

Abstract
Flavobacterium rivuli Ali et al. 2009 emend. Dong et al. 2013 is one of about 100 species in the genus Flavobacterium (family Flavobacteriacae, phylum Bacteroidetes) with a validly published name, and has been isolated from the spring of a hard water rivulet in Northern Germany. Including all type strains of the genus Myroides and Flavobacterium into the 16S rRNA gene sequence phylogeny revealed a clustering of members of the genus Myroides as a monophyletic group within the genus Flavobacterium. Furthermore, F. rivuli WB 3.3-2T and its next relatives seem more closely related to the genus Myroides than to the type species of the genus Flavobacterium, F. aquatile. The 4,489,248 bp long genome with its 3,391 protein-coding and 65 RNA genes is part of the GenomicEncyclopedia ofBacteria andArchaea project. The genome of F. rivuli has almost as many genes encoding carbohydrate active enzymes (151 CAZymes) as genes encoding peptidases (177). Peptidases comprised mostly metallo (M) and serine (S) peptidases. Among CAZymes, 30 glycoside hydrolase families, 10 glycosyl transferase families, 7 carbohydrate binding module families and 7 carbohydrate esterase families were identified. Furthermore, we found four polysaccharide utilization loci (PUL) and one large CAZy rich gene cluster that might enable strain WB 3.3-2T to decompose plant and algae derived polysaccharides. Based on these results we propose F. rivuli as an interesting candidate for further physiological studies and the role of Bacteroidetes in the decomposition of complex polymers in the environment.
Electronic supplementary material
The online version of this article (doi:10.1186/s40793-015-0032-y) contains supplementary material, which is available to authorized users.
Keywords: Carbohydrate active enzyme, Polysaccharide utilization loci, Gram-negative, Non-motile, Aerobic, Hard water rivulet, Flavobacteriaceae, Bacteroidetes, GEBA-KMG I, Myroides
Introduction
Strain WB 3.3-2T (=DSM 21788T = CIP 109865T) is the type strain of Flavobacterium rivuli [1, 22]. The genus Flavobacterium, the type genus [12, 36] of the family Flavobacteriaceae [13], was proposed in the first edition of Bergey’s Manual of Determinative Bacteriology in 1923 [10]. Flavobacteriaceae have been isolated from soil, freshwater, marine and saline environments [13]. However, members of the Cytophaga/Flavobacteria group have been found with greater abundances in rivers and oceans [39], which was attributed to their important role in the decomposition of algal-derived organic matter [24, 39, 70]. F. rivuli WB 3.3-2T has been isolated from a hardwater rivulet in the Harz Mountains, Germany [17]. Therefore, we selected the freshwater strain WB 3.3-2T as a candidate for comparing its polysaccharide decomposition potential with the one of marine Flavobacteriaceae.
Here we present the set of carbohydrate active enzymes, polysaccharide utilization loci and peptidases of strain WB 3.3-2T, together with a summary of its present classification, the set of known phenotypic features and a description of the permanent draft genome sequencing and annotation derived from a culture of strain DSM 21788T.
Organism information
Classification and features
The draft genome of F. rivuli DSM 21788T (ARKJ00000000) has one full-length 16S rRNA gene sequence (Q765_20790, 1415 bp) and one partial 16S rRNA gene sequence (Q765_20790, 594 bp) which were both 100 % identical with the sequence from the original species description (AM934661, NR_115084) [1]. BLAST search revealed the presence of a closely related strain CH1-10 (JX971542, 98.4 %) from a mushroom, two closely related (98.5 %) clone sequences from floor dust (FM872607, FM872591) [69], and two clone sequences from human skin (HM274288, HM269957, 98.2 %).
The next related species was Flavobacteriumsubsaxonensis WB 4.1-42T [1], whereas other affiliations are poorly supported (Fig. 1). In contrast to the original affiliation with the genus Flavobacterium, F. rivuli WB 3.3-2T belongs to a group of Flavobacterium species which seem more closely related to the genus Myroides [71] than to the type species of Flavobacterium, F. aquatile [10, 15, 29] (Fig. 2). However, the backbone of the 16S rRNA gene phylogenetic tree is essentially unresolved. A summary of the classification and general features of F. rivuli WB 3.3-2T is shown in Table 1. Cells of strain WB 3.3-2T are Gram-negative, aerobic to microaerobic, non-motile (flagella are absent) and non-gliding, catalase- and oxidase-positive 0.4–0.6 × 1.5–2.0 μm rods which produce extracellular polymeric substances (EPS) (Fig. 3). Colonies are pearl-white on R2A and CY agars and yellow on TSA and NA agars. Flexirubin pigments are absent. Sparse growth occurs between 4 and 8 °C and no growth was observed above 29 °C; the growth optimum is between 16 and 24 °C. Growth occurs between pH 6.4 and 7.8 (optimum 7.0) and at NaCl concentrations between 0 and 2 % (w/v) with an optimum at 1 % (w/v). Nitrate reduction is negative. The strain hydrolyses aesculin, cellobiose, glycogen, starch, Tween 40 and Tween 80, but not alginate, caseine, cellulose, chitin, DNA and pectin. The tests for β-galactosidase and acid phosphatase are strongly positive. Other physiological properties are available for the API ZYM and API 20NE systems (bioMérieux) and the GN MicroPlate system (Biolog) substrate panels [1]. Maltose and other carbohydrates are assimilated. Properties that can be used for the differentiation from the closely related type strain of F. subsaxonicum are, according to the substrates provided by the GN MicroPlate, positive utilization of acetic acid, α-d-lactose, trehalose and Tween 40, and lack of utilization of l-alanine, l-fucose, α-ketobutyric acid, dl-lactic acid, methyl ß-d-glucoside, l-ornithine, l-rhamnose and l-serine.
Fig. 1.
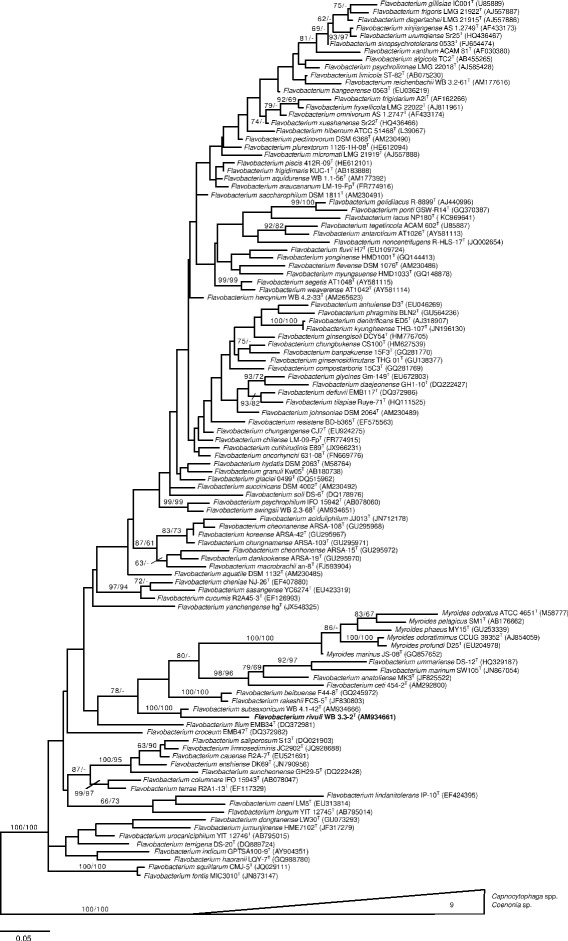
Phylogenetic tree of the genus Flavobacterium and its most closely related genus Capnocytophaga. The tree was inferred from 1,254 aligned characters of the 16S rRNA gene sequence under the maximum likelihood (ML) criterion as previously described [34]. The sequences were aligned using poa [45] and the resulting alignment restricted to its conserved part using Gblocks [20]. The branches are scaled in terms of the expected number of substitutions per site. Numbers adjacent to the branches are support values from 1,000 ML bootstrap replicates (left) and from 1,000 maximum-parsimony bootstrap replicates (right) if larger than 60 % [34]. Acccession numbers of 16S rRNA gene sequences are listed in Acccession numbers of 16S rRNA gene sequences are listed in Additional file 1: suppl. Table 6
Fig. 2.
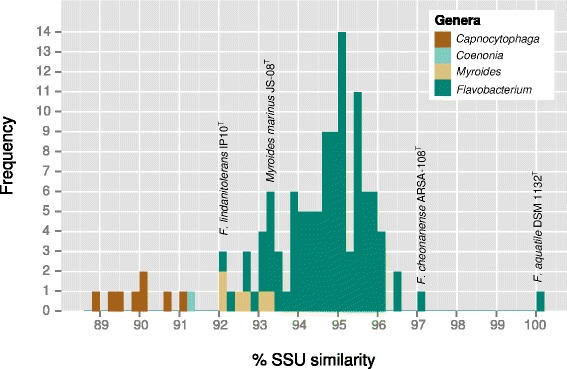
Histogram showing the distribution of pairwise SSU similarities of the type species Flavobacterium aquatile with respect to all other 119 strains in the dataset. Except the genus Myroides, all genera are clearly segregated from each other. Pairwise SSU similarities were calculated using the recommended approach described in [55]. Bars are colored according to genus affiliation. The figure was visualized using the ggplot package [72] for the R statistical framework [63]. Acccession numbers of 16S rRNA gene sequences are listed in suppl. Table 6
Fig. 3.
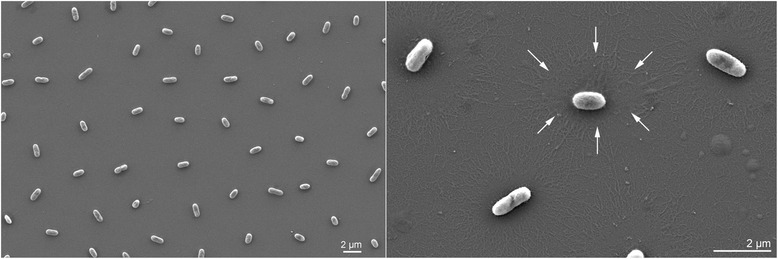
Scanning electron micrograph of F. rivuli WB 3.3-2T (DSM 21788T) showing expression of extracellular polymeric substances, EPS (arrows)
Chemotaxonomic data
Major fatty acids (>5 % of total) are i-C15:0, ai-C15:0, C16:0, C16:0 3-OH, iso-C17 : 0 3-OH and, as main component, summed feature C16 : 1 ω7c and/or iso-C15 : 0 2-OH [1]. Although the original publication indicates that “summed feature 3” is present (C16 : 1 ω7c and/or iso-C15 : 0 2-OH) and is generally explained as “summed features are groups of two or three fatty acids that cannot be separated by GLC using the MIDI System” this is a misrepresentation of information provided by MIDI Inc as well as a failure to further inspect the final results. Re-examination of the original data held in the DSMZ indicates that a single peak is present with an ECL of 15.819, coinciding with the ECL of C16 :1 ω7c in the MIDI Sherlock TSBA40 peak naming table, indicating that C16:1 ω7c is present and iso-C15:0 2-OH is absent. While these differences may appear trivial this information can be linked back to the enzymes (their encoding genes) and biosynthetic pathways leading to the synthesis of these two very different fatty acids as has been pointed out previously by [57, 58]. No data are available on respiratory quinone, peptidoglycan, polar lipid, polyamine and whole-cell sugar composition. The DNA G + C content of the type strain was previously determined as 40.4 mol% [1].
The genera Flavobacterium and Myroides
Figures 1 and 2 give an overview of the phylogenetic relationships of members of the genus Flavobacterium based on the comparison of 16S rRNA gene sequences (see list in Additional file 1: Table S1). In addition members of the genus Myroides are included and members of the genus Capnocytophaga and Coenonia are used as outgroups. Members of the genera Flavobacterium and Myroides form a monophyletic group, but the division of that monophyletic group to produce a monophyletic group including all members of the genus Myroides does not result in members of the genus Flavobacterium forming a monophyletic group. In such cases the genus Flavobacterium may be divided into several monophyletic groups or the group representing members of the genus Flavobacterium and may be described as being paraphyletic. If a genus is to be composed of species that constitute a monophyletic group then the present data suggest at least two alternatives. If one retains the genus Myroides as a monophyletic group then the division of the genus Flavobacterium into several monophyletic groups may need closer investigation, potentially resulting in the creation of several new genera. Alternatively, the fact that a monophyletic group is recovered that includes members of both the genera Flavobacterium and Myroides may be indicative of the inclusion of members of both taxa in a single genus, where the genus name Flavobacterium Bergey et al. [10] has priority over the genus name Myroides Vancanneyt et al. [44, 71]. The type species of the genus Myroides, Myroides odoratus (Stutzer [68]) Vancanneyt et al. [71] was originally named F. odoratum Stutzer [68], i.e. the two names are homotypic synonyms. The lowest 16S rRNA gene sequence pairwise similarity values between the type strain of the type species of the genus Flavobacterium, F. aquatile and other type strains of species considered to be members of the genus Flavobacterium is 92-93 %, close to the 16S rRNA gene sequence pairwise similarity value of 92 % to the type strain of the type species of Myroides, M. odoratum.
Genome sequencing information
Genome project history
F. rivuli DSM 21788T was selected for sequencing on the basis of its phylogenetic position [35, 40], and is part of Genomic Encyclopedia of Type Strains, Phase I: the one thousand microbial genomes project [43], a follow-up of the Genomic Encyclopedia of Bacteria and Archaea pilot project [74], which aims at increasing the sequencing coverage of key reference microbial genomes and to generate a large genomic basis for the discovery of genes encoding novel enzymes [61]. KMG-I is the first of the production phases of the “Genomic Encyclopedia of Bacteria and Archaea: sequencing a myriad of type strains initiative a [42] and a Genomic Standards Consortium project [27]. The genome project is deposited in the Genomes OnLine Database [59] and the permanent draft genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI) using state-of-the-art sequencing technology [49]. A summary of the project information is shown in Table 2.
Table 1.
Classification and general features of F. rivuli WB 3.3-2T in accordance with the MIGS recommendations [26], as developed by [25], List of Prokaryotic names with Standing in Nomenclature [23] and the Names for Life database [31]
| MIGS ID | Property | Term | Evidence code |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [73] | |
| Phylum Bacteroidetes | TAS [2, 41] | ||
| Class Flavobacteriia | TAS [3, 11] | ||
| Order Flavobacteriales | TAS [14, 65] | ||
| Family Flavobacteriaceae | TAS [13, 65] | ||
| Genus Flavobacterium | TAS [12, 36] | ||
| Species Flavobacterium rivuli | TAS [1] | ||
| type strain WB 3.3-2T | TAS [1] | ||
| Gram-stain | negative | TAS [1] | |
| Cell shape | rod-shaped | TAS [1] | |
| Motility | nonmotile | TAS [1] | |
| Sporulation | non-spore forming | TAS [13] | |
| Temperature range | mesophilic (4–29 °C) | TAS [1] | |
| Optimum temperature | 16–24 °C | TAS [1] | |
| pH range; Optimum | 6.4–7.8, 7 | TAS [1] | |
| Carbon source | Carbohydrates, peptides | TAS [1] | |
| MIGS-6 | Habitat | fresh water | TAS [1, 17] |
| MIGS-6.3 | Salinity | 0–2 % | TAS [1] |
| MIGS-22 | Oxygen requirement | obligate aerobe | TAS [1] |
| MIGS-15 | Biotic relationship | free-living | TAS [1, 17] |
| MIGS-14 | Pathogenicity | not reported | NAS |
| MIGS-4 | Geographic location | Harz Mountains, North Germany | TAS [1, 17] |
| MIGS-5 | Sample collection time | 9 June 2005 | TAS [1, 17] |
| MIGS-4.1 | Latitude | 51.758065 | TAS [1, 17] |
| MIGS-4.2 | Longitude | 10.11638 | TAS [1, 17] |
| MIGS-4.4 | Altitude | 273 m | TAS [17] |
Evidence codes - IDA, Inferred from Direct Assay (first time in publication), TAS traceable author statement (i.e., a direct report exists in the literature), NAS, non-traceable author statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). Evidence codes are from the Gene Ontology project [5]
Table 2.
Genome sequencing project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31.1 | Sequencing quality | Level 2: High-Quality Draft |
| MIGS-28.1 | Libraries method | Illumina Std shotgun library |
| MIGS-28.2 | Reads count | 14,972,538 sequencing reads |
| MIGS-29 | Sequencing method | Illumina HiSeq 2000, |
| MIGS-31.2 | Fold coverage | 124.1x |
| MIGS-30 | Assembly method | Velvet v. 1.1.04; ALLPATHS v. r41043 |
| MIGS-32 | Gene calling method | Prodigal, GenePRIMP, IMG-ER |
| NCBI project ID | 182404 | |
| Genbank ID | ARKJ00000000 | |
| Genbank Date of Release | 16-SEP-2013 | |
| IMG object ID | 2519103183 | |
| GOLD ID | Gi11501 | |
| MIGS-13 | Source Material Identifier | DSM 21788 |
| Project relevance | Tree of Life, GEBA-KMG |
Growth Conditions and genomic DNA preparation
A culture of DSM 21788T was grown aerobically in DSMZ medium 830 [4] at 20 °C. Genomic DNA was isolated using Jetflex Genomic DNA Purification Kit (GENOMED 600100) following the standard protocol provided by the manufacturer but modified by an incubation time of 60 min. the incubation on ice overnight on a shaker, the use of additional 50 μl proteinase K, and the addition of 200 μl protein precipitation buffer. DNA is available from DSMZ through the DNA Bank Network [32].
Genome sequencing and assembly
The draft genome of DSM 21788T was generated using the Illumina technology [9]. An Illumina Std. shotgun library was constructed and sequenced using the Illumina HiSeq 2000 platform which generated 14,972,538 reads totaling 2,245.9 Mbp (Table 3). All general aspects of library construction and sequencing performed at the JGI can be found at [21]. All raw sequence data were passed through DUK, a filtering program developed at JGI, which removes known Illumina sequencing and library preparation artifacts (Mingkun L, Copeland A, Han J, DUK. Unpublished). Following steps were performed for assembly: (1) filtered reads were assembled using Velvet [77], (2) 1–3 Kbp simulated paired end reads were created from Velvet contigs using wgsim [46], (3) Sequence reads were assembled with simulated read pairs using Allpaths–LG [33]. Parameters for assembly steps were: 1) Velvet (velveth: 63 –shortPaired and velvetg: –very clean yes –export- Filtered yes –min contig lgth 500 –scaffolding no –cov cutoff 10) 2) wgsim (–e 0 –1 100 –2 100 –r 0 –R 0 –X 0) 3) Allpaths–LG (PrepareAllpathsInputs: PHRED 64 = 1 PLOIDY = 1 FRAG COVERAGE = 125 JUMP COVERAGE = 25 LONG JUMP COV = 50, RunAllpathsLG: THREADS = 8 RUN = std shredpairs TARGETS = standard VAPI WARN ONLY = True OVERWRITE = True). The final draft assembly contained 26 contigs in 23 scaffolds, with three contigs shorter than the threshold used to generate Table 3. The total size of the genome is 4.5 Mbp and the final assembly is based on 560.1 Mbp of data, which provides a 124.1x average coverage of the genome.
Table 3.
Genome statistics
| Attribute | Number | % of Total |
|---|---|---|
| DNA, total number of bases | 4489248 | 100.0 |
| DNA coding number of bases | 3981399 | 88.7 |
| DNA G + C number of bases | 1777758 | 39.6 |
| DNA scaffolds | 23 | 100.0 |
| Genes total number | 4056 | 100.0 |
| Protein coding genes | 3991 | 98.4 |
| RNA genes | 65 | 1.6 |
| rRNA genes | 8 | 0.2 |
| 5S rRNA | 5 | 0.1 |
| 16S rRNA | 1 | <0.1 |
| 23S rRNA | 2 | 0.1 |
| tRNA genes | 48 | 1.2 |
| Other RNA genes | 9 | 0.2 |
| Protein coding genes with function prediction | 2842 | 70.1 |
| without function prediction | 1149 | 28.3 |
| Protein coding genes with COGs | 2570 | 63.4 |
| Protein coding genes with Pfam | 2924 | 72.1 |
| Protein coding genes coding signal peptides | 654 | 16.1 |
| Protein coding genes coding transmembrane proteins | 906 | 22.3 |
| CRISPR repeats | 0 |
Genome annotation
Genes were identified using Prodigal [37] as part of the DOE-JGI genome annotation pipeline [49], followed by manual curation using the JGI GenePRIMP pipeline [60]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) non-redundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro database. These data sources were combined to assert a product description for each predicted protein. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes-Expert Review (IMG-ER) platform [48].
Genome properties
The assembly of the draft genome sequence consists of 23 scaffolds amounting to 4,489,248 bp. The G + C content is 39.6 % (Table 3) which is similar to the G + C content determined by Ali et al. [1] and is within the acceptable range for a microbial species [56]. Of the 4,056 genes predicted, 3,991 were protein-coding genes, and 65 RNAs. The majority of the protein-coding genes (70.1 %) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.
Table 4.
Number of genes associated with the general COG functional categories
| Code | Value | % age | Description |
|---|---|---|---|
| J | 152 | 5.4 | Translation, ribosomal structure and biogenesis |
| A | 1 | 0.1 | RNA processing and modification |
| K | 209 | 7.4 | Transcription |
| L | 138 | 4.9 | Replication, recombination and repair |
| B | 1 | 0.1 | Chromatin structure and dynamics |
| D | 21 | 0.8 | Cell cycle control, cell division, chromosome partitioning |
| Y | 0 | 0.0 | Nuclear structure |
| V | 49 | 1.7 | Defense mechanisms |
| T | 177 | 6.3 | Signal transduction mechanisms |
| M | 237 | 8.4 | Cell wall/membrane/envelope biogenesis |
| N | 10 | 0.4 | Cell motility |
| Z | 0 | 0.0 | Cytoskeleton |
| W | 0 | 0.0 | Extracellular structures |
| U | 50 | 1.8 | Intracellular trafficking, secretion, and vesicular transport |
| O | 111 | 3.9 | Posttranslational modification, protein turnover, chaperones |
| C | 152 | 5.4 | Energy production and conversion |
| G | 201 | 7.2 | Carbohydrate transport and metabolism |
| E | 199 | 7.1 | Amino acid transport and metabolism |
| F | 67 | 2.4 | Nucleotide transport and metabolism |
| H | 124 | 4.4 | Coenzyme transport and metabolism |
| I | 109 | 3.9 | Lipid transport and metabolism |
| P | 133 | 4.7 | Inorganic ion transport and metabolism |
| Q | 48 | 1.7 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 360 | 12.8 | General function prediction only |
| S | 261 | 9.3 | Function unknown |
| - | 1486 | 36.6 | Not in COGs |
Insights from the genome sequence
Comparative genomics
Here we present a brief comparative genomics analysis of F. rivuli DSM 21788T with a selection of its closest phylogenetic neighbour (according to Fig. 1), F. subsaxonicum [1] (NZ_AUGP00000000), other potentially closely related species such as F. filum [66] (NZ_AUDM00000000) and F. beibuense [30] (NZ_JRLV00000000), as well as the genome of the type species of the genus Flavobacterium, F. aquatile [10, 15, 29] (NZ_JRHH00000000). The genomes of these five sequenced Flavobacterium type strains differ significantly in their size: F. rivuli 4.49 Mbp (see above), F. beibuense 3.8 Mbp, F. filum 3.19 Mbp, F. subsaxonicum 4.63 Mbp and F. aquatile 3.49 Mbp. Since these genome sequences have not been sequenced completely yet, the final values might change slightly in future analyses based on complete genome sequences.
An estimate of the overall similarity between F. rivuli and the other strains in this data set was generated with the Genome-to-Genome Distance Calculator (2.0) [6, 7, 53]. It calculates intergenomic distances by comparing two respective genomes to obtain HSPs (high-scoring segment pairs) and, afterwards, infers distances via a set of formulas (1, HSP length/total length; 2, identities/HSP length; 3, identities/total length). The GGDC also reports model-based DDH estimates (digital DDH or dDDH) along with their confidence intervals [53]. Since formula 2 is robust against the use of incomplete genome sequences (see above), it is especially suited for this type of analysis.
The result of this comparison is shown in Table 5 and yields dDDH of below 22 % throughout, which underlines the expected status of distinct species, as inferred from the 16S rRNA sequence similarities. Consequently, with 21.3 % dDDH F. subsaxonicum has the highest similarity to F. rivuli, whereas F. aquatile has the lowest similarity of 18.4 % dDDH. The comparison of F. rivuli with F. aquatile and F. filum reached the lowest value (2 %) regarding the average genome length covered with HSPs. This value was slightly increased (7 %) between F. rivuli and F. beibuense and clearly higher (31 %) with respect to F. subsaxonicum, the closest related species according to Fig. 1. The identity within the HSPs was 77 % on average, whereas the identity over the whole genome was 24 % regarding the comparison of F. rivuli with F. subsaxonicum, and, was even below 10 % regarding the remaining comparisons (Table 5).
Table 5.
Pairwise comparison of F. rivuli with F. filum, F. subsaxonicum, F. beibuense and F. aquatile using the GGDC (Genome-to-Genome Distance Calculator). Digital DDH (dDDH) and the respective confidence intervals (C.I.) are specified for GGDC’s recommended formula 2
| F. rivuli versus | % dDDH | % C.I. dDDH | HSP length/total length [%] | Identities/HSP length [%] | Identities/total length [%] |
|---|---|---|---|---|---|
| F. aquatile | 18.4 | 2.5 | 2 | 76 | 1 |
| F. beibuense | 18.7 | 2.6 | 7 | 76 | 6 |
| F. filum | 19.0 | 2.5 | 2 | 77 | 1 |
| F. subsaxonicum | 21.3 | 2.9 | 31 | 79 | 24 |
Gliding motility
The gliding motility machinery among Bacteroidetes is composed of adhesion-like proteins, an ATP-binding cassette transporter, the PorS secretion system, and additional proteins, as described by McBride and Zhu [51]. In the genome of F. rivuli all genes necessary for gliding motility were identified (Table 6). However, adhesin-like proteins comparable to the ones of F. johnsoniae UW101 were not found, and gliding motility of F. rivuli was not observed previously [1].
Table 6.
Gliding motility-related genes in strain DSM 21788T compared to genes in Flavobacterium strains studied by McBride and Zhu [51]
| F. rivuli DSM 21788T | F. psychrophilum JIP02/86T | F. johnsoniae ATCC 17061T | ||
|---|---|---|---|---|
| Locus tag prefix | F565_ RS01 | FP | Fjoh_ | |
| Gliding motility | – | + | + | |
| Adhesin-like | ||||
| remA | – | 1959 | 0808 | |
| remB | – | 2117 | 1657 | |
| sprB | – | 0016 | 0979 | |
| ATP-binding cassette transporter | ||||
| gldA | 05270 | 0252 | 1516 | |
| gldF | 00760 | 1089 | 2722 | |
| gldG | 00765 | 1090 | 2721 | |
| Additional protein required for gliding | ||||
| gldBa | 13390 | 2069 | 1793 | |
| gldC | 13385 | 2068 | 1794 | |
| gldDa | 18865 | 1663 | 1540 | |
| gldE | 18860 | 1358 | 1539 | |
| gldHa | 10515 | 0024 | 0890 | |
| gldJa | 11845 | 1389 | 1557 | |
| Peptidoprolyl isomerase (Flavobacteriia, protein folding) | ||||
| gldI | 08180 | 1892 | 2369 | |
| PorS secretion system (secretion of RemA/RemB and SprA/SprB) | ||||
| gldKa | 18605 | 1973 | 1853 | |
| gldLa | 18600 | 1972 | 1854 | |
| gldMa | 18595 | 1971 | 1855 | |
| gldNa | 18590 | 1970 | 1856 | |
| sprAa | 06065 | 2121 | 1653 | |
| sprEa | 19150 | 2467 | 1051 | |
| sprTa | 05475 | 0326 | 1466 | |
aessential gliding motility genes after McBride and Zhu [51]
Peptidases
The MEROPS [64] annotation was carried out by searching the sequences against MEROPS 9.10 (access date: 2014.10.16, version: pepunit.lib). F. rivuli processes 177 peptidases the majority of which were 59 metallo (M) and 89 serine (S) peptidases (Table 7 and Additional file 1: Table S2). Furthermore, the F. rivuli genome contained 22 I39, two I87 and one I71 simple peptidase inhibitors (Table 7 and Additional file 1: Table S3).
Table 7.
Peptidases and simple peptidase inhibitors in the genome of strain DSM 21788T
| Peptidase family | M01 | M03 | M12 | M13 | M14 | M16 | M19 | M20 | M23 |
|---|---|---|---|---|---|---|---|---|---|
| Counts | 6 | 2 | 2 | 2 | 8 | 3 | 1 | 5 | 8 |
| Peptidase family | M24 | M28 | M38 | M41 | M42 | M43 | M48 | M50 | M61 |
| Counts | 2 | 2 | 6 | 1 | 1 | 1 | 1 | 1 | 1 |
| Peptidase family | M75 | M79 | M90 | M96 | |||||
| Counts | 2 | 1 | 1 | 2 | |||||
| Peptidase family | S01 | S08 | S09 | S11 | S12 | S14 | S16 | S24 | S26 |
| Counts | 2 | 5 | 31 | 1 | 6 | 3 | 3 | 5 | 1 |
| Peptidase family | S33 | S41 | S46 | S49 | S51 | S54 | S66 | ||
| Counts | 16 | 6 | 3 | 1 | 2 | 3 | 2 | ||
| Peptidase family | C01 | C25 | C26 | C40 | C44 | C56 | C82 | ||
| Counts | 1 | 1 | 8 | 3 | 4 | 4 | 1 | ||
| Peptidase family | N11 | T02 | U32 | U73 | A08 | A28 | |||
| Counts | 1 | 1 | 2 | 1 | 1 | 1 | |||
| Inhibitor family | I39 | I71 | I87 | ||||||
| Counts | 22 | 1 | 2 |
Carbohydrate active enzymes
The CAZyme annotation was a combination of RAPSearch2 search [75, 78] and HMMER scanning [28]. The RAPSearch2 database was created from the protein sequences listed at the CAZy website [18, 47] (access date: 2014.09.18) while the profile HMMs were downloaded from dbCAN [76] (version: dbCAN-fam-HMMs.txt.v3). The outputs of these two program runs were compared and only their intersections were kept (i.e., loci confirmed by both methods). In case of conflicting family assignments, the RAPSearch2 results were preferred.
Overall, in its genome F. rivuli DSM 21788T possess a variety of carbohydrate active enzymes including 94 glycoside hydrolases (GH) belonging to 31 families, 11 carbohydrate binding modules (CBM) belonging to 7 families, 13 carbohydrate esterases (CE) belonging to 8 families, one polysaccharide lyase of family 11 (PL11) and 37 glycosyl transferases belonging to 11 families (Table 8 and Additional file 1: Table S4). The carbohydrate esterases CE2, CE6, CE7, CE12 might act as carboxylic-ester hydrolases (EC 3.1.1.-) and the carbohydrate esterases CE11, CE14 as linear amides (EC 3.5.1.-). The genome of strain DSM 21788T comprised a set of four GH5 and three GH51, for the potential hydrolysis of various cellulose or xylan polysaccharides. The absence of GH50, GH86 (agarose hydrolysis), GH18, GH19, GH20 (chitin hydrolysis) and a gene for alginate lyase (EC 4.2.2.3) corroborate the results of Ali et al. [1] that F. rivuli can not hydrolyze agarose, chitin and alginate, respectively. F. rivuli is equipped with one GH1, five GH5 and three GH30 as potential β-glucosidases and was shown to utilize cellobiose (d-Glc-β(1 → 4)-d-Glc) but not cellulose [1]. Gentobiose (d-Glc-β(1 → 6)-d-Glc) utilization and β-galactosidase activity was shown for F. rivuli [1] which has one GH1, fifteen GH2, eleven GH3 and one GH42 encoded in its genome. Starch was hydrolyzed by F. rivuli [1] presumably by enzyme activity of the four GH13 (α-amylase) and trehalose [1] by four GH13, one GH37, one GH65 (trehalase). The products of starch hydrolysis, maltose and d-glucose, can be utilized by F. rivuli [1]. Melibiose (d-Gal-α(1 → 6)-d-Glc) was metabolized by F. rivuli and α-galactosidase activity was confirmed [1], which might be mediated by the two GH27, two GH36 and five GH97.
Table 8.
Carbohydrate active enzymes (CAZy) in the genome of strain DSM 21788T
| CAZy family | GH1 | GH2 | GH3 | GH5 | GH13 | GH16 | GH23 |
|---|---|---|---|---|---|---|---|
| Counts | 1 | 15 | 11 | 4 | 4 | 2 | 2 |
| CAZy family | GH25 | GH27 | GH28 | GH29 | GH30 | GH31 | GH36 |
| Counts | 1 | 2 | 5 | 2 | 3 | 5 | 2 |
| CAZy family | GH37 | GH39 | GH42 | GH43 | GH51 | GH65 | GH73 |
| Counts | 1 | 3 | 1 | 11 | 3 | 1 | 1 |
| CAZy family | GH78 | GH88 | GH92 | GH95 | GH97 | GH105 | GH106 |
| Counts | 1 | 1 | 2 | 3 | 5 | 4 | 2 |
| CAZy family | GH127 | GH130 | GHa | ||||
| Counts | 1 | 2 | 3 | ||||
| CAZy family | GT2 | GT4 | GT5 | GT9 | GT19 | GT20 | GT28 |
| Counts | 13 | 10 | 1 | 3 | 1 | 1 | 1 |
| CAZy family | GT30 | GT41 | GT51 | GTa | |||
| Counts | 1 | 1 | 4 | 1 | |||
| CAZy family | CBM2 | CBM10 | CBM13 | CBM32 | CBM35 | CBM50 | CBM57 |
| Counts | 1 | 1 | 1 | 1 | 2 | 4 | 1 |
| CAZy family | CE2 | CE4 | CE6 | CE7 | CE11 | CE12 | CE14 |
| Counts | 1 | 1 | 1 | 1 | 1 | 3 | 2 |
| CAZy family | CEa | PL11 | |||||
| Counts | 3 | 1 |
agenes attributed to an enzyme class, but not to a family
Polysaccharide utilization loci
Members of flavobacteria were frequently found in aquatic habitats and play a pivotal role in the remineralization of complex organic matter [24, 39]. The coincidence of (i) a preference for polymeric substrates [39], (ii) the occurrence during algal blooms [62, 70] and (iii) the organization of genes involved in polysaccharide decomposition in polysaccharide utilization loci (PUL) [16, 67], suggests a specialization of Flavobacteriia members towards the utilization of complex organic matter.
In F. rivuli DSM 21788T four PULs were identified consisting of a TonB-dependent receptor, a SusD-like protein and a series of carbohydrate active enzymes (Figs. 4, 5, 6 and 7). The synteny between the identified PULs and 40 currently available Flavobacteriaceae genomes were investigated using MultiGeneBlast [52]. Figure 4 shows one of the PULs being conserved between some strains from the genera Flavobacterium, Cellulophaga, Gramella and Zunongwangia. Kabisch et al. [38] showed that proteins of the same PUL in ‘Gramella forsetii’ KT0803 were specifically expressed when grown on laminarin. The second PUL comprised of three glycosyl transferases, two GH5 and GH43 was found also in F. denitrificans DSM 15936T and F. johnsoniae UW101 [50], but with an additional GH2 (Fig. 5). Two further PULs comprised combinations of GH2, GH3, GH31, GH97 and other glycoside hydrolases and were only partially identical with PULs of other Flavobacterium members (Fig. 6a and b). These PULs potentially enable F. rivuli to decompose hemicellulose or xylose.
Fig. 4.

Synteny between a potentially laminarin-specific PUL of F. rivuli DSM 21788T and other Flavobacteriaceae. Open circles indicate genes which were specifically expressed by ‘Gramella forsetii’ KT0803 when grown on laminarin, as shown by Kabisch et al. [38]. Locus tags are given below both the first and last gene of the loci. Accession numbers in brackets are GenBank accession numbers of the corresponding contig. Investigation of syntenic loci was done using MultiGeneBlast [52]. A description of glycoside hydrolase families (GH) can be seen at the CAZy homepage [18, 47]
Fig. 5.

Synteny between a PUL of F. rivuli DSM 21788T, F. denitrificans DSM 15936T and F. johnsoniae UW101T. Locus tags are given below both the first and last gene of the loci. Accession numbers in brackets are GenBank accession numbers of the corresponding contig. Investigation of syntenic loci was done using MultiGeneBlast [52]. A description of glycoside hydrolase families (GH) and glycoside transferase families (GT) can be seen at the CAZy homepage [18, 47]
Fig. 6.
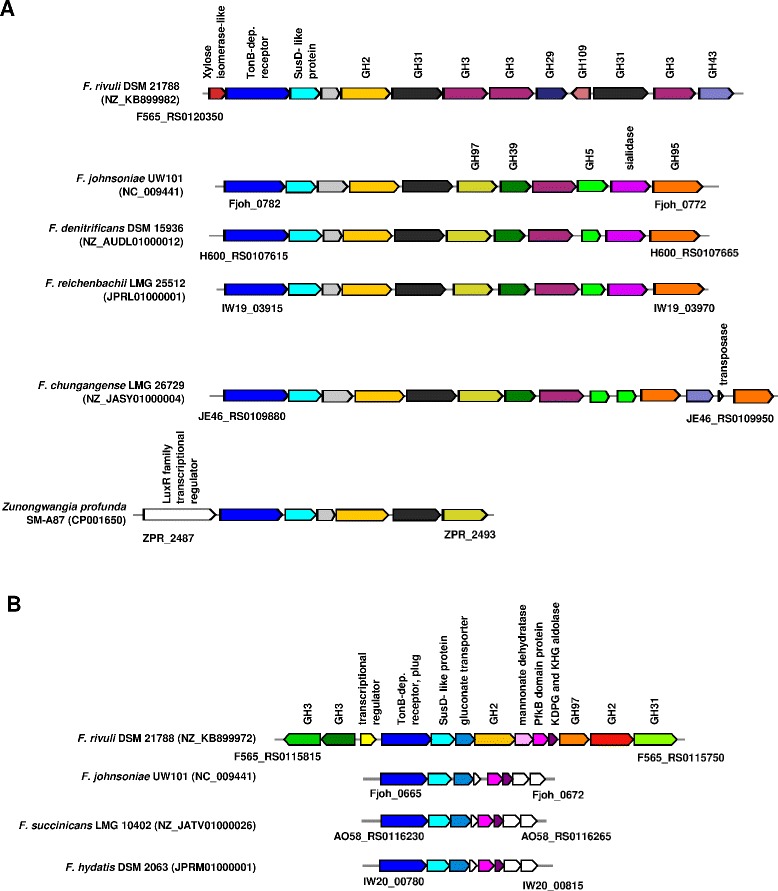
Two PUL of F. rivuli DSM 21788T with low synteny (a, b) to PUL of other Flavobacterium members, potentially mediating the decomposition of hemicellulose or xylose. Locus tags are given below both the first and last gene of the loci. Accession numbers in brackets are GenBank accession numbers of the corresponding contig. Investigation of syntenic loci was done using MultiGeneBlast [52]. A description of glycoside hydrolase families (GH) can be seen at the CAZy homepage [18, 47]
Fig. 7.
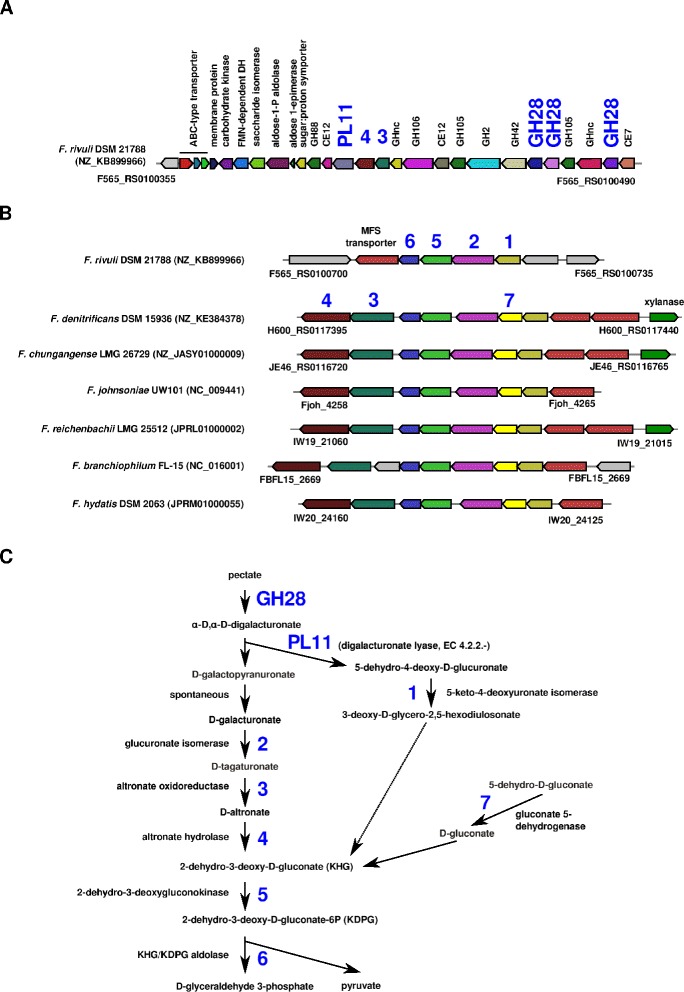
Polygalacturonate decomposition potential in F. rivuli DSM 21788T. a The potentially polygalacturonate specific PUL was found exclusively in F. rivuli DSM 21788T. b Genes for the catabolism of d-galactopyranuronate are colocalized in a gene cluster syntenic between Flavobacterium members. c Enzymes of the pectate decomposition and catabolism pathway. Bold blue numbers indicate the position of enzymes in the pectate catabolism pathway c and their corresponding genes in the gene clusters a, b. Genes in gray encode for hypothetical proteins. Locus tags are given below both the first and last gene of the loci. Accession numbers in brackets are GenBank accession numbers of the corresponding contig. Investigation of syntenic loci was done using MultiGeneBlast [52]. Investigation of pectin degradation pathway was done using the MetaCyc homepage [19]. A description of glycoside hydrolase families (GH) can be seen at the CAZy homepage [18, 47]
In addition to the PULs, F. rivuli DSM 21788T had one large operon-like structure comprising a set of 11 glycoside hydrolases, 3 carbohydrate esterases, one polysaccharide lyase (Fig. 7a), notably three GH28s (exo-poly-α-d-galacturonosidase) and a PL11 (digalacturonate lyase) for the decomposition of a pectate-like polysaccharide (polygalacturonate). Acetyl groups may be split of by CE7 (acetyl xylan esterase) and CE12 (rhamnogalacturonan acetylesterase). Interestingly, this operon additionally includes an altronate hydrolase and an oxidoreductase, which are part of the d-galactopyranuronate catabolic pathway (Fig. 7c), as well as two transporters, an aldose epimerase, a dehydrogenase and a kinase, which may mediate the catabolism of side-chain saccharides such as d-xylose, d-mannose and d-arabinose. In other Flavobacterium species, genes of the d-galactopyranuronate catabolic pathway are all co-located in loci which are syntenic with a gene cluster in F. rivuli (Fig. 7b). However, the gene cluster in F. rivuli did not contain the altronate hydrolase and oxidoreductase. Conclusively, the absence of the two genes of the d-galactopyranuronate catabolic pathway, and thus the ability to utilize polygalacturonate, was possibly compensated by the large CAZy-rich gene cluster.
Conclusion
The high-quality draft genome sequence of the Gram-negative, non-motile F. rivuli WB 3.3-2T (=DSM 21788T) isolated from a spring of a hard water rivulet provided new insights into the polysaccharide-decomposition potential of freshwater Flavobacteriaceae. F. rivuli belongs to a group of deep-branching species within the genus Flavobacterium that might be more closely related to the genus Myroides than to the type species of Flavobacterium,F. aquatile. The present data points towards an unsatisfactory taxonomy irrespective of which interpretation one follows and is largely a result of publishing new species in the genus Flavobacterium without taking into consideration a wider range of species in that genus or including members of the genus Myroides as well as publishing new species within the genus Myroides without taking a larger number of species from the genus Flavobacterium into consideration (including the type species). At the same time all evaluations are primarily based on “phylogenetic data” (i.e., gene sequence data) and genera are often poorly delineated. At first glance it does not appear that this approach will resolve this issue. Bernardet et al. [12] mentioned the clustering of F. rivuli among other Flavobacterium species in groups or possible new genera which have 16S rRNA gene sequence identities below 93 % with the type species F. aquatile of the genus. However, the potentially new genera could not be delineated because different procedures or culture conditions were used to describe common features [12].
The problem of an essentially unresolved backbone in the 16S rRNA gene sequence phylogeny of the Flavobacteriaceae (see above) will most probably be overcome in the near future with the foreseeable increase of publicly available draft genome sequences from large scale projects such as GEBA, which will enable us to infer whole genome sequence based phylogenies with a significantly higher statistical support for the branching topology using genome-based inference methods [54].
The genome of strain F. rivuli WB 3.3-2T (DSM 21788T) comprised 4.48 Mbp on 23 scaffolds and was sequenced as part of the GenomicEncyclopedia ofBacteria andArchaea project. The genome encoded for a great variety of 151 carbohydrate active enzymes and 177 peptidases. The four identified polysaccharide-utilization loci may enable strain WB 3.3-2T to decompose laminarin, hemicellulose and xylose. One gene cluster was identified that may enable strain WB 3.3-2T to decompose pectate-like polysaccharides. This genome in combination with other genomes of the Flavobacteriaceae will give further insights into the evolution and genetic potential of bacteria succeeding in substrate-related niches during polysaccharide decomposition in marine and freshwater habitats.
Acknowledgements
The authors gratefully acknowledge the help of Andrea Schütze, for growing cells of DSM 21788T and of Evelyne Brambilla, for DNA extraction and quality control (both at DSMZ). This work was performed under the auspices of the US Department of Energy’s Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344 Genome analysis was supported by the National Basic Research Program of China (No. 2010CB833801). A.L. was supported in part by Russian Ministry of Science Mega-grant No.11.G34.31.0068 (PI: Stephen J. O’Brien). R.L.H. was supported by the Bundesministerium für Ernährung und Landwirtschaft No. 22016812 (PI: Hans-Peter Klenk).
Additional file
Accession numbers of 16S rRNA gene sequences of Flavobacterium, Myroides, Capnocytophaga and Coenonia type strains used for generating the phylogenetic tree (Fig. 1) and the histogram (Fig. 2). Table S2. Peptidases or homologues in the genome of F. rivuli DSM 21788T. Table S3. Simple peptidases inhibitors in the genome of F. rivuli DSM 21788T. Table S4. Carbohydrate active enzymes (CAZymes) in the genome of F. rivuli DSM 21788T. Table S5. Sulfatases in the genome of F. rivuli DSM 21788T.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RLH, ES, MG, NCK and HPK designed research and project outline. SH performed CAZy and MEROPS analysis. JPMK and RLH performed comparative genomics and 16S rRNA based phylogeny. RLH investigated the CAZymes and PUL. MRO performed electron microscopy. BJT provided the background information on the current taxonomy in relationship to monophyletic groups. RLH, JPMK, BJT and ES drafted the manuscript that was critically reviewed and polished by RLH, BTI, MG and HPK. AL, JH, ST, MH, TBKR, MH, AP, NNI, KM, VM and TW performed genome sequencing, assembly and annotation. All authors read and approved the final manuscript.
References
- 1.Ali Z, Cousin S, Frühling A, Brambilla E, Schumann P, Yang Y, et al. Flavobacterium rivuli sp. nov., Flavobacterium subsaxonicum sp. nov., Flavobacterium swingsii sp. nov. and Flavobacterium reichenbachii sp. nov., isolated from a hard water rivulet. Int J Syst Evol Microbiol. 2009;59:2610–2617. doi: 10.1099/ijs.0.008771-0. [DOI] [PubMed] [Google Scholar]
- 2.Anon Validation List N° 143. Int J Syst Evol Microbiol. 2012;62:1–4. doi: 10.1099/ijs.0.039487-0. [DOI] [Google Scholar]
- 3.Anon Validation List N° 145. Int J Syst Evol Microbiol. 2012;62:1017–1019. doi: 10.1099/ijs.0.043240-0. [DOI] [Google Scholar]
- 4.Anon. List of growth media used at the DSMZ. http://www.dsmz.de/.
- 5.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auch AF, Klenk H-P, Göker M. Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand Genomic Sci. 2010;2:142–148. doi: 10.4056/sigs.541628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Auch AF, von Jan M, Klenk H-P, Göker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci. 2010;2:117–134. doi: 10.4056/sigs.531120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.BAuA 2010 – 2012 update, Classification of bacteria and archaea in risk groups. http://www.baua.de. TRBA 466. 19.
- 9.Bennett S. Solexa Ltd. Pharmacogenomics. 2004;5:433–438. doi: 10.1517/14622416.5.4.433. [DOI] [PubMed] [Google Scholar]
- 10.Bergey DH, Harrison FC, Breed RS, Hammer BW, Huntoon FM. Bergey’s Manual of Determinative Bacteriology 1st edn. Baltimore: Williams and Wilkins Co.; 1923. [Google Scholar]
- 11.Bernardet J-F. Class II. Flavobacteriia class. nov. In: Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig WWWB, editors. Bergey's Manual of Systematic Bacteriology. 2. New York: Springer; 2010. pp. 106–314. [Google Scholar]
- 12.Bernardet JF, Bowman JP, Genus I. Bergey's Manual of Systematic Bacteriology. 2. New York: Springer; 2011. Flavobacterium Bergey, Harrison, Breed, Hammer and Huntoon 1923, 97AL emend. Bernardet, Segers, Vancanneyt, Berthe, Kersters and Vandamme 1996, 139; pp. 112–154. [Google Scholar]
- 13.Bernardet JF, Family I. Flavobacteriaceae Reichenbach 1992b, 327VP (Effective publication: Reichenbach 1989b, 2013.) emend. Bernardet, Segers, Vancanneyt, Berthe, Kersters and Vandamme 1996, 145 emend. Bernardet, Nakagawa and Holmes 2002, 1057. In: Bergey's Manual of Systematic Bacteriology, vol. 4. 2nd ed. New York: The Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes. Springer; 2011. p. 106–314.
- 14.Bernardet JF, Order I. Flavobacteriales ord. nov. In: Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB, editors. Bergey's Manual of Systematic Bacteriology. 2. New York: Springer; 2011. pp. 105–329. [Google Scholar]
- 15.Bernardet JF, Segers P, Vancanneyt M, Berthe F, Kersters K, Vandamme P. Cutting a gordian knot: emended classification and description of the genus Flavobacterium, emended description of the family Flavobacteriaceae, and proposal of Flavobacterium hydatis nom. nov. (basonym, Cytophaga aquatilis Strohl and Tait 1978) Int J Syst Bacteriol. 1996;46:128–148. doi: 10.1099/00207713-46-1-128. [DOI] [Google Scholar]
- 16.Bjursell MK, Martens EC, Gordon JI. Functional genomic and metabolic studies of the adaptations of a prominent adult human gut symbiont, Bacteroides thetaiotaomicron, to the suckling period. J Biol Chem. 2006;281:36269–36279. doi: 10.1074/jbc.M606509200. [DOI] [PubMed] [Google Scholar]
- 17.Brambilla E, Päuker O, Cousin S, Steiner U, Reimer A, Stackebrandt E. High phylogenetic diversity of Flavobacterium spp. isolated from a hardwater creek, Harz Mountains, Germany. Org Divers Evol. 2007;7:145–154. doi: 10.1016/j.ode.2006.03.003. [DOI] [Google Scholar]
- 18.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caspi R, Altman T, Billington R, Dreher K, Foerster H, Fulcher CA, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014;42:D459–D471. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 21.DOE Joint Genome Institute. A DOE office of science user facility of Lawrence Berkeley national laboratory. DOE Jt Genome Inst. http://jgi.doe.gov.
- 22.Dong K, Xu B, Zhu F, Wang G. Flavobacterium hauense sp. nov., isolated from soil and emended descriptions of Flavobacterium subsaxonicum, Flavobacterium beibuense and Flavobacterium rivuli. Int J Syst Evol Microbiol. 2013;63:3237–3242. doi: 10.1099/ijs.0.048652-0. [DOI] [PubMed] [Google Scholar]
- 23.Euzéby JP. List of bacterial names with standing in nomenclature: A folder available on the Internet. Int J Syst Bacteriol. 1997;47:590–592. doi: 10.1099/00207713-47-2-590. [DOI] [PubMed] [Google Scholar]
- 24.Fernández-Gómez B, Richter M, Schüler M, Pinhassi J, Acinas SG, González JM, et al. Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 2013;7:1026–1037. doi: 10.1038/ismej.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Field D, Amaral-Zettler L, Cochrane G, Cole JR, Dawyndt P, Garrity GM, et al. The Genomic Standards Consortium. PLoS Biol. 2011;9:8–10. doi: 10.1371/journal.pbio.1001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Field D, Sterk P, Kottmann R, De Smet JW, Amaral-Zettler L, Cochrane G, et al. Genomic Standards Consortium Projects. Stand Genomic Sci. 2014;9:599–601. doi: 10.4056/sigs.5559608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: The protein families database. Nucleic Acids Res. 2014;42:D222–30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frankland GC, Frankland PF. Über einige typische Mikroorganismen im Wasser und im Boden. Zeitschrift fur Hyg und Infekt Medizinische Mikrobiol Immunol und Virol. 1889;6:373–400. [Google Scholar]
- 30.Fu Y, Tang X, Lai Q, Zhang C, Zhong H, Li W, et al. Flavobacterium beibuense sp. nov., isolated from marine sediment. Int J Syst Evol Microbiol. 2011;61:205–209. doi: 10.1099/ijs.0.018846-0. [DOI] [PubMed] [Google Scholar]
- 31.Garrity G. NamesforLife. BrowserTool takes expertise out of the database and puts it right in the browser. Microbiol Today. 2010;37:9. [Google Scholar]
- 32.Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk H-P, Güntsch A, et al. Bank Network: The start from a German initiative. Biopreserv Biobank. 2011;9:51–55. doi: 10.1089/bio.2010.0029. [DOI] [PubMed] [Google Scholar]
- 33.Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 2011;108:1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Göker M, Cleland D, Saunders E, Lapidus A, Nolan M, Lucas S, et al. Complete genome sequence of Isosphaera pallida type strain (IS1BT) Stand Genomic Sci. 2011;4:63–71. doi: 10.4056/sigs.1533840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Göker M, Klenk H-P. Phylogeny-driven target selection for large-scale genome-sequencing (and other) projects. Stand Genomic Sci. 2013;8:360–374. doi: 10.4056/sigs.3446951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holmes B, Owen RJ. Proposal that Flavobacterium breve be substituted as the type species of the genus in place of Flavobacterium aquatile and emended description of the genus Flavobacterium: status of the named species of Flavobacterium, Request for an Opinion. Int J Syst Bacteriol. 1979;29:416–426. doi: 10.1099/00207713-29-4-416. [DOI] [Google Scholar]
- 37.Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kabisch A, Otto A, König S, Becher D, Albrecht D, Schüler M, et al. Functional characterization of polysaccharide utilization loci in the marine Bacteroidetes ‘Gramella forsetii’ KT0803. ISME J. 2014;8:1492–1502. doi: 10.1038/ismej.2014.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kirchman DL. The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol Ecol. 2002;39:91–100. doi: 10.1111/j.1574-6941.2002.tb00910.x. [DOI] [PubMed] [Google Scholar]
- 40.Klenk HP, Göker M. En route to a genome-based classification of Archaea and Bacteria? Syst Appl Microbiol. 2010;33:175–182. doi: 10.1016/j.syapm.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 41.Krieg NR, Ludwig W, Euzéby J, Whitman WB, Phylum XIV, et al. Bacteroidetes phyl. nov. In: Krieg NR, Staley JT, Brown DR, et al., editors. The Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes. 2. New York: Springer; 2010. pp. 25–469. [Google Scholar]
- 42.Kyrpides NC, Hugenholtz P, Eisen JA, Woyke T, Göker M, Parker CT, et al. Genomic Encyclopedia of Bacteria and Archaea: sequencing a myriad of type strains. PLoS Biol. 2014;12:e1001920. doi: 10.1371/journal.pbio.1001920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kyrpides NC, Woyke T, Eisen JA. Genomic Encyclopedia of Type Strains, Phase I : The one thousand microbial genomes (KMG-I) project. Stand Genomic Sci 2014:628–634. [DOI] [PMC free article] [PubMed]
- 44.Lapage S, Sneath P, Lessel E, Skerman V, Seeliger H, Clark W. International code of nomenclature of bacteria (1990 Revision) Washington, D.C.: American Society for Microbiology; 1992. [PubMed] [Google Scholar]
- 45.Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics. 2002;18:452–464. doi: 10.1093/bioinformatics/18.3.452. [DOI] [PubMed] [Google Scholar]
- 46.Li H. wgsim. 2011: https://github.com/lh3/wgsim.
- 47.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Markowitz VM, Mavromatis K, Ivanova NN, Chen IMA, Chu K, Kyrpides NCIMGER. A system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–2278. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- 49.Mavromatis K, Land ML, Brettin TS, Quest DJ, Copeland A, Clum A, et al. The fast changing landscape of sequencing technologies and their impact on microbial genome assemblies and annotation. PLoS One. 2012;7:e48837. doi: 10.1371/journal.pone.0048837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McBride MJ, Xie G, Martens EC, Lapidus A, Henrissat B, Rhodes RG, et al. Novel features of the polysaccharide-digesting gliding bacterium Flavobacterium johnsoniae as revealed by genome sequence analysis. Appl Env Microbiol. 2009;75:6864–6875. doi: 10.1128/AEM.01495-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McBride MJ, Zhu Y. Gliding motility and Por secretion system genes are widespread among members of the phylum Bacteroidetes. J Bacteriol. 2013;195:270–278. doi: 10.1128/JB.01962-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Medema MH, Takano E, Breitling R. Detecting sequence homology at the gene cluster level with MultiGeneBlast. Mol Biol Evol. 2013;30:1218–1223. doi: 10.1093/molbev/mst025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013;14:60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M. Highly parallelized inference of large genome-based phylogenies. Concurr Comput Pract Exp. 2014;26:1715–1729. doi: 10.1002/cpe.3112. [DOI] [Google Scholar]
- 55.Meier-Kolthoff JP, Göker M, Spröer C, Klenk HP. When should a DDH experiment be mandatory in microbial taxonomy? Arch Microbiol. 2013;195:413–418. doi: 10.1007/s00203-013-0888-4. [DOI] [PubMed] [Google Scholar]
- 56.Meier-Kolthoff JP, Klenk H-P, Göker M. Taxonomic use of DNA G+C content and DNA-DNA hybridization in the genomic age. Int J Syst Evol Microbiol. 2014;64:352–356. doi: 10.1099/ijs.0.056994-0. [DOI] [PubMed] [Google Scholar]
- 57.Montero-Calasanz MDC, Göker M, Rohde M, Spröer C, Schumann P, Busse HJ, et al. Chryseobacterium oleae sp. nov., an efficient plant growth promoting bacterium in the rooting induction of olive tree (Olea europaea L.) cuttings and emended descriptions of the genus Chryseobacterium, C. daecheongense, C. gambrini, C. gleum, C. joostei, C. jejuense, C. luteum, C. shigense, C. taiwanense, C. ureilyticum and C. vrystaatense. Syst Appl Microbiol. 2014;37:342–350. doi: 10.1016/j.syapm.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 58.Montero-Calasanz MDC, Göker M, Rohde M, Spröer C, Schumann P, Busse HJ, et al. Chryseobacterium hispalense sp. nov., a plantgrowth-promoting bacterium isolated from a rainwater pond in an olive plant nursery, and emended descriptions of Chryseobacterium defluvii, Chryseobacterium indologenes, Chryseobacterium wanjuense and Chryseobacterium gregarium. Int J Syst Evol Microbiol. 2013;63:4386–4395. doi: 10.1099/ijs.0.052456-0. [DOI] [PubMed] [Google Scholar]
- 59.Pagani I, Liolios K, Jansson J, Chen IMA, Smirnova T, Nosrat B, et al. The Genomes OnLine Database (GOLD) v. 4: Status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2012;40:D571–9. doi: 10.1093/nar/gkr1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, et al. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods. 2010;7:455–457. doi: 10.1038/nmeth.1457. [DOI] [PubMed] [Google Scholar]
- 61.Piao H, Froula J, Du C, Kim TW, Hawley ER, Bauer S, et al. Identification of novel biomass-degrading enzymes from genomic dark matter: Populating genomic sequence space with functional annotation. Biotechnol Bioeng. 2014;111:1550–1565. doi: 10.1002/bit.25250. [DOI] [PubMed] [Google Scholar]
- 62.Pinhassi J, Sala MM, Havskum H, Peters F, Guadayol O, Malits A, et al. Changes in bacterioplankton composition under different phytoplankton regimens. Appl Env Microbiol. 2004;70:6753–6766. doi: 10.1128/AEM.70.11.6753-6766.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.R Development Core Team . R: a language and environment for statistical computing. 2014. [Google Scholar]
- 64.Rawlings ND, Waller M, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014;42:D503–D509. doi: 10.1093/nar/gkt953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reichenbach H. Order 1. Cytophagales Leadbetter 1974, 99AL. In: Staley JT, Bryant MP, Pfennig N, Holt JG, editors. Bergey’s Manual of Systematic Bacteriology, vol. 3. New York: Springer New York; 1989. pp. 2011–2013. [Google Scholar]
- 66.Ryu SH, Park M, Jeon Y, Lee JR, Park W, Jeon CO. Flavobacterium filum sp. nov., isolated from a wastewater treatment plant in Korea. Int J Syst Evol Microbiol. 2007;57:2026–2030. doi: 10.1099/ijs.0.65138-0. [DOI] [PubMed] [Google Scholar]
- 67.Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, et al. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell. 2010;141:1241–1252. doi: 10.1016/j.cell.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stutzer M. Aussaaten aus den Fäzes des Menschen gelbe Kolonien bildende Bakterien (Gattung Flavobacterium u.a.) In: Stutzer M, Kwaschnina A, editors. Zentralblatt fur Bakteriologie, Parasitenkunde, Infektionskrankheiten und Hygiene. Abteilung I.vol 113. 1929. pp. 219–225. [Google Scholar]
- 69.Täubel M, Rintala H, Pitkäranta M, Paulin L, Laitinen S, Pekkanen J, et al. The occupant as a source of house dust bacteria. J Allergy Clin Immunol. 2009;124:834–840. doi: 10.1016/j.jaci.2009.07.045. [DOI] [PubMed] [Google Scholar]
- 70.Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM, et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science. 2012;336:608–611. doi: 10.1126/science.1218344. [DOI] [PubMed] [Google Scholar]
- 71.Vancanneyt M, Segers P, Torck U, Hoste B, Bernardet J-F, Vandamme P, et al. Reclassification of Flavobacterium odoratum (Stutzer 1929) strains to a new genus, Myroides, as Myroides odoratus comb. nov. and Myroides odoratimimus sp. nov. Int J Syst Bacteriol. 1996;46:926–932. doi: 10.1099/00207713-46-4-926. [DOI] [Google Scholar]
- 72.Wickham H. ggplot2: Elegant graphics for data analysis. 2. New York: Springer; 2009. [Google Scholar]
- 73.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature. 2009;462:1056–1060. doi: 10.1038/nature08656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ye Y, Choi J-H, Tang H. RAPSearch: a fast protein similarity search tool for short reads. BMC Bioinformatics. 2011;12:159. doi: 10.1186/1471-2105-12-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40:W445–W451. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zerbino DR, Velvet BE. Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao Y, Tang H, Ye Y. RAPSearch2: A fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics. 2012;28:125–126. doi: 10.1093/bioinformatics/btr595. [DOI] [PMC free article] [PubMed] [Google Scholar]