

Abstract
AIM: To investigate local corticosterone production and angiotensin-I converting enzyme (ACE) protein expression and their interaction in healthy and inflamed intestine.
METHODS: Acute intestinal inflammation was induced to six weeks old male Balb/c mice by administration of either 3% or 5% dextran sodium sulfate (DSS) in drinking water for 7 d (n = 12 in each group). Healthy controls (n = 12) were given tap water. Corticosterone production and ACE protein shedding were measured from ex vivo incubates of the small and large intestine using EIA and ELISA, respectively. Morphological changes of the intestinal wall were assessed in hematoxylin-eosin stained tissue preparations of jejunum and distal colon. Effects of angiotensin II, captopril and metyrapone on corticosterone production was assessed by incubating pieces of small intestine of healthy mice in the presence of 0.1, 1 or 10 μmol/L angiotensin II, 1, 10 or 100 μmol/L captopril or 1, 10 or 100 μmol/L metyrapone solutions and measuring corticosterone released to the incubation buffer after 90 min (n = 5 in each group).
RESULTS: Both concentrations of DSS induced inflammation and morphological changes in large intestines but not in small intestines. Changes were observed as distortions of the crypt structure, mucosal erosion, immune cell infiltration to the mucosa and submucosal edema. Ex vivo corticosterone production (2.9 ± 1.0 ng/mL vs 2.0 ± 0.8 ng/mL, P = 0.034) and ACE shedding (269.2 ± 97.1 ng/mL vs 175.7 ± 52.2 ng/mL, P = 0.016) were increased in small intestines in 3% DSS group compared to the controls. In large intestine, corticosterone production was increased compared to the controls in both 3% DSS (229 ± 81 pg/mL vs 158 ± 30 pg/mL, P = 0.017) and 5% DSS groups (366 ± 163 pg/mL vs 158 ± 30 pg/mL, P = 0.002). Large intestine ACE shedding was increased in 5% DSS group (41.5 ± 9.0 ng/mL vs 20.9 ± 5.2 ng/mL, P = 0.034). Angiotensin II treatment augmented corticosterone production in small intestine at concentration of 10 μmol/L (0.97 ± 0.21 ng/mg protein vs 0.40 ± 0.09 ng/mg protein, P = 0.036).
CONCLUSION: Intestinal ACE shedding is increased by DSS-induced intestinal inflammation and parallels local corticosterone production. ACE product angiotensin II stimulates corticosterone formation in healthy intestine.
Keywords: Dextran sodium sulfate, Inflammation, Angiotensin-I converting enzyme, Local corticosterone, Intestine
Core tip: Soluble and tissue levels of angiotensin-I converting enzyme (ACE) along with corticosterone production were examined in a dextran sulfate mouse model of intestinal inflammation. Intestine is a site of ACE shedding, which is increased by inflammation. ACE and corticosterone are increased in intestinal incubations of morphologically disrupted and intact parts of the intestine. ACE product Ang II stimulates corticosterone production in small intestine. The results suggest that intestinal Renin-Angiotensin system and glucocorticoids might be counter-regulatory systems in regulation of inflammatory processes in the intestine.
INTRODUCTION
Renin-angiotensin system (RAS) is best known as a regulator of systemic blood pressure. In addition, classic and alternative RAS regulate inflammatory processes in the vasculature[1]. Several components of RAS have been localized in various parts of the gastrointestinal tract but their function is not completely clear[2]. Angiotensin converting enzymes (ACE, ACE2) have been found throughout the human intestine[3-5]. The two types of angiotensin receptors (AT1R, AT2R) have also been detected in rat and human intestine suggesting that ACE aminopeptidases are not only food metabolizing enzymes but also have regulatory functions[2,4-7].
Another recent observation is the formation of adrenocortical glucocorticoid (GC) hormone, corticosterone, in the gut, where the regulation of synthesis is different than in adrenals[8-10]. Intestinal epithelium produces corticosterone to regulate inflammation by the action of tumor necrosis factor (TNF)-α[11]. In kidney and heart, angiotensin II (Ang II) induces TNF-α production[12,13]. Therefore, we hypothesized that these two systems, pro-inflammatory RAS and anti-inflammatory GCs, could play a counter-regulatory role in inflammatory processes of the intestine.
ACE is the central enzyme of classic RAS. ACE is an aminopeptidase, which cleaves two or three C-terminal amino acids from several peptides. The most important substrate for ACE is angiotensin I (Ang I) which is cleaved into pro-inflammatory Ang II. ACE is a membrane-bound enzyme with a short cytosolic C-terminal tail[14]. The extracellular part consists of N-terminal and C-terminal domains which both possess a catalytic site[15]. ACE extracellular domains can be cleaved and released to the circulation by one or more so called ACE sheddase enzymes[15-17]. One of those enzymes is a metalloprotease ADAM9[18]. An analogous shedding mechanism by ADAM17 has been described for ACE2[19,20]. The role of ACE shedding is unclear but it is thought that ACE shedding might be a way to regulate local ACE activity or substrate specificity[15]. Furthermore, ACE shedding has been reported in lung during ischemia/reperfusion and in pulmonary endothelial cells in septic conditions and during LPS treatment in vitro[18,21,22]. Here, we report of ACE shedding outside vasculature from the intestinal tissue in response to DSS-induced inflammation.
Dextran sulfate (DSS)-model of colitis induces inflammation, mucosal erosion and bleeding in mouse colon. There have been several reports of DSS inducing mild inflammatory changes in small intestine histology and biochemical markers up to jejunum[23-25].
The aim of the study was to investigate intestinal corticosterone production and ACE protein expression and their interaction in healthy and inflamed intestine.
MATERIALS AND METHODS
The study was conducted in the Institute of Biomedicine, Pharmacology, at the University of Helsinki. The study was approved by National Animal Experimentation Committee of Finland according to EC Directive 86/609/ECC and Finnish Experimental Animal Act 62/2006.
Animals and colitis model
Six weeks old male balb/c mice weighing 20 g ± 1 g were acclimatized to 12 h light-dark cycle and given water and standard rodent food, 2018 Teklad Global 18% Protein Rodent Diet (Harlan Laboratories, Indianapolis, IN, United States) ad libitum. Three or five percent (w/v) DSS was introduced in the drinking water for 7 d to induce acute colitis (n = 12 in each group). Control group was given tap water. The animals were weighed daily and the weight was calculated in relation to that at the beginning of the experiment. Eight weeks old untreated balb/c male mice were used in ex vivo stimulation experiments.
Sample collection
After 7 d the mice were sacrificed by decapitation. Small and large intestines were excised, opened longitudinally and rinsed with Phosphate Buffered Saline (PBS) (137 mmol/L NaCl, 7.9 mmol/L Na2HPO4, 2.7 mmol/L KCl, 1.5 mmol/L KH2PO4, pH = 7.4). Pieces of jejunum and distal colon were cut out, rinsed in Krebs buffer (119 mmol/L NaCl, 25 mmol/L NaHCO3, 15 mmol/L KCl, 11 mmol/L Glucose, 1.6 mmol/L CaCl2, 1.2 mmol/L KH2PO4, 1.2 mmol/L MgSO4) and fixed in 10% neutral-buffered formalin (Sigma Aldrich, St. Louis, MO, United States) for 24 h. The fixed tissues were washed with PBS and stored in 70% ethanol at +4 °C until paraffin embedding.
Ex vivo tissue incubation
The remaining part from duodenum to mid-jejunum and middle part of colon were sliced and incubated in pre-oxygenated Krebs buffer at 37 °C in gentle agitation. Samples of the buffer were taken after 90 min of incubation and centrifuged at 13000 g. The supernatant was used for the experiments.
In another experiment, the whole small intestine of balb/c mice were cut into pieces which were randomly distributed and incubated in Krebs’ buffer, 0.1, 1 or 10 μmol/L Ang II (Sigma Aldrich) to stimulate corticosterone production, 1, 10 or 100 μmol/L captopril (Sigma Aldrich) to block ACE activity or 1, 10 or 100 μmol/L metyrapone (Sigma Aldrich) to inhibit corticosterone synthesis for 90 min. The supernatants were taken for corticosterone assay and the tissue pieces saved for total protein measurement.
Histological analyses
Formalin-fixed paraffin embedded tissues were sliced and stained with hematoxylin and eosin stain. Crypt and villus structure, erosion of mucous membrane, inflammatory cells and submucosal edema were visually evaluated and compared between treatments.
Immunochemical assays
Corticosterone was measured from ex vivo -incubate samples taken at 90 min of 8 small intestine and large intestine samples using a corticosterone EIA kit (Cayman Chemical, Michigan, MI, United States). ACE protein was measured from ex vivo -incubates and tissue lysates using ELISA (R&D System, Minnesota, MO, United States).
Statistical analysis
Comparisons of corticosterone formation and ACE shedding to controls and the expression analyses were done with Student’s unpaired t-test. Data is presented as mean ± SD in bar graphs. Correlations are presented with Pearson’s correlation coefficient and calculated with GraphPad Prism 5. P values less than 0.05 were considered statistically significant. Statistical analyses were evaluated by a statistics expert. The statistical methods of this study were reviewed by Dr. Oleg Kambur from the University of Helsinki.
RESULTS
Weight development
The weight of the animals, given as percent of the mean initial weight (100%) was similar in all groups from day 1 to day 5, after which the animals in the 5% DSS group begun to lose weight. After 7 d, the animals in 5% DSS group weighed less than controls (93.1% ± 0.8% vs 103.9% ± 0.5%, P < 0.001) or those in 3% DSS group (93.1% ± 0.8% vs 100.5% ± 0.8%, P < 0.001). Animals treated with 3% DSS weighed less compared to controls after 7 d (100.5% vs 103.9%, P = 0.002). The weight loss was accompanied by diarrhea, with blood visible in the 5% DSS group.
Histological analyses
In the large intestine preparations, marked disruption of crypt structure, mononuclear cell infiltration, submucosal edema and mucosal erosion were observed in both 3% and 5% DSS groups but were not present in the control group (Figure 1). Histological changes were similar in both colitis groups. No pathological changes were observed in the small intestines in any group.
Figure 1.
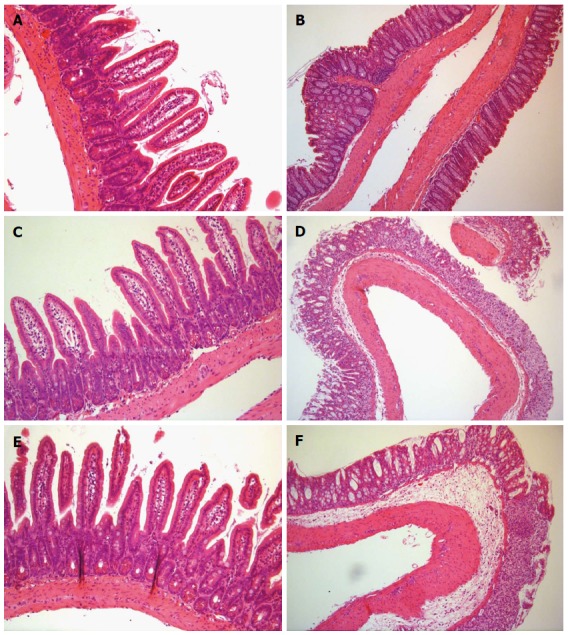
Small and large intestine histology. Hematoxylin eosin-stained tissue preparations of Control (A), 3% DSS (C) and 5% DSS (E) small intestines and Control (B), 3% DSS (D) and 5% DSS (F) large intestines. In D and F, crypt structure is damaged and mucosa is partly eroded. Edema is present in submucosa. Immune cells are present in mucosa and submucosa. Magnification × 20. DSS: Dextran sodium sulfate.
Corticosterone production
Corticosterone production in the ex vivo incubation of the small intestine samples was increased in 3% DSS group compared to control group at 90 min (2.9 ± 1.0 ng/mL vs 2.0 ± 0.8 ng/mL, P = 0.034) (Figure 2A). However, corticosterone levels did not differ significantly from control in 5% DSS-treated mice’s small intestines (2.5 ± 0.9 ng/mL vs 2.0 ± 1.0 ng/mL, P = 0.16). In large intestine, DSS treatment stimulated corticosterone production in both 3% DSS (0.23 ± 0.3 ng/mL vs 0.16 ± 0.1 ng/mL, P = 0.017) and 5% DSS groups (0.37 ± 0.5 ng/mL vs 0.16 ± 0.1 ng/mL, P = 0.002) (Figure 2B). Corticosterone concentration in the incubates of 5% DSS-group’s large intestine samples was higher than in those of 3% DSS group (P = 0.026).
Figure 2.
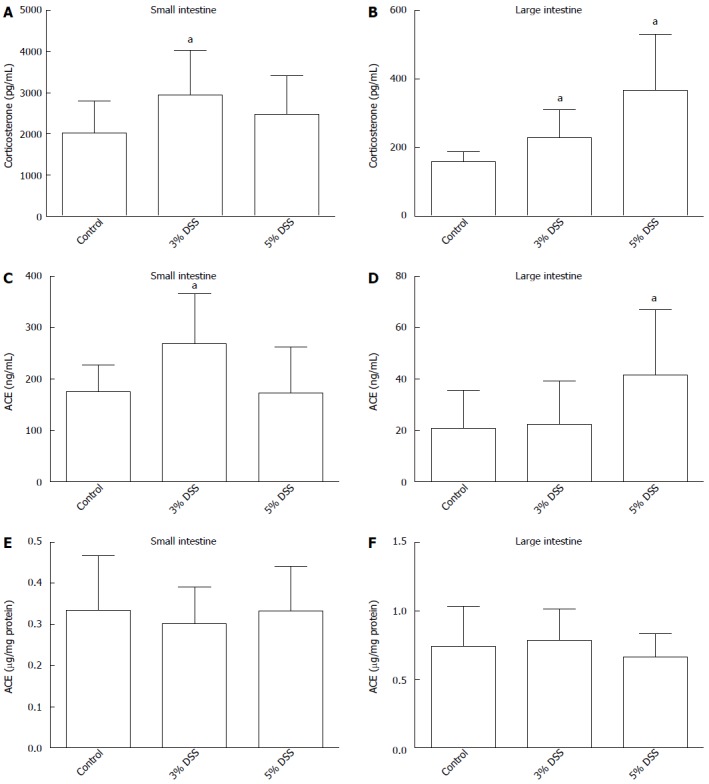
Corticosterone production and angiotensin-I converting enzyme shedding measured from healthy (control) and inflamed (DSS 3% or 5% induced in drinking fluid for 7 d) intestine samples (n = 8) after 90 min at 37 °C. All the conditions were pretested in pilot experiments. Corticosterone production measured from the incubate buffer small intestine (A), large intestine (B). ACE shedding into incubate small intestine (C), large intestine (D). Tissue bound ACE from tissue lysate small intestine (E), large intestine (F). Data expressed as mean ± SD, n = 8 in each column. aP < 0.05 vs control. ACE: Angiotensin-I converting enzyme; DSS: Dextran sodium sulfate.
ACE shedding
ACE protein was present in the supernatants of mouse ex vivo intestinal incubations (Figure 2C and D). ACE concentration was increased in 3% DSS group small intestine supernatants at 90 min compared to control (269.2 ± 90.9 ng/mL vs 175.7 ± 48.8 ng/mL, P = 0.016). In large intestine incubates, ACE concentrations were increased in 5% DSS group compared to control (41.5 ± 23.8 ng/mL vs 20.9 ± 13.7 ng/mL, P = 0.034) but not in 3% DSS group. In tissue lysates, ACE protein concentrations were similar in all groups (Figure 2E and F).
Corticosterone and ACE correlations
Corticosterone and ACE concentrations correlated with each other in both small intestine (Pearson correlation 0.435, 95%CI: 0.169-0.642, P = 0.001) and large intestine (Pearson correlation 0.315, 95%CI 0.033-0.550, P = 0.015) (Figure 3). Similarly, there was a correlation between small and large intestine corticosterone production (Pearson correlation, 0.534, 0.295 to 0.711, P < 0.001) but not in ACE shedding (Pearson correlation 0.027, P = 0.429).
Figure 3.
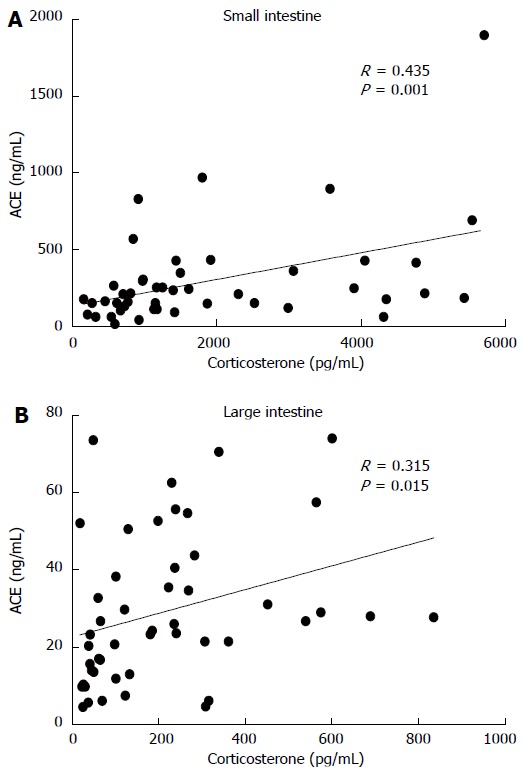
Correlation of angiotensin-I converting enzyme protein and corticosterone in ex vivo intestinal incubations. A: Small intestine, n = 23; B: Large intestine, n = 24.
Effects of angiotensin II, captopril and metyrapone on corticosterone production in healthy tissue
Angiotensin II, a stimulator of adrenal corticosterone synthesis, also increased corticosterone production in small intestines at 10 μmol/L concentration (0.97 ± 0.04 ng/mg protein vs 0.40 ± 0.02 ng/mg protein, P = 0.036), which was the highest concentration tested (Figure 4A). Captopril, an inhibitor of ACE, had no effect on corticosterone production (Figure 4B). Metyrapone, an inhibitor of corticosterone synthesis, unexpectedly increased corticosterone production at the smallest concentration tested, 1 μmol/L (0.95 ± 0.41 ng/mg protein vs 0.40 ± 0.02 ng/mg protein, P = 0.040), but had no effect at higher concentrations (Figure 4C).
Figure 4.
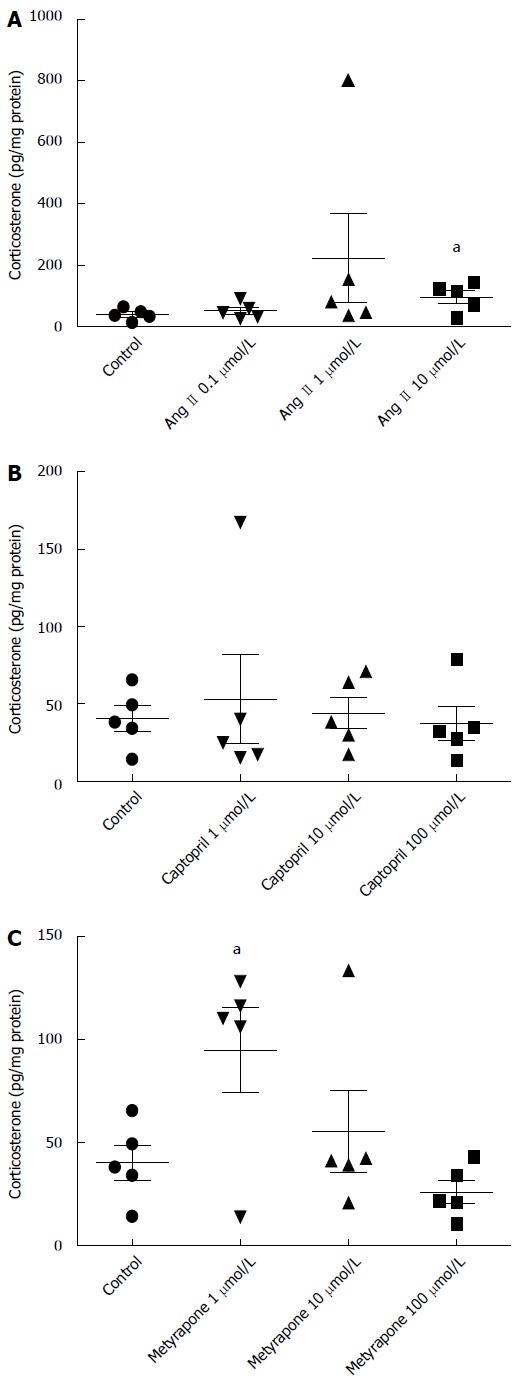
Concentration-response effects of three different concentrations of angiotensin (A), captopril (B) and metyrapone (C) on corticosterone production measured from incubation buffer of small intestine sample after 90 min at 37 °C. Individual values, mean ± SD from (n = 5 in each concentration) are given. aP < 0.05 vs control.
DISCUSSION
Inflammatory bowel diseases (IBDs) are severe clinical ailments which manifest by diarrhea, intestinal bleeding and inflammation. Two major forms of IBD are ulcerative colitis (UC) and Crohn’s disease (CD). In UC, the inflammation is restricted to the colonic mucosa, whereas in CD the inflammation can sporadically affect any part of the intestinal tract and reach through all tissue layers. The causes of IBD are not fully understood but they are thought to be multifactorial and involve hereditary, immunological and environmental factors.
DSS is widely used to induce inflammation of the large intestine as an animal model of IBD[26]. However, it has been reported to induce inflammation in the small intestine as well, mainly in the ileum. In the present study, DSS treatment caused extensive histological damage in the mucosal layer of mouse colon but did not cause any clear histological damage in the jejunum. No appreciable changes were observed in muscularis mucosae, bearing more resemblance to ulcerative colitis than Crohn’s disease.
In the present study, we tested, as far as we know, for the first time the possible association between glucocorticoids and renin-angiotensin system in the gastrointestinal tract. These two systems interact in the adrenal cortex.
Here, we report of inflammation-induced ACE protein shedding from mouse intestine in an ex vivo incubation. ACE shedding from vascular endothelium has been demonstrated earlier by others[15]. Previously, ACE activity has been measured from feces and considered as a sign of cell damage[27]. The current study did not assess whether ACE shedding occurs from the epithelial cells, immune cells or the vascular endothelium. However, due to the handling of the samples and recent reports of an ACE sheddase[18], it is likely the measured protein is in soluble form rather than membrane-bound. Furthermore, in severe colitis, we have observed angiotensinogen expression coinciding with ACE shedding (unpublished data).
Intestinal corticosterone production is increased by inflammation[8]. In line with previous studies, corticosterone production was increased in large intestines of DSS-treated mice. Furthermore, despite no histological changes were observed in small intestines of DSS-treated mice, corticosterone production and ACE shedding were increased in jejuna of DSS-treated mice without marked differences in the tissue ACE levels. This might be due to milder, non-detectable, inflammation of the small intestine.
Since both of these processes, corticosterone production and ACE shedding, were increased by inflammation, we hypothesized that they might interact in the intestine. Indeed, corticosterone production was increased in small intestine tissue incubation by Ang II. However, inhibition of ACE by captopril in vitro did not affect the basal corticosterone production. Further studies will show, whether captopril treatment in vivo has an effect on corticosterone production in inflamed intestine. Interestingly, the in vivo glucocorticoid synthesis inhibitor metyrapone did not inhibit corticosterone synthesis in this in vitro experimental setup but rather increased it at low concentration.
The present study shows that the DSS colitis model can be used to study local angiotensin system in intestinal inflammation. As conclusion, we demonstrate ACE shedding from mouse intestine induced by inflammation. Pro-inflammatory ACE and anti-inflammatory glucocorticoids appeared in incubation buffer even in the small intestine, far from the site of structural damage, further supporting reports of small intestinal manifestations of the DSS model. The results of this study suggest that intestinal RAS and glucocorticoids might be counter-regulatory systems in regulation of inflammatory processes in the intestine.
ACKNOWLEDGMENTS
We are grateful to Anne Reijula at Tissue preparation and histochemistry unit, Medicum, University of Helsinki, Professor Ari Ristimäki for discussions and interpretations of H&E stained tissue slides and Alessandro Accardi for technical assistance.
COMMENTS
Background
Inflammatory bowel diseases (IBD) such as Crohn´s disease and ulcerative colitis are increasing in Western societies. Different animal models (e.g., dextran sulfate sodium, TNBS) have been used to clarify their pathophysiology and pharmacological possibilities for the treatment. Recently, two important regulatory systems (renin-angiotensin-aldosterone and glucocorticoids), generally acting systemically have been described to exist also locally throughout the whole intestine. Because glucocorticoids are one of the main treatments of these inflammatory diseases, the local production may be of importance. Overexpression of renin- angiotensin system in vasculature is regarded pro-inflammatory. Therefore we decided to clarify in the mouse dextran sodium sulfate (DSS)-model, whether these two systems are associated in intestinal inflammation.
Research frontiers
Only few research groups have been interested in local expression and activities of these systems, and if so, only in one of these two items. Therefore the authors believe that the results will have some priority in this field and will stimulate activities of other research teams.
Innovations and breakthroughs
DSS model is rather traditional in pharmacology to study anti-inflammatory agents. The novelties are confirmation of the expression of the two inflammatory players in the intestinal epithelium ex vivo, and observation of association between them. There are differences between the small and large intestine. Furthermore glucocorticoid synthesis in the gut is differently regulated than in the adrenals. The strengths of the study are the diversity of the methods used. The present findings have also given us new ideas for the ongoing studies.
Applications
If intestinal epithelium in chronic inflammation is deficient to produce protective glucocorticoids and shows increased synthesis of pro-inflammatory components of renin-angiotensin system (RAS), the present findings are in agreement with the present clinical practice of use of local glucocorticoids in irritable bowel syndrome (IBS). Whether it would be found a direct mean to stimulate intestinal corticoid synthesis, it would offer a treatment without glucocorticoids' systemic adverse effects. Over-activity of RAS in intestinal inflammation would suggest treatment options for IBS with angiotensin converting enzyme (ACE) inhibitors or angiotensin type 1 receptor blockers.
Terminology
DSS-induced colitis is a model of inflammatory bowel diseases. DSS directly damages intestinal, specifically colonic, mucosa. ACE shedding refers to the enzymatic cleavage of the extracellular domains of ACE protein in which the active enzyme becomes soluble.
Peer-review
Salmenkari et al address the role of corticosterone production and ACE function in modulating intestinal inflammation, using a mouse DSS model of chemically induced IBD. They show that corticosterone and ACE are produced in this model, which could be of interest in vivo.
Footnotes
Supported by Grants from Foundation for Clinical Chemistry Research, Finland (partly).
Institutional review board statement: The study was reviewed and approved by the University of Helsinki, Pharmacology Institutional Review Board.
Institutional animal care and use committee statement: The study was approved by National Animal Experimentation Committee of Finland (ESAVI/6314/04.10.03/2012) according to EC Directive 86/609/ECC and Finnish Experimental Animal Act 62/2006.
Conflict-of-interest statement: The authors declare no conflict of interest.
Data sharing statement: Technical appendix, statistical code, and dataset available from the corresponding author at heikki.vapaatalo@helsinki.fi.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: February 7, 2015
First decision: April 13, 2015
Article in press: July 3, 2015
P- Reviewer: Taylor GA S- Editor: Yu J L- Editor: A E- Editor: Zhang DN
References
- 1.Schiffrin EL. Beyond blood pressure: the endothelium and atherosclerosis progression. Am J Hypertens. 2002;15:115S–122S. doi: 10.1016/s0895-7061(02)03006-6. [DOI] [PubMed] [Google Scholar]
- 2.Garg M, Angus PW, Burrell LM, Herath C, Gibson PR, Lubel JS. Review article: the pathophysiological roles of the renin-angiotensin system in the gastrointestinal tract. Aliment Pharmacol Ther. 2012;35:414–428. doi: 10.1111/j.1365-2036.2011.04971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruneval P, Hinglais N, Alhenc-Gelas F, Tricottet V, Corvol P, Menard J, Camilleri JP, Bariety J. Angiotensin I converting enzyme in human intestine and kidney. Ultrastructural immunohistochemical localization. Histochemistry. 1986;85:73–80. doi: 10.1007/BF00508656. [DOI] [PubMed] [Google Scholar]
- 4.Hirasawa K, Sato Y, Hosoda Y, Yamamoto T, Hanai H. Immunohistochemical localization of angiotensin II receptor and local renin-angiotensin system in human colonic mucosa. J Histochem Cytochem. 2002;50:275–282. doi: 10.1177/002215540205000215. [DOI] [PubMed] [Google Scholar]
- 5.Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sechi LA, Valentin JP, Griffin CA, Schambelan M. Autoradiographic characterization of angiotensin II receptor subtypes in rat intestine. Am J Physiol. 1993;265:G21–G27. doi: 10.1152/ajpgi.1993.265.1.G21. [DOI] [PubMed] [Google Scholar]
- 7.Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006;86:747–803. doi: 10.1152/physrev.00036.2005. [DOI] [PubMed] [Google Scholar]
- 8.Cima I, Corazza N, Dick B, Fuhrer A, Herren S, Jakob S, Ayuni E, Mueller C, Brunner T. Intestinal epithelial cells synthesize glucocorticoids and regulate T cell activation. J Exp Med. 2004;200:1635–1646. doi: 10.1084/jem.20031958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mueller M, Atanasov A, Cima I, Corazza N, Schoonjans K, Brunner T. Differential regulation of glucocorticoid synthesis in murine intestinal epithelial versus adrenocortical cell lines. Endocrinology. 2007;148:1445–1453. doi: 10.1210/en.2006-0591. [DOI] [PubMed] [Google Scholar]
- 10.Mueller M, Cima I, Noti M, Fuhrer A, Jakob S, Dubuquoy L, Schoonjans K, Brunner T. The nuclear receptor LRH-1 critically regulates extra-adrenal glucocorticoid synthesis in the intestine. J Exp Med. 2006;203:2057–2062. doi: 10.1084/jem.20060357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noti M, Corazza N, Tuffin G, Schoonjans K, Brunner T. Lipopolysaccharide induces intestinal glucocorticoid synthesis in a TNFalpha-dependent manner. FASEB J. 2010;24:1340–1346. doi: 10.1096/fj.09-140913. [DOI] [PubMed] [Google Scholar]
- 12.Ferreri NR, Escalante BA, Zhao Y, An SJ, McGiff JC. Angiotensin II induces TNF production by the thick ascending limb: functional implications. Am J Physiol. 1998;274:F148–F155. doi: 10.1152/ajprenal.1998.274.1.F148. [DOI] [PubMed] [Google Scholar]
- 13.Kalra D, Sivasubramanian N, Mann DL. Angiotensin II induces tumor necrosis factor biosynthesis in the adult mammalian heart through a protein kinase C-dependent pathway. Circulation. 2002;105:2198–2205. doi: 10.1161/01.cir.0000015603.84788.47. [DOI] [PubMed] [Google Scholar]
- 14.Hooper NM, Keen J, Pappin DJ, Turner AJ. Pig kidney angiotensin converting enzyme. Purification and characterization of amphipathic and hydrophilic forms of the enzyme establishes C-terminal anchorage to the plasma membrane. Biochem J. 1987;247:85–93. doi: 10.1042/bj2470085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernstein KE, Ong FS, Blackwell WL, Shah KH, Giani JF, Gonzalez-Villalobos RA, Shen XZ, Fuchs S, Touyz RM. A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharmacol Rev. 2013;65:1–46. doi: 10.1124/pr.112.006809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ehlers MR, Chen YN, Riordan JF. Spontaneous solubilization of membrane-bound human testis angiotensin-converting enzyme expressed in Chinese hamster ovary cells. Proc Natl Acad Sci USA. 1991;88:1009–1013. doi: 10.1073/pnas.88.3.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei L, Alhenc-Gelas F, Soubrier F, Michaud A, Corvol P, Clauser E. Expression and characterization of recombinant human angiotensin I-converting enzyme. Evidence for a C-terminal transmembrane anchor and for a proteolytic processing of the secreted recombinant and plasma enzymes. J Biol Chem. 1991;266:5540–5546. [PubMed] [Google Scholar]
- 18.English WR, Corvol P, Murphy G. LPS activates ADAM9 dependent shedding of ACE from endothelial cells. Biochem Biophys Res Commun. 2012;421:70–75. doi: 10.1016/j.bbrc.2012.03.113. [DOI] [PubMed] [Google Scholar]
- 19.Grobe N, Di Fulvio M, Kashkari N, Chodavarapu H, Somineni HK, Singh R, Elased KM. Functional and molecular evidence for expression of the renin angiotensin system and ADAM17-mediated ACE2 shedding in COS7 cells. Am J Physiol Cell Physiol. 2015;308:C767–C777. doi: 10.1152/ajpcell.00247.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel VB, Clarke N, Wang Z, Fan D, Parajuli N, Basu R, Putko B, Kassiri Z, Turner AJ, Oudit GY. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: a positive feedback mechanism in the RAS. J Mol Cell Cardiol. 2014;66:167–176. doi: 10.1016/j.yjmcc.2013.11.017. [DOI] [PubMed] [Google Scholar]
- 21.Hermanns MI, Müller AM, Tsokos M, Kirkpatrick CJ. LPS-induced effects on angiotensin I-converting enzyme expression and shedding in human pulmonary microvascular endothelial cells. In Vitro Cell Dev Biol Anim. 2014;50:287–295. doi: 10.1007/s11626-013-9707-0. [DOI] [PubMed] [Google Scholar]
- 22.Atochina EN, Muzykantov VR, Al-Mehdi AB, Danilov SM, Fisher AB. Normoxic lung ischemia/reperfusion accelerates shedding of angiotensin converting enzyme from the pulmonary endothelium. Am J Respir Crit Care Med. 1997;156:1114–1119. doi: 10.1164/ajrccm.156.4.96-12116. [DOI] [PubMed] [Google Scholar]
- 23.Wildhaber BE, Yang H, Haxhija EQ, Spencer AU, Teitelbaum DH. Intestinal intraepithelial lymphocyte derived angiotensin converting enzyme modulates epithelial cell apoptosis. Apoptosis. 2005;10:1305–1315. doi: 10.1007/s10495-005-2138-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi S, Kawamura T, Kanda Y, Taniguchi T, Nishizawa T, Iiai T, Hatakeyama K, Abo T. Multipotential acceptance of Peyer’s patches in the intestine for both thymus-derived T cells and extrathymic T cells in mice. Immunol Cell Biol. 2005;83:504–510. doi: 10.1111/j.1440-1711.2005.01361.x. [DOI] [PubMed] [Google Scholar]
- 25.Yazbeck R, Howarth GS, Butler RN, Geier MS, Abbott CA. Biochemical and histological changes in the small intestine of mice with dextran sulfate sodium colitis. J Cell Physiol. 2011;226:3219–3224. doi: 10.1002/jcp.22682. [DOI] [PubMed] [Google Scholar]
- 26.Perše M, Cerar A. Dextran sodium sulphate colitis mouse model: traps and tricks. J Biomed Biotechnol. 2012;2012:718617. doi: 10.1155/2012/718617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Letizia C, Picarelli A, De Ciocchis A, Di Giovambattista F, Greco M, Cerci S, Torsoli A, Scavo D. Angiotensin-converting enzyme activity in stools of healthy subjects and patients with celiac disease. Dig Dis Sci. 1996;41:2268–2271. doi: 10.1007/BF02071411. [DOI] [PubMed] [Google Scholar]