

A detailed analysis was performed of the immunomodulatory and proteomic profiles of mesenchymal stromal cells (MSCs) administered to two patients with refractory acute respiratory distress syndrome (ARDS). The respiratory, hemodynamic, and multiorgan failure resolved and multiple pulmonary and systemic inflammation markers decreased. These observations highlight the mechanistic information that can be gained from clinical experience and the value of correlating in vitro potency assessments with clinical effects.
Keywords: Acute respiratory distress syndrome, Pulmonary diseases, Respiratory tract, Stem cells, Cell transplantation, Bone marrow stromal cells, Cellular therapy, Clinical translation
Abstract
Mesenchymal stromal cells (MSCs) have been investigated as a treatment for various inflammatory diseases because of their immunomodulatory and reparative properties. However, many basic questions concerning their mechanisms of action after systemic infusion remain unanswered. We performed a detailed analysis of the immunomodulatory properties and proteomic profile of MSCs systemically administered to two patients with severe refractory acute respiratory distress syndrome (ARDS) on a compassionate use basis and attempted to correlate these with in vivo anti-inflammatory actions. Both patients received 2 × 106 cells per kilogram, and each subsequently improved with resolution of respiratory, hemodynamic, and multiorgan failure. In parallel, a decrease was seen in multiple pulmonary and systemic markers of inflammation, including epithelial apoptosis, alveolar-capillary fluid leakage, and proinflammatory cytokines, microRNAs, and chemokines. In vitro studies of the MSCs demonstrated a broad anti-inflammatory capacity, including suppression of T-cell responses and induction of regulatory phenotypes in T cells, monocytes, and neutrophils. Some of these in vitro potency assessments correlated with, and were relevant to, the observed in vivo actions. These experiences highlight both the mechanistic information that can be gained from clinical experience and the value of correlating in vitro potency assessments with clinical effects. The findings also suggest, but do not prove, a beneficial effect of lung protective strategies using adoptively transferred MSCs in ARDS. Appropriate randomized clinical trials are required to further assess any potential clinical efficacy and investigate the effects on in vivo inflammation.
Significance
This article describes the cases of two patients with severe refractory adult respiratory syndrome (ARDS) who failed to improve after both standard life support measures, including mechanical ventilation, and additional measures, including extracorporeal ventilation (i.e., in a heart-lung machine). Unlike acute forms of ARDS (such in the current NIH-sponsored study of mesenchymal stromal cells in ARDS), recovery does not generally occur in such patients.
Introduction
Acute respiratory distress syndrome (ARDS) is a severe, life-threatening medical condition characterized by widespread uncontrolled inflammation in the lungs associated with the loss of surfactant and impaired pulmonary capillary endothelial barriers, resulting in fluid accumulation in the distal airspaces. A number of etiologies can trigger ARDS, including primary respiratory infections, sepsis, and significant trauma. ARDS continues to result in high mortality and high costs related to often prolonged intensive care unit (ICU) and hospital stays [1–4]. Furthermore, ARDS survivors often have long-term pulmonary, neuromuscular, and cognitive symptoms and a diminished quality of life [2–4]. A number of different pharmacologic therapies have failed to demonstrate benefit, and treatment is currently limited to supportive care and appropriate use of mechanical ventilation and fluid management [5, 6]. The addition of extracorporeal membrane oxygenation (ECMO) has been used in patients with severe ARDS; however, in a recent systematic review and meta-analysis of current evidence, no association with improved outcomes could be demonstrated in adult patients [4]. As such, new therapeutic approaches are needed.
Adoptive transfer of mesenchymal stromal cells (MSCs) ameliorates experimentally induced acute lung injury in preclinical animal models and in ex vivo perfused human lungs [7–15]. Although not completely understood, the mechanisms of MSC actions in acute models of ARDS include the release of paracrine anti-inflammatory and antibacterial peptides and mitochondrial transfer through cell-cell contact with damaged alveolar epithelial cells in the absence of permanent cell engraftment [10, 16–18]. Furthermore, MSCs release extracellular vesicles (EVs), which have reduced inflammation and promote tissue regeneration in different preclinical models [7, 19–21]. Because administration of non-human leukocyte antigen (HLA)-matched allogeneic MSCs has also been demonstrated to be safe and potentially effective in a widening range of clinical applications in both lung diseases and other diseases [6, 8, 22–24], this approach could be beneficial in patients with ARDS. A recent phase I dose-escalation safety study demonstrated the safety of a single i.v. administration of 1–10 million cells per kilogram of MSCs in 9 patients with moderate to severe ARDS, and a phase II efficacy trial is currently under way [25].
However, despite abundant descriptions of in vitro and in vivo MSC actions in preclinical models, correlation of the in vitro MSC release of anti-inflammatory mediators and effects on immune effector cells in clinical applications remains poorly understood [26]. Furthermore, no animal models resembling patients with refractory ARDS who require additional support measures are available. Our incomplete understanding of the mechanisms of action of MSCs has limited the development of relevant in vitro potency assays that can define the MSC populations optimal for clinical use, including the selection of potent cell batches relevant to specific disease indications. More importantly, it has limited the selection of patients likely to benefit from treatment. The treatment of two patients with severe refractory ARDS using MSCs was studied in an attempt to correlate the in vivo actions of the MSCs on lung and systemic inflammation with a detailed investigation of in vitro potency assays, including the effects on inflammatory and immune modulatory cells, and proteomic assessments of the MSCs and EVs released by the MSCs.
Materials and Methods
Ethical Considerations
The production of clinical-grade MSCs was accredited by the Swedish Board of Health and Welfare (approval no. 9.1-57237/2012) and the Medical Products Agency (approval no. 5.9.2-2013-047346). ECMO is a last resort supportive therapy for ARDS. The condition of the two patients in our report had deteriorated with ECMO support; also, because the patients were sedated before MSC administration, informed consent could not be obtained. At a multidisciplinary conference, we decided to use MSCs on a compassionate use basis under the approval of the chief medical officer of the hospital, the hospital ethics committee, and the patients’ relatives.
Isolation and Expansion of Mesenchymal Stromal Cells
A total of 28 ml of bone marrow was aspirated from a 49-year-old HLA-mismatched, third-party, healthy male volunteer. Clinical-grade MSCs were generated under good manufacturing practice (GMP) conditions according to a common protocol elaborated by the European Group for Bone and Marrow Transplantation Developmental Committee, accredited by the Swedish National Board of Health and Welfare [27]. Bone marrow mononuclear cells (146 × 106) were seeded into 175 cm2 flasks (Falcon, Franklin Lakes, NJ, http://www.BD.com) in Dulbecco’s modified Eagle’s medium-low glucose (Life Technologies, Gaithersburg, MD, http://www.lifetechnologies.com) supplemented with lysed human platelets (final concentration, 108 per milliliter). When the cultures were near confluence (>80%), the cells were detached once by treatment with trypsin and EDTA (Invitrogen, Grand Island, NY, http://www.invitrogen.com) and replated once at a density of 4,000 cells per cm2. The MSCs were harvested and cryopreserved in 10% dimethyl sulfoxide (DMSO; WAK-Chemie Medical GmbH, Steinbach, Germany, http://www.wak-chemie.net). After thawing, the cells were washed 3 times in phosphate-buffered saline (PBS) and resuspended in 0.9% saline solution with the addition of 10% AB plasma, to a final concentration of 2 × 106 cells per milliliter. The MSC release criteria for clinical use included the absence of visible clumps, spindle shape morphology, the absence of contamination by pathogens (bacteria and mycoplasma), and viability >95%. The MSCs expressed CD73, CD90, CD105, and HLA-ABC and were negative for CD14, CD31, CD34, CD45, and HLA-DR (supplemental online Fig. 1A).
The total dose administered to the patients was 2 × 106 cells per kilogram of recipient body weight and was determined from previous experience of MSC treatment of graft-vs.-host disease (GVHD) [27]. The dose was divided into 4 syringes of 50 ml, and each syringe was infused over 30 seconds. No visible clumps or cell debris were observed during the infusions.
Clinical Outcome Measures
Blood was collected as standard practice during routine care in the intensive care unit. Bronchoalveolar lavage (BAL) fluid was obtained from lavage performed at intervals as part of the routine clinical care provided to each patient. Blood samples for cell purification were collected in heparinized tubes. Serum and plasma samples for cytokines and extracellular vesicles were collected and frozen immediately after sample preparation and stored at −80°C until analyzed. Chest x-rays (CXRs) and chest computed tomography scans were performed as part of the routine clinical care provided to each patient. Respiratory measurements, including lung volumes and pressure, were obtained from the mechanical ventilation record of each patient. Other standard laboratory measurements were taken as a part of the routine care for each patient and obtained from the patient’s medical records.
BAL Fluid and Serum Analyses
The levels of pro- and anti-inflammatory cytokines in serial samples of BAL fluid and serum were assessed using a multiplex cytokine assay (EMD Millipore, Billerica, MA, http://www.emdmillipore.com) on a Luminex machine (EMD Millipore). In BAL fluid, the surfactant protein B concentration was determined by enzyme-linked immunosorbent assay (ELISA; USCN Life Science Inc., Houston, TX, http://www.uscnk.com). Caspase-cleaved cytokeratin-18 (ccK18) and total K18 were measured using M30-Apoptosense ELISA (Peviva AB, Bromma, Sweden, http://www.peviva.com) and M65 EpiDeath ELISA (Peviva AB), respectively.
Assessment of Circulating MicroRNAs
Inhibitory microRNAs (miRNAs) were isolated from circulating EVs purified from the routine blood samples collected. EV purification and miRNA isolation was performed by Exosome Diagnostic Inc. (New York, NY, http://www.exosomedx.com). In brief, 1.5–2 ml of the blood samples from each time point was purified using the exoRNeasy serum/plasma kit (Qiagen, Hilden, Germany, http://www.qiagen.com). Eluted RNA was processed for microRNA analysis using the low sample input protocol for the TaqMan OpenArray Human MicroRNA Panel (Life Technologies). Megaplex RT Primers, Human Pool A and Human Pool B (Life Technologies), were used for reverse transcription followed by a preamplification step according to the manufacturer’s protocol (95°C for 10 minutes, 55°C for 2 minutes, 72°C for 2 minutes, followed by 16 cycles of 95°C for 15 seconds, 60°C for 4 minutes, followed by 99°C for 10 minutes, followed by 4°C). The preamplified material was diluted 1:20 in Tris-EDTA (pH 8.0) for analysis. The samples were loaded onto OpenArray plates using the standard Accufill protocol (Life Technologies). Amplification was performed according to the protocol established for the TaqMan OpenArray Human MicroRNA panel downloaded for each plate (OpenArray Plate product page; http://www.lifetechnologies.com). A total of 758 miRNAs were assayed, and 200–300 miRNAs gave a detectable Ct value for each time point.
U6 small nuclear RNA (snRNA) is a noncoding small nuclear RNA commonly used for normalization of miRNA [28]. U6 was present in every sample and every subarray and was used to normalize for subarray-to-subarray variations and differences between two Megaplex pools. ΔCt values were calculated for each miRNA by subtracting the U6 Ct value located on the same subarray. The values were corrected for variation of the total RNA amount by subtracting each Ct value by the median Ct value of 128 miRNAs present in all samples in the first patient and by the median Ct of 79 miRNAs present in all samples in the second patient. This procedure was performed for all arrays and time points. Missing values were entered as the highest observed normalized Ct value per patient. Finally, the fold change for every miRNA relative to the normal healthy control sample was calculated.
Mononuclear Cell Collection, Cell Purification, and In Vitro Mixed Lymphocyte Studies
The peripheral blood mononuclear cells (PBMCs) used in the in vitro studies were retrieved from the patients and healthy donors, isolated by density gradient-based centrifugation, and stored in 10% DMSO in liquid nitrogen until additional analysis. For further purification of T cells, a paramagnetic bead-based selection was used (Miltenyi Biotec, Bergisch Gladbach, Germany, http://www.miltenyibiotec.com). The same bone marrow-MSCs used in the patient were cocultured with allogeneic T cells (MSC/T-cell ratio 1:5 and 1:10) for 5 days. The T cells were stimulated with activating anti-CD2/CD3/CD28 antibodies (Miltenyi Biotec) in a 0.5 bead per cell ratio [29]. PBMCs were cultured for 5 days in the presence of MSCs (MSC/PBMC ratio of 1:5 and 1:10), and the monocytic compartment was subsequently analyzed. Polymorphonuclear leukocytes (PMNs) from buffy coats of healthy donors were ultra-purified under endotoxin-free conditions, as previously described [30]. PMNs (>95% purity) and MSCs were cocultured for up to 40 hours in the presence or absence of 100 ng/ml lipopolysaccharide (LPS; Invitrogen, Carlsbad, CA, http://www.invitrogen.com). In all cases, MSCs were plated for 72 hours before the start of cocultures. In selected experiments, MSCs were pretreated with recombinant human interferon-γ (IFN-γ; 10 ng/ml; PeproTech, Rocky Hill, NJ, http://www.peprotech.com) and tumor necrosis factor-α (TNF-α; 15 ng/ml, R&D Systems, Minneapolis, MN, http://www.rndsystems.com) for 48 hours and then cocultured with freshly isolated PMNs. MSC and PMN cocultures were stained with May-Grünwald-Giemsa dye to observe the cell morphology after reciprocal interaction.
In Vitro PMN Assessments
PMNs were stained according to the manufacturer’s instructions with fluorochrome-coupled antibodies, as detailed in supplemental online Table 2. The LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Life Technologies) was used for the exclusion of dead cells for PBMC analysis and propidium iodide (Invitrogen) staining to test the viability of the MSCs. To perform intracellular staining, the cells were treated with the Cytofix/Cytoperm Fixation/Permeabilization Kit (BD Biosciences, San Jose, CA, http://www.bdbiosciences.com). After coculture with MSCs, PMNs were identified on the basis of their typical morphological parameters (forward scatter/side scatter) and their CD45 expression. An annexin-V-FITC staining kit (Miltenyi Biotec) was used to assess the levels of PMN apoptosis. The cells were analyzed using a FACS Canto II cytometer (BD Biosciences) and FlowJo, version 9.5 software (TreeStar, Ashland, OR, http://www.treestar.com).
RNA Preparation and Quantitative Reverse Transcriptase-Polymerase Chain Reaction
Total RNA was extracted (RNeasy Mini Kit; Qiagen) and cDNA prepared (Superscript First Strand Synthesis System; Life Technologies) using a Mastercycler nexus (Eppendorf, Hamburg, Germany, http://www.eppendorf.com). For cDNA transcription, a two-step procedure was performed in line with the manufacturer’s recommendations. In brief, the final reaction was incubated for 10 minutes at 25°C, followed by 60 minutes at 60°C, and terminated by heating at 70°C for 15 minutes. Messenger RNA levels were quantified using quantitative polymerase chain reaction (PCR; Quantitect SYBR Green PCR Kit; Qiagen) on a Rotor Gene Q (Qiagen). Relative gene expression was determined by normalizing the expression to β2-microglobulin using the 2−ΔΔCt method. The following gene-specific primers were used (forward and reverse sequence): indoleamine-2,3-dioxygenase (IDO), 5′-GCATTTTTCAGTGTTCTTCGCATA-3′ and 5′-TCATACACCAGACCGTCTGATAGC-3′; β2-microglobulin, 5′- TGCTGTCTCCATGTTTGATGTATCT-3′ and 5′-TCTCTGCTCCCCACCTCTAAGT-3′.
In Vitro MSC Responses to Inflammatory Stimuli
The MSCs were tested in vitro for their responsiveness to inflammatory stimuli (“MSC licensing”). Culture in the presence of TNF-α (15 ng/ml) or IFN-γ (10 ng/ml) for 48 hours induced the expression of CD54 (ICAM-1), CD106 (VCAM-1), and HLA-ABC and HLA-DR, as assessed by flow cytometry, and IDO mRNA transcripts was measured using quantitative PCR (supplemental online Fig. 1B).
MSC EV Purification
MSCs were cultured in serum-reduced medium (OptiMem; Gibco, Grand Island, NY, http://www.lifetechnologies.com) for 48 hours. The conditioned medium was harvested and spun at 300g for 5 minutes and filtered through 0.2-μm sterile syringe filters to remove cell debris. The EVs were subsequently pelleted by ultracentrifugation at 110,000g for 90 minutes at +4°C. The pellets were resuspended in PBS, pooled, and ultracentrifuged again at 110,000g for 90 minutes at +4 C. The remaining pellet was resuspended in 250 μl of PBS. Nanoparticle tracking analysis was performed using the NS500 (NanoSight, Wiltshire, U.K., http://www.malvern.com) to measure the size distribution of the particles, which is based on the motion and light scatter of nanometer-size particles (Brownian motion) [31]. The number of particles and their movement were recorded using a camera level of 15 and automatic functions for all postacquisition settings except for the detection threshold, which was fixed at 6. The samples were diluted in PBS between 1:500 and 1:2,000 to achieve a particle count between 2 × 108 and 2 × 109 per milliliter. The camera focus was adjusted to make the particles appear as sharp dots. Using the script control function, 5 × 30-second videos were recorded, incorporating a sample advance and a 5-second delay between each recording and analyzed using the NS500 software (NanoSight). The EVs were frozen at −80°C until further analysis.
EV Preparation for Proteomic Assessment
Donor cell pellet and EVs were lysed with 4% SDS, 25 mM HEPES, 1 mM dithiothreitol (DTT). Lysates were heated to 95°C for 5 minutes followed by sonication for 1 minute and centrifugation at 14,000g for 15 minutes. The supernatant was mixed with 1 mM DTT, 8 M urea, and 25 mM HEPES (pH 7.6) and transferred to a centrifugation filtering unit, with a 10-kDa cutoff (Nanosep; Pall Corp., Port Washington, NY, http://www.pall.com), and centrifuged for 15 minutes at 14,000g, followed by another addition of the 8 M urea buffer and centrifugation. Proteins were alkylated by 50 mM indole-3-acetic acid, in 8 M urea and 25 mM HEPES for 10 minutes, centrifuged for 15 minutes at 14,000g, followed by 2 more additions and centrifugation with 8 M urea and 25 mM HEPES. Trypsin (Promega, Madison, WI, http://www.promega.com), at a 1:50 trypsin/protein ratio, was added to the cell lysate in 250 mM urea and 50 mM HEPES and incubated overnight at 37°C. The filter units were centrifuged for 15 minutes at 14,000g, followed by another centrifugation with Milli-Q water (EMD Millipore), and the flow through was collected. The peptides were cleaned using a strata-X-C-cartridge (Phenomenex, Torrance, CA, http://www.phenomenex.com).
Nano Reverse Phase Chromatography Gradient Before Mass Spectrometry Analysis
Before analysis using the Q Exactive mass spectrometer (Thermo Fisher Scientific, San Jose, CA, http://www.thermoscientific.com), the peptides were separated using an Agilent 1200 nano-LC system (Agilent Technologies, Santa Clara, CA, http://www.agilent.com). The samples were trapped on a Zorbax 300SB-C18 (Agilent Technologies) and separated on a NTCC-360/100-5-153 (Nikkyo Technos, Ltd., Tokyo, Japan, http://www.nikkyo-tec.co.jp) column using a gradient of A (5% DMSO, 0.1% formic acid) and B (90% acetonitrile, 5% DMSO, 0.1% formic acid), ranging from 5% to 37% B in 240 minutes with a flow of 0.4 µl/minute. The Q Exactive mass spectrometer (Thermo Scientific, Wilmington, DE, http://www.thermoscientific.com) was operated in a data-dependent manner, selecting the top five precursors for fragmentation by higher energy C-trap dissociation (HCD). The survey scan was performed at 70,000 resolutions from 300 to 1,700 mass-to-charge ratio (m/z), with a maximum injection time of 100 milliseconds and target of 1 × 106 ions. For generation of HCD fragmentation spectra, a maximum ion injection time of 500 milliseconds and automatic gain control of 1 × 105 were used before fragmentation at 30% normalized collision energy, 35,000 resolutions. Precursors were isolated with a width of 2 m/z and put on the exclusion list for 70 seconds. Single and unassigned charge states were rejected from precursor selection.
Peptide, Protein Identification, and Data Analyses
Proteome Discoverer, version 1.4 (Thermo Scientific), with SEQUEST-Percolator was used for protein identification. The precursor mass tolerance was set to 10 ppm and for fragments to 0.02 Da. Oxidized methionine was set as dynamic modification and carbamidomethylation as static modification. Spectra were matched to a combined database of UniProt human (140203) combined with the 250 most abundant proteins from 4-hour nLC-MS/MS analysis of FBS (Bos Taurus, UniProt 140203). The results were filtered to 1% false discovery rate (FDR). Identifications in Bos taurus were considered to originate from FBS and were removed. Gene ontology term enrichment analysis was done using the protein analysis through evolutionary relationships (PANTHER) classification system [32].
Statistical Analysis
The Wilcoxon paired test was used to compare the differences between two different groups. One-way analysis of variance was used to statistically evaluate the difference of sample means among multiple groups. The significance level was set at p ≤ .05.
Results
Clinical Presentation
Patient 1
A 58-year-old man with a history of hypertension was hospitalized 8 days after the onset of high fever, cough, and dyspnea. The CXR demonstrated diffuse bilateral infiltrates and reverse transcription (RT)-PCR of BAL fluid was positive for influenza A H1N1. Oseltamivir (Tamiflu; Roche, Basel, Switzerland, http://www.roche.com) was initiated; however, the patient’s condition deteriorated over the next 2 days, with increased bilateral opacities on the CXR and progressive respiratory failure requiring mechanical ventilation, followed initially by venovenous (VV), and later by venoarterial, ECMO (CARDIOHELP System; Maquet, Rastatt, Germany, http://www.maquet.com) for refractory hypoxemia. He was diagnosed with severe ARDS according to the Berlin criteria (Table 1) [33]. Acute kidney failure developed, requiring continuous renal replacement therapy (CRRT).
Table 1.
ARDS diagnostics according to the Berlin criteria at admission to the ICU
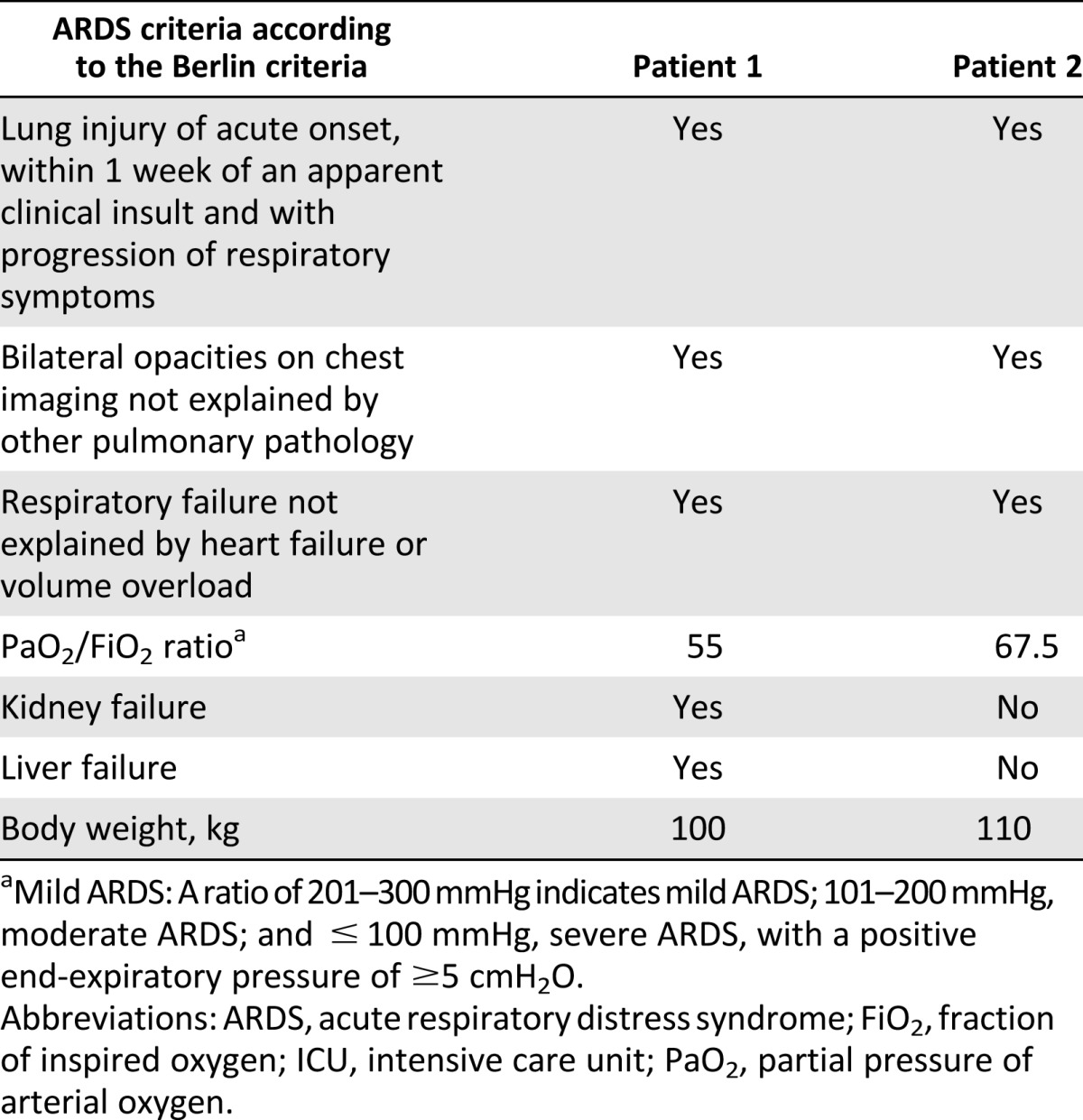
Over the next 4 days, the patient’s condition worsened, with persistent hypoxemia, progressive bilateral infiltrates, and liver failure. At this juncture, after extensive discussions with the family, hospital administration, and ethics board, HLA-mismatched allogeneic bone marrow-derived MSCs, obtained from a healthy volunteer, were systemically infused through a central venous catheter positioned in the right atrium of the heart on a compassionate use basis. The total dose (2 × 106 cells per kilogram of recipient weight) was divided into aliquots of 5 ml of cell suspension given at 6 time points over 20 minutes. To avoid infusion of MSCs into the ECMO circuit and to maximize the delivery of cells to the pulmonary circulation, the ECMO outflow cannula was clamped during each infusion and opened after six heartbeats. ECMO support was maintained for 3 minutes between each MSC infusion. At the time of infusion, the patient had no detectable influenza A H1N1 particles, as assessed by RT-PCR, and had no other detectable active infection.
Patient 2
A 40-year-old man with no previous medical history was admitted to the hospital because of malaise, weight loss, night sweats, gingival hyperplasia, and fever. Acute myeloid leukemia was diagnosed in the bone marrow aspirate, and induction chemotherapy was initiated. Because of caries and dental root abscesses, several teeth were extracted at the initiation of cytotoxic therapy. The following day, the patient became progressively hypoxemic and developed acute respiratory failure requiring intubation and mechanical ventilation. Blood and sputum cultures were negative, and CXR demonstrated bilateral infiltrates. Despite treatment with broad-spectrum antibiotics in the now neutropenic patient, progressive hypercapnia and hypoxia developed over the next 2 days, and VV ECMO was initiated. An echocardiogram demonstrated normal cardiac function. The patient was initially stable with ECMO; however, the clinical course was complicated by severe transfusion-dependent cytotoxic chemotherapy-induced thrombocytopenia. Given the large fluid volume from the transfusions, CRRT was initiated to maintain an appropriate fluid balance and reduce interstitial edema. Fourteen days after initiation of ECMO, the patient’s condition continued to deteriorate, with increasing hypoxia and progressive bilateral infiltrates on CXR. Possible contributing factors included progressive severe ARDS according to the Berlin criteria [33] (Table 1), possibly secondary to an initial infection, although the findings from blood and sputum cultures and viral screens had remained negative. Transfusion-related acute lung injury could also have played a role in the patient’s deterioration. After extensive discussion with the family, hospital administration, and ethics board, HLA-mismatched allogeneic bone marrow-derived MSCs (from the same donor and passage number used for patient 1; 2 × 106 cells per kilogram recipient weight) were administered in the same manner as for the first patient 28 days after the initiation of ECMO.
Clinical Course After Allogeneic MSC Infusion
No obvious adverse events or changes in clinical status were observed in either patient during the MSC infusions, including no alterations in hemodynamic parameters or in oxygen saturation. Serial CXRs demonstrated progressive decreases in pulmonary infiltrates that were evident 24 hours after infusion in both patients (Figs. 1A, 2A). In accordance with local guidelines for patients receiving ECMO support, the positive end-expiratory pressure (PEEP) and peak inspiratory pressure were set and maintained within a defined range while the patient was receiving ECMO support. Concomitant clinical improvements were observed in the tidal volumes and compliances, thus, implying improvement in respiratory dynamics and recovery of the injured lung without manipulation of the ventilator settings. Oxygenation and pulmonary compliance improved in both patients only a few days after MSC administration in the setting of unchanged ECMO settings and PEEP (supplemental online Fig. 2). In patient 1, pulmonary compliance increased from 6 ml/cmH2O before infusion to 20 ml/cmH2O at 2 days, increasing to 44 ml/cmH2O at day 8 (Fig. 1B). In patient 2, pulmonary compliance increased from 20 ml/cmH2O before infusion to 35 ml/cmH2O at day 1, increasing to 73 ml/cmH2O at day 12 (Fig. 2B). The tidal volumes similarly increased; increasing in patient 1 from 100 to 420 ml over a 2-day period, reaching 720 ml at 5 days (Fig. 1C). In parallel with the improvements in respiratory status, patient 1, who was hemodynamically unstable at the time of infusion, stabilized over the first days and was weaned off intravenous pressor agents.
Figure 1.

Clinical characteristics of patient 1 before and after MSC administration. (A): Disease progression and concomitant chest x-rays (CXRs) from the onset of influenza A H1N1 until hospital discharge. As demonstrated, a progression of bilateral opacifications of the lungs occurred after the onset of the influenza infection, and the patient required both mechanical ventilation and ECMO support. By 24 hours after MSC infusion, a decrease had occurred in the pulmonary infiltrates on CXR, followed by progressive improvement until the time of weaning of ECMO and extubation 4 weeks after MSC administration. In parallel with the improvement on the CXRs, both the pulmonary compliance (B) and tidal volumes (C) improved to normal levels within the first 2 days. However, 5 days after MSC administration, the patient developed nosocomial pneumonia with an increased WBC count (D) and new infiltrates on the CXR (A), which resolved within 3 days. The patient’s liver function also improved during the first week, with normalization of serum ALT, AST, and bilirubin levels (E). Abbreviations: ALT, alanine transaminase; AST, aspartate transaminase; ECMO, extracorporeal membrane oxygenation; MSC, mesenchymal stromal cell; WBC, white blood cell.
Figure 2.

Clinical characteristics of patient 2 before and after MSC administration. (A): Progression of acute respiratory distress syndrome both on chest x-rays (CXRs) and serial computed tomographic scans. As demonstrated, a progressive decrease occurred in pulmonary infiltrates starting the first day after MSC infusion. By day 7, the CXR findings had improved back to normal, and the patient was weaned off ECMO the next day and extubated on day 12. Pulmonary compliance (B) and tidal volumes (C) improved to normal levels during the first week. Bone marrow function continued to be reconstituted with resolution of leukopenia (D) and thrombocytopenia (E). Abbreviations: ECMO, extracorporeal membrane oxygenation; MSC, mesenchymal stromal cell; WBC, white blood cell.
At 5 days after MSC administration, patient 1 developed nosocomial pneumonia with fever, an increased white blood cell count (Fig. 1D), and new infiltrates on CXR (Fig. 1A). The blood cultures were positive for coagulase-negative staphylococci. Vancomycin was added to the antibiotic regimen at day 6, with clinical resolution of the pneumonia by day 8. In patient 2, the tidal volumes increased from 221 to 617 ml over a 7-day period (Fig. 2C). The improvements in respiratory status persisted, allowing discontinuation of ECMO support and, subsequently, a progressive decrease in the need for mechanical ventilation (Figs. 1A, 2A). Liver function in patient 1 improved during the first week, with normalization of transaminase and bilirubin levels (Fig. 1E). Kidney function also steadily improved in patient 1, allowing cessation of CRRT. Patient 2 did not develop renal or hepatic failure and the bone marrow function continued to be reconstituted after MSC infusion, with resolution of leukopenia (Fig. 2D) and thrombocytopenia (Fig. 2E). Patient 1 was discharged from the ICU after 46 days and left the hospital at day 69 (Fig. 1A). Two months later, the patient had returned to work and had recovered both physically and mentally, scoring 38.4 on the physical component and 67.4 on the mental component of the SF-36 quality of life assessment (norm-based scale, with an average of 50 for the physical and mental components both). Patient 2 was weaned off ECMO after 8 days, was removed from mechanical ventilation after 12 days, and returned to the general ward 15 days after the MSC infusion (Fig. 2A). The patient then underwent consolidation chemotherapy for acute myeloid leukemia and subsequently could not perform the SF-36 test.
Changes in Pulmonary and Systemic Inflammatory Markers After MSC Infusion
Detailed assessments of lung and systemic inflammation before the MSC infusions demonstrated elevations in some, but not all inflammatory, markers in both patients, suggestive of some degree of ongoing inflammation. In patient 1, MSC administration was subsequently followed by a decrease in a range of inflammatory markers in both BAL fluid and serum. In patient 1, the BAL fluid albumin levels, a marker of alveolar-capillary barrier integrity and permeability, decreased from 2 g/l at the time of MSC infusion to undetectable levels the next day. Interleukin-6 (IL-6) in BAL fluid, together with IL-8 and IFN-γ levels in plasma, similarly decreased during the first days after MSC administration (Fig. 3A, 3B). The plasma IL-6 and IFN-γ levels subsequently increased transiently in conjunction with the hospital-acquired pneumonia, and IL-8 demonstrated a similar response pattern to that in the BAL fluid (Fig. 3A, 3B). Plasma and BAL fluid granulocyte macrophage colony-stimulating factor levels increased in response to both MSC administration and pneumonia (Fig. 3A, 3B). The proinflammatory chemokine inducible protein 10 (IP10) responded similarly to IL-8 in the BAL fluid with an initial rapid decrease after MSC infusion, followed by a small increase as a response to the pneumonia and a decline again at day 7. No significant change in the plasma IL-8 levels was observed (Fig. 3A).
Figure 3.

Inflammatory marker response, lung epithelial recovery, and miRNA response to MSC administration. The cytokine response in BAL (A) and plasma (B) after MSC infusion. The orange line represents the mean values measured in healthy controls (n = 102–106). The area between the 5th and 95th percentile is shown in gray. Red lines, patient 1; black lines, patient 2. (C): Levels of caspase-cleaved cytokeratin-18, indicative of epithelial apoptosis, and uncleaved cytokeratin-18, indicative of total epithelial death in BAL fluid, decreased in both patients within a few hours after MSC administration. (D): In parallel, surfactant protein B increased during the first 3 days after MSC infusion, suggesting recovery of alveolar epithelial function. Several miRNAs related to inflammation demonstrated a significant decline within the first 24 hours after MSC administration ([E], top row). Proinflammatory: purple, miR-409-3P; green, miR-886-5P; orange, miR-324-3P; red, miR-222; blue, miR-125A-5P; violet, miR-339-3P; brown, miR-155. A transient increase in these miRNA levels was observed by day 7 and had returned to normal by day 28. Other inflammation-related miRNAs demonstrated a dichotomous pattern with an initial increase within the first hours to the first day, followed by a decline and another peak by day 27 in patient 1 (red) and day 14 in patient 2 (black), returning to normal by day 28 in both patients ([E], bottom row). Abbreviations: BAL, bronchoalveolar lavage; ccK18, caspase-cleaved cytokeratin-18; d, day; GM-CSF, granulocyte macrophage colony-stimulating factor; h, hour; IFN-γ, interferon-γ; IL-6, interleukin-6; IP10, inducible protein 10; K18, cytokeratin-18; m, month; miR, microRNA; MSC, mesenchymal stromal cell.
In patient 2, BAL was only performed twice because of concern of provoking significant bleeding secondary to severe thrombocytopenia. Changes over time in pro- or anti-inflammatory cytokines, such as were seen in patient 1, could not, therefore, be evaluated in the BAL fluid of patient 2 (Fig. 3A). Nonetheless, no significant increase in proinflammatory cytokines was observed in either BAL fluid or plasma after MSC administration (Fig. 3A, 3B).
Improvement in Markers of Lung Epithelial Cell Recovery
BAL fluid levels of total K18, indicative of total epithelial cell death, and ccK18, indicative of epithelial apoptosis [34], were elevated in both patients. ccK18 was elevated in patient 1 before MSC transfer and decreased, starting a few hours after infusion (Fig. 3C). Despite a transient elevation of ccK18 during the episode of nosocomial pneumonia in patient 1, a progressive decrease in both ccK18 and K18 occurred, suggesting no clear shift between epithelial apoptosis and necrosis. In both patients’ BAL fluids, surfactant protein B levels increased during the first 4 days after MSC administration, further suggesting recovery of alveolar epithelial integrity and function (Fig. 3D).
In Vivo MicroRNA Profiling of Blood Extracellular Vesicles
EVs, small membrane vesicles with a lipid bilayer containing a variety of substances, including inhibitory miRNAs, are secreted by nearly all cells and have been increasingly recognized for roles in cell-cell communication, particularly in the transfer of miRNAs [35]. In both patients, circulating levels of several proinflammatory miRNAs in circulating EVs (miR-409-3P, 886-5P, 324-3P, 222, 125A-5P, 339-3P, 155) that were elevated before MSC administration, demonstrated a significant decline within 24 hours after infusion (Fig. 3E). A transient increase in these miRNA levels was observed by day 7 in patient 1 and had returned to normal by day 28 (Fig. 3E). This coincided with the occurrence of nosocomial pneumonia. Although the proinflammatory miRNA trend was similar between the two patients, the absolute value of each miRNA was much lower in patient 2 than in patient 1. Other miRNAs related to inflammation (miR-638, 328, 26B, 29B, 30D, 27B) demonstrated a dichotomous pattern with an initial increase within the first hours to first day, followed by a decline and another peak by day 27 in patient 1 and day 14 in patient 2, returning to normal by day 28 in both patients (Fig. 3E).
MSC-Mediated Promotion of Regulatory Leukocytes In Vitro
One of the key features of ARDS is the accumulation of neutrophils (PMNs) in the lung microvasculature, interstitium, and alveolar space. In both patients, MSC administration was temporally associated with changes in circulating neutrophil counts. Although the BAL fluid total and differential cell counts could not be assessed in these two patients, we nonetheless believed it of value to assess the MSC effects on neutrophil behavior in vitro, including both survival and activation state. To exclude the indirect effects of other leukocyte populations on both MSC and PMN function, highly purified circulating third-party human PMN preparations were used, obtained as previously described [30].
To mimic in part the local inflammatory lung milieu, ex vivo-expanded MSCs were either unprimed (MSCs) or pretreated (primed MSCs, pMSCs) with 10 ng/ml IFN-γ and 15 ng/ml TNF-α for 48 hours and subsequently used in cocultures with PMNs with or without activation by endotoxin (100 ng/ml LPS) [36]. Compared with unprimed MSCs, primed MSCs upregulated cell surface expression of CD54 (ICAM-1), CD106 (VCAM-1), and HLA-ABC and HLA-DR (supplemental online Fig. 3) and the expression of IDO, a potent mediator of many MSC immune regulatory functions [37] (Fig. 4A). Subsequently, the viability and expression levels of surface markers by control or LPS-stimulated PMNs were investigated after 40 hours of direct coculture with either MSCs or pMSCs. As previously reported, CD16 (FcγR-III) expression can be reliably used as a surrogate marker of PMN viability [30]. As shown in Figure 4B, in the absence of LPS, PMN survival was enhanced only in presence of pMSCs. When LPS was added to the coculture, both resting and pMSCs protected PMNs from apoptosis. Accordingly, the percentage of CD16-positive PMNs matched the percentage of viable PMNs in all culture conditions (Fig. 4C). In parallel, the expression of CD11b and CD54 is typically associated with PMN activation status [38]. As expected, the percentage of CD11b-positive PMNs was higher in the presence of MSCs and further enhanced by LPS treatment (Fig. 4D), although the CD11b relative mean fluorescence intensity (rMFI) did not change significantly, suggesting that more PMNs are becoming activated after MSC or pMSC exposure. In contrast, CD54 rMFI was upregulated by LPS treatment, and this effect was enhanced by coculture with MSCs (Fig. 4E). Overall, the higher PMN survival and activation triggered by MSCs suggests that MSCs might influence the PMN-dependent innate response through functional modifications rather than proapoptotic effects.
Figure 4.

MSC-mediated immunomodulation of leukocytes. (A): In selected experiments, MSCs were exposed to IFN-γ and TNF-α for 48 hours (pMSCs). After priming, IDO expression was substantially upregulated as assessed by quantitative polymerase chain reaction compared with untreated MSCs. (B): Unstimulated MSCs and pMSCs affected viability of control or LPS-stimulated PMNs. (C): CD16 (FcγR-III) expression was used as a marker of PMN viability and matched with the percentage of viable PMNs in all culture conditions. Similarly, CD11b and CD54 expressions were associated with PMN activation status. The percentage of CD11b-positive PMNs was higher in the presence of MSCs and further enhanced by LPS treatment (D), and CD54 relative MFI was upregulated by LPS treatment, and this effect was enhanced by coculture with MSCs (E). The capacity of MSCs to promote the generation of myeloid-derived suppressor cells (MDSCs) was tested within our coculture model with PMNs. Coculture with MSCs led to a marked increase of mature CD11bright/CD16bright/CD62Ldim (N2-type) PMNs resembling granulocytic MDSCs (F). (G): MSC and PMN cocultures were stained with May-Grünwald-Giemsa dye to observe cell morphology after reciprocal interaction for 24 hours. According to the marked increase of mature CD11bright/CD16bright/CD62Ldim (N2-type) PMNs shown at this time by flow cytometry, PMNs displayed a hypersegmented nuclear morphology after coculture with both resting (PMN MSCs at 24 hours) and pMSCs (PMN pMSCs at 24 hours). Similarly, coculturing MSCs with healthy control PBMCs at different ratios promoted CD14+HLA-DRlow monocytes resembling monocytic MDSCs (H). MSCs were also capable of promoting the induction and expansion of immune suppressive CD4+CD25highCD127low TRegs, when cocultured in different ratios with purified healthy control T cells (I). (J): Increased levels of circulating CD4+CD25highCD127low TRegs were also observed up to 20 days after MSC administration (red, patient 1; black, patient 2). (K): This could in part have resulted from increased thymic output, indicated by an increased proportion of CD31+ RTEs among CD45RA+ naïve Tregs in both patients (red lines, patient 1; black lines, patient 2). Bars indicate the SEM. ∗, p ≤ .05; ∗∗, p ≤ .01; ∗∗∗, p ≤ .01. Abbreviations: IDO, indoleamine-2,3-dioxygenase; IFN-γ, interferon-γ; h, hour; LPS, lipopolysaccharide; MFI, mean fluorescence intensity; MSCs, mesenchymal stromal cells; PBMCs, peripheral blood mononuclear cells; PMNs, polymorphonuclear leukocytes; pMSCs, primed MSCs; RTEs, recent thymic emigrants; TNF-α, tumor necrosis factor-α; TRegs, regulatory T cells.
Emerging evidence has suggested that a heterogeneous subpopulation of PMNs and monocytes, termed myeloid-derived suppressor cells (MDSCs), exert immune regulatory effects and are often increased in inflammatory or inflammatory-like conditions such as cancer, where they are postulated to act as an intrinsic negative feedback mechanism [39–41]. Whether they play a role in ARDS has not yet been ascertained; however, preclinical data have pointed toward beneficial effects [42]. Coculturing PMNs with MSCs led to a marked increase in mature CD11bbright/CD16bright/CD62Ldim (N2-type) cells with hypersegmented nuclei, consistent with granulocytic MDSCs [41] (Fig. 4F, 4G). Similarly, coculturing MSCs with healthy control human PBMCs at different ratios promoted CD14+HLA-DRlow monocytes, resembling monocytic MDSCs (Fig. 4H; supplemental online Fig. 3A), as shown previously [43, 44].
The proportion of CD4+CD25highCD127low regulatory T cells (TRegs), a key immune regulatory cell population [45], was also expanded in coculture experiments with PBMCs (Fig. 4I; supplemental online Fig. 3B). In parallel, increased levels of circulating CD4+CD25highCD127low TRegs were observed in both patients’ peripheral blood for up to 20 days after MSC administration (TRegs among CD4+ T cells in healthy controls [n = 11], 3.84% ± 1.60%; TReg range in both patients, 3.34%–17.8%; Fig. 4J), similar to previous findings in MSC-treated patients with graft-vs.-host disease [46]. This finding could have resulted from an elevated thymic output, as indicated by the increased proportion of CD31+ recent thymic emigrants among CD45RA+ naïve TRegs in both patients (Fig. 4K) and/or the MSC-stimulated conversion of conventional T cells in the periphery (Fig. 4I; supplemental online Fig. 3B). The findings from animal models have suggested that recovery from acute lung injury involves upregulation of TRegs [47].
Proteome Characterization of Donor MSCs and Derived Extracellular Vesicles
The ability of the MSCs to ameliorate the diffuse inflammatory processes in ARDS likely results, at least in part, from the release of soluble mediators, including a variety of growth factors, anti-inflammatory cytokines and chemokines, and miRNAs, either directly secreted or delivered by EVs [48–50]. To determine the proteomic profile of the infused MSCs and corresponding EVs, cells were grown in serum-free media for 48 hours. Conditioned medium was collected, and the EVs were isolated by ultracentrifugation according to established protocols with minor changes [51]. Nanoparticle tracking analysis showed a typical EV distribution pattern with particles of a mode size of around 60 nm (supplemental online Fig. 4). Total protein lysates were generated on harvested cells and EVs followed by tryptic digestion into peptides. Digestion was analyzed using nano–reverse-phase chromatography gradient before mass spectrometry (nanoLC-MS/MS) analyses. The nanoLC-MS/MS analysis identified 3,040 proteins in the MSCs and 937 proteins in their EVs (1% FDR; supplemental online Table 1), with a total overlap of 754 proteins (Fig. 5A). The median number of peptide spectrum matches per protein was approximately 10 for cells and 8 for EVs (supplemental online Fig. 5A). Several proteins were only detected in EVs and not in the corresponding cells. This might reflect gradient separation limitations of the cell fraction, resulting in identification of the most abundant part of the proteome by nanoLC-MS/MS, with the potentially less abundant proteins that are packed into EVs missed in cell analyses. A relatively poor correlation of protein abundance was observed between EVs and cells. This indicates that EVs are not merely formed by lysed cells but rather result from selective protein packing into vesicles, corroborating previous reports on EVs derived from other cell types [48] (Fig. 5B; supplemental online Fig. 5B).
Figure 5.

Proteome characterization of donor MSCs and donor-derived EVs. (A): Overlap in protein identification between MSC donor cells and EVs. Proteins were considered identified if they had a quantifiable protein area. (B): Correlation of protein abundance between MSC donor cells and EVs. Protein abundance was estimated by label-free quantification using protein signal intensity (area under peak of identified peptides/protein). (C): Pie chart of Gene Ontology (GO) major biological process categories in MSC donor EVs and donor cells. Percentages were based on GO assignment. (D): Selected GO enrichment terms for donor cells (WCs) and EVs. All enriched GO terms can be found in supplemental online Figure 6A–6E. Abbreviations: EVs, extracellular vesicles; MSCs, mesenchymal stromal cells; WCs, whole cells.
To delineate the potential functions of the identified proteins, GO enrichment analysis was performed using the on-line PANTHER classification system [52]. An overview of the percentage of GO biological processes in donor cells and EVs is illustrated in Figure 5C. Examples of significantly enriched GO categories in EVs and/or cells (p < .001) include metabolic processes such as the tricarboxylic acid cycle and glycolysis pathway and biological processes and pathways such as adhesion and integrin signaling (Fig. 5D). Additional GO enrichment data on biological processes, molecular function, cellular component, protein class, and pathways can be found in the supplemental online data (supplemental online Fig. 6A–6E).
Discussion
We have described the administration of non-HLA-matched allogeneic bone marrow-derived MSCs to 2 consecutive patients with severe refractory ARDS of different underlying etiologies under compassionate use. In both instances, clinical improvement was observed, with both patients achieving discharge from the ICU and hospital. Although causality can only be speculated in this small case series, the clinical outcomes paralleled those observed after MSC administration in a number of preclinical models of acute lung injury [7, 12–15, 19–21]. After MSC infusion, both patients demonstrated improved lung function, including tidal volumes and compliance, and CXRs with concomitant overall progressive clinical improvement. These are provocative clinical observations in patients with severe refractory ARDS, providing additional impetus for larger formal clinical investigations such as the recently published NIH-sponsored phase I trial that demonstrated the safety of systemic administration of allogeneic bone marrow-derived MSCs in 9 patients with moderate to severe ARDS and that has since been expanded to a phase II clinical trial [53, 54].
At the time of MSC infusion, the first patient was critically ill with hemodynamic and respiratory instability and multiorgan failure. Within several hours after MSC administration, with no other supportive measures changed, including no changes in the ECMO or PEEP settings as per the standard clinical protocol, a number of clinical and laboratory parameters improved. This trend continued, with subsequent discontinuation of supportive therapies, resolution of multiorgan failure, discharge from the ICU and, eventually, from the hospital, and a return to excellent functional status. A transient setback with nosocomial (ventilator-associated) pneumonia occurred several days after MSC administration but responded to appropriate antimicrobial treatment. Whether MSC administration increases the risk of infectious complications in ARDS must be assessed in future investigations; however, no such increase has been reported in clinical trials of MSCs given to severely immunoincompetent recipients [22, 27]. In contrast, the findings from a number of recent preclinical studies suggest that MSCs have antimicrobial effects [55–57]. The second patient mainly had severe respiratory failure but nonetheless demonstrated an improvement in clinical status after MSC administration, with subsequent discontinuation of supportive therapies and discharge from the ICU. Similar trends in inflammatory indices were observed, although the chemotherapy-induced bone marrow suppression likely altered the inflammatory pathways, obscuring interpretations of possible in vivo MSC effects.
The range of improvements in the in vivo inflammatory indices was broad, with decreases in multiple markers of inflammation, including markers of epithelial apoptosis, alveolar-capillary fluid leakage, and proinflammatory cytokines, miRNAs, and chemokines in plasma and BAL fluid. These results demonstrated that ongoing inflammation was still present long after the initial inciting events that had provoked the development of ARDS. Each of these inflammatory outcomes reflects altered inflammatory and immune pathways that play a role in the pathogenesis or perpetuation of ARDS. Furthermore, as a sign of recovery of alveolar epithelial function, the surfactant protein B levels increased in both patients’ BAL fluid after MSC administration. This was more pronounced in patient 1 and could potentially be explained by the difference in clinical status at the time of MSC administration. The validity and relevance of the changes in inflammatory indices are suggested by the subsequent development of nosocomial pneumonia in patient 1, with correlating increases in the inflammatory markers that had decreased after MSC administration. Nonetheless, because this was a descriptive hypothesis-generating study, it would be difficult to know what is relevant with respect to measures of inflammatory mediators. Only further clinical and preclinical studies investigating the roles of specific mediators potentially involved in the pathogenesis and/or resolution of ARDS can answer this question.
The dose used in these patients was based on previous use of MSCs in GVHD patients [27]. Until the recently published phase I trial, a paucity of information was available concerning the appropriate dosing in ARDS patients; thus, the dosing used in these 3 patients was an empiric application. The study of systemic MSC administration in patients with chronic obstructive pulmonary disease also used an empiric approach for multiple dosing [24]. However, those were stable outpatients, not critically and acutely ill patients. Moreover, the preclinical studies investigating use of MSCs in models of acute lung injury have virtually all shown beneficial effects after a single dose or administration. As such, multiple dosing of the administration of MSCs was not considered.
The lack of understanding of MSCs’ actions in vivo [26] and the lack of corresponding methods for evaluating their functional quality before application have limited the clinical application of MSCs. Previous investigations of patients receiving allogeneic MSC treatment have suggested that directly on contact with whole blood, such as by i.v. infusion, the cells elicit activation of complement and coagulation cascades (a reaction termed “instant blood-mediated reaction” [IBMIR]) that results in destruction of most of the infused cells and virtually no cell engraftment [58–60]. However, skewing toward a tolerogenic Th2 T-cell repertoire occurs and is most evident in patients as late as 20–30 days after infusion [46, 61]. The events subsequent to the instant IBMIR are unknown in humans; however, previous clinical studies have indicated that in vitro assays can correlate with ex vivo observations and potentially explain MSCs’ clinical efficacy [46]. Accordingly, parallel in vitro studies of the MSCs used in our two patients demonstrated anti-inflammatory actions on relevant inflammatory and immune cells, in particular, monocytes and neutrophils, which play significant roles in ARDS pathogenesis or resolution. Although we did not specifically assess this in vivo in either BAL fluid or serum, promotion of the anti-inflammatory M2 monocyte (macrophage) phenotype is relevant for the treatment of ARDS. The role of TRegs is less clear in ARDS compared with monocytes/macrophages and PMNs, although some data have suggested a role in experimental models of acute lung injury [62]. Nonetheless, the correlation between the induction of TRegs in vitro and increased levels of circulating TRegs in vivo is striking. The role of PMNs in ARDS is complex, and it is unclear at present whether the promotion of PMN survival, activation, and/or increase in MDSC subsets will indicate beneficial clinical effects. Nevertheless, the marked increase in mature CD11bbright/CD16bright/CD62Ldim (N2-type) cells with hypersegmented nuclei found by coculturing PMNs with MSCs is in agreement with published data showing that the onset of granulocytic MDSCs is associated with such an immunological phenotype, which might be considered a surrogate pattern of PMN functional polarization.
As such, the in vitro findings are hypothesis generating and need to be extensively explored in future studies of a larger number of patients with ARDS of diverse etiologies. This also raises an overall consideration regarding whether the decrease in any latent inflammation present in severe persistent ARDS would be beneficial or harmful. A growing body of evidence suggests that failure of resolution of inflammation is an important pathogenic mechanism underlying ARDS [63]. It is perhaps in this regard in which the anti-inflammatory actions of MSCs would be beneficial. Further preclinical and clinical studies of MSCs are needed to fully comprehend the potential role of the timing of MSC administration in ARDS.
In the in vitro experiments, we mimicked in part an inflammatory milieu by pretreating the ex vivo expanded MSCs with IFN-γ and TNF-α. This is an established technique that can be performed to assess MSCs’ immune modulatory potency. To this aim, the MSC Committee of the International Society for Cellular Therapy recently published a working proposal to highlight the need for standardized in vitro methods to support the various MSC-based clinical approaches used to treat immunological diseases [64]. The standard immune plasticity assay based on IFN-γ, with or without TNF-α, is a good model of in vitro priming to assess the immune regulatory properties of MSCs. The use of serum from patients does not meet these criteria for in vitro studies owing to the high variability of the content of biological active molecules secondary to a large number of factors (e.g., comorbidities, treatments, sample collection methods).
None of the standardized in vitro tests, when used alone, can be considered the perfect surrogate and biomarker of in vivo behavior of MSCs. Nevertheless, the combination of different in vitro standardized tests can be useful to decipher the clinical results. Furthermore, the range of biologic mediators present in the inflammatory lung/ARDS environment that might alter or license MSC biology is not yet well understood. A recent study has demonstrated that exposure of MSCs to serum from ARDS patients altered the expression of a range of potential anti-inflammatory mediators and improved efficacy of MSC actions when administered to mice with endotoxin-induced acute lung injury [65]. This suggests that licensing or priming of the MSCs with a standardized set of inflammatory cytokines or perhaps even specific patient serum or BAL fluid before administration might be beneficial in ARDS. This offers new opportunities to investigate and correlate disease-specific in vitro potency measures with clinical actions.
Many of the therapeutic effects of MSCs in ARDS are likely mediated by the release of soluble proteins and EVs containing a range of bioactive macromolecules. Some of these proteins and pathways were identified in the MSCs and EVs that have been suggested as potentially associated with the therapeutic effects in other disease models [66]. This includes pathways associated with platelet-derived growth factor receptor and epidermal growth factor receptor activation, which have been implicated in MSC recruitment [66–68], cell adhesion molecules [69–72], and glycolysis and tricarboxic acid cycle-related proteins and proteins involved in translation and ribosomal components. Although the relevance of these pathways to ARDS is not clear, enrichment of the latter proteins could conceivably complement an energy deficit in the ARDS-injured lung, critical in the restoration of normal surfactant production [73]. Just as with the in vitro MSC effects on the inflammatory and immune cells, these initial findings and potential correlations with the in vivo observations are hypothesis generating and need to be further explored in larger numbers of patients. This baseline characterization of successfully used MSCs and their vesicle components is also important for understanding the molecular determinants of functional cell batches and, in the future, to define biomarkers to select MSCs for clinical use in ARDS.
Our findings contrast with those observed in a recent study of the administration of non-HLA-matched allogeneic adipose-derived MSCs in ARDS patients for whom, although short-term improvement in oxygenation occurred after MSC administration, no improvement in ventilator-free days, ICU-free days, or length of hospital stay was observed [74]. Limited measures of inflammatory outcomes were obtained but demonstrated no significant changes in the circulating levels of IL-6, IL-8, or surfactant protein D (SP-D) after MSC administration. Whether this reflects differences in the adipose vs. bone marrow origin of the MSCs or differences in other aspects of the respective protocols used remains to be determined. To investigate the cytokine response after MSCs infusion in ARDS patients, larger cohorts are needed, as well as an expanded range of inflammatory mediators studied. For example, circulating C-reactive protein was not assessed in the present study. The MSCs used in our study were directly infused after thawing, and the adipose MSCs were thawed and cultured for 24–48 hours in each respective patient’s own serum before infusion. The use of freshly thawed vs. cultured MSCs has been shown to result in different effects in the in vitro mixed lymphocyte reactions [75]; however, it is as yet unclear how these different approaches will affect the potential use of MSCs in ARDS.
Conclusion
Our overall results are congruent with those observed in preclinical models and suggest that MSCs might have clinical efficacy in severe refractory ARDS and, perhaps, ARDS resulting from a wider spectrum of etiologies than those in the two patients in our study. These case studies also highlight the wealth of mechanistic and hypothesis-generating information that can be obtained, even from a few patients, and the critical need to incorporate mechanistic studies and in vitro potency assessments as a part of any clinical study of MSCs in ARDS, as well as in other diseases.
Supplementary Material
Acknowledgments
We thank Per Vikholm and the intensive care unit nurses at the Department of Cardiothoracic Surgery, Uppsala University Hospital, Uppsala, Sweden, for their outstanding work in the care of the patients and for helping us with the study. We also thank Helena Lönnies and Maritha Marcusson Ståhl at the Center of Hematology and Regenerative Medicine, Department of Medicine, Karolinska Institutet, for their help with the preparation of the MSCs and sample analysis. The Cancer Society of Stockholm, the Children’s Cancer Foundation, Karolinska Institutet, Stockholm City Council, the Swedish Cancer Society, the Swedish Research Council, the Swedish Society of Medicine, the Tobias Foundation, VINNOVA, and The Swedish Heart and Lung Foundation supported this study.
Author Contributions
O.E.S., K.-H.G., K.L.B.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, clinical care of patients; D.M., N.H., M.K., D.J.W.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; M.D., L.H.-J., P.S., P.-O.J., H.H., M.M.: collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, clinical care of patients; G.B., H.J.J., R.J., S.R., M.C., S.E.A., O.P.B.W., J.Z.N., J.S., C.R., T.K., J.L., O.R.F., R.S., O.K.: collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
O.E.S., M.C., and K.-H.G. are cofounders and owners of Islet One AB, have uncompensated employment, and have compensated intellectual property rights and ownership interest. S.R. is a minority co-owner of Biolamina and has compensated intellectual property rights and ownership interest. J.S. is founder and co-owner of Exosome Diagnostics and has compensated employment, intellectual property rights, and ownership interest. C.R. and T.K. are compensated employees of Exosome Diagnostics. D.J.W. has compensated research funding from Athersys Inc. and United Therapuetics. The other authors indicated no potential conflicts of interest.
References
- 1.Rincon F, Ghosh S, Dey S, et al. Impact of acute lung injury and acute respiratory distress syndrome after traumatic brain injury in the United States. Neurosurgery. 2012;71:795–803. doi: 10.1227/NEU.0b013e3182672ae5. [DOI] [PubMed] [Google Scholar]
- 2.Angus DC, Clermont G, Linde-Zwirble WT, et al. Healthcare costs and long-term outcomes after acute respiratory distress syndrome: A phase III trial of inhaled nitric oxide. Crit Care Med. 2006;34:2883–2890. doi: 10.1097/01.CCM.0000248727.29055.25. [DOI] [PubMed] [Google Scholar]
- 3.Walkey AJ, Summer R, Ho V, et al. Acute respiratory distress syndrome: epidemiology and management approaches. Clin Epidemiol. 2012;4:159–169. doi: 10.2147/CLEP.S28800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zampieri FG, Mendes PV, Ranzani OT, et al. Extracorporeal membrane oxygenation for severe respiratory failure in adult patients: A systematic review and meta-analysis of current evidence. J Crit Care. 2013;28:998–1005. doi: 10.1016/j.jcrc.2013.07.047. [DOI] [PubMed] [Google Scholar]
- 5.Wilson JG, Matthay MA. Mechanical ventilation in acute hypoxemic respiratory failure: A review of new strategies for the practicing hospitalist. J Hosp Med. 2014;9:469–475. doi: 10.1002/jhm.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 7.Lee JW, Zhu Y, Matthay MA. Cell-based therapy for acute lung injury: Are we there yet? Anesthesiology. 2012;116:1189–1191. doi: 10.1097/ALN.0b013e3182567fc5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss DJ. Concise review: Current status of stem cells and regenerative medicine in lung biology and diseases. Stem Cells. 2014;32:16–25. doi: 10.1002/stem.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toonkel RL, Hare JM, Matthay MA, et al. Mesenchymal stem cells and idiopathic pulmonary fibrosis: Potential for clinical testing. Am J Respir Crit Care Med. 2013;188:133–140. doi: 10.1164/rccm.201207-1204PP. [DOI] [PubMed] [Google Scholar]
- 10.Lee JW, Fang X, Gupta N, et al. Allogeneic human mesenchymal stem cells for treatment of E. coli endotoxin-induced acute lung injury in the ex vivo perfused human lung. Proc Natl Acad Sci USA. 2009;106:16357–16362. doi: 10.1073/pnas.0907996106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curley GF, Ansari B, Hayes M, et al. Effects of intratracheal mesenchymal stromal cell therapy during recovery and resolution after ventilator-induced lung injury. Anesthesiology. 2013;118:924–932. doi: 10.1097/ALN.0b013e318287ba08. [DOI] [PubMed] [Google Scholar]
- 12.Mei SH, McCarter SD, Deng Y, et al. Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Med. 2007;4:e269. doi: 10.1371/journal.pmed.0040269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu J, Qu J, Cao L, et al. Mesenchymal stem cell-based angiopoietin-1 gene therapy for acute lung injury induced by lipopolysaccharide in mice. J Pathol. 2008;214:472–481. doi: 10.1002/path.2302. [DOI] [PubMed] [Google Scholar]
- 14.Iyer SS, Torres-Gonzalez E, Neujahr DC, et al. Effect of bone marrow-derived mesenchymal stem cells on endotoxin-induced oxidation of plasma cysteine and glutathione in mice. Stem Cells Int. 2010;2010:868076. doi: 10.4061/2010/868076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Danchuk S, Ylostalo JH, Hossain F, et al. Human multipotent stromal cells attenuate lipopolysaccharide-induced acute lung injury in mice via secretion of tumor necrosis factor-α-induced protein 6. Stem Cell Res Ther. 2011;2:27. doi: 10.1186/scrt68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mei SH, Haitsma JJ, Dos Santos CC, et al. Mesenchymal stem cells reduce inflammation while enhancing bacterial clearance and improving survival in sepsis. Am J Respir Crit Care Med. 2010;182:1047–1057. doi: 10.1164/rccm.201001-0010OC. [DOI] [PubMed] [Google Scholar]
- 17.Gupta N, Su X, Popov B, et al. Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J Immunol. 2007;179:1855–1863. doi: 10.4049/jimmunol.179.3.1855. [DOI] [PubMed] [Google Scholar]
- 18.Vallabhaneni KC, Haller H, Dumler I. Vascular smooth muscle cells initiate proliferation of mesenchymal stem cells by mitochondrial transfer via tunneling nanotubes. Stem Cells Dev. 2012;21:3104–3113. doi: 10.1089/scd.2011.0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kordelas L, Rebmann V, Ludwig AK, et al. MSC-derived exosomes: A novel tool to treat therapy-refractory graft-versus-host disease. Leukemia. 2014;28:970–973. doi: 10.1038/leu.2014.41. [DOI] [PubMed] [Google Scholar]
- 20.Lai RC, Arslan F, Lee MM, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res (Amst) 2010;4:214–222. doi: 10.1016/j.scr.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Xie RL, Gordon J, et al. Control of mesenchymal lineage progression by microRNAs targeting skeletal gene regulators Trps1 and Runx2. J Biol Chem. 2012;287:21926–21935. doi: 10.1074/jbc.M112.340398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Bahr L, Sundberg B, Lonnies L, et al. Long-term complications, immunologic effects, and role of passage for outcome in mesenchymal stromal cell therapy. Biol Blood Marrow Transplant. 2012;18:557–564. doi: 10.1016/j.bbmt.2011.07.023. [DOI] [PubMed] [Google Scholar]
- 23.Lalu MM, McIntyre L, Pugliese C, et al. Safety of cell therapy with mesenchymal stromal cells (SafeCell): A systematic review and meta-analysis of clinical trials. PLoS One. 2012;7:e47559. doi: 10.1371/journal.pone.0047559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiss DJ, Casaburi R, Flannery R, et al. A placebo-controlled, randomized trial of mesenchymal stem cells in COPD. Chest. 2013;143:1590–1598. doi: 10.1378/chest.12-2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson JG, Liu KD, Zhuo H, et al. Mesenchymal stem (stromal) cells for treatment of ARDS: A phase 1 clinical trial. Lancet Respir Med. 2015;3:24–32. doi: 10.1016/S2213-2600(14)70291-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bianco P, Cao X, Frenette PS, et al. The meaning, the sense and the significance: Translating the science of mesenchymal stem cells into medicine. Nat Med. 2013;19:35–42. doi: 10.1038/nm.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le Blanc K, Frassoni F, Ball L, et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: A phase II study. Lancet. 2008;371:1579–1586. doi: 10.1016/S0140-6736(08)60690-X. [DOI] [PubMed] [Google Scholar]
- 28.Schaefer A, Jung M, Miller K, et al. Suitable reference genes for relative quantification of miRNA expression in prostate cancer. Exp Mol Med. 2010;42:749–758. doi: 10.3858/emm.2010.42.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mougiakakos D, Jitschin R, Johansson CC, et al. The impact of inflammatory licensing on heme oxygenase-1-mediated induction of regulatory T cells by human mesenchymal stem cells. Blood. 2011;117:4826–4835. doi: 10.1182/blood-2010-12-324038. [DOI] [PubMed] [Google Scholar]
- 30.Cassatella MA, Mosna F, Micheletti A, et al. Toll-like receptor-3-activated human mesenchymal stromal cells significantly prolong the survival and function of neutrophils. Stem Cells. 2011;29:1001–1011. doi: 10.1002/stem.651. [DOI] [PubMed] [Google Scholar]
- 31.Dragovic RA, Gardiner C, Brooks AS, et al. Sizing and phenotyping of cellular vesicles using nanoparticle tracking analysis. Nanomedicine (Lond) 2011;7:780–788. doi: 10.1016/j.nano.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ranieri VM, Rubenfeld GD, Thompson BT, et al. Acute respiratory distress syndrome: The Berlin definition. JAMA. 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 34.Luft T, Conzelmann M, Benner A, et al. Serum cytokeratin-18 fragments as quantitative markers of epithelial apoptosis in liver and intestinal graft-versus-host disease. Blood. 2007;110:4535–4542. doi: 10.1182/blood-2006-10-049817. [DOI] [PubMed] [Google Scholar]
- 35.Collino F, Deregibus MC, Bruno S, et al. Microvesicles derived from adult human bone marrow and tissue specific mesenchymal stem cells shuttle selected pattern of miRNAs. PLoS One. 2010;5:e11803. doi: 10.1371/journal.pone.0011803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krampera M. Mesenchymal stromal cell “licensing”: A multistep process. Leukemia. 2011;25:1408–1414. doi: 10.1038/leu.2011.108. [DOI] [PubMed] [Google Scholar]
- 37.Francois M, Romieu-Mourez R, Li M, et al. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol Ther. 2012;20:187–195. doi: 10.1038/mt.2011.189. [DOI] [PubMed] [Google Scholar]
- 38.Pelletier M, Micheletti A, Cassatella MA. Modulation of human neutrophil survival and antigen expression by activated CD4+ and CD8+ T cells. J Leukoc Biol. 2010;88:1163–1170. doi: 10.1189/jlb.0310172. [DOI] [PubMed] [Google Scholar]
- 39.Mougiakakos D, Jitschin R, von Bahr L, et al. Immunosuppressive CD14+HLA-DRlow/neg IDO+ myeloid cells in patients following allogeneic hematopoietic stem cell transplantation. Leukemia. 2013;27:377–388. doi: 10.1038/leu.2012.215. [DOI] [PubMed] [Google Scholar]
- 40.Lança T, Silva-Santos B. The split nature of tumor-infiltrating leukocytes: Implications for cancer surveillance and immunotherapy. OncoImmunology. 2012;1:717–725. doi: 10.4161/onci.20068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pillay J, Kamp VM, van Hoffen E, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest. 2012;122:327–336. doi: 10.1172/JCI57990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poe SL, Arora M, Oriss TB, et al. STAT1-regulated lung MDSC-like cells produce IL-10 and efferocytose apoptotic neutrophils with relevance in resolution of bacterial pneumonia. Mucosal Immunol. 2013;6:189–199. doi: 10.1038/mi.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bernardo ME, Fibbe WE. Mesenchymal stromal cells: Sensors and switchers of inflammation. Cell Stem Cell. 2013;13:392–402. doi: 10.1016/j.stem.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 44.Chen HW, Chen HY, Wang LT, et al. Mesenchymal stem cells tune the development of monocyte-derived dendritic cells toward a myeloid-derived suppressive phenotype through growth-regulated oncogene chemokines. J Immunol. 2013;190:5065–5077. doi: 10.4049/jimmunol.1202775. [DOI] [PubMed] [Google Scholar]
- 45.Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: A jack of all trades, master of regulation. Nat Immunol. 2008;9:239–244. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jitschin R, Mougiakakos D, Von Bahr L, et al. Alterations in the cellular immune compartment of patients treated with third-party mesenchymal stromal cells following allogeneic hematopoietic stem cell transplantation. Stem Cells. 2013;31:1715–1725. doi: 10.1002/stem.1386. [DOI] [PubMed] [Google Scholar]
- 47.Pietropaoli A, Georas SN. Resolving lung injury: A new role for Tregs in controlling the innate immune response. J Clin Invest. 2009;119:2891–2894. doi: 10.1172/JCI40880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.EL Andaloussi S, Mäger I, Breakefield XO, et al. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12:347–357. doi: 10.1038/nrd3978. [DOI] [PubMed] [Google Scholar]
- 49.Yu B, Zhang X, Li X. Exosomes derived from mesenchymal stem cells. Int J Mol Sci. 2014;15:4142–4157. doi: 10.3390/ijms15034142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu YG, Feng XM, Abbott J, et al. Human mesenchymal stem cell microvesicles for treatment of Escherichia coli endotoxin-induced acute lung injury in mice. Stem Cells. 2014;32:116–125. doi: 10.1002/stem.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 52.Blake JA, Dolan M, Drabkin H, et al. Gene Ontology annotations and resources. Nucleic Acids Res. 2013;41:D530–D535. doi: 10.1093/nar/gks1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matthay MA. Human mesenchymal stem cells for acute respiratory distress syndrome (START) NCT01775774. Bethesda, MD: U.S. National Library of Medicine, 2013. Available at https://clinicaltrials.gov/ct2/show/NCT01775774.
- 54.Matthay MA. Human mesenchymal stem cells for acute respiratory distress syndrome (START), NCT0209764. Bethesda, MD: U.S. National Library of Medicine, 2014. Available at https://clinicaltrials.gov/ct2/show/NCT02097641.
- 55.Gupta N, Krasnodembskaya A, Kapetanaki M, et al. Mesenchymal stem cells enhance survival and bacterial clearance in murine Escherichia coli pneumonia. Thorax. 2012;67:533–539. doi: 10.1136/thoraxjnl-2011-201176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krasnodembskaya A, Song Y, Fang X, et al. Antibacterial effect of human mesenchymal stem cells is mediated in part from secretion of the antimicrobial peptide LL-37. Stem Cells. 2010;28:2229–2238. doi: 10.1002/stem.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krasnodembskaya A, Samarani G, Song Y, et al. Human mesenchymal stem cells reduce mortality and bacteremia in gram-negative sepsis in mice in part by enhancing the phagocytic activity of blood monocytes. Am J Physiol Lung Cell Mol Physiol. 2012;302:L1003–L1013. doi: 10.1152/ajplung.00180.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moll G, Jitschin R, von Bahr L, et al. Mesenchymal stromal cells engage complement and complement receptor bearing innate effector cells to modulate immune responses. PLoS One. 2011;6:e21703. doi: 10.1371/journal.pone.0021703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moll G, Rasmusson-Duprez I, von Bahr L, et al. Are therapeutic human mesenchymal stromal cells compatible with human blood? Stem Cells. 2012;30:1565–1574. doi: 10.1002/stem.1111. [DOI] [PubMed] [Google Scholar]
- 60.von Bahr L, Batsis I, Moll G, et al. Analysis of tissues following mesenchymal stromal cell therapy in humans indicates limited long-term engraftment and no ectopic tissue formation. Stem Cells. 2012;30:1575–1578. doi: 10.1002/stem.1118. [DOI] [PubMed] [Google Scholar]
- 61.Mougiakakos D, Machaczka M, Jitschin R, et al. Treatment of familial hemophagocytic lymphohistiocytosis with third-party mesenchymal stromal cells. Stem Cells Dev. 2012;21:3147–3151. doi: 10.1089/scd.2012.0214. [DOI] [PubMed] [Google Scholar]
- 62.Sun J, Han ZB, Liao W, et al. Intrapulmonary delivery of human umbilical cord mesenchymal stem cells attenuates acute lung injury by expanding CD4+CD25+ Forkhead Boxp3 (FOXP3)+ regulatory T cells and balancing anti- and pro-inflammatory factors. Cell Physiol Biochem. 2011;27:587–596. doi: 10.1159/000329980. [DOI] [PubMed] [Google Scholar]
- 63.Levy BD, Serhan CN. Resolution of acute inflammation in the lung. Annu Rev Physiol. 2014;76:467–492. doi: 10.1146/annurev-physiol-021113-170408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krampera M, Galipeau J, Shi Y, et al. Immunological characterization of multipotent mesenchymal stromal cells—The International Society for Cellular Therapy (ISCT) working proposal. Cytotherapy. 2013;15:1054–1061. doi: 10.1016/j.jcyt.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 65.Bustos ML, Huleihel L, Meyer EM, et al. Activation of human mesenchymal stem cells impacts their therapeutic abilities in lung injury by increasing interleukin (IL)-10 and IL-1RN levels. Stem Cells Translational Medicine. 2013;2:884–895. doi: 10.5966/sctm.2013-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim HS, Choi DY, Yun SJ, et al. Proteomic analysis of microvesicles derived from human mesenchymal stem cells. J Proteome Res. 2012;11:839–849. doi: 10.1021/pr200682z. [DOI] [PubMed] [Google Scholar]
- 67.Fiedler J, Etzel N, Brenner RE. To go or not to go: Migration of human mesenchymal progenitor cells stimulated by isoforms of PDGF. J Cell Biochem. 2004;93:990–998. doi: 10.1002/jcb.20219. [DOI] [PubMed] [Google Scholar]
- 68.Ball SG, Shuttleworth CA, Kielty CM. Vascular endothelial growth factor can signal through platelet-derived growth factor receptors. J Cell Biol. 2007;177:489–500. doi: 10.1083/jcb.200608093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Veevers-Lowe J, Ball SG, Shuttleworth A, et al. Mesenchymal stem cell migration is regulated by fibronectin through α5β1-integrin-mediated activation of PDGFR-β and potentiation of growth factor signals. J Cell Sci. 2011;124:1288–1300. doi: 10.1242/jcs.076935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.He J, Baum LG. Galectin interactions with extracellular matrix and effects on cellular function. Methods Enzymol. 2006;417:247–256. doi: 10.1016/S0076-6879(06)17017-2. [DOI] [PubMed] [Google Scholar]
- 71.Chan J, O’Donoghue K, Gavina M, et al. Galectin-1 induces skeletal muscle differentiation in human fetal mesenchymal stem cells and increases muscle regeneration. Stem Cells. 2006;24:1879–1891. doi: 10.1634/stemcells.2005-0564. [DOI] [PubMed] [Google Scholar]
- 72.Kishore R, Qin G, Luedemann C, et al. The cytoskeletal protein ezrin regulates EC proliferation and angiogenesis via TNF-alpha-induced transcriptional repression of cyclin A. J Clin Invest. 2005;115:1785–1796. doi: 10.1172/JCI22849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eckle T, Brodsky K, Bonney M, et al. HIF1A reduces acute lung injury by optimizing carbohydrate metabolism in the alveolar epithelium. PLoS Biol. 2013;11:e1001665. doi: 10.1371/journal.pbio.1001665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zheng G, Huang L, Tong H, et al. Treatment of acute respiratory distress syndrome with allogeneic adipose-derived mesenchymal stem cells: A randomized, placebo-controlled pilot study. Respir Res. 2014;15:39. doi: 10.1186/1465-9921-15-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.François M, Copland IB, Yuan S, et al. Cryopreserved mesenchymal stromal cells display impaired immunosuppressive properties as a result of heat-shock response and impaired interferon-γ licensing. Cytotherapy. 2012;14:147–152. doi: 10.3109/14653249.2011.623691. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.