

Abstract
Chromaffin cells are catecholamine-producing cells derived from neural crest tissue. Chromaffin tumors (ChT) are rare tumors arising from these cells and are divided into pheochromocytoma (PCC) arising from adrenal tissue and paraganglioma (PGL) arising from extra-adrenal ganglia. Previously, ∼10% were believed to be hereditary, but advances in genome sequencing has shown roughly 35% of apparently sporadic tumors have a hereditary component. In this review we describe both classic and newly discovered hereditary ChT syndromes and provide recommendations for genetic testing. In many cases the genes associated with these conditions are linked to common kidney cancer pathways familiar to urologic oncologists.
Keywords: pheochromocytoma, paraganglioma, genetic testing, extra-adrenal pheochromocytoma, carotid body tumor, adrenal nodule
Introduction
Chromaffin cells are catecholamine-producing cells derived from neural crest tissue and are present in ganglia throughout the body. These neural crest cells are found in the adrenal medulla and in extra-adrenal sympathetic and parasympathetic ganglia. Chromaffin tumors are rare tumors that arise from these cells and are divided into two types by the most recent WHO definition. By the most recent WHO definition, pheochromocytoma is defined as a tumor arising from the adrenal tissue while paraganglioma arises from extra-adrenal tissue.1
Hereditary pheochromocytoma was once thought to represent a small fraction (∼10%) of these tumors.2 While many patients are diagnosed with a classic hereditary syndrome due to a known family history, some patients may have a newly diagnosed tumor of hereditary origin by representing a founder mutation, having an incomplete knowledge of family history, having uncertain family lineage due to adoption, or having incomplete penetrance that obscures a family history. Besides the traditional, well-described hereditary syndromes associated with pheochromocytoma/paraganglioma, the expansion of whole genome sequencing technologies has led to the identification of a number of new genes associated with the development of these tumors. Recent studies estimate that roughly 35% of apparently sporadic pheochromocytomas and paragangliomas actually have a hereditary component.3-5 An awareness of the genetic basis of these tumors is necessary in order to appropriately recognize who may benefit from genetic analysis. Identification of patients affected with these familial syndromes is critical; as several of these conditions are associated with aggressive forms of malignancy. With close screening and surveillance of at risk individuals, pheochromocytomas/paragangliomas may be cured or better treated if diagnosed at an early stage. Besides benefit to the individual, their family members can be tested to determine mutation status and if affected, undergo appropriate and effective screening.
Awareness of the classic and newly described syndromes is the key to successful diagnosis. Particular attention is paid to the patients' past medical history, keeping in mind the potential for a heterogeneous presentation (Online Table 1). Physical examination should include dermatologic, head and neck, ophthalmologic, and a neurologic evaluation.
In this review we discuss the traditional hereditary pheochromocytoma/paraganglioma syndromes as well as the recently identified syndromes, indications for genetic testing and testing indications. Many of the genes associated with these pheochromocytoma/paraganglioma syndromes are closely related and several are linked to common kidney cancer pathways.
Background
The incidence of pheochromocytoma/paraganglioma is quite low, with less than 2000 cases each year in the United States, with an annual incidence rates of 0.8 per 100,000 person-years.6 For paraganglioma, despite representing only about 20% of chromafin tumors, urologic surgeons should also be familiar with their management as 85% of these tumors are located in the abdomen and pelvis.7
Despite the WHO dividing chromaffin tumors into two types, many experts still split paragangliomas into head and neck paragangliomas and extra-adrenal pheochromocytomas due to frequent differences in catecholamine secretion and their origin from either parasympathetic or sympathetic nervous system, respectively. The extra-adrenal ganglia can be part of both nervous systems; an important distinction as they differ in both location and symptomatology. The sympathetic- derived paragangliomas are most commonly located in the retroperitoneum, and almost always produce catecholamines (optimally detected by metanephrines, O-methylated catecholamine metabolites). Therefore, it is not uncommon to see these tumors mentioned as extra-adrenal pheochromocytomas. The most common retroperitoneal location of these tumors is alongside the infra-renal aorta, near the inferior mesenteric artery and above the aortic bifurcation (also known as the organ of Zuckerkandl).8 The parasympathetic derived paragangliomas are usually functionally silent and commonly occur in the head and neck region. These are commonly referred to as head and neck paragangliomas. These tumors commonly arise from the carotid body and from cervical branches of the 9th and 10th cranial nerves (glossopharyngeal and vagus nerves).
Clinical Presentation
Patients with symptomatic pheochromocytomas and paragangliomas frequently report hypertension, sweats, palpitations, anxiety, headaches, tachycardia, and tremors. However, with current imaging, 20-30% of these tumors are incidentally detected.9 Pheochromocytomas can be found in approximately 1/5000 patients evaluated for hypertension and in the workup of 1/20 adrenal incidentalomas. While baseline catecholamines may be elevated, at least about half of patients can present with episodic surges of high blood pressure, tachycardia and other catecholamine-related symptoms and signs induced by stress, physical exertion, trauma, surgery, general anesthesia, and childbirth. The most common symptom, hypertension, and can be persistent or paroxysmal, depending on catecholamine levels at rest and during stimulation. Left untreated, pheochromocytomas and paragangliomas can be fatal due to catecholamine-induced cardiovascular morbidity, with complications including malignant hypertension, stroke, heart failure, and fatal arrhythmias.10 A subset of patients with pheochromocytomas or paragangliomas can develop metastatic disease, which is most often fatal.
The parasympathetic head and neck paragangliomas generally do not produce catecholamines, and the symptoms and presentation are quite different from those traditionally associate with pheochromocytoma. Patients with head and neck paragangliomas may present with a palpable neck mass, however, increased utilization of cross sectional imaging had lead to more incidental detection. With the improved understanding of hereditary basis of chromaffin tumors, germline mutation testing is recommended as well as screening of at-risk relatives.
Clinical Evaluation
The initial workup for a pheochromocytoma/paraganglioma includes biochemical testing to document excessive catecholamine production (Online Figure 1). Clinicians have the option of performing either, or both, plasma or urinary testing.11 The optimal testing strategy remains to be determined, as the sensitivity and specificity of each test varies. Many clinicians choose to screen patients with what is currently the most sensitive test, plasma free metanephrines.12 There is complexity in evaluating the results of biochemical testing, and there are a number of pitfalls which can potentially lead to erroneous conclusions. While in most patients with a pheochromocytoma there is a several-fold elevation in catecholamines, minor increases frequently represent a false positive result. Patient factors (stress and illness) and dietary factors (caffeine) can significantly alter testing results. Medications must be also reviewed prior to testing, as several can falsely raise catecholamine levels.13 These medications must be held prior to testing to avoid a false positive result. The biochemical diagnosis of pheochromocytoma is not be based on the measurement of plasma or urinary catecholamines, but on their metabolites, metanephrines, that are less prone to variations due to a patient's physical activity, diet, kidney function, or medication.14 Nevertheless, metanephrines can be often elevated due to antihypertensive or antidepressant medications.13 Nevertheless, although exceptions exist, plasma metanephrine elevations over 4 times above the upper reference limit are almost always associated with the presence of a tumor.13 In situations where there is a high clinical suspicious of pheochromocytoma/paraganglioma and plasma metanephrine is not found to be elevated 4 times above the upper reference limit, the clonidine suppression test coupled with the measurement of plasma normetanephrine can be helpful.15 It is important to note that the clonidine suppression test is not helpful in the diagnosis of epinephrine-producing (metanephrine-secreting) tumors, commonly seen in patients with multiple endocrine neoplasia type 2 (MEN2) and neurofibromatosis type 1 (NF1).
Once a biochemical analysis confirms excess catecholamine production, the next step is localization of the tumor. Either abdominal MRI or CT scans are effective anatomical imaging studies to aid localization in a patient with a biochemically suspected pheochromocytoma. Both imaging modalities have excellent sensitivity and specificity (∼90%) for the detection of visceral pheochromocytoma/paraganglioma.16-18 When the results are inconclusive, the functional imaging test, the 123I-MIBG scintigraphy, can visualize uptake of radiopharmaceutical into tumor cells. While this modality, which allows imaging of the whole body, has excellent specificity, it has suboptimal sensitivity. As the sensitivity of the 123I-MIBG scitnigraphy in some of the hereditary syndromes as well as in some patients with malignant pheochromocytoma/paraganglioma is less than ideal, new PET imaging modalities, include the use of 18F-fluorodopamine or 18F-fluorodopa, are emerging.19-22
Prior to offering definitive management for a newly diagnosed pheochromocytoma/paraganglioma, a medical and family history are obtained and a thorough physical exam is performed. If a catecholamine or metanephrine excess is found, alpha or beta adrenoceptor blockade recommended (phenoxybenzamine, as an alpha adrenoceptor blocker, is preferable at our institution).23 Beta adrenoceptor blocker is recommended if tachycardia is present. Alpha methyl-p-tyrosine (Demser), as a catehcolamine synthesis blocker, can be used as a part of preoperative management or in patients with signs and symptoms of catecholamine excess.23,24
Prior to surgical management, genetic testing is recommended. The current expense of performing all available tests can approach several thousand dollars; therefore knowledge of the clinical and biochemical phenotypic associated with the specific familial syndromes associated with the development of pheochromocytoma/paraganglioma can expedite the clinical evaluation as well as reduce the costs of genetic testing significantly.25,26
Pheochromocytoma and Paraganglioma Syndromes
vonHippel-Lindau
Von Hippel-Lindau (VHL) is an autosomal dominant hereditary condition characterized by the development of tumors in a number of organs, including retinal angiomas, central nervous system hemangioblastomas, pancreatic cysts and neuroendocrine tumors, renal cell carcinomas, epididymal cystadenomas, and pheochromocytoma/paraganglioma (Figure 1). The incidence of this disorder is approximately 1:35,000 individuals. Linkage analysis in families affected with VHL localized the region of interest to 3p.27 Subsequently the VHL gene was identified determined to be associated with the development of both von Hippel-Lindau and most cases of sporadic clear cell kidney cancer.28-31 Hypoxia inducible factor (HIF) is targeted for proteosomal degradation by the VHL complex.32,1 HIF requires modification for binding at specific prolyl residues, a process regulated by prolyl hydroxylases.33,34
Figure 1. a-f: VHL patients with pheochromocytoma or paraganglioma.
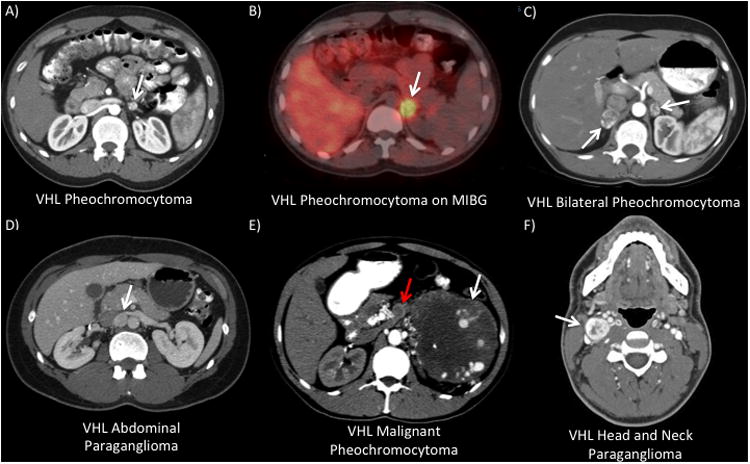
A) and B) A CT and MIBG scan from a 22 year-old male with a prior history of a right total adrenalectomy. He presented with headaches and was found with 3 left adrenal lesions, the largest measuring 1.4 cm. He underwent a robotic left partial adrenalectomy removing 4 nodules and post-operative he had preserved adrenal function. C) 46 year-old female who was found with bilateral pheochromocytomas. She underwent a bilateral laparoscopic partial adrenalectomy and had preservation of adrenal function. D) CT scan from a 25 year-old female with history of a right total adrenalectomy who presented with headaches and hypertension. She was found with a 1.5 cm interaortocaval paraganglioma that excised laparoscopically. E) 47 year-old male previously undiagnosed with VHL who presented with flank pain, hypertension, and anxiety. He was found with a 22 cm left adrenal mass with associated adrenal vein tumor thrombus (red arrow). On metastatic workup he was found with several bone metastasis. He underwent a left total adrenalectomy, thrombectomy, and distal pancreatectomy/splenectomy. F) 22 male with VHL who presented with anxiety, hypertension, and palpatations and was found with bilateral pheochromocytomas and a head and neck paraganglioma. After staged laparoscopic partial adrenalectomies, he had a right-sided neck dissection with excision of a 3 cm paraganglioma located at the carotid bifurcation.
Pheochromocytoms occurs in about 25% of patients with VHL and are most often benign. Pheochromocytomas tend to occur in specific families and genotype-phenotype correlations have demonstrated that missense mutations in VHL predispose to pheochromocytoma, referred to as type 2 VHL.35 Type I VHL, often characterized by germline deletions, insertions, and nonsense mutations of the VHL gene, uncommonly is associated with the development of pheochromocytoma.35
Multiple Endocrine Neoplasia (MEN2)
The multiple endocrine neoplasia syndrome 2 (MEN2) is a hereditary cancer syndrome occurring in approximately 1:40,000 individuals and is characterized by tumors derived from neural ectoderm tissue. The MEN2 syndrome can be subdivided into a MEN2a and MEN2b based on differing phenotype. The majority (>90%) of patients with MEN syndrome fall into the MEN2a type, in which patients are at risk for the development of medullary thyroid cancer, hyperparathyroidism and pheochromocytoma. Roughly 50% of MEN2a patients develop pheochromocytoma, which is frequently is bilateral (Figure 2). The MEN2b phenotype, which is less common, may include medullary thyroid cancer and pheochromocytoma as well as mucosal neuromas and a marfanoid body habitus. The pheochromocytomas associated with both MEN2 phenotypes are generally benign.
Figure 2. a-f: Patients with various hereditary pheochromocytoma or paraganglioma syndromes.
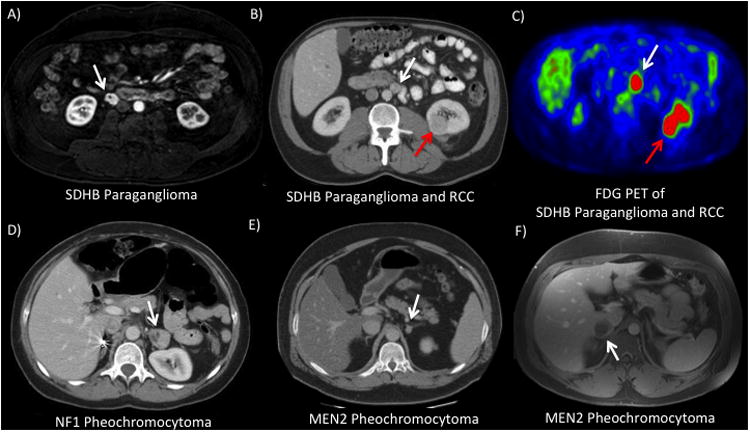
A) 53 year-old male with SDHB who presented with uncontrolled hypertension. He was found on workup with a 2.0 cm paracaval paraganglioma that was managed with laparoscopic excision. CT (B) and FDG-PET (C) scans from a 63 year-old male who underwent screening for SDHB and was found with an incidental 3 cm left renal mass and a 1.5 cm peri-aortic paraganglioma. He subsequently underwent a left robotic partial nephrectomy and paraganglioma resection. (Patient previously reported in Ricketts et. al -citation 53) D) 51 year-old female with NF1 and a previous right total adrenalectomy presented with hypertension. She was found with two left adrenal lesions (3.2 and 1.2 cm) and underwent a left laparoscopic partial adrenalectomy. E) 43 year-old male with MEN2a who presented with tachycardia and hypertension and was found with bilateral pheochromocytoma (left lesion shown). He underwent a bilateral laparoscopic partial adrenalectomy and has remained off adrenal replacement. F) 30 year-old male with MEN2a who presented with flushing and anxiety and was identified with a 4 cm right adrenal mass. He subsequently underwent a right laparoscopic partial adrenalectomy.
The gene responsible for both MEN2 phenotypes was mapped to 10q11.2 and later found to be the RET proto-oncogene.36,37 This gene encodes a trans-membrane tyrosine kinase and is responsible for multiple signaling pathways and is important for neuroendocrine cell development. There is a strong genotype-phenotype correlation between the mutation type in RET and the clinical presentation (MEN2a vs MEN2b).38
Neurofibromatosis 1
Neurofibromatosis 1 (NF1) is an autosomal dominant disease first recognized over 150 years ago by von Recklinghausen. This disorder is one of the most recognized hereditary disorders with an incidence of 1:3500 individuals. NF1 is characterized by neurofibromas, benign peripheral nerve tumors throughout the body. Neurofibromas can occur in either large, deep nerves (plexiform) under the skin or associated with cutaneous peripheral nerve that are quite noticeable. Cutaneous manifestations are common and include freckling in several regions and multiple café au lait spots (large light-brown pigmented birthmarks). Other parts of the syndrome include ocular abnormalities such as Lisch nodules in the iris and optic nerve tumors, as well as skeletal dysplasia. NF1 has historically been thought to have a 5% incidence of pheochromocytoma, however, a recent series reports an incidence as high as 15% (Figure 2).39,40 NF1 associated paragangliomas are uncommon, however the rate of malignancy may be as high as 10%.41
The gene for NF1 was identified in the late 1990's. Initial mapping studies positioned the gene responsible for NF1 to 17p11.2.42 The gene, NF1, was later cloned and is now known to behave like a classical tumor suppressor gene.43,44 Loss of NF1 function leads to dysregulated Ras signaling and tumorigenesis.45 NF1 is a critical negative regulator of TSC2 and mTOR, and loss of function actives downstream effectors such as S6K.46
As NF1 has a relatively high incidence of spontaneous mutation (50%), patients may not present with any family history. While NF1 patients with a family history may be diagnosed within the first few years of life, it is not uncommon for those with a new mutation to be diagnosed later in life. The NF1 clinical diagnosis is frequently made based on NIH diagnostic criteria. Genetic testing for the NF1 gene is costly and sometimes not practical due to its size (60 exons) and lack of mutation “hot-spots”.
Succinate Dehydrogenase Subunit Syndromes
Hereditary Paraganglioma (PGL1-5) is a group of diseases that are associated with the development of pheochromocytoma and/or paraganglioma (including head and neck paraganglioma) (Figure 2). Head and neck paragangliomas are most commonly located in the carotid body or the vagus nerve. Several mapping studies linked the suspected genes to different chromosomal regions (1q21, 11q13, and 11q21).47 Over the past decade the genetics of this syndrome have been elucidated with identification of mutations of the genes encoding the succinate dehydrogenase (SDH) subunits.
The SDH enzyme is an inner mitochondrial membrane enzyme critical to both the tricarboxylic acid cycle (TCA) and complex 2 of the electron transport chain. This enzyme is made up of four subunits (SDHA, SDHB, SDHC, and SDHD) and catalyzes the oxidation of succinate to fumarate. Mutations in specific subunits of this enzyme complex are a common cause of the hereditary paraganglioma/pheochromocytoma syndromes. Mutations in the SDHB and SDHD genes have been the most well characterized of the SDH deficiencies. The PGL1 gene, identified as SDHD, was first identified in 2000 and is associated with both paraganglioma and pheochromocytoma.48 The PGL4 gene, identified as SDHB, was later identified in 2001 and is also linked to the development of paraganglioma and pheochromocytoma.49 Subsequently it was found that patients with germline succinate dehydrogenase B/C/D mutations are also at risk for the development of an aggressive form of kidney cancer (SDH-RCC).50-53 The majority of patients with germline SDHD mutations are at risk for the development of multiple chromafin tumors, with the most common site being head and neck paraganglioma (>79%).50 Patients with germline SDHB gene mutations less commonly are found to develop multiple tumors, however they can be distributed in all locations, most commonly in the abdomen.54,55 Malignancy is reported in two thirds of SDHB-associated tumors.50,54,56
A third loci, the PGL3 gene, was linked to 1q23.3 and appeared only to be associated with PGL only.57 This gene was determined to be SDHC, also part of the SDH enzyme complex.47 Tumors in SDHC patients are more frequently solitary and more commonly located in the carotid body than the other SDH tumors.58 The incidence of malignancy with SDHC associated paragangliomas is low.59,60
SDHA gene mutation can be associated with Leigh's disease, a severe, early onset encephalopathy. Recently, several reports have identified SDHA mutations in patients with functional paraganglioma and pheochromocytoma.61,62
The product of the SDHA gene is known to undergo post-translational modification by SDHAF2. The gene for this protein, SDHAF2, has been found to be altered in a kindred with a hereditary head and neck paraganglioma.63 Without SHDA modification, succinate dehydrogenase cannot function properly leading to the development to this hereditary syndrome. To date only one kindred has been found despite screening for large cohorts of sporadic and hereditary paraganglioma and pheochromocytoma patients.64
Recently Described Pheochromocytoma and Paraganglioma Syndromes
The genetics of chromafin tumors continues to be elucidated and in the past few years a handful of new genes have been linked to familial and sporadic cases. (Online Table 2, Figure 3). A PHD2 mutation has been recently reported in a patient with recurrent abdominal paraganglioma and polycythemia.65 A newly described tumor suppressor gene, KIF1Bβ, has been found to be a downstream effector of one of the prolyl hydroxylases, PHD3.66 Germline mutation in KIF1Bβ has been described in a large family with neural crest tumors and non-neural tumors including several bilateral pheochromocytomas.67
Figure 3. The high incidence of a hereditary cause of pheochromocytoma due to germline mutations in patients with presumed sporadic pheochromocytoma.
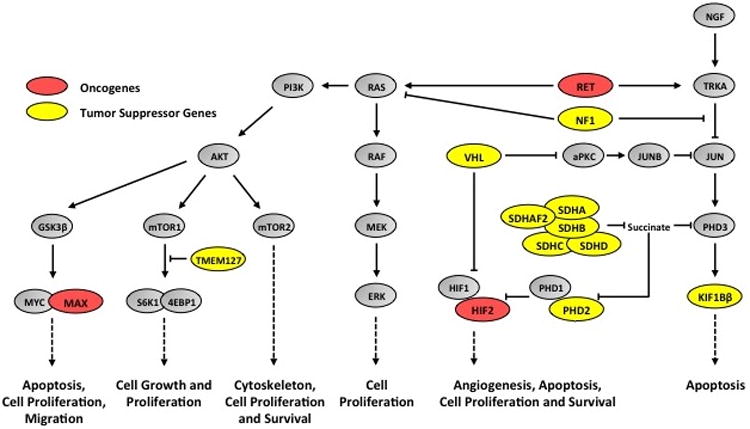
Common pathways associated with the development of pheochromocytoma and paraganglioma. Alterations of numerous pathways via the loss of tumor suppressor genes (red) or the activation of an oncogene (green) can occur in the tumorigenesis of PCC and PGL. The HIF pathway is artificially activated (pseudohypoxia) by mutation and loss of VHL, SDHA/B/C/D, SDHAF2, and PHD2. The JUN apoptosis pathway is down regulated by mutation and loss of VHL, SDHA/SDHB/SDHC/SDHD, SDHAF2, KIF1Bβ, NF1, and RET oncogene activity, which also upregulates the AKT pathway, the mTOR pathway, and the MYC transcription factor and its targets. The mTOR1 pathway is also specifically upregulated by mutation and loss of TMEM127 and the MYC transcription factor is over activated by mutation and loss of MAX.
Adapted from Neumann HP, Bausch B, McWhinney SR, et al: Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 346:1459-66, 2002.
MYC associated factor X (MAX) is a transcription factor that can dimerize with members of the Myc family.68 While MYC can be amplified an act as an oncogene involved in cancer such as neuroblastoma, MAX can interact with members of the MYC family to serve as a transcriptional suppressor.69 Whole genome sequencing identified germline mutations in MAX to be responsible for several familial cases of pheochromocytoma. In patients with suspected familial cases but no identifiable mutations in known genes, MAX mutations were found in 8.5% of cases.70
Linkage analysis of a family with a hereditary pheochromocytoma mapped a susceptibility locus at 2q11.71 Exon sequencing of this region demonstrated a germline mutation at a gene known as TMEM127. Germline mutations were found in 30% of familial and about 3% of sporadic pheochromocytomas. An international, multi-center group subsequently confirmed this finding and identified a 2% frequency of mutation in patients with non-syndromic/familial, unilateral PCC.72 Studies indicate that TMEM127 may be a negative regulator of mTORC1 and loss of protein function activates downstream pathways such as S6K and 4EBP1.73
Several of the pheochromocytoma/paraganglioma syndromes, including VHL and the SDH, are associated with alteration of regulation of hypoxia-inducible pathways. Recently 2 patients at the National Institutes of Health with paraganglioma were found to have somatic gain of function mutation in HIF2a.74
Genetic Testing
The consensus from the First International Symposium on Pheochromocytoma was that while genetic testing should be considered in patients with a chromafin tumors, the current costs may preclude testing of some patients for all recognized genes.11 Clinicians must be very thoughtful in selecting genetic testing and for which particular genes. In patients who do not present with a known hereditary condition, clinicians should look for the additional characteristics of one of the common associated syndromes, and if present, select for the likely affected gene (Online Table 1). Several clinical and familial characteristics, including personal or family history of pheochromocytoma or paraganglioma, age of diagnosis less than 40, the presence of head and neck paraganglioma or bilateral adrenal or multi-focal tumors, and the presence of a malignant or non-functional paraganglioma should raise suspicion for a possible hereditary component. Those patients should be considered for genetic screening.7,75,76,54
Once the decision is made for genetic testing, the next step is to determine what tests to order. While a number of new genes have been identified in the past few years, Clinical Laboratory Improvement Amendment (CLIA) testing is currently only available for certain genes. Of the available tests performed in CLIA-certified labs, well thought specific algorithms regarding a step-wise order of testing will significantly reduce cost.77
Several of the conditions mentioned above, including MEN2A/B, NF1, and VHL, are frequently associated with other commonly recognized clinical phenotypes, such as the presence of an adrenergic (MEN2 and NF1) or noradrenergic (VHL) phenotype. As mentioned above the NF1 is an extremely large gene and sequencing can be very costly; therefore a clinical diagnosis and the findings of elevated epinephrine or metanephrine often suffices, as de novo NF1 mutations are common. Head and neck paragangliomas are more commonly associated with the SDH syndromes and, therefore, initial testing for these genes is often considered. The incidence of malignancy is much higher with SDHB mutations, therefore, if a pheochromocytoma appears to be invasive or associated with metastatic disease, this gene should be considered for testing.78 As described above, the biochemical evaluation often guides the genetic workup as the metabolite secretion profiles differ with various hereditary syndromes.79 Differential expression of the rate-limiting enzymes may be responsible for differences in the production of noradrenergic vs adrenergic biochemical phenotypes.31 Patients affected with VHL-associated pheochromocytomas often are found to have isolated elevations in normetanephrine (metabolite of norepinephrine), while patients with MEN2 and NF1 associated pheochromocytomas may have elevations in metanephrine (metabolite of epinephrine) with or without elevated normetanephrine.25,79 The majority of SDHB and SDHD-associated chromafin tumors will have elevated levels of dopamine metabolite methoxytyramine, either alone or in addition to elevated normetanephrines.25 Using catecholamine information often provides useful guidance for narrowing the suspected genetic alteration (Online Figure 2).
Surgical Management
The surgical management of a paraganglioma and pheochromocytoma has traditionally involved complete excision/total adrenalectomy. In patients with bilateral, multifocal or hereditary pheochromocytoma syndromes, partial adrenalectomy has emerged as an option to preserve adrenal function.80-85 Patients that have permanent need for adrenal replacement with steroids and mineralocorticoids face long-term consequences, such as hypertension, diabetes, osteoporosis, and weight/body habitus changes. To spare patients of these life-altering side effects, consideration for alternatives to total adrenalectomy are considered in selected patietns. Observation of asymptomatic PCC associated with VHL has been reported and appears to be a viable option in select patients.86 The incidence of malignancy in hereditary conditions such as VHL, NF1, and MEN2 is low and several studies have reported excellent functional and oncologic outcome for managing these patients with this approach.80,84,87 In patients with smaller tumors (<4 cm) in a solitary adrenal gland, adrenal function can generally be maintained.88 Laparoscopic and robotic partial adrenalectomy have been described and may decrease the morbidity of open surgery.89,90
In patients without a prior diagnosis of a hereditary syndrome, genetic testing prior to surgery may be usefjul. For younger patients, and those with bilateral/multifocal disease, knowledge of a genetic component may alter surgical management in favor of consideration of partial adrenalectomy. However, findings such as germline SDHB mutation could lead recommendation of total adrenalectomy due to the higher incidence of malignancy.
Conclusions
Chromaffin tumors are rare tumors derived from neural crest tissue and are divided into two categories, pheochromocytomas and paragangliomas. Well described hereditary syndromes such as VHL, NF1, and MEN2 are a common cause of these tumors however, recently more than a half dozen potential novel pheochromocytoma and paraganglioma syndromes have been reported. Currently 35% of “sporadic” pheochromocytomas are found to have a hereditary component, therefore clinicians who manage these patients need to be aware of the potential genetic alterations. Germline mutation testing can be vital not only for the individual patient's care, but may also allow screening and early detection of disease in at-risk family members
Supplementary Material
References
- 1.DeLellis R, Lloyd R, P H, C E. Pathology and genetics of tumors of endocrine organs. Lyon, Fr: 2004. [Google Scholar]
- 2.Elder EE, Elder G, Larsson C. Pheochromocytoma and functional paraganglioma syndrome: no longer the 10% tumor. J Surg Oncol. 2005;89:193. doi: 10.1002/jso.20177. [DOI] [PubMed] [Google Scholar]
- 3.Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 4.Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de PB, Chabre O, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23:8812. doi: 10.1200/JCO.2005.03.1484. [DOI] [PubMed] [Google Scholar]
- 5.Jiang S, Dahia PL. Minireview: the busy road to pheochromocytomas and paragangliomas has a new member, TMEM127. Endocrinology. 2011;152:2133. doi: 10.1210/en.2011-0052. [DOI] [PubMed] [Google Scholar]
- 6.Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc. 1983;58:802. [PubMed] [Google Scholar]
- 7.Erlic Z, Rybicki L, Peczkowska M, Golcher H, Kann PH, Brauckhoff M, et al. Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients. Clin Cancer Res. 2009;15:6378. doi: 10.1158/1078-0432.CCR-09-1237. [DOI] [PubMed] [Google Scholar]
- 8.Hayes WS, Davidson AJ, Grimley PM, Hartman DS. Extraadrenal retroperitoneal paraganglioma: clinical, pathologic, and CT findings. AJR Am J Roentgenol. 1990;155:1247. doi: 10.2214/ajr.155.6.2173385. [DOI] [PubMed] [Google Scholar]
- 9.Kopetschke R, Slisko M, Kilisli A, Tuschy U, Wallaschofski H, Fassnacht M, et al. Frequent incidental discovery of phaeochromocytoma: data from a German cohort of 201 phaeochromocytoma. Eur J Endocrinol. 2009;161:355. doi: 10.1530/EJE-09-0384. [DOI] [PubMed] [Google Scholar]
- 10.Pacak K, Linehan WM, Eisenhofer G, Walther MM, Goldstein DS. Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann Intern Med. 2001;134:315. doi: 10.7326/0003-4819-134-4-200102200-00016. [DOI] [PubMed] [Google Scholar]
- 11.Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007;92:4069. doi: 10.1210/jc.2007-1720. [DOI] [PubMed] [Google Scholar]
- 12.Lenders JW, Pacak K, Walther MM, Linehan WM, Mannelli M, Friberg P, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA. 2002;287:1427. doi: 10.1001/jama.287.11.1427. [DOI] [PubMed] [Google Scholar]
- 13.Neary NM, King KS, Pacak K. Drugs and pheochromocytoma--don't be fooled by every elevated metanephrine. N Engl J Med. 2011;364:2268. doi: 10.1056/NEJMc1101502#SA1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eisenhofer G. Free or total metanephrines for diagnosis of pheochromocytoma: what is the difference? Clin Chem. 2001;47:988. [PubMed] [Google Scholar]
- 15.Eisenhofer G, Goldstein DS, Walther MM, Friberg P, Lenders JW, Keiser HR, et al. Biochemical diagnosis of pheochromocytoma: how to distinguish true- from false-positive test results. J Clin Endocrinol Metab. 2003;88:2656. doi: 10.1210/jc.2002-030005. [DOI] [PubMed] [Google Scholar]
- 16.Lumachi F, Tregnaghi A, Zucchetta P, Cristina MM, Cecchin D, Grassetto G, et al. Sensitivity and positive predictive value of CT, MRI and 123I-MIBG scintigraphy in localizing pheochromocytomas: a prospective study. Nucl Med Commun. 2006;27:583. doi: 10.1097/00006231-200607000-00006. [DOI] [PubMed] [Google Scholar]
- 17.Maurea S, Cuocolo A, Reynolds JC, Tumeh SS, Begley MG, Linehan WM, et al. Iodine-131-metaiodobenzylguanidine scintigraphy in preoperative and postoperative evaluation of paragangliomas: comparison with CT and MRI. J Nucl Med. 1993;34:173. [PubMed] [Google Scholar]
- 18.Timmers HJ, Eisenhofer G, Carrasquillo JA, Chen CC, Whatley M, Ling A, et al. Use of 6-[18F]-fluorodopamine positron emission tomography (PET) as first-line investigation for the diagnosis and localization of non-metastatic and metastatic phaeochromocytoma (PHEO) Clin Endocrinol (Oxf) 2009;71:11. doi: 10.1111/j.1365-2265.2008.03496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Timmers HJ, Chen CC, Carrasquillo JA, Whatley M, Ling A, Havekes B, et al. Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2009;94:4757. doi: 10.1210/jc.2009-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mackenzie IS, Gurnell M, Balan KK, Simpson H, Chatterjee K, Brown MJ. The use of 18-fluoro-dihydroxyphenylalanine and 18-fluorodeoxyglucose positron emission tomography scanning in the assessment of metaiodobenzylguanidine-negative phaeochromocytoma. Eur J Endocrinol. 2007;157:533. doi: 10.1530/EJE-07-0369. [DOI] [PubMed] [Google Scholar]
- 21.King KS, Chen CC, Alexopoulos DK, Whatley MA, Reynolds JC, Patronas N, et al. Functional imaging of SDHx-related head and neck paragangliomas: comparison of 18F-fluorodihydroxyphenylalanine, 18F-fluorodopamine, 18F-fluoro-2-deoxy-D-glucose PET, 123I-metaiodobenzylguanidine scintigraphy, and 111In-pentetreotide scintigraphy. J Clin Endocrinol Metab. 2011;96:2779. doi: 10.1210/jc.2011-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoegerle S, Nitzsche E, Altehoefer C, Ghanem N, Manz T, Brink I, et al. Pheochromocytomas: detection with 18F DOPA whole body PET--initial results. Radiology. 2002;222:507. doi: 10.1148/radiol.2222010622. [DOI] [PubMed] [Google Scholar]
- 23.Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007;92:4069. doi: 10.1210/jc.2007-1720. [DOI] [PubMed] [Google Scholar]
- 24.Perry RR, Keiser HR, Norton JA, Wall RT, Robertson CN, Travis W, et al. Surgical management of pheochromocytoma with the use of metyrosine. Ann Surg. 1990;212:621. doi: 10.1097/00000658-199011000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eisenhofer G, Lenders JW, Timmers H, Mannelli M, Grebe SK, Hofbauer LC, et al. Measurements of Plasma Methoxytyramine, Normetanephrine, and Metanephrine as Discriminators of Different Hereditary Forms of Pheochromocytoma. Clin Chem. 2011;57:1. doi: 10.1373/clinchem.2010.153320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eisenhofer G, Pacak K, Huynh TT, Qin N, Bratslavsky G, Linehan WM, et al. Catecholamine metabolomic and secretory phenotypes in phaeochromocytoma. Endocr Relat Cancer. 2011;18:97. doi: 10.1677/ERC-10-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hosoe S, Brauch H, Latif F, Glenn GM, Daniel L, Bale S, et al. Localization of the von Hippel-Lindau disease gene to a small region of chromosome 3. Genom. 1990;8:634. doi: 10.1016/0888-7543(90)90249-t. [DOI] [PubMed] [Google Scholar]
- 28.Latif F, Tory K, Gnarra JR, Yao M, Duh FM, Orcutt ML, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 29.Gnarra JR, Tory K, Weng Y, Schmidt LS, Wei MH, Li H, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nature Genetics. 1994;7:85. doi: 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- 30.Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res. 2008;14:4726. doi: 10.1158/1078-0432.CCR-07-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moore LE, Nickerson ML, Brennan P, Toro JR, Jaeger E, Rinsky J, et al. Von Hippel-Lindau (VHL) inactivation in sporadic clear cell renal cancer: Associations with germline VHL polymorphisms and etiologic risk factors. PLoS Genet. 2011;7:1. doi: 10.1371/journal.pgen.1002312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 33.Min JH, Yang H, Ivan M, Gertler F, Kaelin WG, Jr, Pavletich NP. Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296:1886. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- 34.Ivan M, Haberberger T, Gervasi DC, Michelson KS, Gunzler V, Kondo K, et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci U S A. 2002;99:13459. doi: 10.1073/pnas.192342099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M, et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat. 1995;5:66. doi: 10.1002/humu.1380050109. [DOI] [PubMed] [Google Scholar]
- 36.Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet. 1993;2:851. doi: 10.1093/hmg/2.7.851. [DOI] [PubMed] [Google Scholar]
- 37.Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363:458. doi: 10.1038/363458a0. [DOI] [PubMed] [Google Scholar]
- 38.Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel RF, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA. 1996;276:1575. [PubMed] [Google Scholar]
- 39.Zinnamosca L, Petramala L, Cotesta D, Marinelli C, Schina M, Cianci R, et al. Neurofibromatosis type 1 (NF1) and pheochromocytoma: prevalence, clinical and cardiovascular aspects. Arch Dermatol Res. 2011;303:317. doi: 10.1007/s00403-010-1090-z. [DOI] [PubMed] [Google Scholar]
- 40.Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM. von Recklinghausen's disease and pheochromocytomas. J Urol. 1999;162:1582. [PubMed] [Google Scholar]
- 41.Walther MM, Keiser HR, Choyke PL, Rayford W, Lyne JC, Linehan WM. Management of hereditary pheochromocytoma in von Hippel-Lindau kindreds with partial adrenalectomy. J Urol. 1999;161:395. [PubMed] [Google Scholar]
- 42.Barker D, Wright E, Nguyen K, Cannon L, Fain P, Goldgar D, et al. Gene for von recklinghausen neurofibromatosis is in the pericentromeric region of chromosome 17. Science. 1987;236:1100. doi: 10.1126/science.3107130. [DOI] [PubMed] [Google Scholar]
- 43.Upadhyaya M, Cheryson A, Broadhead W, Fryer A, Shaw DJ, Huson S, et al. A 90 kb DNA deletion associated with neurofibromatosis type 1. J Med Genet. 1990;27:738. doi: 10.1136/jmg.27.12.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wallace MR, Marchuk DA, Andersen LB, Letcher R, Odeh HM, Saulino AM, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181. doi: 10.1126/science.2134734. [DOI] [PubMed] [Google Scholar]
- 45.Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356:713. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- 46.Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci U S A. 2005;102:8573. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26:268. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- 48.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 49.Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292:943. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- 51.Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peczkowska M, Morrison CD, et al. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet. 2004;74:153. doi: 10.1086/381054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ricketts C, Woodward ER, Killick P, Morris MR, Astuti D, Latif F, et al. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst. 2008;100:1260. doi: 10.1093/jnci/djn254. [DOI] [PubMed] [Google Scholar]
- 53.Ricketts CJ, Shuch B, Vocke CD, Metwalli AR, Bratslavsky G, Middelton L, et al. Succinate dehydrogenase kidney cancer: an aggressive example of the Warburg effect in cancer. J Urol. 2012;188:2063. doi: 10.1016/j.juro.2012.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de PB, Chabre O, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23:8812. doi: 10.1200/JCO.2005.03.1484. [DOI] [PubMed] [Google Scholar]
- 55.Benn DE, Gimenez-Roqueplo AP, Reilly JR, Bertherat J, Burgess J, Byth K, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91:827. doi: 10.1210/jc.2005-1862. [DOI] [PubMed] [Google Scholar]
- 56.Brouwers FM, Eisenhofer G, Tao JJ, Kant JA, Adams KT, Linehan WM, et al. High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab. 2006;91:4505. doi: 10.1210/jc.2006-0423. [DOI] [PubMed] [Google Scholar]
- 57.Niemann S, Steinberger D, Muller U. PGL3, a third, not maternally imprinted locus in autosomal dominant paraganglioma. Neurogenetics. 1999;2:167. doi: 10.1007/s100480050078. [DOI] [PubMed] [Google Scholar]
- 58.Schiavi F, Boedeker CC, Bausch B, Peczkowska M, Gomez CF, Strassburg T, et al. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA. 2005;294:2057. doi: 10.1001/jama.294.16.2057. [DOI] [PubMed] [Google Scholar]
- 59.Niemann S, Muller U, Engelhardt D, Lohse P. Autosomal dominant malignant and catecholamine-producing paraganglioma caused by a splice donor site mutation in SDHC. Hum Genet. 2003;113:92. doi: 10.1007/s00439-003-0938-0. [DOI] [PubMed] [Google Scholar]
- 60.Schiavi F, Boedeker CC, Bausch B, Peczkowska M, Gomez CF, Strassburg T, et al. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA. 2005;294:2057. doi: 10.1001/jama.294.16.2057. [DOI] [PubMed] [Google Scholar]
- 61.Burnichon N, Briere JJ, Libe R, Vescovo L, Riviere J, Tissier F, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011. doi: 10.1093/hmg/ddq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Korpershoek E, Favier J, Gaal J, Burnichon N, van GB, Oudijk L, et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab. 2011;96:E1472. doi: 10.1210/jc.2011-1043. [DOI] [PubMed] [Google Scholar]
- 63.Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kunst HP, Rutten MH, de Monnink JP, Hoefsloot LH, Timmers HJ, Marres HA, et al. SDHAF2 (PGL2-SDH5) and hereditary head and neck paraganglioma. Clin Cancer Res. 2011;17:247. doi: 10.1158/1078-0432.CCR-10-0420. [DOI] [PubMed] [Google Scholar]
- 65.Ladroue C, Carcenac R, Leporrier M, Gad S, Le HC, Galateau-Salle F, et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med. 2008;359:2685. doi: 10.1056/NEJMoa0806277. [DOI] [PubMed] [Google Scholar]
- 66.Schlisio S, Kenchappa RS, Vredeveld LC, George RE, Stewart R, Greulich H, et al. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. 2008;22:884. doi: 10.1101/gad.1648608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yeh IT, Lenci RE, Qin Y, Buddavarapu K, Ligon AH, Leteurtre E, et al. A germline mutation of the KIF1B beta gene on 1p36 in a family with neural and nonneural tumors. Hum Genet. 2008;124:279. doi: 10.1007/s00439-008-0553-1. [DOI] [PubMed] [Google Scholar]
- 68.Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251:1211. doi: 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- 69.Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, et al. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci U S A. 2000;97:3260. doi: 10.1073/pnas.97.7.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Comino-Mendez I, Gracia-Aznarez FJ, Schiavi F, Landa I, Leandro-Garcia LJ, Leton R, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. 2011;43:663. doi: 10.1038/ng.861. [DOI] [PubMed] [Google Scholar]
- 71.Dahia PL, Hao K, Rogus J, Colin C, Pujana MA, Ross K, et al. Novel pheochromocytoma susceptibility loci identified by integrative genomics. Cancer Res. 2005;65:9651. doi: 10.1158/0008-5472.CAN-05-1427. [DOI] [PubMed] [Google Scholar]
- 72.Yao L, Schiavi F, Cascon A, Qin Y, Inglada-Perez L, King EE, et al. Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas. JAMA. 2010;304:2611. doi: 10.1001/jama.2010.1830. [DOI] [PubMed] [Google Scholar]
- 73.Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010;42:229. doi: 10.1038/ng.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhuang Z, Yang C, Lorenzo F, Merino M, Fojo T, Kebebew E, et al. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med. 2012;367:922. doi: 10.1056/NEJMoa1205119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Timmers HJ, Pacak K, Huynh TT, Abu-Asab M, Tsokos M, Merino MJ, et al. Biochemically silent abdominal paragangliomas in patients with mutations in the succinate dehydrogenase subunit B gene. J Clin Endocrinol Metab. 2008;93:4826. doi: 10.1210/jc.2008-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pacak K, Eisenhofer G, Ahlman H, Bornstein SR, Gimenez-Roqueplo AP, Grossman AB, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005. Nat Clin Pract Endocrinol Metab. 2007;3:92. doi: 10.1038/ncpendmet0396. [DOI] [PubMed] [Google Scholar]
- 77.Erlic Z, Rybicki L, Peczkowska M, Golcher H, Kann PH, Brauckhoff M, et al. Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients. Clin Cancer Res. 2009;15:6378. doi: 10.1158/1078-0432.CCR-09-1237. [DOI] [PubMed] [Google Scholar]
- 78.Brouwers FM, Eisenhofer G, Tao JJ, Kant JA, Adams KT, Linehan WM, et al. High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab. 2006;91:4505. doi: 10.1210/jc.2006-0423. [DOI] [PubMed] [Google Scholar]
- 79.Eisenhofer G, Walther MM, Huynh TT, Li ST, Bornstein SR, Vortmeyer A, et al. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab. 2001;86:1999. doi: 10.1210/jcem.86.5.7496. [DOI] [PubMed] [Google Scholar]
- 80.Walther MM, Keiser HR, Choyke PL, Rayford W, Lyne JC, Linehan WM. Management of hereditary pheochromocytoma in von Hippel-Lindau kindreds with partial adrenalectomy. J Urol. 1999;161:395. [PubMed] [Google Scholar]
- 81.Birnbaum J, Giuliano A, Van Herle AJ. Partial adrenalectomy for pheochromocytoma with maintenance of adrenocortical function. J Clin Endocrinol Metab. 1989;69:1078. doi: 10.1210/jcem-69-5-1078. [DOI] [PubMed] [Google Scholar]
- 82.Benhammou JN, Boris RS, Pacak K, Pinto PA, Linehan WM, Bratslavsky G. Functional and oncologic outcomes of partial adrenalectomy for pheochromocytoma in patients with von Hippel-Lindau syndrome after at least 5 years of followup. J Urol. 2010;184:1855. doi: 10.1016/j.juro.2010.06.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pavlovich CP, Linehan WM, Walther MM. Partial adrenalectomy in patients with multiple adrenal tumors. Curr Urol Rep. 2001;2:19. doi: 10.1007/s11934-001-0021-0. [DOI] [PubMed] [Google Scholar]
- 84.Diner EK, Franks ME, Behari A, Linehan WM, Walther MM. Partial adrenalectomy: the National Cancer Institute experience. Urology. 2005;66:19. doi: 10.1016/j.urology.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 85.Walther MM, Herring J, Choyke PL, Linehan WM. Laparoscopic partial adrenalectomy in patients with hereditary forms of pheochromocytoma. J Urol. 2000;164:14. [PubMed] [Google Scholar]
- 86.Robles J, Venkatesan A, Pacak K, Eisenhofer G, Landers J, Linehan WM, Bratslavsky G. Active Surveillance of Pheo in Von Hippel-Lindau (VHL) Patients: Evaluation of Safety and Growth Rates. Journal of Urology Supplement. 2010 May 29; [Google Scholar]
- 87.Edstrom E, Grondal S, Norstrom F, Palmer M, Svensson KA, Widell H, et al. Long term experience after subtotal adrenalectomy for multiple endocrine neoplasia type IIa. Eur J Surg. 1999;165:431. doi: 10.1080/110241599750006659. [DOI] [PubMed] [Google Scholar]
- 88.Sanford TH, Storey BB, Linehan WM, Rogers CA, Pinto PA, Bratslavsky G. Outcomes and timing for intervention of partial adrenalectomy in patients with a solitary adrenal remnant and history of bilateral phaeochromocytomas. BJU Int. 2011;107:571. doi: 10.1111/j.1464-410X.2010.09568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Asher KP, Gupta GN, Boris RS, Pinto PA, Linehan WM, Bratslavsky G. Robot-assisted laparoscopic partial adrenalectomy for pheochromocytoma: the National Cancer Institute technique. Eur Urol. 2011;60:118. doi: 10.1016/j.eururo.2011.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Boris RS, Gupta G, Linehan WM, Pinto PA, Bratslavsky G. Robot-assisted laparoscopic partial adrenalectomy: initial experience. Urology. 2011;77:775. doi: 10.1016/j.urology.2010.07.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.