

Summary
The present work describes the chemopreventive activities of combinations of indole-3-carbinol (I3C) and silibinin (Sil) against inflammation-driven lung tumorigenesis induced by the tobacco smoke carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and enhanced by lipopolysaccharide (LPS), a potent inflammatory agent and a constituent of tobacco smoke.
Abstract
Chronic inflammation is an important risk factor for lung cancer. Therefore, identification of chemopreventive agents that suppress inflammation-driven lung cancer is indispensable. We studied the efficacy of combinations of indole-3-carbinol (I3C) and silibinin (Sil), 20 µmol/g diet each, against mouse lung tumors induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and driven by lipopolysaccharide (LPS), a potent inflammatory agent and constituent of tobacco smoke. Mice treated with NNK + LPS developed 14.7±4.1 lung tumors/mouse, whereas mice treated with NNK + LPS and given combinations of I3C and Sil had 7.1±4.5 lung tumors/mouse, corresponding to a significant reduction of 52%. Moreover, the number of largest tumors (>1.0mm) was significantly reduced from 6.3±2.9 lung tumors/mouse in the control group to 1.0±1.3 and 1.6±1.8 lung tumors/mouse in mice given I3C + Sil and I3C alone, respectively. These results were paralleled by significant reductions in the level of proinflammatory and procarcinogenic proteins (pSTAT3, pIκBα and COX-2) and proteins that regulate cell proliferation (pAkt, cyclin D1, CDKs 2, 4, 6 and pRB). Further studies in premalignant bronchial cells showed that the antiproliferative effects of I3C + Sil were higher than the individual compounds and these effects were mediated by targeting cyclin D1, CDKs 2, 4 and 6 and pRB. I3C + Sil suppressed cyclin D1 by reducing its messenger RNA level and by enhancing its proteasomal degradation. Our results showed the potential lung cancer chemopreventive effects of I3C + Sil in smokers/former smokers with chronic pulmonary inflammatory conditions.
Introduction
Lung cancer is the major cause of cancer-related death in the USA and the world at large (1). Despite significant advances in the diagnosis and therapy of lung cancer during the last decades, the 5 year survival rate of lung cancer patients is still under 20%. One promising approach to reduce the mortality rate of lung cancer is the use of chemopreventive agents in high risk populations. Although tobacco smoke accounts for ~90% of lung cancer, only 10–15% of smokers develop lung cancer. It is not clear why some smokers develop lung cancer, whereas most smokers do not. Epidemiological studies have identified additional risk factors that could increase lung cancer incidence among smokers. For instance, smokers with chronic obstructive pulmonary disease (COPD), the single most important risk factor for lung cancer after smoking, have been found to have a 4- to 6-fold higher risk of developing lung cancer compared with smokers without COPD (2,3). Therefore, for efficient chemoprevention of lung cancer, it is critical to target both tobacco-carcinogen-induced cellular alterations and pulmonary inflammation.
Owing to their time-tested safety, dietary agents and medicinal plants are the most ideal candidates for cancer chemoprevention. Among these compounds are indole-3-carbinol (I3C), the breakdown product of glucobrassicins, which are found at high concentrations in commonly consumed Brassica vegetables, and silibinin (Sil), a constituent of Silybum marianum (milk thistle) used commonly for the treatment of liver diseases. Consistent reports from preclinical studies have shown that both I3C and Sil suppress the development of cancer in several organs, including lung cancer, and possess anti-inflammatory effects (4,5). In the present study, we assessed the chemopreventive efficacies of I3C and Sil, alone or in combination, against chronic inflammation-driven mouse lung tumorigenesis. For this, first, mice were treated with the tobacco smoke carcinogen 4-(methylnitro-samino)-1-(3-pyridyl)-1-butanone (NNK). Beginning 1 week after carcinogen treatment until termination of the study, mice were treated with the inflammatory agent lipopolysaccharide (LPS), which is contained in substantial amounts in mainstream and sidestream cigarette smoke (6,7), and received I3C and/or Sil in the diet.
Materials and methods
Chemicals
I3C and Sil were purchased from LKT (Minneapolis, MN) and Sigma (St Louis, MO), respectively. NNK was synthesized as described elsewhere (8). Anti-phospho-STAT3, anti-total STAT3, anti-phospho-Akt, anti-total Akt, anti-cyclin D1, anti-CDK2, anti-CDK4, anti-CDK6, anti-Mcl-1, anti-phospho-IκBα, anti-total IκBα, anti-Cox-2, anti-phospho-RB and goat anti-rabbit IgG or mouse IgG secondary antibody were acquired from Cell Signaling Technology (Beverly, MA). Anti-p21 and anti-p27 were purchased from Santa Cruz Biotechnology (Dallas, TX). Mouse diets (AIN-93G and AIN-93M) were purchased from Harlan Teklad (Madison, WI). These diets are standard diets for lung tumorigenesis studies in A/J mice. The AIN-93G diet, high in protein and fat, was used to support rapid growth of the mice until 8 weeks of age. AIN-93G diet was then replaced by AIN-93M diet, a low-protein and low-fat diet, which is recommended for adult maintenance.
Tumor bioassay
Six-week-old female A/J mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and housed in the specific-pathogen-free animal quarters of Research Animal Resources, University of Minnesota Academic Health Center. After 1 week of acclimatization, mice were randomized into six treatment groups (10 mice/group for groups 1 and 2 and 20 mice/group for the rest of the groups) and treated as follows: mice in group 1 received physiological saline solution, the vehicle control. Mice in group 2 were intranasally treated with LPS, under isofluorane anesthesia, at a dose of 5 µg/mouse in 50 μl of phosphate-buffered saline. The endotoxin was administered once a week for 22 weeks. Mice in groups 3–6 were treated with a single dose of NNK (100mg/kg, i.p.) and LPS (as in group 2) and maintained on control diet (group 3) or diet supplemented with I3C (20 μmol/g diet, group 4), Sil (20 μmol/g diet, group 5) or combination of I3C and Sil (group 6) (Figure 1A). To determine the relative lung tumorigenicity of NNK + LPS, two additional groups of mice were treated with a single dose of NNK (100mg/kg, i.p.) or two doses of NNK (at an interval of a week) without LPS. The chemopreventive agents were administered beginning 1 week after treatment with the carcinogen until the end of the study. Food consumption was measured three times a week, and body weights were determined weekly throughout the study. At week 25, the mice were euthanized with an overdose of carbon dioxide. The lungs were harvested, and tumors on the surface of the lung counted, and their sizes determined under a dissecting microscope. Left lung lobes were preserved in 10% buffered formalin for histopathological analyses. The remaining lung lobes were either stored at −80°C for western immunoblotting studies or kept in RNAlater solution for RNA isolation. The experimental design for the tumor bioassay is shown in Figure 1A.
Figure 1.

Inhibition of NNK + LPS-induced lung tumors by I3C + Sil. (A) Experimental design of the study. Groups of mice were treated with a single dose of NNK (100mg/kg body weight, by i.p. injection) and, beginning 1 week after carcinogen treatment until the end of the study, received LPS (5 µg/mouse, weekly) by intranasal instillation and I3C and/or Sil in the diet. For the purpose of comparison, groups of mice also received one or two doses (at an interval of a week) of NNK without LPS. The study was terminated at week 25. (B) LPS increased the multiplicity of NNK-induced lung tumors and I3C + Sil significantly reduced these effects. During necropsy, lung tumors on the surface of the lung were counted under a stereo microscope and the number of lung tumors in mice treated with a single dose of NNK without LPS was compared with that from mice treated with two doses of NNK without LPS or a single dose of NNK and LPS. Also, to determine the chemopreventive efficacy of I3C and/or Sil, the tumor number in mice treated with a single dose of NNK and LPS was compared with that from mice treated the same agents and given the chemopreventive agents in the diet. *P < 0.05. (C) LPS enhanced the growth of NNK-induced lung tumors and I3C alone and I3C + Sil significantly reduced these effects. During necropsy, the size of lung tumors was categorized into three classes (<0.5, 0.5–1.0 and >1mm) and the differential effects of the chemopreventive agents on these groups of tumors was determined. *P < 0.05 (D) I3C alone and I3C + Sil significantly reduced the progression of lung adenoma to lung adenoma with dysplasia as determined by histopathological analysis of the lung tumors following established criteria. *P < 0.05, NNK + LPS group versus NNK + LPS + I3C or NNK + LPS + I3C + Sil group. (E) Photomicrograph of adenoma and adenoma with dysplasia. Note fairly uniform size of cells and nuclei in adenoma in contrast to the presence of cells with large hyperchromatic nuclei (arrows) in an adenoma with dysplasia.
Histopathological analysis of lung tumors
Formalin-fixed lung tissues were embedded in paraffin using standard methods and serially sectioned. Histology slides were prepared from two sections, 400–600 µm apart (depending on the thickness and degree of inflation of each lung), and stained with hematoxylin and eosin prior to counting and classifying tumors in the sections using light microscopy. Lung tumors were classified as adenomas or adenomas with dysplasia based on the recommendations of the Mouse Models of Human Cancers Consortium (9) and our previous reports (10,11). An adenoma was considered to be an adenoma with dysplasia when a discrete focus of cells was found in the adenoma that had hyperchromatic nuclei and nuclear size greater than those found in the predominant cell type in the adenoma. Adenocarcinomas were not observed in this study.
Cells and cell culture
Bronchial premalignant cell lines (1799 and 1198 cells) were kindly provided by Dr Klein-Szanto (Fox Chaser Cancer Center, Philadelphia, PA) in 2010. Upon receiving the cell lines, the authenticity of the cells was determined by short tandem repeat analysis technology (Cell ID™ System, Promega, Madison, WI) and screened for mycoplasma contamination. Our laboratory has never grown HeLa cells, which are incriminated in cell line contamination. We routinely carry out mycoplasma screening of all of our cell lines. Cell line 1799 was developed from BEAS-2B cells explanted along with beeswax pellets into rat tracheas that had been denuded of bronchial epithelium and further transplanted into the dorsal subcutaneous tissues of nude mice (12). Cell line 1198 was developed in a similar manner except that the beeswax pellets contained cigarette smoke condensate. All bronchial cells were maintained in keratinocyte serum-free medium with recommended supplements (Life Technologies, Gaithersburg, MD) in a humidified atmosphere containing 5% CO2. The cells were of the same passage number when used for the study.
Cell proliferation assay
Cell proliferation was determined using the methylthiazoletetrazolium (MTT; Biotium, Hayward, CA) assay as follows. Bronchial 1799 and 1198 cells were plated on a 24-well plate at a density of 20000 cells/well, grown for 24h and treated with I3C and/or Sil (0–20 μM) for 48h followed by methylthiazoletetrazolium treatment (40 μl per well) for 4h. Subsequently, culture media were aspirated, 300 μl of dimethyl sulfoxide was added to each well and absorbance was read at 570nm with a plate reader. Each treatment with I3C and/or Sil was carried out in triplicate and the assays were repeated three times on different days.
Western blot analysis of mouse lung tissues and cell lines
For the analysis of mouse lung tissues, aliquots of normal lungs (vehicle control or LPS-treated mice) or microdissected tumors (carcinogen-treated mice) from three mice were homogenized in a 1× RIPA buffer containing protease and phosphatase inhibitors. For the assay with 1799 and 1198 bronchial cells, ~1×106 cells were suspended in the RIPA buffer containing protease and phosphatase inhibitors on ice for 1h. Subsequently, tissue or cell lysates were centrifuged (14000g for 25min at 4°C), the supernatants collected, aliquoted and stored at −80°C. Twenty micrograms protein from cell lysates or 50 μg protein from tissue lysates were electrophoresed on a 4–12% Novex Tris–glycine gel (Invitrogen, Carlsbad, CA), and transferred to a polyvinylidene difluoride membrane (Bio-Rad). After blocking with 5% non-fat powdered milk in Tris-buffered saline containing 0.05% Tween 20, the membrane was incubated with appropriate primary antibodies at 4°C overnight. Subsequently, the membrane was washed with Tris-buffered saline containing 0.05% Tween 20 and incubated with horseradish peroxidase-conjugated secondary antibody for 1h at room temperature. The protein-antibody complexes were detected by enhanced chemiluminescence (ECL kit) in accordance with the manufacturer’s directions (Pierce, Rockford, IL). Biotinylated protein ladder (Cell Signaling Technology) was loaded into the first-left lane of each gel and used as the molecular weight marker to detect the correct target band. As positive control, cell lysates from A549 were loaded in the last lane. All membranes were stripped and reprobed with anti-β-actin (1:1000) to check for differences in the amount of protein loaded in each lane. For each protein, at least three western assays were carried out. For quantitative determination of protein levels, densitometry measurements of western blot bands were performed using digitalized scientific software program UN-SCAN-IT software (Silk Scientific, Orem, UT).
STAT3- and NF-κB-DNA binding assay
To determine STAT3 and NF-κB binding to DNA, we used the TransAM transcription factor assay (Active Motif, CA), a non-radioactive transcription factor enzyme-linked immunosorbent assay kit that facilitates the study of transcription factor activation, which is an alternative method to radioisotopic EMSA detection methods. Briefly, 10 μg of nuclear protein extract prepared from mouse lung tissues were diluted in complete lysis buffer and added into each well coated with oligonucleotide containing STAT3 or NF-κB consensus binding site. STAT3 or NF-κB subunit proteins in nuclear extract bind to the oligonucleotide under the assay conditions and were detected by using a primary antibody specific to the proteins followed by incubation with horseradish peroxidase conjugated secondary antibody and colorimetric reading at 450nm. Wild-type consensus oligonucleotide was used as a competitor to prevent STAT3 or NF-κB binding to the probe immobilized on the plate. For the specificity of the assay, nuclear protein extracts (10 µg) from interleukin-6-treated HepG2 cells and TPA-treated Jurkat cells were used as positive controls for STAT3 or NF-κB assays, respectively. These experiments were performed with triplicate samples. Error bars represent standard deviation.
Quantitative reverse transcription–PCR analysis of tumor necrosis factor alpha, interleukin-6 (IL-6) and interleukin 17-a (IL-17a) in mouse lungs and cyclin D1 in bronchial cells
Total RNA was extracted from mouse lung tissues, 1799 and 1198 premalignant bronchial cells using the miRNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s instruction. The purity and integrity of total RNA were confirmed by Nanodrop and Agilent Bioanalyzer. The first-strand complementary DNA was synthesized by using QuantiTect Reverse Transcription Kit (Qiagen, Valencia, CA) with one microgram of RNA in 20 µl reaction. The first-strand complementary DNA mixture was further diluted to 100 µl with nuclease-free water and stored at −20°C until use.
Quantitative reverse transcription–PCR was performed by Applied Biosystems 7500 fast real-time PCR system with 96-Well Block Module (Life Technologies, Carlsbad, CA) using QuantiTect SYBR Green PCR Kit (Qiagen, Valencia, CA) and the following gene-specific primers: the forward primers for mouse tumor necrosis factor alpha (TNF-α), IL-7, IL-17a and β-actin were 5′-TATGGCTCAGGGTCCAACTC-3′, 5′-ACGGCCTTCCCTACTTCACA-3′, 5′-TCCCTCTGTGATCTGGGAAG-3′ and 5′-CGTGCGTGACATCAAAGAGAA-3′, respectively, whereas the respective reverse primers were 5′-CTCCCTTTGCAGAACTCAGG-3′, 5′-CATTTCCACGATTTCCCAGA-3′, 5′-AGCATCTTCTCGACCCTGAA-3′ and 5′-TGGATGCCACAGGATTCCAT-3′. The forward and reverse primers for human cyclin D1 were 5′-CCTGGTGAACAAGCTCAAGT-3′ and 5′-GTGTTTGCGGATGATCTGTTTG-3′, respectively, whereas for the internal control, β-actin, the respective forward and reverse primers were 5′-ACTCTTCCAGCCTTCCTTCC-3′ and 5′-GTACTTGCGCTC AGGAGGAG-3′. Twenty-five nanogram of complementary DNA sample was added to a 20 µL reaction and the final concentration of each primer was 0.5 µM. For the PCR amplification, a program of initial denaturation at 95°C for 15min, followed by 40 cycles consisting of denaturation at 94°C for 15 s, annealing at 55°C for 30 s and elongation at 70°C for 34 s was used. All samples were normalized to an internal control gene, β-actin (Actb). Comparative Ct method was used to assess the relative gene expression. Values were expressed as relative units compared with the vehicle control mouse lung tissue and the respective standard errors of the mean.
Statistical analyses
Data for tumor bioassay, western immunoblotting, EMSA and methylthiazoletetrazolium are reported as mean ± SD of triplicate determinations. Between-group comparisons were performed using one-way analysis of variance and two-tailed t-test in Graphpad Prism 4 software (Graphpad, La Jolla, CA). Cytokine data are reported as mean ± SEM for each group and between-group comparisons were performed using Kruskal–Wallis test in SAS 9.3 (SAS Institute, Cary, NC). *P < 0.05.
Results
Combinatorial treatment with I3C and Sil suppresses the multiplicity and size of NNK+ LPS-induced lung tumors
As part of the tumor bioassay, we measured food consumption and body weight of the mice three times a week and once a week, respectively. There was no difference in the food consumption rate of control mice and mice in the treatment groups. The body weight of mice given I3C or I3C + Sil in the diet was ~3–5% less than that of the control group, but this difference was not statistically significant. Also, LPS-treated mice lost a significant amount of weight immediately after treatment, but the lost weight was regained completely 3–4 days after treatment.
To determine if chronic inflammation enhances NNK-induced lung tumorigenesis, we compared the lung tumor burden of mice treated with a single dose of NNK (100mg/kg) and the inflammatory agent LPS with that of mice given a single dose or two doses of NNK without LPS. As shown in Figure 1B, mice treated with one dose of NNK and chroncically exposed to LPS developed 14.7±4.1 tumors/mouse, which was significantly higher than the tumor multiplicity observed in mice treated with a single dose of NNK without LPS (4.8±3.8 tumors/mouse), but less than the tumor multiplicity induced by two doses of NNK (24.1±6.7 tumors/mouse). To determine if the average size of the lung tumors differs among the three groups, the tumors were classified in to three size categories: <0.5, 0.5–1 and >1mm. The frequency of the biggest tumors (>1mm) was significantly higher in mice treated with a single dose of NNK and given LPS compared with the group that received two doses of NNK (Figure 1C). The lung tumor multiplicity of mice treated with LPS alone was similar to that of the group treated with the vehicle control (0.11±0.3 tumors/mouse, data not shown).
To assess the inhibitory effects of combinations of I3C and Sil against NNK + LPS-induced lung tumors, we compared the multiplicity and size of lung tumors observed in mice treated with NNK + LPS and maintained on control diet with that of groups of mice treated with NNK + LPS and given diets supplemented with I3C and/or Sil. As shown in Figure 1B, mice treated with NNK + LPS and fed the control diet had 14.7±4.1 tumors/mouse, whereas mice treated with NNK + LPS and given the combination of I3C and Sil had only 7.1±4.5 tumors/mouse, corresponding to a significant 52% reduction in lung tumor multiplicity (P < 0.01). The lung tumor multiplicity of mice given I3C or Sil was not significantly different from that of the control group (15.0±11.1 and 12.5±5.2 tumors/mouse for mice whose diets were supplemented with I3C and Sil, respectively). Combinatory treatment with I3C and Sil or I3C alone significantly reduced the growth of lung tumors (Figure 1C). In mice given I3C and Sil and I3C alone, the multiplicity of the largest tumors (>1mm) was significantly reduced to 1.0±1.3 and 1.6±1.8 tumors/mouse (P < 0.01) compared with 6.3±2.9 tumors/mouse in the control group. Sil alone did not significantly reduce the multiplicity of the largest tumors.
Malignant progression of lung adenomas was suppressed by combinatory treatment with I3C and Sil
To determine if the chemopreventive agents suppress malignant progression of lung adenoma, lung tissue sections were scored for adenoma, adenoma with dysplasia and adenocarcinoma, following established protocols (9). As depicted in Figure 1D, there was no difference in the average number of dysplastic adenomas between the groups treated with a single dose of NNK and LPS and two doses of NNK without LPS. Combinatory treatment with I3C and Sil and I3C alone significantly reduced the frequency of dysplastic adenomas in mice treated with a single dose of NNK and LPS. A similar trend was observed in the group treated with Sil alone but the reduction was not significant. Neither I3C + Sil nor I3C or Sil alone reduced the number of lung adenomas. Representative images of lung adenoma and dysplastic adenoma lesions are shown in Figure 1E.
Combinatory treatment with I3C and Sil significantly reduced the expression of TNF-α and IL-6 in lung tumor tissues
Inflammation is mediated by a variety of soluble factors, including cytokines and therefore we determined the expression of TNF-α, IL-6 and IL-17a, cytokines known to have a pivotal role in enhancing tumorigenesis. First, we determined if treatment with NNK, LPS or NNK + LPS increased the expression of TNF-α, IL-6 and IL-17a in lung tissues. As shown in Table 1, compared with the expression in the control group, levels of TNF-α, IL-6 and IL-17a were significantly increased in the groups treated with LPS or NNK + LPS, but not NNK. Then, we assessed the effect of I3C and/or Sil on NNK + LPS-induced expression of TNF-α, IL-6 and IL-17a. Combinatory treatment with I3C and Sil significantly reduced NNK + LPS-induced expression of TNF-α and IL-6 to the level observed in the control group. Although not significant, a similar trend was also observed for IL-17a. On the contrary, in mice fed with I3C alone or Sil alone, levels of the three cytokines were similar to that observed in the NNK + LPS group.
Table 1.
Expression of IL-6, TNF-α and IL-17a in lung tissues of mice treated with vehicle control, NNK, LPS, NNK + LPS or NNK + LPS and I3C and/or Sila
| Vehicle control (n = 3) | NNK (n = 3) | LPS (n = 3) | NNK/LPS (n = 3) | NNK/LPS/Sil (n = 3) | NNK/LPS/I3C (n = 3) | NNK/LPS/Sil/ I3C (n = 3) | |
|---|---|---|---|---|---|---|---|
| IL-6 | 1.00±0.09 | 0.51±0.14 | 4.17±1.48b | 3.61±1.20b | 7.03±2.63 | 3.33±2.15 | 0.45±0.22c |
| TNF-α | 1.00±0.18 | 0.84±0.33 | 14.77±3.36b | 7.90±3.48b | 14.83±4.39 | 12.70±8.29 | 1.79±0.53c |
| IL-17a | 1.00±0.32 | 1.07±0.24 | 71.70±40.92b | 100.19±61.13b | 74.98±40.23 | 42.81±27.94 | 11.71±8.98 |
aFemale A/J mice were treated with NNK and/or LPS as described in the materials and methods section and maintained on control or I3C and/or Sil-supplemented diet. At week 25, mice were euthanized with an overdose of carbon dioxide, lung tissue harvested, and normal lung tissue or dissected tumors were kept in RNAlater and used for QRT-PCR-based analysis of IL-6, TNFα, and IL17a expression. Levels of the cytokines in the NNK, LPS and NNK + LPS groups were compared with that of the control group using Kruskal-Wallis test. Likewise, levels of the cytokines in the NNK + LPS + I3C and/or Sil groups were compared with that of the NNK + LPS group.
b P < 0.05, compared to vehicle control.
c P < 0.05, compared to the NNK + LPS group.
Combinatory treatment with I3C and Sil suppressed levels of inflammation- and cell growth-related proteins in NNK + LPS-induced lung tumors
Since combinations of I3C and Sil significantly reduced the number and size of inflammation-driven lung tumors, we sought to determine if the chemopreventive agents suppress inflammation- and cell-proliferation-related proteins in mouse lung tumors. As depicted in Figure 2A, lung tumors from NNK + LPS-treated mice exhibited significantly increased phosphorylation of STAT3 and IκBα, and expression of COX2, all of which are critical players in inflammation and tumorigenesis. NNK + LPS also significantly increased levels of cell cycle regulating proteins cyclin D1, CDK2 and phosphorylated Rb (Figure 2B). We also observed that, compared with the vehicle group, the mice treated with LPS alone had significantly increased levels of phosphorylated STAT3, phosphorylated IκBα, CDK2 and phosphorylated Rb (Figure 2A and B).
Figure 2.
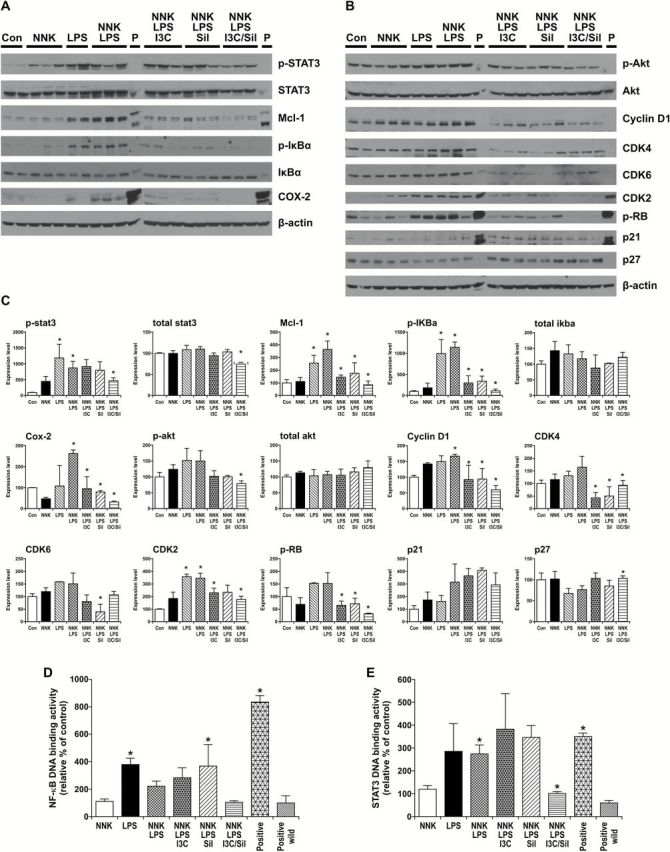
Effects of NNK + LPS on inflammation- and cell proliferation-related proteins and modulation of these effects by I3C and/or Sil. (A, B) Mouse lung tissue levels of proinflammatory and procell growth proteins were determined by western immunoblotting as described in Materials and methods. (C) Quantification of the western blot results. Densitometry measurements of western blot bands were performed using digitalized scientific software program UN-SCAN-IT software. *P < 0.05, NNK, LPS or NNK + LPS group versus vehicle control group or NNK + LPS + Sil, NNK + LPS + I3C or NNK + LPS + I3C + Sil group versus NNK + LPS group. Effects of NNK and/or LPS on NF-κB-DNA binding (D) and STAT3-DNA binding (E) and reversal of these effects by I3C + Sil as determined by ELISA-based EMSA assays (Active motif). *P < 0.05, LPS or NNK + LPS group versus vehicle control group or NNK + LPS + I3C + Sil group versus NNK + LPS group.
Supplementation of mouse diets with combinations of I3C and Sil significantly attenuated NNK + LPS-induced phosphorylated STAT3, total STAT3, phosphorylated IκBα, phosphorylated Akt, COX2, Mcl-1, cyclin D1, CDK2, CDK4 and phosphorylated Rb, but increased the expression of p27 (Figure 2A and B). Also, administration of I3C alone or Sil alone suppressed the expression of most of these proteins, although their effects were in general lower than that of I3C + Sil. Quantitative data on the effect of the chemoprevnetive agents on inflammation- and cell growth-related proteins are shown in Figure 2C. Combinations of I3C and Sil failed to cause cleavage of poly (ADP ribose) polymerase, which is considered as a marker of apoptosis (data not shown).
In order to further assess activation of the proinflammatory transcription factors STAT3 and NF-κB by NNK + LPS and if I3C and/or Sil modulates these effects, we performed STAT3- and NF-κB-DNA binding assays using the TransAM transcription factor assay kit. As depicted in Figure 2D, compared with the level in the vehicle group, the percentage of NF-κB-DNA binding was significantly increased by 3.8- and 2.2-fold in nuclear fractions of lung tissues from mice treated with LPS and NNK + LPS, respectively, whereas the level in the NNK group was similar to the vehicle group. Dietary administration of I3C + Sil to NNK + LPS-treated mice significantly reversed NF-κB-DNA binding to the control level; I3C alone and Sil alone failed to modulate NF-κB-DNA binding. Similarly, LPS and NNK + LPS increased STAT3-DNA binding activity by 2.9- and 2.7-fold, respectively, and I3C + Sil significantly reduced NNK + LPS-induced STAT3-DNA binding to the level in the vehicle group, but I3C alone or Sil alone failed to modulate STAT3-DNA binding (Figure 2E).
Combinations of I3C and Sil, but not I3C or Sil alone, caused antiproliferative effects and suppressed the expression of cyclin D1 and related proteins in premalignant bronchial 1799 and 1198 cells
In order to assess if the in vivo effects of I3C and Sil could be reproduced in cell line models, we determined the antiproliferative effects of the drugs in 1799 and 1198 premalignant bronchial cells and modulation of cell cycle-related proteins. Similar to our observation in the mouse model of lung cancer, exposure of 1799 and 1198 cells to the combination of I3C and Sil for 48h significantly reduced the proliferation of both cell lines in a dose-dependent manner (Figure 3A). In particular, whereas I3C alone or Sil alone did not exhibit significant antiproliferative effects even at a concentration of 20 µM, I3C + Sil, 10 µM of each, significantly reduced the viability of 1799 and 1198 cells by 62 and 57%, respectively. Consistent with their growth inhibitory effects, combinations of I3C and Sil suppressed the expression of cyclin D1, CDK2, CDK4, CDK6 and phosphorylated Rb, whereas I3C or Sil alone failed to do so (Figure 3B). Furthermore, exposure of 1799 and 1198 cells to I3C + Sil, 10 µM each, for different time periods showed time-dependent suppression of cyclin D1, CDK2, CDK4, CDK6, phospho-Rb and phospho Akt. In particular, the level of cyclin D1 was reduced as early as 12h after treatment, further reduced at 24h and the effect persisted until 48h (Figure 3C).
Figure 3.
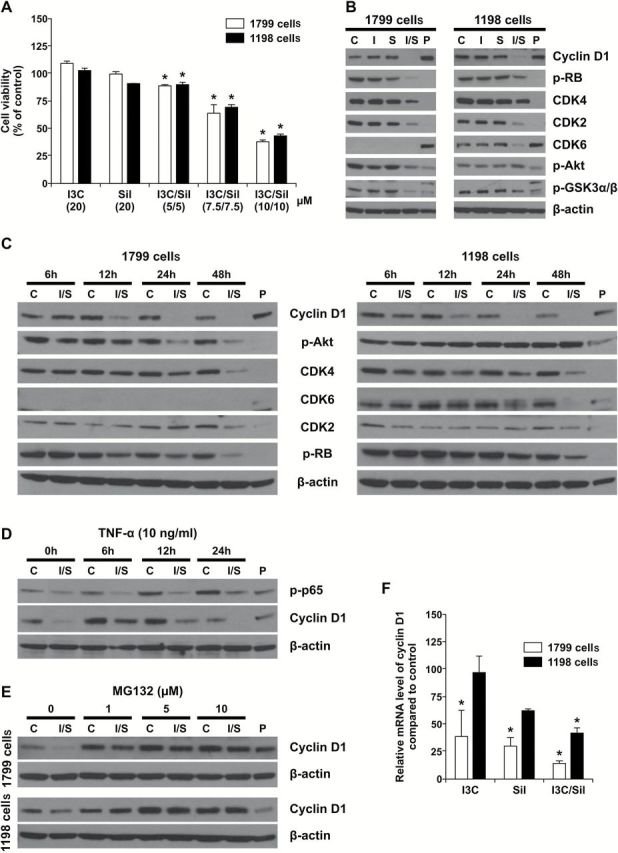
In vitro chemopreventive activities of I3C + Sil. (A) I3C + Sil suppressed the proliferation of premalignant bronchial 1799 and 1198 cells in a dose-dependent manner. Bronchial 1799 and 1198 cells were exposed to I3C and/or Sil for 48h and the proliferation of the cells was determined by methylthiazoletetrazolium assay as described in Materials and methods. *P < 0.05, compared with the control group. (B) Suppressive effects of I3C + Sil on cell cycle regulating proteins. Bronchial 1799 and 1198 cells were exposed to I3C and/or Sil for 48h, cell lysates prepared and the protein levels of cyclin D1, CDKs 2, 4 and 6, pRb and pAkt were determined by western immunoblotting. (C) Time-dependent effects of I3C and/or Sil on the expression of cyclin D1, CDKs 2, 4 and 6, pRb and pAkt. Bronchial cells were exposed to I3C and/or Sil for different periods of time and levels of the proteins were determined as in Figure 3B. (D) Cotreatment of 1198 cells with I3C + Sil and TNF-α attenuated the effects of I3C + Sil on cyclin D1 expression. Bronchial 1998 cells were exposed to 10 μM each of I3C and Sil for 12h and then treated with 10ng/ml TNF-α for 6, 12 and 24h. Cells were harvested at the indicated time, and whole cell extracts were prepared and used for western blotting analysis. (E) MG132, a proteasomal inhibitor, blocks I3C + Sil-induced downregulation of cyclin D1 protein in premalignant bronchial 1799 and 1198 cells. Bronchial 1799 and 1198 cells were pretreated with the proteasomal inhibitor MG132 at a concentration of 1, 5 or 10 μM for 1h and then exposed to 10 μM each of I3C and Sil for 12h. Whole cell extracts were prepared and examined by western blot analysis using cyclin D1 antibody. (F) I3C + Sil downregulates the expression of cyclin D1 mRNA in premalignant bronchial 1799 and 1198 cells. Cells were treated with 10 μM each of I3C and Sil for 48h, total RNA was prepared and the mRNA level of cyclin D1 was measured by quantitative reverse transcription–PCR as described in Materials and methods. *P < 0.05, compared with the control group.
Combinations of I3C and Sil suppressed TNF-α-induced and NF-κB-mediated overexpression of cyclin D1
NF-κB is known to regulate the expression of several genes, including cyclin D1. To examine if I3C + Sil suppresses NF-κB-mediated expression of cyclin D1, premalignant bronchial 1799 cells were treated with 10 μM each of I3C and Sil for 12h in keratinocyte serum-free medium and then 10ng/ml TNF-α (R&D systems) was added for 6, 12 and 24h. Cells were harvested at the indicated time, whole cell extracts prepared and analyzed by western blotting for the expression of phospho-NF-κB and cyclin D1. As expected, TNF-α increased phosphorylation of NF-κB in a time-dependent manner and I3C + Sil attenuated the level of phospho-NF-κB (Figure 3D). Cyclin D1 expression increased 6h after treatment with TNF-α, remained high until 12h, but decreased to the control level at 24h. I3C + Sil partially reduced TNF-α-induced cyclin D1 at 6 and 12h but completely abolished it at 24h (Figure 3D).
Decreased gene expression and enhanced proteasomal degradation contribute to suppression of cyclin D1 protein levels by I3C + Sil in 1799 and 1198 bronchial cells
To determine the ability of MG132, a proteasome 26S inhibitor, to block I3C and/or Sil-induced degradation of cyclin D1, 1799 and 1198 cells were pretreated with 1, 5 and 10 μM of the inhibitor for 1h and then exposed to I3C and Sil, 10 μM of each, for 12h. Subsequently, whole cell extracts were prepared and examined by western blot analysis using cyclin D1 antibody. As shown in Figure 3E, in the absence of MG132, I3C + Sil-induced degradation of cyclin D1 in both cell lines. However, pretreatment of the cells with MG132 completely abolished the effect of the chemopreventive agents even at the lowest concentration, indicating that proteasomal degradation was involved in I3C + Sil-induced disappearance of cyclin D1.
To determine if combinatory treatment with I3C and Sil suppresses cyclin D1 protein levels by suppressing the synthesis of its messenger RNA (mRNA), 1799 and 1198 cells were treated with I3C and/or Sil for 48h, RNA was prepared and analyzed by quantitative reverse transcription–PCR. As depicted in Figure 3F, combinatory treatment with I3C and Sil significantly reduced the mRNA levels of cyclin D1 in both cell lines, suggesting that decreased RNA synthesis of cyclin D1 contributes to the reduction in the protein level of the gene.
Discussion
In the present study, we showed that combinations of I3C and Sil reduced the multiplicity, size and progression of inflammation-driven lung tumors in mice and these effects were associated with suppression of the inflammatory cytokines TNFα and IL-6 and the proinflammatory, pro-proliferative and pro-survival proteins NF-κB, STAT3, Akt and COX-2. Similarly, in in vitro studies with premalignant bronchial 1799 and 1198 cells, I3C + Sil inhibited the growth of the cells and reduced the level of cyclin D1 and related proteins.
Tobacco smoke-associated chronic inflammation of the airways and lung parenchyma play a central role in the development of COPD (13), which is known to be a very important risk factor of lung cancer in smokers (2,3). Molecular players in COPD-driven lung cancer include NF-κB, STAT3, COX-2 and PI3K/Akt (14,15). The transcription factors NF-κB and STAT3, which are commonly activated by proinflammatory cytokines in the bronchial epithelium and inflammatory cells of the lower airways of COPD patients, regulate the transcription of several cancer-related genes, including cyclin D1, COX-2, MMP-9, VEGF and antiapoptosis genes (16–18). Thus, NF-κB and STAT3 are considered to be potential links between chronic inflammation and cancer. Also, COX-2, the rate-limiting enzyme in the production of proinflammatory prostaglandins, is commonly overexpressed in the lung tissues of COPD patients (19,20) and it promotes tumorigenesis by enhancing abnormal expression of epithelial growth factors, epithelial and microvascular proliferation, resistance to apoptosis and suppression of antitumor immunity (21). The PI3K/Akt signaling pathway, which is activated by a broad range of inflammatory cytokines, growth factors and cigarette smoke constituents and known to be the earliest alteration in the development of lung cancer (22), contributes to the development of COPD and lung cancer by enhancing the proliferation and survival of inflammatory cells and altered epithelial cells, expression and activation of inflammatory mediators, inflammatory cell recruitment, immune cell function, airway remodeling and corticosteroid insensitivity (23,24). Moreover, persistent activation of Akt secondary to somatic mutations in regulatory oncogenes, such as PTEN, is believed to be the reason why COPD-related inflammation does not resolve after cessation of smoking (23).
Drugs commonly employed for the prevention of inflammation-driven lung cancer include inhaled corticosteroids (25,26), selective COX-2 inhibitors (27,28) and signaling pathway modulators such as inhibitors of NF-κB, STAT3 and PI3K/Akt (29). However, whereas inhaled glucocorticoids are immunosuppressive and their anticancer effects are inconsistent (25,26), there is a concern regarding the cardiovascular system toxicity of selective COX-2 inhibitors (27,28). Similarly, the potential side effects of selective NF-κB, STAT3 and PI3K/Akt inhibitors have hindered the clinical use of these drugs as lung cancer chemopreventive agents (16,30). We showed in the present study that combinations of I3C and Sil significantly suppressed activation of NF-κB, STAT3 and Akt and expression of COX-2. In particular, simultaneous targeting of NF-κB and STAT3 could block the cooperation between these proinflammatory pathways in promoting the development and progression of cancer (31). Since cyclin D1 is a common downstream effector of NF-κB, STAT3 and Akt (32) and overexpression of cyclin D1 is a key abnormality in lung carcinogenesis (33–35), we also examined NNK + LPS-induced deregulation of cyclin D1-CDK-Rb axis, a hub for the regulation of the G1–S phase transition in the cell cycle, and its modulation by I3C and/or Sil. Indeed, protein levels of cyclin D1, CDK2, CDK4, CDK6 and phospho-Rb were increased in lung tumor tissues of NNK + LPS-treated mice, but I3C + Sil reduced their expressions although the effects were not uniformly significant. On the other hand, I3C + Sil increased the expression of p27, a tumor suppressor protein that regulates progression from G1 to S phase by binding and inhibiting cyclin-dependent kinases (36). Similar results were found in in vitro assays with premalignant bronchial 1799 and 1198 cells. These findings are consistent with previous reports in which both I3C and Sil were found to have suppressive effects on members of cyclin D1-CDK-Rb pathway, but increased levels of the CDK inhibitors p21 and p27 in several cancer cell lines (4,5). Further studies on the potential mechanisms responsible for the regulation of cyclin D1 by I3C + Sil indicated that both proteasomal degradation and suppression of mRNA synthesis are involved although it was not clear whether the latter effect results from destabilization of cyclin D1 mRNA or inhibition of its synthesis.
The dose levels of I3C and Sil administered in the present studies are equivalent to the amounts of the drugs used in previous mouse lung cancer chemoprevention studies (37–41) and human clinical trials (42,43). However, the efficacies of the drugs observed in the present study were weaker than what had been reported in previous mouse lung cancer chemoprevention studies, irrespective of the model used. For instance, in our previous study, I3C alone (10 μmol/g diet) and Sil alone (7 μmol g diet) reduced the multiplicity of NNK-induced lung tumors by 43% and 36%, respectively (11), whereas in the present study, neither I3C nor Sil reduced the multiplicity of NNK + LPS-induced lung tumors although administered at a dose level 2-fold higher than that given in the earlier study. The reasons for this discrepancy remain to be determined. One possibility is that NNK + LPS-induced lung tumors exhibit more complex genetic alterations, compared with lung tumors induced in classical carcinogen models, which could make them less responsive to preventive/therapeutic agents (44).
Overall, our results indicate that combined administration of I3C and Sil is a promising approach for the chemoprevention of inflammation-associated lung cancer in high risk populations. In particular, given that 50% of newly diagnosed lung cancer patients have been shown to have COPD (3) and no safe and effective chemopreventive agents are currently available against lung cancer, the combination of I3C and Sil is an attractive candidate for clinical lung cancer chemoprevention trials. Currently, further studies are going on to assess the efficacy of I3C + Sil to inhibit malignant progression of inflammation-driven lung tumors when administered after the formation of lung adenoma.
Funding
National Cancer Institute/National Institute of Health (NCI/NIH) grant (RO1CA166615-01 to F.K.).
Acknowledgement
We want to express our indebtedness to B.Carlson for his help in formatting the figures.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- COPD
chronic obstructive pulmonary disease
- I3C
indole-3-carbinol
- LPS
lipopolysaccharide
- mRNA
messenger RNA
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- Sil
silibinin
- TNF-α
tumor necrosis factor alpha
References
- 1. Siegel R., et al. (2014) Cancer statistics, 2014. CA Cancer J. Clin., 64, 9–29. [DOI] [PubMed] [Google Scholar]
- 2. Mannino D.M., et al. (2003) Low lung function and incident lung cancer in the United States: data from the first NHANES follow-up. Arch. Intern.Med., 163, 1475–1480. [DOI] [PubMed] [Google Scholar]
- 3. Young R.P., et al. (2009) COPD prevalence is increased in lung cancer independence of age, gender and smoking history. Eur. Respir. J., 34, 380–386. [DOI] [PubMed] [Google Scholar]
- 4. Aggarwal B.B., et al. (2005) Molecular targets and anticancer potential of indole-3-carbinol and its derivatives. Cell Cycle, 4, 1201–1215. [DOI] [PubMed] [Google Scholar]
- 5. Agarwal R., et al. (2006) Anticancer potential of silymarin: from bench to bed side. Anticancer Res., 26, 4457–4498. [PubMed] [Google Scholar]
- 6. Hasday J.D., et al. (1999) Bacterial endotoxin is an active component of cigarette smoke. Chest, 115, 829–835. [DOI] [PubMed] [Google Scholar]
- 7. Larsson L., et al. (2012) Microbiological components in mainstream and sidestream cigarette smoke. Tob. Induc. Dis., 10, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hecht S.S., et al. (1983) Effects of alpha-deuterium substitution on the mutagenicity of 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Carcinogenesis, 4, 305–310. [DOI] [PubMed] [Google Scholar]
- 9. Nikitin A.Y., et al. (2004) Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res., 64, 2307–2316. [DOI] [PubMed] [Google Scholar]
- 10. Kassie F., et al. (2010) Inhibition of lung carcinogenesis and critical cancer-related signaling pathways by N-acetyl-S-(N-2-phenethylthiocarbamoyl)-l-cysteine, indole-3-carbinol and myo-inositol, alone and in combination. Carcinogenesis, 31, 1634–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dagne A., et al. (2011) Enhanced inhibition of lung adenocarcinoma by combinatorial treatment with indole-3-carbinol and silibinin in A/J mice. Carcinogenesis, 32, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Klein-Szanto A.J., et al. (1992) A tobacco-specific N-nitrosamine or cigarette smoke condensate causes neoplastic transformation of xenotransplanted human bronchial epithelial cells. Proc. Natl Acad. Sci. USA, 89, 6693–6697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Milara J., et al. (2012) Tobacco, inflammation, and respiratory tract cancer. Curr. Pharm. Des., 18, 3901–3938. [DOI] [PubMed] [Google Scholar]
- 14. Houghton A.M. (2013) Mechanistic links between COPD and lung cancer. Nat. Rev. Cancer, 13, 233–245. [DOI] [PubMed] [Google Scholar]
- 15. Di Stefano A., et al. (2002) Increased expression of nuclear factor-kappaB in bronchial biopsies from smokers and patients with COPD. Eur. Respir. J., 20, 556–563. [DOI] [PubMed] [Google Scholar]
- 16. Caramori G., et al. (2004) Anti-inflammatory inhibitors of IkappaB kinase in asthma and COPD. Curr. Opin. Investig. Drugs, 5, 1141–1147. [PubMed] [Google Scholar]
- 17. Qu P., et al. (2009) Stat3 downstream genes serve as biomarkers in human lung carcinomas and chronic obstructive pulmonary disease. Lung Cancer, 63, 341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ruwanpura S.M., et al. (2012) Deregulated Stat3 signaling dissociates pulmonary inflammation from emphysema in gp130 mutant mice. Am. J. Physiol. Lung Cell. Mol. Physiol., 302, L627–L639. [DOI] [PubMed] [Google Scholar]
- 19. Chen Y., et al. (2008) Enhanced levels of prostaglandin E2 and matrix metalloproteinase-2 correlate with the severity of airflow limitation in stable COPD. Respirology, 13, 1014–1021. [DOI] [PubMed] [Google Scholar]
- 20. Xaubet A., et al. (2004) Cyclooxygenase-2 is up-regulated in lung parenchyma of chronic obstructive pulmonary disease and down-regulated in idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffuse Lung Dis., 21, 35–42. [PubMed] [Google Scholar]
- 21. Mao J.T., et al. (2005) Chemoprevention strategies with cyclooxygenase-2 inhibitors for lung cancer. Clin. Lung Cancer, 7, 30–39. [DOI] [PubMed] [Google Scholar]
- 22. Tsao A.S., et al. (2003) Increased phospho-AKT (Ser(473)) expression in bronchial dysplasia: implications for lung cancer prevention studies. Cancer Epidemiol. Biomarkers Prev., 12, 660–664. [PubMed] [Google Scholar]
- 23. Bozinovski S., et al. (2006) Akt in the pathogenesis of COPD. Int. J. Chron. Obstruct. Pulmon. Dis., 1, 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ito K., et al. (2007) Therapeutic potential of phosphatidylinositol 3-kinase inhibitors in inflammatory respiratory disease. J. Pharmacol. Exp. Ther., 321, 1–8. [DOI] [PubMed] [Google Scholar]
- 25. Parimon T., et al. (2007) Inhaled corticosteroids and risk of lung cancer among patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med., 175, 712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Calverley P.M., et al. (2007) Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N. Engl. J. Med., 356, 775–789. [DOI] [PubMed] [Google Scholar]
- 27. Kim E.S., et al. (2010) Biological activity of celecoxib in the bronchial epithelium of current and former smokers. Cancer Prev. Res. (Phila)., 3, 148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mao J.T., et al. (2011) Lung cancer chemoprevention with celecoxib in former smokers. Cancer Prev. Res. (Phila)., 4, 984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adcock I.M., et al. (2011) Chronic obstructive pulmonary disease and lung cancer: new molecular insights. Respiration, 81, 265–284. [DOI] [PubMed] [Google Scholar]
- 30. John A., et al. (2010). Phosphatidylinositol 3-kinase isoforms as targets in respiratory Disease. Ther. Adv. Resp. Dis., 3, 19–34. [DOI] [PubMed] [Google Scholar]
- 31. Witzel I.I., et al. (2010) Regulation of cyclin D1 gene expression. Biochem. Soc. Trans., 38(Pt 1), 217–222. [DOI] [PubMed] [Google Scholar]
- 32. Ratschiller D., et al. (2003) Cyclin D1 overexpression in bronchial epithelia of patients with lung cancer is associated with smoking and predicts survival. J. Clin. Oncol., 21, 2085–2093. [DOI] [PubMed] [Google Scholar]
- 33. Gautschi O., et al. (2007) Cyclin D1 in non-small cell lung cancer: a key driver of malignant transformation. Lung Cancer, 55, 1–14. [DOI] [PubMed] [Google Scholar]
- 34. Ayed A.K., et al. (2006) Prognostic significance of cyclin D1 expression in resected stage I, II non-small cell lung cancer in Arabs. Interact. Cardiovasc. Thorac. Surg., 5, 47–51. [DOI] [PubMed] [Google Scholar]
- 35. Surh Y.J., et al. (2010) Breaking the NF-κB and STAT3 alliance inhibits inflammation and pancreatic tumorigenesis. Cancer Prev. Res. (Phila)., 3, 1379–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fero M.L., et al. (1996) A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell, 85, 733–744. [DOI] [PubMed] [Google Scholar]
- 37. Yu Z., et al. (2006) Indole-3-carbinol in the maternal diet provides chemoprotection for the fetus against transplacental carcinogenesis by the polycyclic aromatic hydrocarbon dibenzo[a,l]pyrene. Carcinogenesis, 27, 2116–2123. [DOI] [PubMed] [Google Scholar]
- 38. Benninghoff A.D., et al. (2013) The role of estrogen receptor β in transplacental cancer prevention by indole-3-carbinol. Cancer Prev. Res. (Phila)., 6, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kassie F., et al. (2008) Dose-dependent inhibition of tobacco smoke carcinogen-induced lung tumorigenesis in A/J mice by indole-3-carbinol. Cancer Prev. Res. (Phila)., 1, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Singh R.P., et al. (2006) Effect of silibinin on the growth and progression of primary lung tumors in mice. J. Natl Cancer Inst., 98, 846–855. [DOI] [PubMed] [Google Scholar]
- 41. Ramasamy K., et al. (2011) Silibinin prevents lung tumorigenesis in wild-type but not in iNOS-/- mice: potential of real-time micro-CT in lung cancer chemoprevention studies. Clin. Cancer Res., 17, 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reed G.A., et al. (2005) A phase I study of indole-3-carbinol in women: tolerability and effects. Cancer Epidemiol. Biomarkers Prev., 14, 1953–1960. [DOI] [PubMed] [Google Scholar]
- 43. Flaig T.W., et al. (2010) A study of high-dose oral silybin-phytosome followed by prostatectomy in patients with localized prostate cancer. Prostate, 70, 848–855. [DOI] [PubMed] [Google Scholar]
- 44. Colotta F., et al. (2009) Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis, 30, 1073–1081. [DOI] [PubMed] [Google Scholar]