parentheses are references.
In the present study, PCR was performed directly from clinical sample.
RCA requires amplification of the target first.
Other detection methods can be used.
—, data not available.
DISCUSSION
In the present study, we investigated the performances of three molecular diagnostic methods, viz. PCR and two isothermal DNA amplification techniques, for mycetoma caused by M. mycetomatis. With all methods, and for the first time, we were able to detect the fungal DNA directly from clinical specimens.
The M. mycetomatis-specific PCR was first described in 2004 by Ahmed et al. (17). With the recent description of new sibling species of Madurella similar to M. mycetomatis (31), the specificity of this method might be uncertain. Therefore, after amplification of DNA from clinical specimens, PCR products were sequenced. Since the PCR and sequencing are expensive and time-consuming, the two isothermal amplification techniques were developed as alternative detection methods.
Using M. mycetomatis-specific LAMP primers, we successfully amplified DNA from strains of different origins. The reaction was performed at a constant temperature and in a simple heating block, but it has also been reported that LAMP can be performed in a pocket warmer, which explores the potential application of the method in poor resource settings (32). To simplify the technique, reduce contamination risk, and increase the stability of LAMP reagents, the method was commercialized in ready-to-use kits (21). Despite the high sensitivity, specificity, and analytical accuracy of LAMP, use of the kit is recommended because the risk of cross-contamination remains a concern.
LAMP offers operational advantages over direct PCR in that the result can easily be detected by visual inspection after adding various noninhibitory dyes or a double-stranded DNA (dsDNA)-binding dye (19, 20, 33, 34). Furthermore, detection of turbidity produced from precipitation of insoluble magnesium pyrophosphate can be used for both visual endpoint reading and real-time detection (33). LAMP has been combined with chromatographic lateral-flow (LF) dipstick using labeled primers to simplify the reading (35–37). In the present study, LAMP results were easily detected in gel electrophoresis and by the naked eye after addition of SYBR green.
The LAMP primers developed in this study were designed to target a distinctive region in the rDNA ITS of M. mycetomatis. These primers were found to be specific; however, when high concentrations of DNA were used, some nonspecific amplification was detected. The reason for this amplification remains unexplained, since a wide range of parameters related to both the primer and the target sequence can influence the reaction (38). Therefore, we recommend a maximum of 10 ng of DNA from pure cultures. This is 20 times higher than the lowest detection limit of M. mycetomatis LAMP, which was <0.5 ng of DNA.
In this study, RPA was evaluated as an alternative isothermal amplification method with potential for use in low-technology laboratory settings. Specific RPA primers were based on the ribosomal ITS region, not only because of the in silico specificity but also because of the high copy number of the ribosomal operon (18). RPA was performed using reagents that were provided as dry pellets, and thus no additional instrumentation was required except for a heating block. Incubation using human body temperature has been described, with the reaction tubes simply being incubated in the axilla with a bandage (39). Detection of amplified RPA products mostly relies on a specially designed probing system that ensures specific amplification and increases accuracy (25, 26). In the present study, a basic detection system with agarose gel electrophoresis was used to minimize the cost and to evaluate the method with standard laboratory resources.
This is the first study describing the application of molecular techniques for direct detection of M. mycetomatis in clinical specimens. Previously, this was hampered by difficult DNA isolation due to the compactness and melanized nature of M. mycetomatis grains. We overcame this by bead beating with metal beads followed by application of a Qiagen extraction kit. Starting from purified DNA, with all three methods tested, we were able to identify the cause of black-grain eumycetoma in 10 patients as M. mycetomatis infection. When comparing the practical applicability of the three methods, we found that RPA is the easiest to perform, and the reagents are very stable. Future development of the technique is required, particularly in the application of probe-based detection and LF dipsticks, which are currently being implemented for several viral, bacterial, and parasitic infections (26–29, 40).
The present study confirms that culture-free diagnostic methods can be accurate and can be used successfully for human eumycetoma diagnosis. The isothermal techniques introduced here are simple, rapid, and cost-effective. The only limitation is that multiplex systems need to be developed for detection of multiple species. In the present version, the technique is most useful in areas where a single causative agent is preponderant. Furthermore, expansion of the current protocol to include other Madurella species is required to overcome the false-negative results with non-M. mycetomatis causative agents.
Supplementary Material
ACKNOWLEDGMENTS
We thank Francoise Dromer for her comments on the manuscript and for providing some clinical isolates.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.01544-15.
REFERENCES
- 1.van de Sande WWJ. 2013. Global burden of human mycetoma: a systematic review and meta-analysis. PLoS Negl Trop Dis 7:e2550. doi: 10.1371/journal.pntd.0002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahgoub ES, Murray IG. 1973. Mycetoma. William Heinemann, Medical Books, London, United Kingdom. [Google Scholar]
- 3.Ahmed AO, van Leeuwen W, Fahal A, van de Sande W, Verbrugh H, van Belkum A. 2004. Mycetoma caused by Madurella mycetomatis: a neglected infectious burden. Lancet Infect Dis 4:566–574. doi: 10.1016/S1473-3099(04)01131-4. [DOI] [PubMed] [Google Scholar]
- 4.van de Sande WW, Mahgoub el S, Fahal AH, Goodfellow M, Welsh O, Zijlstra E. 2014. The mycetoma knowledge gap: identification of research priorities. PLoS Negl Trop Dis 8:e2667. doi: 10.1371/journal.pntd.0002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fahal A, Mahgoub el S, El Hassan AM, Abdel-Rahman ME, Alshambaty Y, Hashim A, Hago A, Zijlstra EE. 2014. A new model for management of mycetoma in the Sudan. PLoS Negl Trop Dis 8:e3271. doi: 10.1371/journal.pntd.0003271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fahal A, Mahgoub ES, Hassan AME, Abdel-Rahman ME. 2015. Mycetoma in the Sudan: an update from the Mycetoma Research Centre, University of Khartoum, Sudan. PLoS Negl Trop Dis 9:e0003679. doi: 10.1371/journal.pntd.0003679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van de Sande WW, Fahal AH, Goodfellow M, Mahgoub el S, Welsh O, Zijlstra EE. 2014. Merits and pitfalls of currently used diagnostic tools in mycetoma. PLoS Negl Trop Dis 8:e2918. doi: 10.1371/journal.pntd.0002918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fahal AH. 2006. Mycetoma—clinicopathological monograph, 1st ed Khartoum University Press, Khartoum, Sudan. [Google Scholar]
- 9.Fahal AH, Suliman SH. 1994. Clinical presentation of mycetoma. Sudan Med J 32:46–66. [Google Scholar]
- 10.McGinnis MR. 1996. Mycetoma. Dermatol Clin 14:97–104. doi: 10.1016/S0733-8635(05)70329-6. [DOI] [PubMed] [Google Scholar]
- 11.Chalmers AJ, Archibald RG. 1918. The classification of mycetomas. J Trop Med Hyg 21:121–123. [Google Scholar]
- 12.Hospenthal DR. 2010. Agents of mycetoma, p 3281–3285. In Mandell GL, Bennett JE, Dolin R. (ed), Principles and practice of infectious diseases, 7th ed Elsevier Churchill Livingstone, Philadelphia, PA. [Google Scholar]
- 13.Ahmed SA, van de Sande WW, Stevens DA, Fahal A, van Diepeningen AD, Menken SB, de Hoog GS. 2014. Revision of agents of black-grain eumycetoma in the order Pleosporales. Persoonia 33:141–154. doi: 10.3767/003158514X684744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmed SA, de Hoog GS, Stevens DA, Fahal AH, van de Sande WW. 2015. In vitro antifungal susceptibility of coelomycete agents of black grain eumycetoma to eight antifungals. Med Mycol 53:295–301. doi: 10.1093/mmy/myu098. [DOI] [PubMed] [Google Scholar]
- 15.Ahmed AO, Desplaces N, Leonard P, Goldstein F, De Hoog S, Verbrugh H, van Belkum A. 2003. Molecular detection and identification of agents of eumycetoma: detailed report of two cases. J Clin Microbiol 41:5813–5816. doi: 10.1128/JCM.41.12.5813-5816.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desnos-Ollivier M, Bretagne S, Dromer F, Lortholary O, Dannaoui E. 2006. Molecular identification of black-grain mycetoma agents. J Clin Microbiol 44:3517–3523. doi: 10.1128/JCM.00862-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmed AO, Mukhtar MM, Kools-Sijmons M, Fahal AH, de Hoog S, van den Ende BG, Zijlstra EE, Verbrugh H, Abugroun ES, Elhassan AM, van Belkum A. 1999. Development of a species-specific PCR-restriction fragment length polymorphism analysis procedure for identification of Madurella mycetomatis. J Clin Microbiol 37:3175–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahmed SA, van den Ende BHGG, Fahal AH, van de Sande WWJ, de Hoog GS. 2014. Rapid identification of black grain eumycetoma causative agents using rolling circle amplification. PLoS Negl Trop Dis 8:e3368. doi: 10.1371/journal.pntd.0003368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. 2000. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 28:E63. doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mori Y, Kanda H, Notomi T. 2013. Loop mediated isothermal amplification (LAMP): recent progress in research and development. Infect Chemother 19:404–411. doi: 10.1007/s10156-013-0590-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mori Y, Notomi T. 2009. Loop-mediated isothermal amplification (LAMP): a rapid, accurate, and cost-effective diagnostic method for infectious diseases. J Infect Chemother 15:62–69. doi: 10.1007/s10156-009-0669-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomita N, Mori Y, Kanda H, Notomi T. 2008. Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat Protoc 3:877–882. doi: 10.1038/nprot.2008.57. [DOI] [PubMed] [Google Scholar]
- 23.Mitarai S, Okumura M, Toyota E, Yoshiyama T, Aono A, Sejimo A, Azuma Y, Sugahara K, Nagasawa T, Nagayama N, Yamane A, Yano R, Kokuto H, Morimoto K, Ueyama M, Kubota M, Yi R, Ogata H, Kudoh S, Mori T. 2011. Evaluation of a simple loop-mediated isothermal amplification test kit for the diagnosis of tuberculosis. Int J Tuberc Lung Dis 15:1211–1217. doi: 10.5588/ijtld.10.0629. [DOI] [PubMed] [Google Scholar]
- 24.Fu S, Qu G, Guo S, Ma L, Zhang N, Zhang S, Gao S, Shen Z. 2011. Applications of loop-mediated isothermal DNA amplification. Appl Biochem Biotechnol 163:845–850. doi: 10.1007/s12010-010-9088-8. [DOI] [PubMed] [Google Scholar]
- 25.Piepenburg O, Williams CH, Stemple DL, Armes NA. 2006. DNA detection using recombination proteins. PLoS Biol 4:e204. doi: 10.1371/journal.pbio.0040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rohrman BA, Richards-Kortum RR. 2012. A paper and plastic device for performing recombinase polymerase amplification of HIV DNA. Lab Chip 12:3082–3088. doi: 10.1039/c2lc40423k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boyle DS, Lehman DA, Lillis L, Peterson D, Singhal M, Armes N, Parker M, Piepenburg O, Overbaugh J. 2013. Rapid detection of HIV-1 proviral DNA for early infant diagnosis using recombinase polymerase amplification. mBio 4:pii=e00135-13. doi: 10.1128/mBio.00135-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyle DS, McNerney R, Teng Low H, Leader BT, Pérez-Osorio AC, Meyer JC, O'Sullivan DM, Brooks DG, Piepenburg O, Forrest MS. 2014. Rapid detection of Mycobacterium tuberculosis by recombinase polymerase amplification. PLoS One 9:e103091. doi: 10.1371/journal.pone.0103091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kersting S, Rausch V, Bier FF, von Nickisch-Rosenegk M. 2014. Rapid detection of Plasmodium falciparum with isothermal recombinase polymerase amplification and lateral flow analysis. Malar J 13:99. doi: 10.1186/1475-2875-13-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Möller EM, Bahnweg G, Sandermann H, Geiger HH. 1992. A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Res 20:6115–6116. doi: 10.1093/nar/20.22.6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Hoog GS, van Diepeningen AD, Mahgoub el S, van de Sande WW. 2012. New species of Madurella, causative agents of black-grain mycetoma. J Clin Microbiol 50:988–994. doi: 10.1128/JCM.05477-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ablordey A, Amissah DA, Aboagye IF, Hatano B, Yamazaki T, Sata T, Ishikawa K, Katano H. 2012. Detection of Mycobacterium ulcerans by the loop mediated isothermal amplification method. PLoS Negl Trop Dis 6:e1590. doi: 10.1371/journal.pntd.0001590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mori Y, Nagamine K, Tomita N, Notomi T. 2001. Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem Biophys Res Commun 289:150–154. doi: 10.1006/bbrc.2001.5921. [DOI] [PubMed] [Google Scholar]
- 34.Almasi MA, Dehabadi SMH, Moradi A, Eftekhari Z, Ojaghkandi MA, Aghaei S. 2013. Development and application of loop-mediated isothermal amplification assay for rapid detection of Fusarium oxysporum f. sp lycopersici. Plant Pathol Microbiol 4:177. [Google Scholar]
- 35.Arunrut N, Seetang-Nun Y, Phromjai J, Panphut W, Kiatpathomchai W. 2011. Rapid and sensitive detection of Laem-Singh virus by reverse transcription loop-mediated isothermal amplification combined with a lateral flow dipstick. J Virol Methods 177:71–74. doi: 10.1016/j.jviromet.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 36.Roskos K, Hickerson AI, Lu HW, Ferguson TM, Shinde DN, Klaue Y, Niemz A. 2013. Simple system for isothermal DNA amplification coupled to lateral flow detection. PLoS One 8:e69355. doi: 10.1371/journal.pone.0069355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaewphinit T, Arunrut N, Kiatpathomchai W, Santiwatanakul S, Jaratsing P, Chansiri K. 2013. Detection of Mycobacterium tuberculosis by using loop-mediated isothermal amplification combined with a lateral flow dipstick in clinical samples. Biomed Res Int 2013:926230. doi: 10.1155/2013/926230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kimura Y, de Hoon MJ, Aoki S, Ishizu Y, Kawai Y, Kogo Y, Daub CO, Lezhava A, Arner E, Hayashizaki Y. 2011. Optimization of turn-back primers in isothermal amplification. Nucleic Acids Res 39:e59. doi: 10.1093/nar/gkr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crannell ZA, Rohrman B, Richards-Kortum R. 2014. Equipment-free incubation of recombinase polymerase amplification reactions using body heat. PLoS One 9:e112146. doi: 10.1371/journal.pone.0112146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahmed A, van der Linden H, Hartskeerl RA. 2014. Development of a recombinase polymerase amplification assay for the detection of pathogenic Leptospira. Int J Environ Res Public Health 11:4953–4964. doi: 10.3390/ijerph110504953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Massire C, Buelow DR, Zhang SX, Lovari R, Matthews HE, Toleno DM, Ranken RR, Hall TA, Metzgar D, Sampath R, Blyn LB, Ecker DJ, Gu Z, Walsh TJ, Hayden RT. 2013. PCR followed by electrospray ionization mass spectrometry for broad-range identification of fungal pathogens. J Clin Microbiol 51:959–966. doi: 10.1128/JCM.02621-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pryce TM, Palladino S, Kay ID, Coombs GW. 2003. Rapid identification of fungi by sequencing the ITS1 and ITS2 regions using an automated capillary electrophoresis system. Med Mycol 41:369–381. doi: 10.1080/13693780310001600435. [DOI] [PubMed] [Google Scholar]
- 43.Oriero EC, Jacobs J, Van Geertruyden JP, Nwakanma D, D'Alessandro U. 2015. Molecular-based isothermal tests for field diagnosis of malaria and their potential contribution to malaria elimination. J Antimicrob Chemother 70:2–13. doi: 10.1093/jac/dku343. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.