

Abstract
AIM: To test the hypothesis that introduction of antisense TβR I and TβR II eukaryotic expressing plasmids into a rat model of immunologically induced liver fibrosis might block the action of TGF-β1 and halt the progression of liver fibrosis.
METHODS: RT-Nest-PCR and gene recombination techniques were used to construct rat antisense TβR I and TβR II recombinant plasmids which could be expressed in eukaryotic cells. The recombinant plasmids and empty vector (pcDNA3) were encapsulated by glycosyl-poly-L-lysine and then transducted into rats of pig serum-induced liver fibrosis model. Expression of exogenously transfected gene was assessed by Northern blot, and hepatic expressions of TβR I and TβR II were evaluated by RT-PCR and Western blot.We also performed ELISA for serum TGF-β1, hydroxyproline of hepatic tissues, immunohistoche-mistry for collagen types I and III, and VG staining for pathological study of the liver tissues.
RESULTS: The exogenous antisense TβR I and TβR II plasmids could be well expressed in vivo, and block mRNA and protein expression of TβR I and TβR II in the fibrotic liver at the level of mRNA respectively. These exogenous plasmid expressions reduced the level of TGF-β1 (antisense TβR I group 23.998 ± 3.045 ng/mL, antisense TβR II group 23.156 ± 3.131 ng/mL, disease control group 32.960 ± 3.789 ng/mL; F = 38.19, 36.73, P < 0.01). Compared with disease control group, the contents of hepatic hydroxyproline (antisense TβR I group 0.169 ± 0.015 mg/g liver, antisense TβR II group 0.167 ± 0.009 mg/g liver, disease control group 0.296 ± 0.026 mg/g liver; F = 14.39, 15.48, P < 0.01) and the deposition of collagen types I and III decreased in the two antisense treatment groups (antisense TβR I group, collagen type I 669.90 ± 50.67, collagen type III 657.29 ± 49.48; antisense TβR II group, collagen type I 650.26 ± 51.51, collagen type III 661.58 ± 55.28; disease control group, collagen type I 1209.44 ± 116.60, collagen type III 1175.14 ± 121.44; F = 15.48 to 74.89, P < 0.01). Their expression also improved the pathologic classification of liver fibrosis models (compared with disease control group, χ2 = 17.14, 17.24, P < 0.01). No difference was found in the level of TGF-β1, the contents of hepatic hydroxyproline and collagen types I and III and pathologic grade between pcDNA3 control group and disease control group or between the two antisense treatment groups (F = 0.11 to 1.06, χ2 = 0.13 to 0.16, P > 0.05).
CONCLUSION: Antisense TβR I and TβR II recombinant plasmids have certain reverse effects on liver fibrosis and can be used as possible candidates for gene therapy.
INTRODUCTION
Liver fibrosis is a common sequel to diverse liver injuries. In the formation of liver fibrosis and cirrhosis, synthesis of collagen increases and its degradation decreases. It has been thought that liver fibrosis can be reversed and liver cirrhosis is irreversible[1-5]. Profound studies have been conducted on the treatment of liver fibrosis. However, this disease is still lack of efficient therapy[6-11]. Searching for a new therapy seems very important.
In the formation of liver fibrosis and cirrhosis, many cytokines produce marked effects through autocrine and paracrine[1,2,5]. Molecular mechanisms involved in fibrogenesis reveal that transforming growth factorβ (TGF-β), especially TGF-β1, plays a pivotal role[12-16]. Signaling by TGF-β occurs through a family of transmembranes and ser/thr kinase receptors. Both components of the receptor complex, known as receptor I (TβR I) and receptor II (TβR II) are essential for signal transduction[17,18]. So in theory, blockage of TGF-β signal transduction by inhibiting the expression of TβR I and/or TβR II may have therapeutic effects on liver fibrosis.
At present, gene therapy for liver fibrosis targeting TGF-β mainly includes inhibiting the expression of TGF-β1 (for instance, antisense TGF-β1 RNA) and using deficient TβR II[19-21]. But therapeutic researches which target TβR I or use antisense TβR II as a therapeutic tool have not been reported. In the present experiments, we constructed antisense TβR I and TβR II eukaryotic expressing plasmids and performed in vivo transfection. We aimed to test the hypothesis that introduction of these two exogenous plasmids into a rat model of immunologically induced liver fibrosis might block the action of TGF-β1 and halt the progression of liver fibrosis.
MATERIALS AND METHODS
Construction of recombinant plasmid
Nested primers were designed and synthesized according to rat TβR I and TβR II cDNA sequences (GenBank)[22,23]. The length of amplified PCR products was anticipated to be 470 bp, 606 bp (Figure 1). Total RNA was extracted from normal rat liver with Trizol reagent (GIBCO, USA) according to the manufacturer’s directions. RT-Nest-PCR was used to construct TβR I and TβR II cDNA fragments. Samples were heated at 94 °C for 7 min and subjected to 32 PCR cycles of denaturation at 94 °C for 1 min, annealing at 55 °C for 1 min, extension at 72 °C for 1 min, followed by a final extension at 72 °C for 5 min. After separation, reclaim and purification, the PCR products of TβR I and TβR II were connected with T vector (Promega, USA) and then transferred into JM-109 strain. PT/TβR I and PT/TβR II were successfully constructed after IPTG/X-gal screening. The target fragments were cut and inserted reversely into eukaryotic expressing plasmid pcDNA3 (Invitrogen, CA), and then transferred into JM-109 strain again. By using enzyme-cutting identification (TβR I: EcoRI and XhoI; TβR II: EcoRI and BamHI) and DNA autosequencing (PE377 Auto sequencer, USA), the successful constructions of antisense TβR I and TβR II eukaryotic expressing plasmids were proved (Figure 2).
Figure 1.

Primer pairs for PCR reactions.
Figure 2.
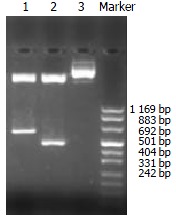
Enzyme-cutting identification of the recombinant plasmids, Lane 1: pANTI-RII, Lane 2: pANTI-RI, Lane 3: pcDNA3.
Experimental protocols
Forty-six male Sprague-Dawley rats (weighing 100 to 120 g) provided by Experimental Animal Center, Zhongshan Hospital, Fudan University, were randomly divided into the following 5 groups: 10 in experimental liver fibrosis model induced by pig-serum as disease control group (M), 10 in antisense TβR I plasmid transfection as treatment group (A), 10 in antisense TβR II plasmid transfection as treatment group (B), 10 in pcDNA3 transfection as pcDNA3 control group (C) and 6 in normal control group (N). Animals in the normal control group received 0.5 mL of NS twice weekly by intraperitoneal injection for 8 wk. Rats in the other 4 groups received 0.5 mL of pig-serum twice weekly by intraperitoneal injection for 8 wk[24]. Among groups A, B and C, the recombinant antisense TβR I and TβR II plasmids and empty vector (pcDNA3) of 100 mg each time were encapsulated by glycosyl-poly-L-lysine (G-PLL) and then transducted into rats of liver fibrosis model via caudal vein every 2 wk respectively. The molecular ratio of galactose and poly-L-lysine was 15:28 and the average molecular weight of G-PLL was 8.5 ku. All experimental rats were sacrificed at the end of the 8th wk. The middle lobes of the livers were removed and specimens were fixed in Carnoy’s fixative (glacial acetic acid, chloroform, and ethanol at a volume ratio of 1:3:6) and then embedded in paraffin for histological analysis. The remaining tissue was quickly partitioned and immediately stored in liquid nitrogen and then frozen below -70 °C. After that the rats were humanely killed.
RNA isolation and Northern blot analysis for exogenous gene and hepatic TβR I and TβR II expression
For Northern blot analysis, 30 mg of total tissue RNAs was separated by electrophoresis on a 1% denaturing agarose gel, transferred to a Hybond-N membrane (Amersham, UK) and fixed by baking for 2 h at 80 °C. The probe for pcDNA3 according to its special T7 promoter sequence (the oligonucleotide fragment: 5'-CAGAGGGATATCACTCAGCATAAT-3'), which was to detect exogenous gene expression, was labeled with [α-32P]dATP using DNA tailing kit (Roche, Germany). TβR I and TβR II cDNA probes were labeled with [α-32P]dCTP using a high prime DNA labeling kit (Roche, Germany) to detect rat TβR I and TβR II expression. Blots were pre- hybridized for at least 3 h at 42 °C, and then hybridized for 20 h at 42 °C. Auto radiographs were exposed for indicated times to Kodak films at -70 °C for 7 d. As an internal standard (loading control) the blots were re-hybridized with [α-32P] β-actin.
RT-PCR analysis for TβR I and TβR II mRNA expression
Total RNA of 1 mg from each sample was reversely transcribed and amplified using an RT-PCR kit (ShengNeng-BoCai Biotechnology Co. Shanghai, China). The RT-PCR reaction contained 12.5 pmol each primer (TβR I: sense sequence 5’-TCACTAGATCGCCCTTTCAT-3’; antisense sequence 5’-GATAATCCGACACCAACCAC-3’, a product of 355 bp; TβR II: sense sequence 5’-CCACGACCCCAAGTTCACCT-3’, antisense sequence 5’-TGGGCAGCAGTTCCGTATTG-3’, a product of 428 bp) for detecting the TβR I and TβR II mRNA expression in rat liver tissues. Additionally primer pairs (sense sequence 5’- TGGGACGATATGGAGAAGAT -3’; antisense sequence 5’- ATTGCCGATAGTGATGACCT -3’) were used for amplifying the expression of β-actin (a product of 521 bp) as internal control. Samples were placed in a thermocycler with the incubation program at 37 °C for 60 min, at 90 °C for 5 min, then 30 cycles at 94 °C for 45 s, at 54 °C for 45 s, and at 72 °C for 1 min, and a final extension at 72 °C for 5 min. Products of RT-PCR were electrophoresed on a 20 g/L agarose gel to show the amplified bands. The areas under curve of the bands were calculated by Photo-Treater (Tanon GIS-1000, China). The ratio of the objective band and β-actin (control) represented the relative value of expression of the objective band.
Western blot for hepatic TβR I and TβR II expression
Lysates from 50 mg rat liver tissues were prepared with RIPA buffer [1 × PBS, 10 g/L NP40, 5 g/L sodium deoxycholate, 1 g/L SDS, 10 mmol/L PMSF (in isopropanol), 36 mg/mL aprotinin and 1mmol/L sodium orthovanadate]. Tissue lysates were centrifuged at 3000 r/min for 10 min at 4 °C, and protein concentrations were determined by BCA protein assay (Pierce, IL). Protein samples (100 mg) were heated for 5 min at 100 °C and separated on 120 g/L SDS-PAGE and then transferred to PVDF membranes (Schleicher&Schuell, Germany) in Tris-glycine buffer (pH 8.5) plus 200 mL/L methanol. The membranes were blocked overnight in 50 mL/L non-fat dried milk in Tris-buffer containing 1 g/L Tween-20 and then washed with Tris-buffer. The blots were incubated overnight at 4 °C with rabbit TβR I, TβR II and β-actin polyclonal IgG (Santa Cruz, USA) diluted 1:200 in Tris-buffer. The blots were washed and then incubated with AP-conjugated secondary antibodies (Santa Cruz, USA) at 1:500 dilution for 2 h at room temperature. The protein bands were visualized with BCIP/NBT (DAKO, USA) system.
ELISA assay of serum TGF-β1
Assay of serum TGF-β1 content was performed with double antibody ABC-ELISA method according to the previous reports of Ueno et al[21] and Roth et al[25].
Hepatic hydroxyproline (OH-Pro) content
Hydroxyproline content was determined by the previous method with some modification[26]. A total of 500 mg of liver tissues was hydrolyzed in 6 mol/L HCl solution at 120 °C for 24 h. The hydroxyproline content of the liver was expressed as micrograms per gram of wet weight.
Immunohistochemistry of hepatic collagen types I and II
After dewaxed with xylene and rehydrated through a graded alcohol series, sections were digested with 4 g/L trypsin. The sections were incubated with rabbit polyclonal antibodies against types I or II collagen (diluted 1:100) at 37 °C for 60 min and then overnight at 4 °C. Then the sections were incubated with EnvisionTM secondary antibodies of biotinylated sheep anti-rabbit IgG (DAKO, USA) at 37 °C for 30 min and then with the substrate solution (3,3-diaminobenzidine tetrahydrochloride in H2O2 in Tris buffer pH7.4) for 10 min, followed by counterstaining with hematoxylin. The sections were washed 3 times with 0.01 mol/L PBS (pH7.4) after each step. For control staining, PBS was used instead of the primary antibody. The slides were then analyzed with an image analyzing system (LeiCA-Q500IW System, Germany) to obtain the integral light density and the average area of positive staining. The multiplication of both parameters represented the relative contents of hepatic collagen types I and II.
Histology
Sections from paraffin-embedded blocks were dewaxed and stained with Van Gieson method. Fibrosis was scored according to the classification which graded liver fibrosis into seven degrees (0-VI).
Statistical analysis
All values were expressed as mean ± SD. Differences between groups were analyzed by one-way ANOVA (SPSS10.0 software) and the pathological grading of fibrosis of each group was analyzed by trend analysis and chi-square test (EPI5.0 software). P < 0.05 was considered statistically significant.
RESULTS
General situation of animals
No animal died during the experimental period. No significant difference was found in the levels of ALT (51 ± 9 U/L vs 53 ± 8 U/L) and Cr (91 ± 13 mmol/L vs 92 ± 14 mmol/L) between pcDNA3 control group and disease control group (P > 0.05). No significant difference was found in the height or weight of rats among antisense TβR I, antisense TβR II and pcDNA3 control groups (P > 0.05).
Expression of exogenous gene and TβR I, TβR II gene in liver tissues by Northern blot
The exogenous gene expression could be detected in transfection groups (antisense TβR I, antisense TβR II and pcDNA3 control groups) by Northern blot, but not in disease control group (Figure 2). The expression of TβR I mRNA was much higher in pcDNA3 control group and disease control group than in antisense TβR I group. The expression of TβR II was also much higher in pcDNA3 control group and disease control group than in antisense TβR II group at mRNA level (Figure 3).
Figure 3.
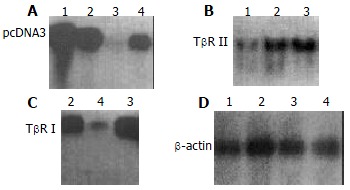
Expression of exogenous transfected gene assessed by Northern blot, A: Hybridization using 32P labeled oligo-nucleotide probe which targets T7 promoter; B: Hybridization by using 32P labeled TβR II cDNA probe; C: Hybridization by using 32P labeled TβR I cDNA probe; D: β-actin probe used as control. Lane 1: Antisense TβR II group; Lane 2: pcDNA3 con-trol group; Lane 3: Disease control group; Lane 4: Antisense TβR I group.
Hepatic TβR I and TβR II mRNA expression by RT-PCR
The expression of antisense TβR I RNA induced a decreased mRNA level of TβR I in antisense TβR I group (1.039 ± 0.110) compared with pcDNA3 control group (1.453 ± 0.112) and disease control group (1.472 ± 0.099) (P < 0.01). The expression of antisense TβR II RNA also induced a decreased mRNA level of TβR II in antisense TβR II group (0.805 ± 0.105) compared with pcDNA3 control group (1.743 ± 0.151) and disease control group (1.798 ± 0.139) (P < 0.01). No difference was found in the level of TβR I and TβR II mRNA between pcDNA3 control and disease control groups (P > 0.05).
Hepatic TβR I and TβR II protein expression by Western blot
The expression of TβR I protein was decreased in antisense TβR I group and the expression of TβR II protein was also decreased in antisense TβR II group when compared with pcDNA3 control group and disease control group, but they were still slightly higher than those in normal control group. The expressions of TβR I and TβR II protein in pcDNA3 control group were similar to those in disease control group (Figure 4).
Figure 4.
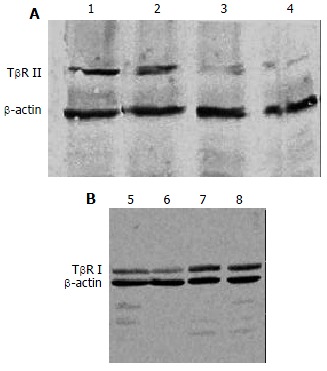
Hepatic protein expression of TβR I, TβR II assessed by Western blot, A: Hepatic protein expression of TβR II; B: Hepatic protein expression of TβR I; Lanes 1,8: Disease control group; Lanes 2,7: pcDNA3 control group; Lane 3: Antisense TβR II group; Lanes 4,6: Normal control group; Lane 5: Antisense TβR I group.
ELISA assay of serum TGF-β1
The level of serum TGF-β1 was reduced in antisense TβR I group (23.998 ± 3.045 ng/mL) and antisense TβR II group (23.156 ± 3.131 ng/mL) compared with pcDNA3 control group (32.275 ± 1.884 ng/mL) and disease control group (32.960 ± 3.789 ng/mL), but was still higher than that in normal control group (14.338 ± 2.421 ng/mL) (P < 0.01). No significant difference was found between antisense TβR I and antisense TβR II groups (P > 0.05).
Effects of antisense TβR I and TβR II plasmid transfection on hepatic hydroxyproline and collagen types I and II content
The hepatic hydroxyproline content and the accumulation of hepatic collagen types I and III in antisense TβR I and antisense TβR II groups were significantly decreased compared with disease control and pcDNA3 control groups (P < 0.01). No statistical difference was found between pcDNA3 control and disease control groups or between antisense TβR I and antisense TβR II groups (P > 0.05). Hepatic hydroxyproline content and accumulation of collagen in these four groups were significantly higher than those in normal control group (P < 0.01) (Table 1).
Table 1.
Effects of transfected plasmid on the content of hepatic hydroxyproline and deposition of collagen types I and III (mean ± SD)
| Group | Number | Hydroxyproline (mg/g liver) | Collagen I | Collagen III |
| Disease control group | 10 | 0.296 ± 0.026d | 1209.44 ± 116.60d | 1175.14 ± 121.44d |
| Antisense TβR II group | 10 | 0.167 ± 0.009bd | 650.26 ± 51.51bd | 661.58 ± 55.28bd |
| Antisense TβR I group | 10 | 0.169 ± 0.015d | 669.90 ± 50.67d | 657.29 ± 49.48d |
| pcDNA3 control group | 10 | 0.284 ± 0.025d | 1205.70 ± 110.11d | 1149.89 ± 91.61d |
| Normal control group | 6 | 0.128 ± 0.004 | 449.33 ± 35.95 | 421.15 ± 20.15 |
P < 0.01 vs disease control group and pcDNA3 control group,
P < 0.01 vs normal control group.
Effects of transfected plasmid on patho-histology
The fibrosis grade of antisense TβR I and antisense TβR II groups alleviated significantly compared with disease control and pcDNA3 control groups (P < 0.01). All these four groups had significantly higher fibrosis grades when compared to normal control group (P < 0.01). No significant difference was found between disease control and pcDNA3 control groups or between antisense TβR I and antisense TβR II groups (P > 0.05) (Table 2).
Table 2.
Pathological grade of liver fibrosis at the 8th week
| Group |
Grade |
Total | ||||||
| 0 | I | II | III | IV | V | VI | ||
| Disease control groupd | 0 | 0 | 0 | 1 | 4 | 4 | 1 | 10 |
| Antisense TβR II groupdb | 1 | 7 | 2 | 0 | 0 | 0 | 0 | 10 |
| Antisense TβR I groupdb | 1 | 6 | 3 | 0 | 0 | 0 | 0 | 10 |
| pcDNA3 control groupd | 0 | 0 | 0 | 1 | 5 | 3 | 1 | 10 |
| Normal control group | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 6 |
P < 0.01 vs disease control group and pcDNA3 control group,
P < 0.01 vs normal control group.
DISCUSSION
Pig serum-induced rat hepatic fibrosis is a model that shows an intense immune response to the administration of heterologous serum. Histologically, the changes were characterized by mononuclear cell infiltration and fibrotic response in the periportal area, followed by the septum formation connecting portal tract with central veins without hepatocyte injury[24].
In the formation of liver fibrosis and cirrhosis, a number of cytokines could produce marked effects through autocrine and paracrine[25,26]. Molecular mechanisms involved in fibrogenesis revealed that transforming growth factorβ (TGF-β), especially TGF-β1, played a pivotal role in the regulation of the production, degradation and accumulation of extracellular matrix (ECM)[1,21,27]. Secreted as a latent precursor, TGF-β is activated at sites of injury. Active TGF-β binds to specific, high-affinity receptors present on most cells, initiating a signaling cascade that results in biological effects. Signaling by TGF-β occurs through a family of transmembranes and ser/thr kinase receptors. Both components of the receptor complex, known as receptor I (TβR I) and receptor II (TβR II) are essential for signal transduction. Signaling is initiated by binding of TGF-β to the TβR II. Once bound, the TGF-β/TβR II complex then recruits TβR I into a heteromeric complex. Within the heteromeric complex, the kinase domain of type II receptor could transphosphorylate and activate type I receptor kinase, which then functioned to propagate the signal to downstream targets[21-23]. At present, gene therapy for liver fibrosis targeting TGF-β receptor mainly used adenoviral vectors expressing a dominant-negative type II TGF-β receptor or an adenovirus expressing an entire ectodomain of human TGF-β type II receptor fused to Fc portion of human IgG to block the TGF-β signal transduction[19,21].
Antisense technique has been used to inhibit the target genes and proteins expressed in experimental rat hepatic fibrosis[7]. We used RT-Nest-PCR and gene recombination techniques to construct rat antisense TβR I and TβR II recombinant plasmids successfully which could be expressed in eucaryotic cells. We transducted the exogenous plasmid into the fibrotic liver based on our previous research on gene target therapy. The exogenous antisense TβR I and TβR II recombinant plasmids were well expressed in vivo, and could block the mRNA and protein expression of TβR I and TβR II in the fibrotic liver induced by pig serum.
In our study, serum TGF-β1 analysis showed the expression of TGF-β1 was decreased after the expression of TβR I and TβR II was blocked. Our results were similar to the previous report that used deficient TβR II in treating experimental liver fibrosis. It was indicated that the production of active TGF-β1 was inhibited after the expression of TGF-β receptors was blocked and the signal transduction of TGF-β was also inhibited.
Collagen types I and III constitute the main components of increased ECM in liver fibrosis. It has been proposed that degradation of collagen types I and III is very important in the reversion of liver fibrosis[5,28,29]. In our study, the recombinant plasmid could be delivered to liver by G-PLL and expressed in the tissue of liver. In addition, there was a significant decrease of collagen deposition after the recombinant antisense TβR I and TβR II plasmids was transduced into fibrotic liver through mensuring the hepatic hydroxyproline content and immunohistochemistry of collagen types I and III. This suggested that the recombinant plasmids could increase the degrading capacity of collagen types I and III, decrease the deposition of ECM, and probably reverse hepatic fibrosis. In the data analysis, there was a significant difference in fibrosis grades between the disease control group and the recombinant plasmid transfected group. It showed that the antisense TβR I and TβR II plasmids had significant ameliorative effects on liver fibrosis.
No difference was found in the level of TGF-β1, the contents of hepatic hydroxyproline and collagen types I and III and the pathologic grade between empty plasmid control and disease control groups or between the two antisense treatment groups. It indicated that the signal transduction of TGF-β could be inhibited by blocking the expression of either TβR I or TβR II and the effects of antisense TβR I and antisense TβR II plasmids on experimental liver fibrosis were similar. Both TβR I and TβR II were essential for signal transduction.
In summary, our results demonstrate that TGF-β1 plays an important role in liver fibrosis development especially in degradation of collagens. Antisense TβR I and antisense TβR II recombinant plasmids have certain reverse effects on liver fibrosis and could be used as possible candidates for gene therapy. As a new therapeutic means, gene therapy has a long way to go, especially in transductive efficiency, gene targeting, stability and adverse effects.
Footnotes
Edited by Zhu LH and Wang XL Proofread by Xu FM
References
- 1.Alcolado R, Arthur MJ, Iredale JP. Pathogenesis of liver fibrosis. Clin Sci (Lond) 1997;92:103–112. doi: 10.1042/cs0920103. [DOI] [PubMed] [Google Scholar]
- 2.Olaso E, Friedman SL. Molecular regulation of hepatic fibrogenesis. J Hepatol. 1998;29:836–847. doi: 10.1016/s0168-8278(98)80269-9. [DOI] [PubMed] [Google Scholar]
- 3.Huang ZG, Zhai WR, Zhang YE, Zhang XR. Study of heteroserum-induced rat liver fibrosis model and its mechanism. World J Gastroenterol. 1998;4:206–209. doi: 10.3748/wjg.v4.i3.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benyon RC, Iredale JP. Is liver fibrosis reversible. Gut. 2000;46:443–446. doi: 10.1136/gut.46.4.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neubauer K, Saile B, Ramadori G. Liver fibrosis and altered matrix synthesis. Can J Gastroenterol. 2001;15:187–193. doi: 10.1155/2001/870205. [DOI] [PubMed] [Google Scholar]
- 6.Lieber CS. Prevention and treatment of liver fibrosis based on pathogenesis. Alcohol Clin Exp Res. 1999;23:944–949. [PubMed] [Google Scholar]
- 7.Rockey DC. Gene therapy for hepatic fibrosis-bringing treatment into the new millennium. Hepatology. 1999;30:816–818. doi: 10.1002/hep.510300335. [DOI] [PubMed] [Google Scholar]
- 8.Cheng ML, Wu YY, Huang KF, Luo TY, Ding YS, Lu YY, Liu RC, Wu J. Clinical study on the treatment of liver fibrosis due to hepatitis B by IFN-alpha (1) and traditional medicine preparation. World J Gastroenterol. 1999;5:267–269. doi: 10.3748/wjg.v5.i3.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang LT, Zhang B, Chen JJ. Effect of anti-fibrosis compound on collagen expression of hepatic cells in experimental liver fibrosis of rats. World J Gastroenterol. 2000;6:877–880. doi: 10.3748/wjg.v6.i6.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Du B, You S. Present situation in preventing and treating liver fibrosis with TCM drugs. J Tradit Chin Med. 2001;21:147–152. [PubMed] [Google Scholar]
- 11.Murphy F, Arthur M, Iredale J. Developing strategies for liver fibrosis treatment. Expert Opin Investig Drugs. 2002;11:1575–1585. doi: 10.1517/13543784.11.11.1575. [DOI] [PubMed] [Google Scholar]
- 12.Bachem MG, Meyer D, Melchior R, Sell KM, Gressner AM. Activation of rat liver perisinusoidal lipocytes by transforming growth factors derived from myofibroblastlike cells. A potential mechanism of self perpetuation in liver fibrogenesis. J Clin Invest. 1992;89:19–27. doi: 10.1172/JCI115561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okuno M, Moriwaki H, Imai S, Muto Y, Kawada N, Suzuki Y, Kojima S. Retinoids exacerbate rat liver fibrosis by inducing the activation of latent TGF-beta in liver stellate cells. Hepatology. 1997;26:913–921. doi: 10.1053/jhep.1997.v26.pm0009328313. [DOI] [PubMed] [Google Scholar]
- 14.Pinzani M, Marra F, Carloni V. Signal transduction in hepatic stellate cells. Liver. 1998;18:2–13. doi: 10.1111/j.1600-0676.1998.tb00120.x. [DOI] [PubMed] [Google Scholar]
- 15.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 16.Bissell DM, Roulot D, George J. Transforming growth factor beta and the liver. Hepatology. 2001;34:859–867. doi: 10.1053/jhep.2001.28457. [DOI] [PubMed] [Google Scholar]
- 17.Wrana JL, Attisano L, Wieser R, Ventura F, Massagué J. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 18.Willis SA, Zimmerman CM, Li LI, Mathews LS. Formation and activation by phosphorylation of activin receptor complexes. Mol Endocrinol. 1996;10:367–379. doi: 10.1210/mend.10.4.8721982. [DOI] [PubMed] [Google Scholar]
- 19.Qi Z, Atsuchi N, Ooshima A, Takeshita A, Ueno H. Blockade of type beta transforming growth factor signaling prevents liver fibrosis and dysfunction in the rat. Proc Natl Acad Sci USA. 1999;96:2345–2349. doi: 10.1073/pnas.96.5.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin JS, Song YH, Kong XJ, Li B, Liu NZ, Wu XL, Jin YX. Preparation and identification of anti-transforming growth factor beta1 U1 small nuclear RNA chimeric ribozyme in vitro. World J Gastroenterol. 2003;9:572–577. doi: 10.3748/wjg.v9.i3.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ueno H, Sakamoto T, Nakamura T, Qi Z, Astuchi N, Takeshita A, Shimizu K, Ohashi H. A soluble transforming growth factor beta receptor expressed in muscle prevents liver fibrogenesis and dysfunction in rats. Hum Gene Ther. 2000;11:33–42. doi: 10.1089/10430340050016139. [DOI] [PubMed] [Google Scholar]
- 22.Choi ME, Kim EG, Huang Q, Ballermann BJ. Rat mesangial cell hypertrophy in response to transforming growth factor-beta 1. Kidney Int. 1993;44:948–958. doi: 10.1038/ki.1993.336. [DOI] [PubMed] [Google Scholar]
- 23.Bassing CH, Yingling JM, Howe DJ, Wang T, He WW, Gustafson ML, Shah P, Donahoe PK, Wang XF. A transforming growth factor beta type I receptor that signals to activate gene expression. Science. 1994;263:87–89. doi: 10.1126/science.8272871. [DOI] [PubMed] [Google Scholar]
- 24.Tsukamoto H, Matsuoka M, French SW. Experimental models of hepatic fibrosis: a review. Semin Liver Dis. 1990;10:56–65. doi: 10.1055/s-2008-1040457. [DOI] [PubMed] [Google Scholar]
- 25.Roth S, Schurek J, Gressner AM. Expression and release of the latent transforming growth factor beta binding protein by hepatocytes from rat liver. Hepatology. 1997;25:1398–1405. doi: 10.1002/hep.510250616. [DOI] [PubMed] [Google Scholar]
- 26.Sakaida I, Hironaka K, Uchida K, Suzuki C, Kayano K, Okita K. Fibrosis accelerates the development of enzyme-altered lesions in the rat liver. Hepatology. 1998;28:1247–1252. doi: 10.1002/hep.510280512. [DOI] [PubMed] [Google Scholar]
- 27.Takiya S, Tagaya T, Takahashi K, Kawashima H, Kamiya M, Fukuzawa Y, Kobayashi S, Fukatsu A, Katoh K, Kakumu S. Role of transforming growth factor beta 1 on hepatic regeneration and apoptosis in liver diseases. J Clin Pathol. 1995;48:1093–1097. doi: 10.1136/jcp.48.12.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Branch AD. A hitchhiker's guide to antisense and nonantisense biochemical pathways. Hepatology. 1996;24:1517–1529. doi: 10.1002/hep.510240634. [DOI] [PubMed] [Google Scholar]
- 29.Brenner DA, Waterboer T, Choi SK, Lindquist JN, Stefanovic B, Burchardt E, Yamauchi M, Gillan A, Rippe RA. New aspects of hepatic fibrosis. J Hepatol. 2000;32:32–38. doi: 10.1016/s0168-8278(00)80413-4. [DOI] [PubMed] [Google Scholar]