

Abstract
Progressive supranuclear palsy (PSP) is a neurodegenerative syndrome that is clinically characterized by progressive postural instability, supranuclear gaze palsy, parkinsonism and cognitive decline. Pathologically, diagnosis of PSP is based on characteristic features, such as neurofibrillary tangles, neutrophil threads, tau-positive astrocytes and their processes in basal ganglia and brainstem, and the accumulation of 4 repeat tau protein. PSP is generally recognized as a sporadic disorder; however, understanding of genetic background of PSP has been expanding rapidly. Here we review relevant publications to outline the genetics of PSP. Although only small number of familial PSP cases have been reported, the recognition of familial PSP has been increasing. In some familial cases of clinically probable PSP, PSP pathologies were confirmed based on NINDS neuropathological diagnostic criteria. Several mutations in MAPT, the gene that causes a form of familial frontotemporal lobar degeneration with tauopathy, have been identified in both sporadic and familial PSP cases. The H1 haplotype of MAPT is a risk haplotype for PSP, and within H1, a sub-haplotype (H1c) is associated with PSP. A recent genome-wide association study on autopsyproven PSP revealed additional PSP risk alleles in STX6 and EIF2AK3. Several heredodegenerative parkinsonian disorders are referred to as PSP-look-alikes because their clinical phenotype, but not their pathology, mimics PSP. Due to the fast development of genomics and bioinformatics, more genetic factors related to PSP are expected to be discovered. Undoubtedly, these studies will provide a better understanding of the pathogenesis of PSP and clues for developing therapeutic strategies.
Keywords: Progressive supranuclear palsy, Genetics, MAPT, Microtubule-associated protein tau, Familial progressive supranuclear palsy
INTRODUCTION
Since the first description of progressive neurodegenerative cases with supranuclear gaze palsy and bulbar palsy, progressive supranuclear palsy (PSP; formerly, Richardson’s syndrome) has been established as a neurodegenerative disorder with characteristic clinicopathological features [1]. Clinically PSP is diagnosed according to the NINDS-SPSP criteria [2], where falls and supranuclear gaze palsy are the most important features. Definitive diagnosis of PSP is based on pathological criteria [3]. Accumulation of 4 repeat tau protein in neurons and glia in basal ganglia and the brainstem is a characteristic feature of PSP [4]. The clinical phenotypic spectrum of PSP has broadened during the last decade [5-11]. After a seminal article published by Williams et al. [5] introduced the concept of PSP-parkinsonism and PSP-Richardson’s syndrome, subtypes of PSP have been recognized in pathologically confirmed PSP cases [12]. These include pure akinesia with gait freezing [13], corticobasal syndrome [6], progressive non-fluent aphasia [14], primary lateral sclerosis [8], and a behavioral variant of frontotemporal dementia [15]. Differential burden and distribution of tau pathologies between cortices, and basal ganglia and brainstem are related to atypical phenotypes [4]. Likewise, although PSP has been generally recognized as a sporadic disorder, understanding of the genetic background of PSP has been expanded.
This review will cover the genetic aspects of PSP, focusing on family history, single gene mutation causes of PSP, common risk alleles or haplotypes and PSP-mimicking disorders with known causative genes.
FAMILIAL PSP
Traditionally, PSP has been considered as a sporadic condition. The prevalence of PSP is reportedly 5–6 cases/100,000 [16-19]. The incidence of PSP increases steeply with age, from 1.7 at 50 to 59 years to 14.7 at 80 to 99 years [20]. It was not long ago that family history of PSP and other neurodegenerative conditions was investigated systematically in PSP [21]. Using a structured questionnaire studying family history, Donker Kaat et al. [21] found that 57 (33%) out of 172 patients with PSP had at least one first-degree relative who had dementia or parkinsonism, which is higher compared to those for controls from the Rotterdam study (25%) [odds ratio (OR) 1.5, 95% confidence interval (CI) 1.02–2.13]. In patients with PSP, first-degree relatives with parkinsonism were more commonly observed compared to controls (OR 3.9, 95% CI 1.99–7.61). Seven percent of the patients with PSP fulfilled the criteria for an autosomal dominant mode of transmission.
Recently, Fujioka et al. [32] reviewed 59 cases out of 19 families with familial PSP [21-31]. While not all reported patients with familial PSP fulfilled the NINDS-SPSP criteria for the diagnosis of clinically probable PSP in which supranuclear ophthalmoplegia was observed in 76.3% of the cases and falls were in reported in 44.1% of the cases, there were pathologically confirmed cases [32]. In a retrospective study reviewing autopsy results and medical records of 375 pathologically confirmed PSP cases, excluding those carrying a MAPT mutation, family history of PSP was observed in 3% of the cases, whereas family history of PSP, parkinsonism or dementia was observed in 15% of the cases [33]. PSP cases with a family history showed atypical features more often, and the H1 haplotype was overrepresented in familial PSP compared to sporadic PSP.
SINGLE-GENE MUTATION AS A CAUSE OF PSP
Cases of familial or sporadic PSP caused by single-gene mutation, which includes the MAPT and LRRK2 genes, have been reported.
MAPT mutations in PSP
Tau protein, which is encoded by MAPT, is a microtubule-binding protein that is believed to be involved in assembly and stabilization of microtubules [34,35]. Neuropathological changes in tau protein accumulation is observed in Alzheimer’s disease, PSP, corticobasal degeneration (CBD), Pick’s disease and argyrophilic grain disease [36]. There are imperfect amino acid repeats in exons 9, 10, 11, and 12 which constitute the microtubule-binding region in tau protein. Alternative messenger ribonucleic acid splicing of MAPT in the human adult brain produces 6 isoforms of the tau protein. They differ from one another by the presence or absence of exons 2 or 3 in the amino-terminal half and inclusion, or not, of exon 10 in the carboxy-terminal half [35]. Exclusion of exon 10 leads to the production of 3-repeat isoforms, whereas its inclusion leads to 4-repeat isoforms. The expression levels of 3-repeat and 4-repeat isoforms are similar in normal adult human cerebral cortex, whereas those may change in neurodegenerative tauopathies [35].
Mutation of MAPT was identified as a causative gene for familial tauopathies, which have several different clinicopathological diagnoses [37-39]. These familial tauopathies caused by mutations of MAPT (FTDP-17T/MAPT) are characterized by frontal lobe dysexecutive symptoms, disinhibition, dementia, parkinsonism, amyotrophy or supranuclear gaze palsy [40] and are highly variable within or between families. FTDP-17T/MAPT may present with a variety of clinical syndromes such as frontotemporal lobar degeneration with parkinsonism, a behavioral variant of frontotemporal dementia, Pick disease, PSP-like, CBD-like, or neurodegeneration with overlapping features. Biochemical analysis of insoluble tau is also variable, which shows predominance of 3-repeat, or 4-repeat, or 3- and 4-repeat at a similar level [35].
The first MAPT mutation found in PSP was a p.R5L mutation in a sporadic case whose brain showed globose neurofibrillary tangles, neutrophil thread, and tufted astrocytes, which were stained with antitau antibodies in basal ganglia and brainstem [41]. Biochemical analysis of insoluble aggregates from subcortical regions showed predominance for four-repeat tau, similar to PSP. Additionally, a functional study revealed that the p.R5L allele of MAPT alters the ability of tau to promote microtubule assembly. Although pathological confirmation was not performed in all published cases of familial or sporadic PSP with a MAPT mutation, there are cases of PSP with a MAPT mutation diagnosed either clinically or pathologically [21,31]. Because the number of PSP cases included are small and studies were not systematically performed [21,41-43], the prevalence of a MAPT mutation in PSP cannot be estimated.
Currently, more than 40 different MAPT mutations have been shown to cause FTDP-17T/MAPT in individuals from over 100 families, while in PSP, 10 different MAPT mutations have been reported (Figure 1) [44-47]. Interestingly, except for the R5L mutation, all other variants (p.L284R, p.S285R, p.delN296, p.N296N, p.P301L, p.G303V, p.S305S, IVS10+3, and IVS10+6) are located in exon 10 and its splicing region. Seven mutations are in coding regions and the other two mutations are in the splicing region of exon 10. Patients with p.L284R or p.S285R MAPT mutations presented with only the PSP phenotype; however, both PSP and FTDP-17T/MAPT phenotypes were recognized in the other mutations. Given that PSP is 4-repeat tauopathy, mutations in MAPT exon 10 or its splicing regions may enhance production of 4 repeat tau isoforms [48,49] or alter conformation of tau protein and affect its degradation process.
Figure 1.
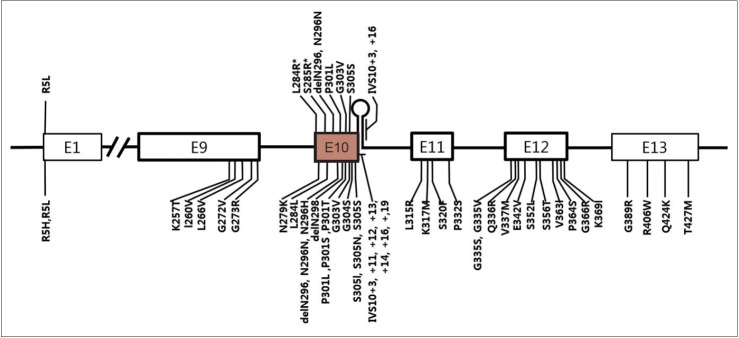
A schematic diagram showing exons of MAPT and locations of mutations. Mutations discovered in patients with progressive supranuclear palsy (PSP) or PSP-like phenotypes were marked in the upper half of the diagram and those with frontotemporal lobar degeneration presentation were marked in the lower half. E1 to E13 refer to number of exons. Note all mutations except for p.R5L are located in exon 10 or in stem-loop structure at the boundary between exon 10 and the intron following exon 10. Two mutations (p.L284R and p.S285R; marked with asterisk) were found only in patients with PSP phenotype.
LRRK2 mutations and PSP
Patients with levodopa-responsive parkinsonism in western Nebraska kindred in whom a LRRK2 (PARK8) R1441C mutation was discovered, pleomorphic pathology was reported in the autopsied brain [50]. Within this family, affected members showed Lewy body Parkinson’s disease, diffuse Lewy body disease, nigral degeneration without distinctive histopathology, or PSP-like pathology. The affected family members with PSP-like pathology had supranuclear palsy, neuronal loss in the substantia nigra, and tau pathologies (globose neurofibrillary tangles, tufted astrocytes, and oligodendroglial coiled bodies) without Lewy body or Lewy neuritis.
In an analysis of the LRRK2 mutation in familial or sporadic PD from Crete by Spanaki et al. [51], two parkinsonian brothers were identified with a R1441H mutation in LRRK2. In both siblings, parkinsonism began as typical levodopa-responsive parkinsonism followed by a relatively rapid deterioration after a decade. The brothers later showed postural instability, supranuclear gaze palsy, bulbar dysfunction, dementia and other symptoms/signs, which are typical for PSP. However, neuropathological studies of these cases were not reported.
Contrary to these two families suggesting that mutations in LRRK2 may be a genetic cause of PSP, other studies failed to detect a LRRK2 mutation in patients with PSP, although only small number of PSP cases were included for a LRRK2 mutation screen. Screening for LRRK2 mutations were negative in clinically diagnosed PSP patients of Caucasian origin [52,53] or of Asian origin [54]. A similar study of definite PSP cases in whom diagnosis was confirmed by neuropathology also reported negative results [55,56]. Taken together, these results suggest that mutations of LRRK2 in patients with PSP are rare.
Other locus for familial PSP
Currently, only one PSP locus has been identified by linkage analysis. Ros et al. [28] identified a PSP locus from a linkage analysis of a large Spanish family in which PSP was inherited in autosomal dominant fashion [26-28]. Clinical phenotypes were typical of PSP, although a minority of patients showed atypical features. Pathological diagnosis of PSP was confirmed according to the NINDS pathological criteria [27]. Haplotype analysis around a chromosomal region with a maximum multipoint LOD score identified a 3.4 cM candidate disease locus between markers D1S238 and D1S2823 on chromosome 1q31.1 [28]. However, no further progress has been published on the gene responsible [57]. Recently, familial PSP with a definite PSP pathology but negative for a MAPT mutation has been published, but no locus has been reported [21,31].
GENETIC RISK FACTORS ASSOCIATED WITH PSP
MAPT haplotypes and sub-haplotypes
Conrad et al. [58] identified an association of an intronic dinucleotide repeat polymorphic marker in MAPT with PSP, but not with other tauopathies such as Alzheimer’s disease or parkinsonism-dementia complex of Guam. Single nucleotide polymorphisms (SNPs) in an extended haplotype of 1.3 Mbp covering MAPT were found to be in complete linkage disequilibrium. Moreover, a common haplotype (H1 or MAPT H1-clade) was significantly over-represented in patients with PSP [59-61]. The identification of the H1 haplotype conferring a risk for PSP was consistently replicated in Caucasian populations. Although underlying molecular mechanisms conferred by the H1 haplotype have not been identified, supporting evidence in cell models is that the H1 haplotype enhanced MAPT expression [62]. The H2 haplotype that is present exclusively in Caucasians was found to be inversion polymorphism [63]. Using haplotype tagging SNPs spanning MAPT, Pittman et al. [64] identified sub-haplotypes within H1. They found that a common sub-haplotype (MAPT H1c) on the background of the MAPT H1-clade is overrepresented in PSP. MAPT H1c was also found overrepresented in patients with PSP-parkinsonism compared to controls [9]. The MAPT H1-clade was also found associated with an increased risk of PD, however, MAPT H1c is not involved [65,66]. MAPT H1c was associated with increased MAPT expression, especially the 4 repeat-containing transcript [67].
Genome-wide association study in PSP
To find additional PSP risk variants other than the MAPT H1 haplotype, a genome-wide association (GWA) study was performed using a pooled-DNA approach in 288 PSP patients from the US or Canada diagnosed based on neuropathology and 344 age- and sex-matched normal controls [68]. By assessing differences in allelic frequency, based on ranking differences in probe intensities for more than 500,000 SNPs between two cohorts, this study confirmed the MAPT H1 haplotype as a risk for PSP. A second major locus on chromosome 11p12-p11 showed evidence of association at allelic (p < 0.001), genotypic (p < 0.001), and haplotypic (p < 0.0001) levels. The DNA damage-binding protein-2 and lysosomal acid phosphatase-2 genes located in a single haplotype block of this locus were suggested as candidates for conferring risk of PSP.
Höeglinger et al. [69] carried out a GWA study in two stages. Stage 1, a discovery cohort consists of 1,114 autopsy-confirmed PSP cases and 3,247 controls of European ancestry. SNPs that passed a cutoff (p ≤ 10-3) in Stage 1 were genotyped in the second stage (an independent set of validation cohort), which consisted of 1,051 PSP cases that were diagnosed clinically and 3,560 controls. Joint analysis of stage 1 and 2 revealed that SNPs in STX6, MOBP, MAPT, and EIF2AK3 were associated with PSP. In the MAPT H1 region, after controlling H1/H2, some SNPs remained associated with the maximum falling in MAPT (rs242557). STX6 encodes syntaxin 6, a SNARE class protein that is involved in intracellular membrane trafficking along endocytic and secretory transport pathways [70]. A recent study showed that the GWA SNP rs1411478 in STX6 is a strong expression quantitative trait locus with significantly lower expression of STX6 in the white matter of carriers of the risk allele [71]. EIF2AK3 encodes PERK, a component of the endoplasmic reticulum (ER) unfolded protein response (UPR). When excess unfolded proteins accumulate in the ER, PERK is activated by dimerization and trans-autophosphorylation, which in turn phosphorylates eukaryotic translation-initiation factor 2α (eIF2α). eIF2α inhibits global translation, which allows ER to clear unfolded proteins. In the affected regions in PSP, the UPR was activated [72]. MOBP encodes MOBP protein, which is produced by oligodendrocytes and is present in the major dense line of central nervous system myelin. Regions of high MOBP expression overlap with those affected in PSP, suggesting that dysfunction of myelin or oligodendrocytes is involved in the PSP pathogenesis. Genotype-expression correlation analysis showed that SNP alleles across the entire H1/H2 inversion and flanking regions show strong correlation with MAPT expression. However, after controlling for H1/H2, all significant genotype-expression correlation for MAPT disappeared [69]. CBD is a neurodegenerative disorder with tauopathy that mimics PSP. Recently, a GWA study for CBD was reported to share common risk variants with PSP, which included rs1768208 at MOBP and rs242557 in MAPT H1c [73].
HEREDITARY NEURODEGENERATIVE DISORDERS MIMICKING PSP: PSP-LOOK-ALIKES
The expression of “look-alikes” in neurodegenerative disorders was first coined by Dr. Marsden in describing five cases whose symptoms and signs on clinical grounds were compatible with typical CBD, but who turned out not to have CBD, but rather, other disorders, such as periventricular ischemic lesion with multiple infarction, sudanophilic leurkodystrophy, Pick disease and PSP in postmortem examination [74]. As current understanding of the genetics and molecular pathology of neurodegenerative disorders has been advancing, the complicated relationship of convergence and divergence among clinical manifestation, pathology and causative genes has been recognized [75,76]. Differentiation of a hereditary neurodegenerative disease from a similar phenotypic syndrome may be important with regard to different prognoses and genetic counseling, as well as potential therapeutic and research implications [77,78]. “PSP-look-alikes” refer to hereditary neurodegenerative disorders that clinically mimic PSP, showing parkinsonism and early supranuclear gaze palsy, but that are not compatible with neuropathology of PSP. Recently, PSP-look-alikes were extensively reviewed by Stamelou et al. [79] PSP-look-alike disorders include frontotemporal lobar degeneration caused by mutations in PGRN, C9orf72, CHMP2B or FUS, Perry syndrome (mutations in DCTN1), Kufor-Rakeb disease (mutations in ATP13A2), Niemann-Pick type C (mutations in NPC1), Gaucher disease (mutations in GBA), mitochondrial disorders, and familial Creutzfeldt-Jakob disease (mutations in PRNP).
CONCLUSIONS
Our understanding of the genetic background of PSP has been expanding rapidly. This knowledge will not only help improve accurate diagnosis of PSP and other PSP-look-alike disorders, but will also provide insight into the pathogenesis, toward the eventually development of therapeutic strategies.
Acknowledgments
This research was supported by the basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2013R1A1A4A01007783).
Footnotes
Conflicts of Interest
The authors have no financial conflicts of interest.
REFERENCES
- 1.Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol. 1964;10:333–359. doi: 10.1001/archneur.1964.00460160003001. [DOI] [PubMed] [Google Scholar]
- 2.Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47:1–9. doi: 10.1212/wnl.47.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy) Neurology. 1994;44:2015–2019. doi: 10.1212/wnl.44.11.2015. [DOI] [PubMed] [Google Scholar]
- 4.Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol. 2010;23:394–400. doi: 10.1097/WCO.0b013e32833be924. [DOI] [PubMed] [Google Scholar]
- 5.Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain. 2005;128(Pt 6):1247–1258. doi: 10.1093/brain/awh488. [DOI] [PubMed] [Google Scholar]
- 6.Tsuboi Y, Josephs KA, Boeve BF, Litvan I, Caselli RJ, Caviness JN, et al. Increased tau burden in the cortices of progressive supranuclear palsy presenting with corticobasal syndrome. Mov Disord. 2005;20:982–988. doi: 10.1002/mds.20478. [DOI] [PubMed] [Google Scholar]
- 7.Josephs KA, Boeve BF, Duffy JR, Smith GE, Knopman DS, Parisi JE, et al. Atypical progressive supranuclear palsy underlying progressive apraxia of speech and nonfluent aphasia. Neurocase. 2005;11:283–296. doi: 10.1080/13554790590963004. [DOI] [PubMed] [Google Scholar]
- 8.Josephs KA, Katsuse O, Beccano-Kelly DA, Lin WL, Uitti RJ, Fujino Y, et al. Atypical progressive supranuclear palsy with corticospinal tract degeneration. J Neuropathol Exp Neurol. 2006;65:396–405. doi: 10.1097/01.jnen.0000218446.38158.61. [DOI] [PubMed] [Google Scholar]
- 9.Williams DR, Pittman AM, Revesz T, Lees AJ, de Silva R. Genetic variation at the tau locus and clinical syndromes associated with progressive supranuclear palsy. Mov Disord. 2007;22:895–897. doi: 10.1002/mds.21393. [DOI] [PubMed] [Google Scholar]
- 10.Han HJ, Kim H, Park JH, Shin HW, Kim GU, Kim DS, et al. Behavioral changes as the earliest clinical manifestation of progressive supranuclear palsy. J Clin Neurol. 2010;6:148–151. doi: 10.3988/jcn.2010.6.3.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Josephs KA, Eggers SD, Jack CR, Jr, Whitwell JL. Neuroanatomical correlates of the progressive supranuclear palsy corticobasal syndrome hybrid. Eur J Neurol. 2012;19:1440–1446. doi: 10.1111/j.1468-1331.2012.03726.x. [DOI] [PubMed] [Google Scholar]
- 12.Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol. 2009;8:270–279. doi: 10.1016/S1474-4422(09)70042-0. [DOI] [PubMed] [Google Scholar]
- 13.Williams DR, Holton JL, Strand K, Revesz T, Lees AJ. Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Mov Disord. 2007;22:2235–2241. doi: 10.1002/mds.21698. [DOI] [PubMed] [Google Scholar]
- 14.Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR, et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology. 2006;66:41–48. doi: 10.1212/01.wnl.0000191307.69661.c3. [DOI] [PubMed] [Google Scholar]
- 15.Hassan A, Parisi JE, Josephs KA. Autopsy-proven progressive supranuclear palsy presenting as behavioral variant frontotemporal dementia. Neurocase. 2012;18:478–488. doi: 10.1080/13554794.2011.627345. [DOI] [PubMed] [Google Scholar]
- 16.Golbe LI, Davis PH, Schoenberg BS, Duvoisin RC. Prevalence and natural history of progressive supranuclear palsy. Neurology. 1988;38:1031–1034. doi: 10.1212/wnl.38.7.1031. [DOI] [PubMed] [Google Scholar]
- 17.Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet. 1999;354:1771–1775. doi: 10.1016/s0140-6736(99)04137-9. [DOI] [PubMed] [Google Scholar]
- 18.Nath U, Ben-Shlomo Y, Thomson RG, Morris HR, Wood NW, Lees AJ, et al. The prevalence of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) in the UK. Brain. 2001;124(Pt 7):1438–1449. doi: 10.1093/brain/124.7.1438. [DOI] [PubMed] [Google Scholar]
- 19.Kawashima M, Miyake M, Kusumi M, Adachi Y, Nakashima K. Prevalence of progressive supranuclear palsy in Yonago, Japan. Mov Disord. 2004;19:1239–1240. doi: 10.1002/mds.20149. [DOI] [PubMed] [Google Scholar]
- 20.Bower JH, Maraganore DM, McDonnell SK, Rocca WA. Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology. 1997;49:1284–1288. doi: 10.1212/wnl.49.5.1284. [DOI] [PubMed] [Google Scholar]
- 21.Donker Kaat L, Boon AJ, Azmani A, Kamphorst W, Breteler MM, Anar B, et al. Familial aggregation of parkinsonism in progressive supranuclear palsy. Neurology. 2009;73:98–105. doi: 10.1212/WNL.0b013e3181a92bcc. [DOI] [PubMed] [Google Scholar]
- 22.David NJ, Mackey EA, Smith JL. Further observations in progressive supranuclear palsy. Neurology. 1968;18:349–356. doi: 10.1212/wnl.18.4.349. [DOI] [PubMed] [Google Scholar]
- 23.Mata M, Dorovini-Zis K, Wilson M, Young AB. New form of familial Parkinson-dementia syndrome: clinical and pathologic findings. Neurology. 1983;33:1439–1443. doi: 10.1212/wnl.33.11.1439. [DOI] [PubMed] [Google Scholar]
- 24.Ohara S, Kondo K, Morita H, Maruyama K, Ikeda S, Yanagisawa N. Progressive supranuclear palsy-like syndrome in two siblings of a consanguineous marriage. Neurology. 1992;42:1009–1014. doi: 10.1212/wnl.42.5.1009. [DOI] [PubMed] [Google Scholar]
- 25.Brown J, Lantos P, Stratton M, Roques P, Rossor M. Familial progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 1993;56:473–476. doi: 10.1136/jnnp.56.5.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Yébenes JG, Sarasa JL, Daniel SE, Lees AJ. Familial progressive supranuclear palsy. Description of a pedigree and review of the literature. Brain. 1995;118(Pt 5):1095–1103. doi: 10.1093/brain/118.5.1095. [DOI] [PubMed] [Google Scholar]
- 27.Rojo A, Pernaute RS, Fontán A, Ruíz PG, Honnorat J, Lynch T, et al. Clinical genetics of familial progressive supranuclear palsy. Brain. 1999;122(Pt 7):1233–1245. doi: 10.1093/brain/122.7.1233. [DOI] [PubMed] [Google Scholar]
- 28.Ros R, Gómez Garre P, Hirano M, Tai YF, Ampuero I, Vidal L, et al. Genetic linkage of autosomal dominant progressive supranuclear palsy to 1q31.1. Ann Neurol. 2005;57:634–641. doi: 10.1002/ana.20449. [DOI] [PubMed] [Google Scholar]
- 29.Gazeley S, Maguire JA. Familial progressive supranuclear palsy. Clin Neuropathol. 1996;15:215–220. [PubMed] [Google Scholar]
- 30.Tetrud JW, Golbe LI, Forno LS, Farmer PM. Autopsyproven progressive supranuclear palsy in two siblings. Neurology. 1996;46:931–934. doi: 10.1212/wnl.46.4.931. [DOI] [PubMed] [Google Scholar]
- 31.Fujioka S, Sanchez Contreras MY, Strongosky AJ, Ogaki K, Whaley NR, Tacik PM, et al. Three sib-pairs of autopsyconfirmed progressive supranuclear palsy. Parkinsonism Relat Disord. 2015;21:101–105. doi: 10.1016/j.parkreldis.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujioka S, Van Gerpen JA, Uitti RJ, Dickson DW, Wszolek ZK. Familial progressive supranuclear palsy: a literature review. Neurodegener Dis. 2014;13:180–182. doi: 10.1159/000354975. [DOI] [PubMed] [Google Scholar]
- 33.Fujioka S, Algom AA, Murray ME, Strongosky A, Soto-Ortolaza AI, Rademakers R, et al. Similarities between familial and sporadic autopsy-proven progressive supranuclear palsy. Neurology. 2013;80:2076–2078. doi: 10.1212/WNL.0b013e318294b2eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goedert M. Neurofibrillary pathology of Alzheimer’s disease and other tauopathies. Prog Brain Res. 1998;117:287–306. doi: 10.1016/s0079-6123(08)64022-4. [DOI] [PubMed] [Google Scholar]
- 35.Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta. 2005;1739:240–250. doi: 10.1016/j.bbadis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 36.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 37.Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 38.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 39.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park HK, Chung SJ. New perspective on parkinsonism in frontotemporal lobar degeneration. J Mov Disord. 2013;6:1–8. doi: 10.14802/jmd.13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H, et al. An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol. 2002;52:511–516. doi: 10.1002/ana.10340. [DOI] [PubMed] [Google Scholar]
- 42.Kim HJ, Jeon BS, Yun JY, Seong MW, Park SS, Lee JY. Screening for MAPT and PGRN mutations in Korean patients with PSP/CBS/FTD. Parkinsonism Relat Disord. 2010;16:305–306. doi: 10.1016/j.parkreldis.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 43.Ogaki K, Li Y, Takanashi M, Ishikawa K, Kobayashi T, Nonaka T, et al. Analyses of the MAPT, PGRN, and C9orf72 mutations in Japanese patients with FTLD, PSP, and CBS. Parkinsonism Relat Disord. 2013;19:15–20. doi: 10.1016/j.parkreldis.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 44.Goedert M, Spillantini MG. Tau mutations in frontotemporal dementia FTDP-17 and their relevance for Alzheimer’s disease. Biochim Biophys Acta. 2000;1502:110–121. doi: 10.1016/s0925-4439(00)00037-5. [DOI] [PubMed] [Google Scholar]
- 45.Rohrer JD, Paviour D, Vandrovcova J, Hodges J, de Silva R, Rossor MN. Novel L284R MAPT mutation in a family with an autosomal dominant progressive supranuclear palsy syndrome. Neurodegener Dis. 2011;8:149–152. doi: 10.1159/000319454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goedert M, Ghetti B, Spillantini MG. Frontotemporal dementia: implications for understanding Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006254. doi: 10.1101/cshperspect.a006254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat. 2012;33:1340–1344. doi: 10.1002/humu.22117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skoglund L, Viitanen M, Kalimo H, Lannfelt L, Jönhagen ME, Ingelsson M, et al. The tau S305S mutation causes frontotemporal dementia with parkinsonism. Eur J Neurol. 2008;15:156–161. doi: 10.1111/j.1468-1331.2007.02017.x. [DOI] [PubMed] [Google Scholar]
- 49.Stanford PM, Halliday GM, Brooks WS, Kwok JB, Storey CE, Creasey H, et al. Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: expansion of the disease phenotype caused by tau gene mutations. Brain. 2000;123(Pt 5):880–893. doi: 10.1093/brain/123.5.880. [DOI] [PubMed] [Google Scholar]
- 50.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 51.Spanaki C, Latsoudis H, Plaitakis A. LRRK2 mutations on Crete: R1441H associated with PD evolving to PSP. Neurology. 2006;67:1518–1519. doi: 10.1212/01.wnl.0000239829.33936.73. [DOI] [PubMed] [Google Scholar]
- 52.Hernandez D, Paisan Ruiz C, Crawley A, Malkani R, Werner J, Gwinn-Hardy K, et al. The dardarin G 2019 S mutation is a common cause of Parkinson’s disease but not other neurodegenerative diseases. Neurosci Lett. 2005;389:137–139. doi: 10.1016/j.neulet.2005.07.044. [DOI] [PubMed] [Google Scholar]
- 53.Madzar D, Schulte C, Gasser T. Screening for LRRK2 R1441 mutations in a cohort of PSP patients from Germany. Eur J Neurol. 2009;16:1230–1232. doi: 10.1111/j.1468-1331.2009.02702.x. [DOI] [PubMed] [Google Scholar]
- 54.Tan EK, Skipper L, Chua E, Wong MC, Pavanni R, Bonnard C, et al. Analysis of 14 LRRK2 mutations in Parkinson’s plus syndromes and late-onset Parkinson’s disease. Mov Disord. 2006;21:997–1001. doi: 10.1002/mds.20875. [DOI] [PubMed] [Google Scholar]
- 55.Ross OA, Whittle AJ, Cobb SA, Hulihan MM, Lincoln SJ, Toft M, et al. Lrrk2 R1441 substitution and progressive supranuclear palsy. Neuropathol Appl Neurobiol. 2006;32:23–25. doi: 10.1111/j.1365-2990.2006.00693.x. [DOI] [PubMed] [Google Scholar]
- 56.Gaig C, Ezquerra M, Martí MJ, Valldeoriola F, Muñoz E, Lladó A, et al. Screening for the LRRK2 G2019S and codon-1441 mutations in a pathological series of parkinsonian syndromes and frontotemporal lobar degeneration. J Neurol Sci. 2008;270:94–98. doi: 10.1016/j.jns.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 57.Pastor P. Familial neurodegeneration in progressive supranuclear palsy: more frequent than expected? Neurology. 2009;73:86–87. doi: 10.1212/WNL.0b013e3181aa2a71. [DOI] [PubMed] [Google Scholar]
- 58.Conrad C, Andreadis A, Trojanowski JQ, Dickson DW, Kang D, Chen X, et al. Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann Neurol. 1997;41:277–281. doi: 10.1002/ana.410410222. [DOI] [PubMed] [Google Scholar]
- 59.Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, et al. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999;8:711–715. doi: 10.1093/hmg/8.4.711. [DOI] [PubMed] [Google Scholar]
- 60.Pastor P, Ezquerra M, Perez JC, Chakraverty S, Norton J, Racette BA, et al. Novel haplotypes in 17q21 are associated with progressive supranuclear palsy. Ann Neurol. 2004;56:249–258. doi: 10.1002/ana.20178. [DOI] [PubMed] [Google Scholar]
- 61.Pittman AM, Myers AJ, Duckworth J, Bryden L, Hanson M, Abou-Sleiman P, et al. The structure of the tau haplotype in controls and in progressive supranuclear palsy. Hum Mol Genet. 2004;13:1267–1274. doi: 10.1093/hmg/ddh138. [DOI] [PubMed] [Google Scholar]
- 62.Kwok JB, Teber ET, Loy C, Hallupp M, Nicholson G, Mellick GD, et al. Tau haplotypes regulate transcription and are associated with Parkinson’s disease. Ann Neurol. 2004;55:329–334. doi: 10.1002/ana.10826. [DOI] [PubMed] [Google Scholar]
- 63.Rademakers R, Melquist S, Cruts M, Theuns J, Del-Favero J, Poorkaj P, et al. High-density SNP haplotyping suggests altered regulation of tau gene expression in progressive supranuclear palsy. Hum Mol Genet. 2005;14:3281–3292. doi: 10.1093/hmg/ddi361. [DOI] [PubMed] [Google Scholar]
- 64.Pittman AM, Myers AJ, Abou-Sleiman P, Fung HC, Kaleem M, Marlowe L, et al. Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J Med Genet. 2005;42:837–846. doi: 10.1136/jmg.2005.031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vandrovcova J, Pittman AM, Malzer E, Abou-Sleiman PM, Lees AJ, Wood NW, et al. Association of MAPT haplotypetagging SNPs with sporadic Parkinson’s disease. Neurobiol Aging. 2009;30:1477–1482. doi: 10.1016/j.neurobiolaging.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 66.Ezquerra M, Pastor P, Gaig C, Vidal-Taboada JM, Cruchaga C, Muñoz E, et al. Different MAPT haplotypes are associated with Parkinson’s disease and progressive supranuclear palsy. Neurobiol Aging. 2011;32:547. doi: 10.1016/j.neurobiolaging.2009.09.011. e11-e16. [DOI] [PubMed] [Google Scholar]
- 67.Myers AJ, Pittman AM, Zhao AS, Rohrer K, Kaleem M, Marlowe L, et al. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis. 2007;25:561–570. doi: 10.1016/j.nbd.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 68.Melquist S, Craig DW, Huentelman MJ, Crook R, Pearson JV, Baker M, et al. Identification of a novel risk locus for progressive supranuclear palsy by a pooled genomewide scan of 500,288 single-nucleotide polymorphisms. Am J Hum Genet. 2007;80:769–778. doi: 10.1086/513320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Höglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43:699–705. doi: 10.1038/ng.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jung JJ, Inamdar SM, Tiwari A, Choudhury A. Regulation of intracellular membrane trafficking and cell dynamics by syntaxin-6. Biosci Rep. 2012;32:383–391. doi: 10.1042/BSR20120006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ferrari R, Ryten M, Simone R, Trabzuni D, Nicolaou N, Hondhamuni G, et al. Assessment of common variability and expression quantitative trait loci for genome-wide associations for progressive supranuclear palsy. Neurobiol Aging. 2014;35:1514. doi: 10.1016/j.neurobiolaging.2014.01.010. e1-e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stutzbach LD, Xie SX, Naj AC, Albin R, Gilman S, PSP Genetics Study Group et al. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol Commun. 2013;1:31. doi: 10.1186/2051-5960-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kouri N, Ross OA, Dombroski B, Younkin CS, Serie DJ, Soto-Ortolaza A, et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun. 2015;6:7247. doi: 10.1038/ncomms8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bhatia KP, Lee MS, Rinne JO, Revesz T, Scaravilli F, Davies L, et al. Corticobasal degeneration look-alikes. Adv Neurol. 2000;82:169–182. [PubMed] [Google Scholar]
- 75.Klein C, Schneider SA, Lang AE. Hereditary parkinsonism: Parkinson disease look-alikes--an algorithm for clinicians to “PARK” genes and beyond. Mov Disord. 2009;24:2042–2058. doi: 10.1002/mds.22675. [DOI] [PubMed] [Google Scholar]
- 76.Karageorgiou E, Miller BL. Frontotemporal lobar degeneration: a clinical approach. Semin Neurol. 2014;34:189–201. doi: 10.1055/s-0034-1381735. [DOI] [PubMed] [Google Scholar]
- 77.Halliday GM, Song YJ, Creasey H, Morris JG, Brooks WS, Kril JJ. Neuropathology in the S305S tau gene mutation. Brain. 2006;129(Pt 3):E40. doi: 10.1093/brain/awh720. [DOI] [PubMed] [Google Scholar]
- 78.Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80:496–503. doi: 10.1212/WNL.0b013e31827f0fd1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stamelou M, Quinn NP, Bhatia KP. “Atypical” atypical parkinsonism: new genetic conditions presenting with features of progressive supranuclear palsy, corticobasal degeneration, or multiple system atrophy-a diagnostic guide. Mov Disord. 2013;28:1184–1199. doi: 10.1002/mds.25509. [DOI] [PubMed] [Google Scholar]