

This review presents an introduction to the process development challenges of cell therapies and describes some of the tools available to address production issues. A summary is provided of what should be considered to efficiently advance a cellular therapy from the research stage through clinical trials and finally toward commercialization.
Keywords: Process development, Mesenchymal stem cells, Cellular therapy, T cell, Pluripotent stem cells
Abstract
The development of robust and well-characterized methods of production of cell therapies has become increasingly important as therapies advance through clinical trials toward approval. A successful cell therapy will be a consistent, safe, and effective cell product, regardless of the cell type or application. Process development strategies can be developed to gain efficiency while maintaining or improving safety and quality profiles. This review presents an introduction to the process development challenges of cell therapies and describes some of the tools available to address production issues. This article will provide a summary of what should be considered to efficiently advance a cellular therapy from the research stage through clinical trials and finally toward commercialization. The identification of the basic questions that affect process development is summarized in the target product profile, and considerations for process optimization are discussed. The goal is to identify potential manufacturing concerns early in the process so they may be addressed effectively and thus increase the probability that a therapy will be successful.
Significance
The present study contributes to the field of cell therapy by providing a resource for those transitioning a potential therapy from the research stage to clinical and commercial applications. It provides the necessary steps that, when followed, can result in successful therapies from both a clinical and commercial perspective.
Introduction
As a growing number of cellular therapies advance through clinical trials toward approval, the need for robust and well-characterized methods of production has become increasingly important. In order for a cell therapy to be successful from a clinical or commercial perspective, patients must be treated with a consistent, safe, and effective cell product, regardless of the cell type or application. Developing these products can be challenging from many perspectives, including manufacturing, regulatory, distribution, testing, and delivery. Process development aims to gain efficiency and drive down costs while maintaining or improving quality. It applies to all process elements such as cell isolation, cell characterization, optimization of cell culture media, scale-up, and removal of impurities.
In many respects, the growth pattern of the cell therapy industry may resemble that of the biotherapeutics industry. Over the course of more than 20 years, the development of biologic drugs has gone from theory to blockbuster status, enabled in large part by the success of process development resulting in large-scale production of viable and commercially successful products [1]. In contrast, there are notable examples of clinically promising molecules in this industry that have failed to reach the commercialization stage because of the inability to manufacture the product through a robust and economical process [2]. This highlights the need for attention to manufacturing of complex cell therapies to support commercial success.
The purpose of this review is to provide a level of awareness of the process development challenges of cell therapies for those new to the field with little process development experience, for academic researchers looking to advance into the clinic, or for scientists who may have process development experience with unrelated product types but who are doubtful that it can be applied to cell therapies. Whether a cell therapy strategy is aimed at treating multiple patients from central lots of cells from qualified donors (an “off-the-shelf allogeneic” model) or consists of using the patient’s own cells as the source (an autologous model), some common factors must be considered to ensure success. In both of these approaches, it is necessary to sufficiently characterize the therapeutic cell to define properties that confirm acceptable production and thereby reduce the risk of incorporating anomalies or shifts in cell function that might compromise safety or efficacy. This, along with robust, reproducible manufacturing processes, will help achieve consistency from dose to dose. Common considerations of robust, reproducible manufacturing processes include the use of well-defined raw materials and methods under Good Manufacturing Practice (GMP), which is necessary for regulatory approval and batch consistency. In addition to scientific and quality considerations, it is critical to control the cost of goods (COGs) because complex, labor-intensive manufacturing and testing processes that often use expensive raw materials can exceed a cell product’s reimbursable value. Consequently, costs that are competitive with both cellular and noncellular therapeutics are indispensable to the widespread adoption of cell therapies. Furthermore, it is necessary to achieve a scale of production that can fulfill product demand. “Scale-out” for an autologous product requires the ability to carry out a process of small size many times, whereas “scale-up” for an unmatched allogeneic process requires the ability to carry out a process of large size only a few times. A recent article [3] examined and presented the issues specific to autologous cell manufacturing. The focus of this review will be directed at allogeneic models, but many of the principles discussed are applicable to both autologous and allogeneic products.
In the following sections, we provide a summary of what should be considered to drive a therapy from the research stage to full regulatory approval and commercialization. A critical focus is continuous work toward a full understanding of the cell (or cell population) of interest and the processes used to manufacture the product. Many of the critical factors that characterize a process or product will be established throughout development and may not be defined from the beginning. How will the cells be expanded and manufactured while ensuring retention of potency? How will the product be stored and administered? What lot size (the number of cells per production batch) does the market dictate? What characteristics define the cell product of interest and what assays measure them? These are some of the basic questions that affect process development in the target product profile (TPP). Once the TPP is determined (discussed in “A Vision for Process Development”), the goal is to develop streamlined, scalable processes that can maintain comparability throughout clinical development. Quality by design (QbD) strategies and classical design of experiment (DoE) approaches, for example, can be applied to optimize cell culture media, reagents, and process parameters to create a robust, reproducible, and scalable process. Product specifications at the laboratory scale have manufacturing process implications; therefore, it is imperative to begin identifying potential scale-up pain points early in the process so that smooth transitions can be made as a therapy advances. Furthermore, the difficulty of implementing changes increases as product development through clinical trials proceeds. If potential manufacturing concerns are addressed early in the process, significant manpower and money will be saved later, making it more likely that a therapy will be commercially successful. Figure 1 shows a graphical representation of the relationship and interaction of the various process steps. Once the potential therapy is defined, the TPP is generated in the context of the necessary process attributes. Each step is dependent on the other parts of the workflow and continually feeds back into the TPP document.
Figure 1.
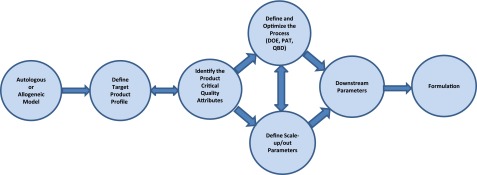
Workflow representation of process steps. Abbreviations: DOE, design of experiment; PAT, process analytical technology; QBD, quality by design.
The TPP is initiated according to a framework depending on the type of therapy or indication targeted. The critical quality attributes are then identified and used as the basis to design process development activities (incorporating the strategies of DoE, process analytical technology [PAT], or potentially QbD) aimed at optimizing manufacturing conditions and downstream parameters. The design and process optimization steps have an interactive relationship that is continually being refined.
A Vision for Process Development
The TPP is a document that serves as an excellent tool for aligning manufacturing process requirements with product specifications of a commercially relevant product. The TPP includes a vision for all known elements of the product to provide a roadmap for development. These elements encompass all product aspects including clinical, regulatory, and manufacturing components. This discussion focuses on the aspects that affect process development. The phrase, “Beginning with the end in mind,” taken from the U.S. Food and Drug Administration guidelines for TPPs [4], describes the thought process of preparing for and achieving a successful outcome. Some examples of product or process elements that are addressed in the TPP can be found in Table 1.
Table 1.
Examples of elements of the target product profile
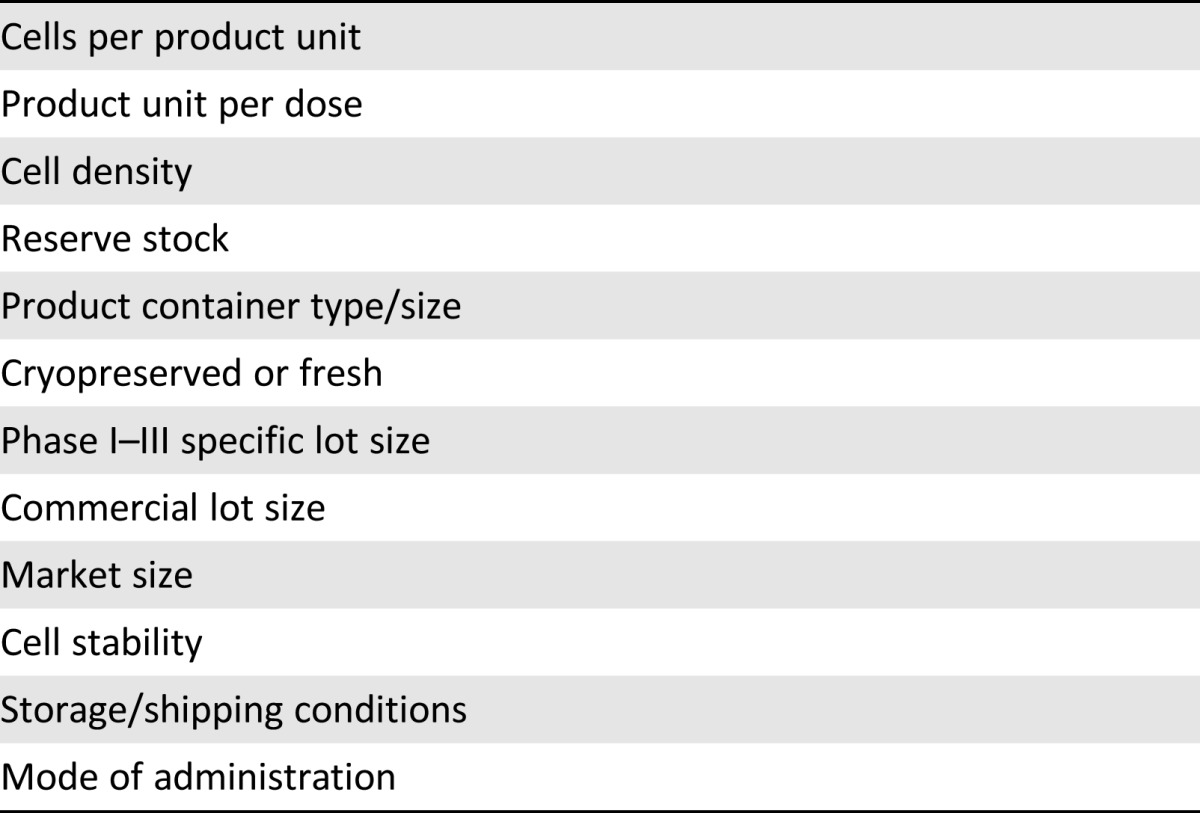
Each element is defined at three levels. First, what is minimally acceptable for a commercial product? Second, what would a good target specification be? Third, what would the best case scenario or “ideal” specification be? Definitions will become clear as knowledge emerges throughout clinical development, but some specifications can be solidified much earlier than others. It is important to identify the potential ranges for certain product characteristics to identify challenges in meeting product specifications that could arise. The TPP should be treated as a “living document” that can be continually updated and reviewed in response to emerging knowledge. In this way, although total cells per dose will not likely be determined until the end of a phase II dose-escalation trial, it is likely that the total product volume will be easier to specify because there are usually technical limitations depending on mode of delivery. The long-term goal will be ideal dosing that maximizes efficacy and minimizes COGs; however, determining an early estimate of the effective number of cells per dose will help identify the target process scale, the volume in which doses should be packaged, and the minimal lot sizes to be produced. Judiciously evaluation of these parameters across technical, regulatory, and commercial perspectives is critical for success.
An example of applying these principles to process decisions is shown in the case study. The goal of the case study is to improve the production of a hypothetical cell product, Celiofix, to enable large-scale production in a closed process. A TPP is generated, and the potential effects on product quality of the proposed process development steps are considered and ranked. The outcome is a plan that addresses potential risks and outlines a path to optimize the process.
TPP Case Study
Celiofix is a ready-to-use, unmatched allogeneic cell therapy product for treatment of peripheral artery disease that is given as 15 intramuscular injections of 1 ml each. The product is in development stages between phase I and phase II. Celiofix is packed in cryobags and stored frozen in the vapor phase of a liquid nitrogen freezer. The company Celiofix Ltd. wishes to scale up the manufacturing and move to 6-ml (nominal) cryovials, which will allow a closed and automated filling system and easier handling at the clinical sites.
In order to choose the vials and filling system and to direct the process development of the packaging change, the following TPP table (Table 2) was designed including the relevant parameters.
Table 2.
Case study target product profile
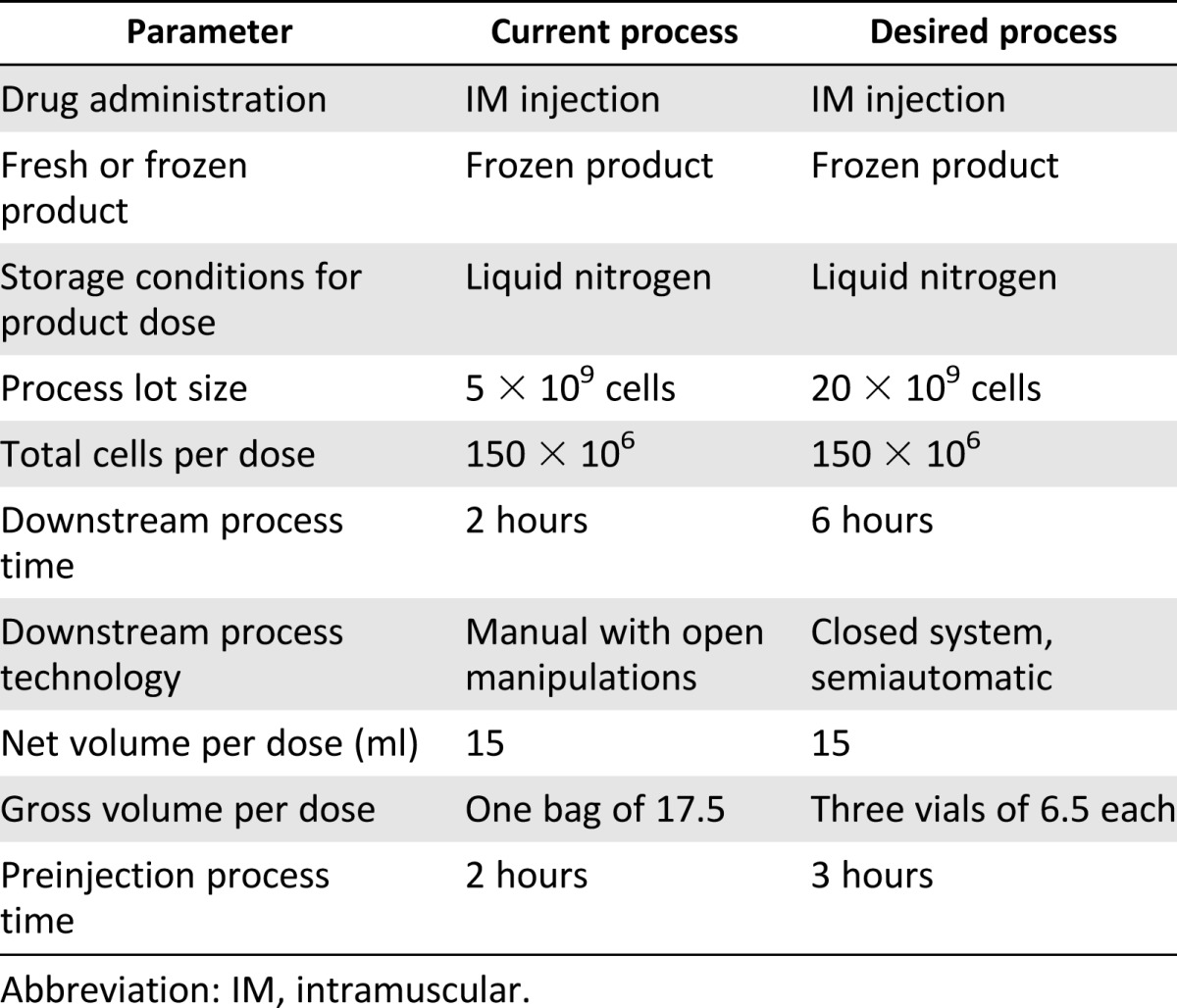
The TPP analysis indicates that scaling up the process will increase the batch size fourfold, which in turn will require fourfold more cells to be processed prior to cryopreservation. Furthermore, the storage conditions and product unit size limit the possible vials and filling systems, resulting in 6-ml aseptic vials. The choice of vials led to a fill volume change from a net 15 ml in one cryobag to a minimum of three vials. Testing of dead volume in the vials and losses in the syringe nozzle and needle showed that in order to deliver a net volume of 5 ml per vial, a total of 6.5 ml gross fill must be achieved. Since that is above the vendor's validated fill volume, additional validation must be performed to ensure that the performance of the vial is maintained at the higher fill volume. There are various scalable automatic vial filling systems that support from 3 vials per minute to 300 vials per minute, with a similar scaling of capital investment. For the given batch size of more than 100 vials, the fill time for these systems would range from less than 1 minute to nearly 45 minutes. As cells remain alive during the filling process, the chosen filling system must fill the vials within a time that does not harm the cells. Therefore, the stability of cells during hold times while filling must be characterized to define such limits. Furthermore, the change from one bag to three vials might affect the preinjection process time after thaw, requiring a similar stability characterization post-thaw.
The next step was to prepare a risk analysis on the change and its effect on the product. The outcome of the risk analysis (Table 3) is the process development plan. The risks were tested and the outcome defined the technologies that can be used for the process. Based on the risk assessment, a process development program was designed and executed and the TPP was established.
Table 3.
Process development plan
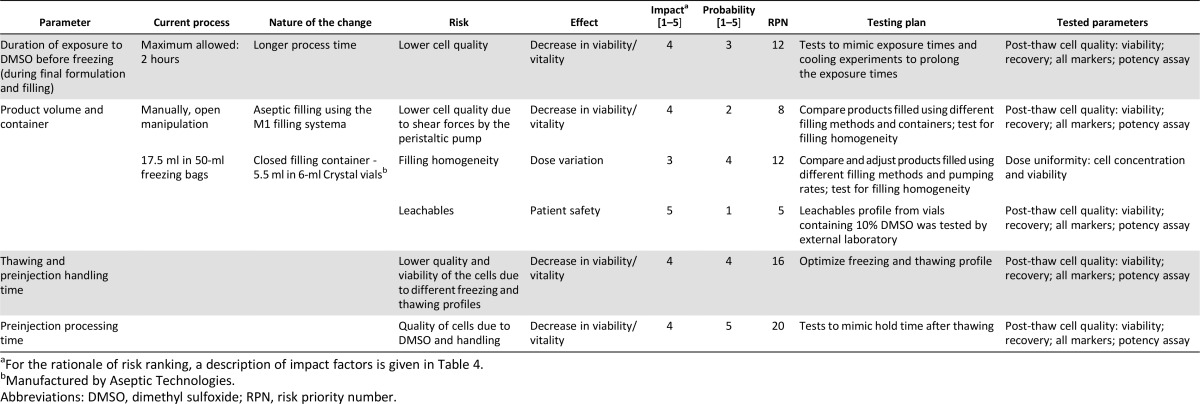
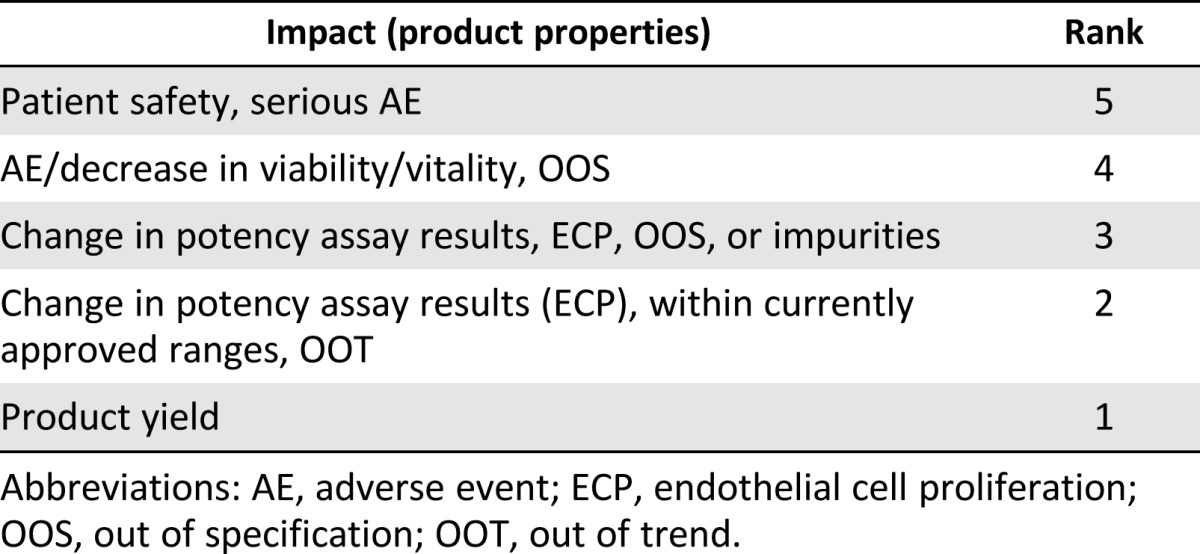
Table 4.
Ranking scale
The TPP strategy defined the critical attributes of the product, which in turn defined the applicable technologies that can be tested to fit the relevant need. Once the optional technologies were chosen, the risk assessment methodology allowed definition of the critical quality aspects to be tested, leading to the development of a design of experiment study. The outcome of the experiments allowed the selection of the correct technologies to be used, resulting in a process that supports the product needs and maintains the product target profile. In the above case study, the TPP criteria such as transition to 5-ml vials led to the use of the 6.5-ml vial to ensure the net 5-ml volume fill as required. Another example from this case study is the definition of the needed post-thaw time and then testing to make sure the final formulation can support it. If the outcome had been that the formulation did not support it, then that outcome would have led to testing of additional formulations. If the development process were not guided and defined by the TPP endpoints, an endpoint such as stability post-thaw might not have supported the needs of the product. The TPP strategy should be a life-cycle strategy that should be constantly revisited and updated.
Process Characterization
Process characterization is the foundation for development of a robust, optimized, and reproducible process. It encompasses the accumulation of scientific data throughout the product life cycle that aims not only to demonstrate that a process can consistently produce a quality product (i.e., process development and validation) but also to guide process design toward the achievement of a consistent process. In order to ensure the quality of the product and process, a holistic risk-based approach has evolved, termed “quality by design.” QbD means designing and developing manufacturing processes during the product development stage to consistently ensure a predefined level of quality at the end of the manufacturing process [5]. The first step that a developer takes to design and develop a product under QbD is to define the desired product performance and identify the critical quality attributes (CQAs) of the product. Based on the definition and analysis of CQAs, a process can be designed that considers quality attributes and aims to control the sources of variability that can affect the end product [6]. A thorough characterization of process parameters allows the designer to select set points that best preserve the CQAs. Although this methodology has been used in the pharmaceutical and biologic industries, it has not yet been fully applied to the cell therapy field. The inherent variability and undefined nature of biological systems may limit the implementation of QbD, but as products and processes become more characterized, the concept will become useful to drive efficiencies.
Process analytical technology is defined as a system for designing, analyzing, and controlling manufacturing through in-process measurements of critical quality and performance attributes with the goal of ensuring final product quality [7]. As such, the goal of PAT is to enhance understanding and to control the manufacturing process to build product quality into the design process. A key aspect of PAT is the requirement to monitor important characteristics and process variables. In a cell therapy process, this can become challenging because of the difficulty of fully characterizing a living cell and obtaining relevant data in real time. As a process becomes more defined and repeatable over time, it may be possible to collect enough relevant data sets to build trends that may be used to incorporate PAT in a cell therapy manufacturing workflow.
Strategies for gathering useful, meaningful process characterization data require alignment with both the intended application for the data and the underlying science of the product and process. A key component of such strategies is the selection of reductionist or multivariate approaches to experimental design [8]. Reductionist approaches, in which parameters are varied individually while all others are held constant, are commonly used in scientific studies. They have the advantage of straightforward interpretation and design and lend themselves to systems in which the number of possible experimental conditions or replicates is limited. A drawback of the reductionist approach is that parameter interactions, in which the degree of product impact of one parameter is a function of another parameter, are not captured, although such interactions are common in biological systems. Consequently, the response to a particular parameter will be valid only at the interacting parameter set points of the characterization experiment. Adding multiple growth factors to a culture system, for example, may have synergistic effects at specific concentrations; testing these components in an individual manner will not identify such effects. In contrast, properly designed multivariate approaches address interacting parameters via concurrent experimental variation. A DoE approach, for example, can be used to screen multiple cell culture media components for significant effects. As shown in Figure 2, cell growth results can be analyzed to determine optimal predictive media conditions. Although this example focuses on one response (cell growth), additional responses such as specific cell surface marker expression, viability, or cell function can be analyzed individually or collectively to determine significant effects and tradeoffs.
Figure 2.

Example of a design of experiment (DoE) approach for medium development. A DoE strategy was used to design and formulate variant serum-free media that were tested for the ability to support bone marrow-derived mesenchymal stromal cell growth. An array of media was formulated according to a two-level factorial design testing four experimental components (components 1–4). The DoE layout shows relative concentrations of each component in which “−1” equals a low concentration (or, in some cases, no component added) and 1 equals a high concentration. Cells were allowed to grow for 5 days, and counts of viable cells were recorded. The data were analyzed for statistical significance, and the optimal predicted formulation for cell growth was identified. The values for components 3 and 4 were set at 1, and the effects of components 1 and 2 are shown graphically.
These multivariate approaches not only characterize interactions between multiple parameters but also can be used to identify or rule out interactions. A drawback of multivariate approaches is that experimental designs that appropriately address interactions via sufficient cross conditions with sufficient numbers of replicates for statistical significance can greatly exceed the logistically possible number of experimental conditions. Furthermore, the impact of many parameters is manifested only at scale, at which the ability to run large numbers of conditions is limited. Strategic statistical designs allowing fewer conditions than prescribed by a full factorial design can be used to mitigate such limitations [9]. Given the advantages and drawbacks of reductionist and multivariate experimental approaches, a proper strategy involves a pragmatic combination of both based on considerations of the science, the need, and logistical limitations.
It is important to note that process characterization experiments, like experiments of any type, must be designed and interpreted using appropriate statistical methods. For reductionist designs, the application of appropriate statistical tests to evaluate the significance of a conclusion has become routine for most scientists. For multivariate experiments, statistical evaluations of significance become more complex because of the combinations of conditions and the greater number of comparisons (and thus useful information) that are made [8]. Process engineers with basic statistical knowledge can perform these analyses using software suites designed for experimental support purposes. It is important to note that automated calculations performed by software produce relevant analyses only if applied properly. Equally important to statistical analysis of experiments is statistical design of experiments. An effective design will set up an experiment such that its analysis will generate the statistically significant comparisons and conclusions defined in its objectives. The definition of appropriate conditions, controls, and replicates form the foundation of an effective design. The statistical software used to analyze data can also be used to design experiments.
The data generated by properly designed and analyzed process characterization experiments can ultimately result in improved design of robust processes and confidence in process performance. These ends are achieved through application of process characterization data toward process definition: the specification of parameter set points and acceptable ranges. The product characteristics measured as a function of varied levels of a parameter or combination of parameters, the response surface, is a useful tool for process definition. It is important to note that a response surface, like any characterization, requires a sufficient amount of data to achieve statistical significance. A response surface allows the process engineer to define parameter ranges in which the product characteristics will be maintained within desired ranges and thus to design a process that is robust and reproducible. The slope of the response surface is an important factor in choosing parameter set points. Preferred parameter set points are in ranges in which product characteristics are minimally affected by parameter variation. This introduces robustness, that is, the minimization of risk that process parameter variation or deviations would affect the product. The process engineer will also consider parameter ranges that can be realistically run by both the staff and the equipment. This analysis requires the balance of response surfaces of all relevant product attributes. Ultimately, a balance of multiple product attribute responses, operational and equipment capabilities, and pragmatism will result in process definition that supports the production of a consistent product.
Process Scalability
For an allogeneic cell therapy for which large numbers and doses of cells are needed from each batch, industrial scalability of the process is dependent on product cost and quality. The key cell characteristic that enables such economies of scale is the ability to be cultured to high population doublings (minimally 25) while retaining function or potency but without senescence, slowing of growth rates, differentiation, or loss of phenotype. It is important to note, however, that a scale-up of cell culture requires a scale-up on downstream unit operations such as washing and filling to accommodate the increased cell yield. Cell banks that are used as seeds for repeated batches should be generated to meet these important characteristics to increase the success of the product.
The therapeutic dose of cells per patient, the number of patients that will be treated per year, the stability of the cell product, the product life cycle, the market size, and other considerations will determine the size and frequency of production batches (or lots) that need to be manufactured to meet the market demand. As cell demand increases, suspension bioreactor-based approaches provide a good option for providing homogeneous control and scalability, provided that the cell characteristics are maintained. The advantage of a scale-up approach is that bioreactors of increasing size to produce increasing numbers of cells can be controlled and operated with a small number of skilled staff, achieving economies of scale; however, significant process development is required to transform static processes to suspension cultures. An example for adherent cells is the choice and optimization of solid attachment substrates (microcarriers or other structures) that are suspended in culture and can provide very large surface areas for cell growth [10]. Both single-use and reusable bioreactors are available at escalating scales. To achieve economies of scale, both require greater capital investment that small-scale processes; however, there may be significant cleaning and sterilization validation investment costs for equipment and associated instruments for reusable bioreactors. A key challenge of scaling up a cell culture process is that the cells must retain their key quality attributes with respect to identity, potency, purity, and safety. Consequently, compared with bioprocesses in which stable cell lines are used to produce a secondary product, an additional focus during scaling of cell-based therapies is to ensure that the cell does not undergo any quality changes, such as differentiation, during expansion and that the final cell product must be comparable to available functional data.
The environment in which the cells are grown can have an impact on cell behavior, metabolism, and differentiation state. In manufacturing and scale-up, changes in factors can affect the cells. Among these many factors are physicochemical environment (including pH, dissolved oxygen, temperature, and process time), shear stress, concentration of metabolites within the culture, surface composition and geometry, and cell density [11]. In order to successfully scale up a cell culture process, the design space of critical process parameters (e.g., the mass, force and energy balances) can be defined at the small scale, and then these critical parameters can be set within the defined design space during the scale-up to preserve the cell attributes. This must be well validated at large scale to establish consistency.
Cell Culture Process Parameters
Whether the cells are proliferating, differentiating or being conditioned in culture, the supply of fresh nutrients, oxygen, signals from growth factors and serum, and removal of waste must be maintained. When increasing the scale of a culture system, it is important to make sure that the distribution of nutrients is reasonably homogeneous within the system. Heterogeneities in the system, and thus in the cell microenvironment, may lead to the creation of populations with different growth, phenotype, or potency within the culture. Furthermore, growth gradients lead to inefficient use of the surface area and thus limit cell yields. Another cell culture parameter is the removal of toxic or cell growth-inhibiting products of metabolism. Studies have shown that as culture scale increases or as cultures become more efficient by achieving high cell densities, the accumulation of waste products must be managed because of higher generation rates and lower mass transfer rates [12]. When culturing with static systems, there are few options beyond medium exchange to address waste produce management. Consequently, a reduction in cell growth rate is expected as the scale of the system increases unless surface area-normalized medium volumes and gas exchange interfaces remain constant. Fed-batch and perfusion systems, in which fresh medium is applied during culture and spent medium is removed (in the case of perfusion), have been used in bioreactor-based systems to overcome the accumulation of products of metabolism and replenish consumed components [13]. A balance between delivery of required nutrients and the removal of waste, governed by the perfusion rate and even material distribution, is predicted by mass balance calculations based on specific consumption and generation rates and cell densities. To minimize the costs associated with resultant medium consumption, the perfusion rate is often set near the minimum amount to maintain nutrients and eliminate waste at steady state, as predicted by the mass balance approach [14].
pH plays a critical role in influencing cell growth and metabolism. In traditional planar platforms, the control of pH is passively achieved by the medium composition (typically bicarbonate buffer systems) and CO2 gas exchange from the outside environment (incubator or warm room) to the gas phase inside the flask or multitray. Dissolved oxygen is also a critical parameter for cell growth. The most common approaches are to maintain the oxygen concentration experienced by the cells in the culture at either saturated conditions or at the in vivo physiological conditions from which the cells have originated. Static suspensions or nonsparged systems depend on surface mass transfer for the supply of gasses. When using a controlled growth system such as a bioreactor, the oxygen, CO2, and pH are controlled by continuous aseptic monitoring and by responding to maintain set points by actively applying gases in the quantity and with the composition needed.
Unlike most biological products produced by cell cultures, many cell therapy processes are dependent on the use of serum; however, elimination of serum and other animal-derived components from the medium should be a high priority due to concerns about disease transmission, variability (quality and cost), and the lack of availability [15]. Consequently, there is an industry trend to move toward serum-free, xeno-free (containing no nonhuman animal derived raw materials), and animal origin-free raw materials for producing cells. Alternatives such as GMP quality growth factors or small molecules could replace the function of serum while maintaining the desired cell properties, but these components should also be checked for function, consistency, safety, availability, and cost.
Downstream Processing
Harvesting of adherent cells typically involves both enzymatic digestion with chelation and shear force. High mechanical shear forces or enzymatic activities will ensure high yields but will result in low cell viability; therefore, balancing these parameters to allow efficient harvesting with minimal impact on viability is a major challenge. This is affected by the geometry of the vessel, the cell shear sensitivity, and the mode of mixing [16].
Most of today’s process technologies for cell concentration and washing (defined in this review as “downstream processing”) are derived from batch centrifugation used in traditional cell culture research laboratories and thus are not suitable for efficient mass production of cells. In addition, they do not meet the needed GMP standards. These GMP standards, which include the need for aseptic procedures in systems closed to the surrounding environment and controlled processes, present challenges for the downstream processes for cell therapies. The key to surmounting these challenges is designing the downstream process based on understanding of the cell product and the critical process parameters and by applying scalable technologies.
Two approaches for GMP volume reduction and cell washing are continuous centrifugation (manual or automated) and tangential flow filtration (TFF). When choosing between these methods, one should consider the cell numbers, shear exposure, process time, and culture volume that will be needed to supply development, clinical trial, and commercial efforts. Similar to the case with upstream processes, one should also consider a scale-up versus a scale-out approach, depending on the number of cells that a commercial-scale process will eventually need to produce. It is also critical to determine which methods give the desired cell viability, characteristics, and yields, and this should be done early on in the process development phase.
The use of automated centrifugation systems, such as have been used in the blood processing and apheresis sectors, can be adapted for downstream processing of cell therapy products when larger cell numbers (>108) and cell culture volumes (2–10 liters) are being processed. These systems also have the advantage of being fully closed to the environment, further minimizing risk of contamination. Although most single-use cell-washing systems are not scalable to larger volumes (>10–200 liters), they can be scaled out with the use of multiple units. Alternatively, KBI Biopharma (Durham, NC, http://www.kbibiopharma.com) has developed a technology called the kSep centrifuge. In this technology, cells are continuously pumped toward the center of a rotating chamber. Through the balance of the resulting centrifugal and fluid flow forces acting in opposite directions, the kSep centrifuge retains the cells as a concentrated fluidized bed under a continuous flow of medium or buffer [17]. This allows washing of the cells and concentrating them with minimal stress using a closed and automated system.
Recently, the use of TFF devices have been reported [18] as a scalable means of cell concentration and washing (diafiltration). In these devices, cells are recirculated along membranes in either a cassette or hollow fiber format. During recirculation, the cells remain in the lumen of the membranes (retentate), whereas fluid pressures force spent media, enzymes, or other components below the size cutoff of the membrane to pass through the filter into the permeate stream for removal. When designing a downstream process based on the TFF systems, it is important to consider the effect of pressure and shear stress on the cells. In order to have a good control over the process-critical parameters, calibrated pumps and pressure sensors must be implemented for accurate flow rate measurements. In addition, biomass monitors can be used to ensure that appropriate cell densities are achieved. TFF devices have the advantage of being readily scalable by increasing the membrane surface area in the device through increases in the number or the length of fibers. It is important to note that increasing fiber length changes the system dynamics and is not as straightforward as increasing the number of fibers. TFF systems are available as presterilized, single-use systems that maintain a fully closed configuration and that can be used for volumes in the range of 5 to >200 liters.
Final Formulation and Filling
Once the cells have been harvested, the clock begins to tick; cells are maintained outside of their ideal environment without nutrition or suitable conditions and will lose viability and activity. The sensitivity of the cells to these stressful conditions depends on the cell product and is a consideration in the design of the downstream process. After the downstream steps produce a concentrated, washed cell suspension, the product to be cryopreserved is formulated by determining the cell concentration and adding the final formulation additives to the suspension to achieve the desired concentrations of all components (including cells). Opening the container for the sampling and supplementation increases the risk of contamination of the product at its final stage, so closed-system manipulations are preferred. Furthermore, because lot size and thus filling scale increases, maintaining a homogenous cell suspension and relatively short process time will be difficult. Consequently, a vast amount of effort is invested in developing appropriate mixing vessels capable of closed-system sampling and cell quality monitoring. Most of the final formulation technologies are custom made per product to accommodate cell type-specific needs. The final product unit and filling step duration is a function of lot size and the number of cells per product unit. Larger lots will require automated filling systems to decrease process duration and maintain cell quality. The use of new closed-system plastic vials from multiple vendors coupled with traditional pharmaceutical fill-line automation can enable the processing of lot sizes in the several hundred to several thousand product units per lot using the same scalable technology. Cells that are cryopreserved are typically maintained in the vapor phase in a liquid nitrogen freezer at −196°C and thawed rapidly in a 37°C water bath. A dramatic temperature change is very stressful to most materials; therefore, the containers are very limited to small infusion bags, plastic tubes with screw-on caps, or the vials patented by Aseptic Technologies (Isnes, Belgium, http://www.aseptictech.com). A disadvantage of bags is related to handling because of their fragility and sensitivity when frozen. In particular, the cells freeze in a thin layer, making them very sensitive to temperature changes that might occur during shipment or prior to the intended thawing. The plastic screw-on cap vials are commonly used in cell culturing and preservation for research purposes, but they require opening the cap for filling and for extracting the cells, introducing contamination risk. Commonly used septum vials do not survive the liquid nitrogen freeze-thaw cycle because the differences in materials of the vial and the septum react differently to the temperature, resulting in leaks or breaks in the glass. Aseptic Technologies developed a unique technology in which a thermoplastic septum vial is preclosed and can survive the freeze cycle without leaking. In order to fill the vial, a needle is inserted through the thermoplastic septum, the cells are pumped in, and the thermoplastic septum is resealed using a laser beam.
Product Comparability Through Process Changes
As a product advances although development, the manufacturing process evolves to improve product quality, scalability, and cost [19, 20]. The product must remain comparable throughout process evolution to maintain support from the in vitro, preclinical, and clinical data establishing product safety and efficacy. To establish product comparability, it is necessary to “demonstrate that the manufacturing change does not affect safety, identity, purity, or potency” [21]. Demonstrating product comparability, however, can be challenging, given cell therapy complexity and the need to achieve consistent results from multiple predictive assays for determining safety or toxicity, identity, and efficacy. Even with meaningful assay data correlated to safety or toxicity and efficacy, it is difficult to disprove the possibility of changes in cell characteristics beyond those tested. Furthermore, “omic” analyses, which address cell characteristics in totality, generate enormous amounts of data that present equally mammoth hurdles for data analysis and interpretation. In contrast to analyses focused on a small number of measurements that do not rule out the possibility of changes in untested characteristics, omic analyses are likely to reveal changes for which the impact to cell comparability may not be meaningful; therefore, it is important to try to define the critical quality attributes that are suspected to be relevant to the mechanism of action and safety of the product and to minimize changes that affect these attributes. Ultimately, the most trusted practice for maintaining product comparability through process evolution is to maintain process consistency. The common paradigm is that if the process remains unchanged throughout process evolution, the product must remain unchanged. Conversely, an inference of this paradigm is that a change in the process introduces a risk of product change. This paradigm, if applied broadly, becomes restrictive to the point of impracticality.
The described approaches can be combined into a logical risk-based strategy for demonstrating product comparability through a manufacturing process change. This strategy determines the burden of data required to demonstrate product comparability based on the severity and likelihood of product impact resulting from the process change. This model can be applied through a framework for assessing the risk incurred from a given process change and the scientific rationale for product characterization that balances that risk. The cornerstone for assessing risk and formulating a characterization rationale is an analysis of the cell experience. In particular, this involves consideration of the conditions experienced by the cell before and after a change. In the few cases in which a process change causes no change in the cell experience, the process change does not introduce a risk of compromising product comparability. Accordingly, the change would likely be implemented without prior experimental data, and any unlikely gross product differences would be detected in routine release testing of production batches. An example of such a process change might be a pipette vendor switch in which the pipette geometry and contact materials are consistent. In the case of an extreme process change in which there is a fundamental change in the cell experience, and thus a high risk of a large product change is expected, the burden of data are massive. In this case, it is likely that all studies, including clinical studies, would be repeated as if the product were new. Examples of such extreme process changes in which the product would not be comparable might be altering the type of tissue from which the cell product is derived or a medium reformulation in which serum is eliminated and growth factor identities are drastically altered.
The burden of data needed to rationally support product comparability through a process change is defined not by the amount of data but rather by the inferences that can be supported by the data. The baseline requirement is the nonmicrobiology release tests for the cell therapy product; these tests are designed to confirm the product identity, viability, potency, and purity. Additional characterization to more rigorously confirm these product qualities might fulfill higher burdens of data. These characterizations might include additional markers for identity that might not be linked directly to the product function, additional mechanisms of action, or multiplex analyses that more thoroughly probe the product. Although such attributes might not be deemed to have the highest impact in the continuum of product attribute criticality, their consistency increases confidence in product comparability through a process change. To this end, it is good practice to develop and apply a large set of analytical tools that ultimately produces product and process understanding. In addition to the untargeted characterization approaches described, a useful data-generation strategy is the use of measurements that address reasonable hypotheses about process change effects on cells. A change in the cryopreservation process, for example, might imply cell damage. Accordingly, sensitive assays assessing cell viability or damage would address this inference and thus be required as appropriate supplemental information. Depending on the change, it is sometimes necessary to develop new assays to support the change.
When large burdens of data are required, it may be desirable to implement multiple changes simultaneously. Although this approach affords data-generation efficiency, the drawback is an increased risk of failure or product impact. Regardless, it is usually good practice to implement changes as early in the development process as possible to minimize risk to the product and its development program. Because there is some subjectivity in the risk-based approach to process changes, it is advisable to consult regulatory authorities before implementing higher risk changes.
Discussion
A growing number of cell therapies demonstrate great potential to be the source of revolutionary treatments for a variety of diseases [22]. The pathway of therapeutic development is a very long and arduous process. Numerous challenges include regulatory, manufacturing, and economic concerns that must be continuously addressed. Process development-related causes for commercial failures include high costs of goods, poor product characterization, process inconsistencies, and production limitations. Process innovation and development tools can enable strategies to overcome these challenges.
Because a potential therapy demonstrates efficacy in the early stages of development, it is natural to begin envisioning widespread access and treatment of patients throughout the world. Preparation to meet the targeted demand must start as early as possible in the development path, or many years, millions of dollars, and many opportunities may be lost. Scale-up processes can provide the opportunity for process optimization toward higher cell yield, reduced cost of goods, and higher quality of cells [23]. A key aspect to get alignment early on between the technical teams and the commercial teams is the expected commercial demand for a successful product and what the yearly cell production needs will be for a commercial product. It will be critical to outline manufacturing platforms, for both the upstream cell culture and the downstream processing, that can be used in clinical trials but that are also scalable to commercially relevant lot sizes without taking undue comparability risks.
Author Contributions
A.C., T.B., L.R., J.R., K.N., H.B., S.O., and O.K.: conception and design, manuscript writing.
Disclosure of Potential Conflicts of Interest
A.C. has compensated employment with Thermo Fisher Scientific, Inc. T.B. has compensated employment with Celgene Cellular Therapeutics. S.O. has compensated employment with Bioprocessing Technology Institute, A*STAR, Singapore. O.K. has compensated employment with Pluristem Therapeutics Inc. L.R. has compensated employment with Pluristem Therapeutics Inc. J.R. has compensated employment with RoosterBio Inc. K.N. has compensated employment with Novartis Pharma. H.B. has compensated employment with Pall Corporation.
References
- 1.Shire SJ, Combotz W, Bechtold-Peters K, et al. Current Trends in Monoclonal Antibody Development and Manufacturing. New Uork, NY: Springer; 2010. [Google Scholar]
- 2.Jones S, McKee S, Levine H. Emerging challenges in cell therapy manufacturing: Solutions from monoclonal antibodies. Bioprocess Int. 2012;10(suppl):4–7. [Google Scholar]
- 3.Eaker S, Armant M, Brandwein H, et al. Concise review: Guidance in developing commercializable autologous/patient-specific cell therapy manufacturing. Stem Cells Translational Medicine. 2013;2:871–883. doi: 10.5966/sctm.2013-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guidance for industry and review staff: Target product profile — a strategic development process tool. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm080593.pdf. Accessed July 20, 2015.
- 5.Rathore AS. Roadmap for implementation of quality by design (QbD) for biotechnology products. Trends Biotechnol. 2009;27:546–553. doi: 10.1016/j.tibtech.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 6.Riley BS, Li X. Quality by design and process analytical technology for sterile products--where are we now? AAPS PharmSciTech. 2011;12:114–118. doi: 10.1208/s12249-010-9566-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rathore AS, Bhambure R, Ghare V. Process analytical technology (PAT) for biopharmaceutical products. Anal Bioanal Chem. 2010;398:137–154. doi: 10.1007/s00216-010-3781-x. [DOI] [PubMed] [Google Scholar]
- 8.Lee K-M, Gilmore DF. Statistical experimental design for bioprocess modeling and optimization analysis: Repeated-measures method for dynamic biotechnology process. Appl Biochem Biotechnol. 2006;135:101–116. doi: 10.1385/abab:135:2:101. [DOI] [PubMed] [Google Scholar]
- 9.Mandenius C-F, Brundin A. Bioprocess optimization using design-of-experiments methodology. Biotechnol Prog. 2008;24:1191–1203. doi: 10.1002/btpr.67. [DOI] [PubMed] [Google Scholar]
- 10.Nienow AW, Rafiq QA, Coopman K, et al. A potentially scalable method for the harvesting of hMSCs from microcarriers. Biochem Eng J. 2014;85:79–88. [Google Scholar]
- 11.Yourek G, McCormick SM, Mao JJ, et al. Shear stress induces osteogenic differentiation of human mesenchymal stem cells. Regen Med. 2010;5:713–724. doi: 10.2217/rme.10.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park B-G, Chun J-M, Lee C-J, et al. Development of high density mammalian cell culture system for the production of tissue-type plasminogen activator. Biotechnol Bioprocess Eng. 2000;5:123–129. [Google Scholar]
- 13.Clincke MF, Mölleryd C, Zhang Y, et al. Very high density of CHO cells in perfusion by ATF or TFF in WAVE bioreactor™. Part I. Effect of the cell density on the process. Biotechnol Prog. 2013;29:754–767. doi: 10.1002/btpr.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y. Approaches to optimizing animal cell culture process: Substrate metabolism regulation and protein expression improvement. Adv Biochem Eng Biotechnol. 2009;113:177–215. doi: 10.1007/10_2008_19. [DOI] [PubMed] [Google Scholar]
- 15.Brindley DA, Davie NL, Culme-Seymour EJ, et al. Peak serum: Implications of serum supply for cell therapy manufacturing. Regen Med. 2012;7:7–13. doi: 10.2217/rme.11.112. [DOI] [PubMed] [Google Scholar]
- 16.Leung HW, Chen A, Choo ABH, et al. Agitation can induce differentiation of human pluripotent stem cells in microcarrier cultures. Tissue Eng Part C Methods. 2011;17:165–172. doi: 10.1089/ten.TEC.2010.0320. [DOI] [PubMed] [Google Scholar]
- 17.Ko H-F, Bhatia R. Evaluation of single-use fluidized bed centrifuge system for mammalian cell harvesting. Biopharm Int. 2012;25:34–40. [Google Scholar]
- 18.Pattasseril J, Varadaraju H, Lock L, et al. Downstream technology landscape for large-scale therapeutic cell processing. Bioprocess Int. 2013;11(suppl):38–47. [Google Scholar]
- 19.Bardy J, Chen AK, Lim YM, et al. Microcarrier suspension cultures for high-density expansion and differentiation of human pluripotent stem cells to neural progenitor cells. Tissue Eng Part C Methods. 2013;19:166–180. doi: 10.1089/ten.TEC.2012.0146. [DOI] [PubMed] [Google Scholar]
- 20.Goh TK, Zhang ZY, Chen AK et al. Microcarrier culture for efficient expansion and osteogenic differentiation of human fetal mesenchymal stem cells. Biores Open Access 2013;2:84–97. [DOI] [PMC free article] [PubMed]
- 21.Demonstration of comparability of human biological products, including therapeutic biotechnology-derived products. Available at http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm122879.htm. Accessed July 31, 2015.
- 22.Mason C, Brindley DA, Culme-Seymour EJ, et al. Cell therapy industry: Billion dollar global business with unlimited potential. Regen Med. 2011;6:265–272. doi: 10.2217/rme.11.28. [DOI] [PubMed] [Google Scholar]
- 23.Rowley J, Abraham E, Campbell A, et al. Meeting lot-size challenges of manufacturing adherent cells for therapy. Bioprocess Int. 2012;10(suppl):16–22. [Google Scholar]