

Abstract
Purpose
To evaluate the progression of the earliest stage of disease in ABCA4-associated retinal degenerations (RDs).
Methods
Near-infrared excited reduced-illuminance autofluorescence imaging was acquired across the retina up to 80 degrees eccentricity in 44 patients with two ABCA4 alleles. The eccentricity of the leading disease front (LDF) corresponding to the earliest stage of disease was measured along the four meridians. A mathematical model describing the expansion of the LDF was developed based on 6 years of longitudinal follow-up.
Results
The extent of LDF along the superior, inferior, and temporal meridians showed a wide spectrum from 3.5 to 70 degrees. In patients with longitudinal data, the average centrifugal expansion rate was 2 degrees per year. The nasal extent of LDF between the fovea and ONH ranged from 4.3 to 16.5 degrees and expanded at 0.35 degrees per year. The extent of LDF beyond ONH ranged from 19 to 75 degrees and expanded on average at 2 degrees per year. A mathematical model fit well to the longitudinal data describing the expansion of the LDF.
Conclusions
The eccentricity of the LDF in ABCA4-RD shows a continuum from parafovea to far periphery along all four meridians consistent with a wide spectrum of severity observed clinically. The model of progression may provide a quantitative prediction of the LDF expansion based on the age and eccentricity of the LDF at a baseline visit, and thus contribute significantly to the enrollment of candidates appropriate for clinical trials planning specific interventions, efficacy outcomes, and durations.
Keywords: lipofuscin, melanin, atrophy, photoreceptors, autofluorescence, Stargardt disease, disease progression
Centrifugal expansion rate of the earliest ABCA4 disease stages were studied in patients followed for 6 years. A predictive model of progression was developed based on the age and the eccentricity of the leading disease front.
Hereditary retinal degenerations (RDs) are caused by mutations of genes primarily or preferentially expressed in photoreceptor or RPE cells.1 One of the most common molecular causes of RD is due to mutations in the ABCA4 gene,2–7 which encodes the critical enzyme ABCR in rod and cone outer segments.8 ABCR acts as an active transporter of all-trans-retinal (atRAL) by flipping N-retinylidene-phosphatidylethanolamine from the extracellular to cytoplasmic leaflet of internal disc membranes within photoreceptor outer segments.9,10 Mutations in ABCA4 cause the accumulation of toxic bisretinoid adducts of atRAL within photoreceptors and the RPE.11–13 The disease process in ABCA4-RD is thought to initiate near the center of the macula resulting in loss of visual acuity.14–21 Over decades, there is greater loss of vision as the disease progresses to loss of a greater proportion of macular photoreceptors and RPE. Importantly, however, ABCA4-RD is not limited to the macular regions of the retina; there is often progression from the macula to extramacular involvement.14,18
ABCA4-RDs are currently not treatable, but multiple promising therapeutic avenues are being pursued to protect photoreceptors and RPE from disease progression. Proposed interventions include slowing of the visual cycle kinetics,22–29 augmentation of ABCR function by gene therapy,30–32 reduction of vitamin A dimerization,33,34 removal of lipofuscin from the RPE35,36 and stabilization of stressed photoreceptor cells by nutritional and pharmacologic approaches.17,37–39 ABCA4-RD, like nearly all other retinal degenerative conditions in humans, shows an intraretinal distribution of varied disease stages from normal photoreceptors and RPE to complete degeneration. Ideally a successful treatment should halt the progression at all stages of disease existing within the eye at the time of the intervention. However, in preclinical studies thus far, it has been easier to prevent the onset of ABCA4 disease23,24,26,30,31,33,37 as opposed to arresting or reversing later stages of disease.34,36 Better understanding of the spatial distribution of disease stages in ABCA4-RD eyes, and the spatiotemporal progression over serial follow-up would allow selection of patients and retinal locations for treatments, and informed choice of efficacy outcomes.
Here, we examine the initiation of disease by following its leading front in ABCA4-RD patients longitudinally over an average of 6 years. Data obtained in patients with a large spectrum of disease severity from fovea-only maculopathies to retina-wide involvement over a range of ages from first to sixth decade of life are used to develop a model of spatiotemporal disease progression in ABCA4-RD. The model reliably predicts the expansion of the leading disease front (LDF) in four cardinal meridians and can be useful for estimating clinical trial duration as a function of the disease stage chosen for patient enrollment.
Methods
Subjects
The study population consisted of 44 eyes of 44 patients (mean age = 28.4, range, ages 7–61 years at the first visit) with a clinical diagnosis within the spectrum of Stargardt disease or cone–rod dystrophy caused by ABCA4 mutations (Supplementary Table S1). A complete eye examination was performed in all subjects, including best-corrected ETDRS visual acuity. Informed consent was obtained for all subjects; procedures followed the Declaration of Helsinki and were approved by the institutional review board. Visual function results from 20 of the patients have been previously reported; identical patient designations (P4–P66; Supplementary Table S1) are used to enable comparison with the previous work.18 Seventeen of 20 patients had more recent follow-up reported here but not previously. Molecular studies in patients and their family members were performed as previously reported.7
Retinal Imaging
En face imaging was performed with a confocal scanning laser ophthalmoscope (HRA2 or Spectralis HRA; Heidelberg Engineering, Heidelberg, Germany) to obtain near-infrared reduced-illuminance autofluorescence imaging (NIR-RAFI)16,21 between October 2005 and April 2015. For HRA2 (visits between 2005 and 2011), NIR-RAFI laser (790 nm) was at the manufacturer's standard calibration of 2.6 mW at the cornea and an instrument sensitivity setting of 95% was used the majority of the time (rare exceptions included recordings at 93% and 94% settings). Starting in 2007, the ‘automatic real time' (ART) feature of the acquisition software was used to average multiple frames with intensity normalization turned off. Starting in 2011, Spectralis HRA was used with laser output setting of 100% and detector sensitivity of either 87% or 105%. Spectralis HRA+optical coherence tomography (OCT) is much more commonly available in retina clinics and could potentially be used by other investigators to perform NIR-RAFI albeit with lower signal-to-noise ratio compared with the Spectralis HRA used in the current work. In all patients, the 30° lens was used; in the subset of visits performed in 2009 and later, additional imaging with the 55° lens was obtained to extend coverage to further in the periphery. In addition, short-wavelength (SW)-excited RAFI16 was performed with the 30° lens mostly at and near the macular region. All images were acquired with the high speed mode wherein either 30° × 30° square or 55° diameter circular fields were sampled onto 768 × 768 pixels. The dilated pupil of the subject was aligned with the optical axis of the instrument (head position stabilized with chin and forehead rests) in a room with dimmed ambient lights. Wide-field image montage was assembled by manually specifying corresponding retinal landmark pairs in overlapping segments using custom-written software (MATLAB 6.5; MathWorks, Natick, MA, USA). Images from left eyes are displayed as equivalent right eyes for ease of comparability. Cross-sectional imaging was performed with a spectral-domain (SD) OCT system (RTVue-100; Optovue, Inc., Fremont, CA, USA).
The extent of abnormal NIR-RAFI signal was determined along four cardinal meridians by a procedure named ‘windmill analysis' (Supplementary Fig. S1). First, OCT imaging was registered to NIR-RAFI to locate the anatomical fovea. Next, four wedges starting at the anatomical fovea and extending along each cardinal meridian were overlaid digitally onto the wide-angle NIR-RAFI composite images. The tip angle of each wedge was 30° and was demarcated into small areas of interest, which incremented in 1° steps up to 9° eccentricity along superior, inferior, and temporal meridians, and up to 15° eccentricity along the nasal meridian; thereafter, the areas of interest incremented in 2° steps (Supplementary Fig. S1). For each area of interest along each wedge, the coverage of local NIR-RAFI heterogeneity was determined manually by a single grader (Supplementary Fig. S1). The percent coverage of local heterogeneity was plotted as a function of eccentricity and the furthest eccentricity demonstrating a transition to a criterion threshold of 10% coverage was defined as the LDF detectable by NIR-RAFI.
Results
Earliest Stages of Disease in ABCA4-RD
Short wavelength–RAFI (or its higher light level equivalent fundus autofluorescence [FAF] imaging) of the macula is a readily available and a convenient method for evaluating natural lipofuscin accumulation disturbances described in ABCA4-RD. The RAFI method provides the same information content while reducing the patient discomfort from bright lights and minimizing the likelihood of disease acceleration in light-sensitive retinas.16,21 Short wavelength–RAFI imaging of the macula can demonstrate the extent of the lipofuscin disturbance in early macular disease such as P12 and P11 (Fig. 1A). The central lesion tends to expand over time as demonstrated by P64 and P47, and eventually fills the entire macula as demonstrated by P90 (Fig. 1A).
Figure 1.

The retinal location of the LDF in the wide spectrum of severity encountered in ABCA4-RD. (A) Short-wavelength excited RAFI of the macula in a healthy subject and five ABCA4-RD patients selected to represent increasingly wider expansion of disease from the fovea. The apparent boundaries of the diseased area are visible for P12, P11, and P64, but extend beyond the field of view of imaging for P47 and P90. (B) Ultra–wide-angle NIR excited RAFI (main images) demonstrate similarities and differences in the extent of the LDF in P72 and P49 in whom the macular SW-RAFI (insets) appear to show similar disease severity. Square region on NIR-RAFI delimits the location of the SW-RAFI shown. (C, D) Comparison of SW-RAFI and NIR-RAFI in two representative patients P58 and P55. Shown on the right are OCTs along the vertical meridian crossing the fovea, and quantitative classification of the OCT layers in terms of ONL thickness, ISe existence, and rod-outer segment (ROS) thickness. Also shown on the right are rod-mediated sensitivity loss (RSL) and cone-mediated sensitivity loss (CSL) along the vertical meridian.18 All data are classified into normal (white) and abnormal (black) regions. Vertical dashed lines show the location of the data shown on the right. Horizontal dashed lines delimit the greatest extent of abnormality detectable within the data shown in the right panels. Relevant layers and boundaries on the OCT are painted for visibility (ONL = blue; ISe = yellow; RPE = orange). All images are shown as equivalent right eyes and individually contrast stretched to allow visibility of features.
Wide-angle lipofuscin imaging beyond the macula is possible, however, longer imaging times with bright lights repeated to cover overlapping regions across the retina adds to patient discomfort and negates any advantages provided by RAFI methods. Ultra-wide imaging methods can be used but the lower spatial resolution in the macular region makes it difficult to follow central progression. An alternative method for evaluating RPE disturbances takes advantage of the autofluorescence of melanin-based pigments with NIR-RAFI.16,21,40,41 Near-infrared–RAFI appears as a dim red light and can be performed across the retina with no patient discomfort. Wide-angle NIR-RAFI of P72 and P49 (Fig. 1B, main panels) demonstrate how two patients of similar age and with similar macular SW-RAFI appearance (Fig. 1B, insets) can have substantially different distribution of disease across and within the retina. Both patients show extended regions of signal with spatial heterogeneity that transitions to brighter signal and spatial homogeneity in the midperipheral regions. The extent of the heterogeneity is similar in the nasal and inferior directions but substantially different in the temporal retina. Superior retina also shows substantial differences except for a small island of heterogeneity in P72 along the vertical meridian that is at the same eccentricity as the extent of disease in P49 (Fig. 1B, right).
The transition from spatial heterogeneity to homogeneity apparent on NIR-RAFI either matches or leads the earliest abnormalities detectable on other modalities as demonstrated with two examples. P58 at age 62 represents patients with a maculopathy that shows a central region of signal loss with SW- and NIR-RAFI, which is consistent with end stage of disease with total atrophy of the photoreceptor and RPE cells (Fig. 1C). Surrounding the region of end stage disease is an annular region of heterogeneity mostly with brighter spots on SW-RAFI and mostly darker spots on NIR-RAFI implying abnormalities in RPE pigmentation in small clusters. Photoreceptor structure (outer nuclear layer [ONL] and OS thickness and inner-segment ellipsoid [ISe] detectability) tends to be abnormal in this annular region. The transition from atrophy to detectable RPE and photoreceptor structure represents the trailing disease front, which is expected to expand peripherally over time. Surrounding the heterogenous annulus is a region of relative homogeneity on SW-RAFI and NIR-RAFI corresponding to normal-appearing photoreceptor structure and RPE pigmentation. The transition from heterogeneity to homogeneity likely represents the LDF.
P55 at age 55 represents patients with abnormal regions extending beyond the macula (Fig. 1D). The central region of RPE atrophy is smaller than P58, but there is a larger annulus of heterogeneity extending well beyond the vascular arcades. The region of heterogeneity is mostly associated with abnormal photoreceptor structure. Toward the peripheral edges of the annular region of heterogeneity, photoreceptor structure becomes normal suggesting an LDF substantially separated from the trailing front. In the remainder of the current work, the LDF in ABCA4-RD will be estimated using wide angle NIR-RAFI, which was obtained at far enough eccentricities to allow visibility of a clear transition from a heterogeneous appearance to a homogeneous and brighter appearance.
Expansion of the LDF in ABCA4-RD
Qualitatively, NIR-RAFI in ABCA4-RD shows a region of heterogeneity that often appears darker than normal with rare brighter spots intermingled. The LDF was defined quantitatively along the four cardinal meridians as the eccentricity at which retinal loci with heterogeneity cross the criterion threshold of 10% spatial coverage (Supplementary Fig. S1). Analyses were performed in 44 patients with two ABCA4 variants likely to cause the retinal disease (Supplementary Table S1). The segregation of alleles could be confirmed in 37 patients. Included in the analyses were a subset of 26 patients (ages at first visit: 7–55 years, mean, 27.3 years; median, 24.8 years) with long-term serial data available (mean follow-up interval: 5.9 years; range, 2.2–9.1 years; median, 5.4 years). Some patients, like P34, demonstrated modest expansion of the LDF by 2° to 6.5° over 9 years (Fig. 2A), whereas others, like P74, showed much greater expansion up to 19° over 7 years of follow-up (Fig. 2B).
Figure 2.

Long-term progression of the LDF as determined from wide-angle NIR-RAFI performed serially in ABCA4-RD along the superior, inferior, and temporal meridians. (A) Representative progression in a patient (P34) with a central macular lesion; NIR-RAFI recorded over a 9-year interval at ages 30 and 39. (B) Representative progression in a patient (P74) with a lesion extending well beyond the macula; NIR-RAFI recorded over a 7-year interval at ages 22 and 29. (C–E) Eccentricity of the LDF in ABCA4-RD as a function of age along the superior (C), inferior (D), and temporal (E) meridians. Symbols connected by lines represent serial follow-up in the same eye at two visits.
Initially, quantitative analyses were performed along superior, inferior, and temporal meridians; the complexities of the nasal meridian will be considered separately below. The LDF along the superior meridian ranged from 3.9° to 69.8° (Fig. 2C). Along the inferior meridian the LDF ranged from 3.5° to 52.3° (Fig. 2D), whereas in the temporal retina the range was 4.6° to 64.4° (Fig. 2E). These data illustrate the wide spectrum of LDF results encountered in ABCA4-RD in the current work. In the subset of patients with longitudinal data, younger ages tended to display steeper progression than older ages (Figs. 2C–E, connected symbols). The average rates of expansion were 1.64°, 1.10°, and 1.52°/y along the superior, inferior, and temporal meridians, respectively. Among the three nonnasal meridians, the largest rate of expansion tended to be more along superior and temporal meridians as compared with the inferior meridian (36%, 41% vs. 23% of the eyes, respectively). When only the meridian with the largest rate of expansion was considered for each patient, the average rate of expansion was 2.0°/y.
Emergence and Progression of Early Disease in the Nasal Retina
Nasal to the fovea, human retinas have a natural discontinuity due to the optic nerve head (ONH), an approximately 5° diameter round structure centered on average 16° nasal and 2° superior to the fovea. The ONH consists of nerve fibers leading from the ganglion cells across the retina to the central nervous system and does not have photoreceptors or RPE. We have previously shown that an approximately 2°-wide annulus centered approximately 4° from the center of the ONH is protected from the progressive retinal degeneration in ABCA4-RD.15 The underlying reason for this protection remains unknown to date. Due to the combination of the ONH and the parapapillary preservation, determination of the disease progression in the nasal meridian is more complex than the other three meridians. In all patients, there is an inner nasal LDF defined along the meridian between the fovea and ONH. In a subset of patients with extramacular disease, there is also an outer nasal LDF located nasal to the ONH (Supplementary Fig. S1).
P82 represented patients where at the first visit there was only an inner nasal LDF and no evidence of disease peripheral to the ONH; 3.5 years later, the inner nasal LDF had expanded modestly by 0.23°/y and there was surprising emergence of local heterogeneity beyond the ONH, which was not contiguous with the central disease (Fig. 3A). P32 represented the next stage where punctate abnormalities apparent beyond the ONH at the first visit progressed to a bona fida outer nasal LDF 5 years later (Fig. 3B). P52 was representative of the patients with both inner and outer nasal LDFs at the first visit that both expanded and merged over 8 years to result in an appearance of parapapillary preservation within a wide expanse of disease (Fig. 3C). And P72 showed an example of substantial expansion of the outer nasal LDF over a 5-year follow-up period (Fig. 3D).
Figure 3.

Serial wide-angle NIR-RAFI demonstrates different progression patterns observed along the nasal meridian in representative ABCA4-RD patients (A–D) and summary of all patients (E, F). (A) De novo appearance of outer nasal disease over 4 years in P82. (B) Formation of LDF in the outer nasal region over 5 years in P32. (C, D) Expansion of the LDF in the outer nasal region over 8 and 5 years, in P52 and P72, respectively. All eyes are shown as equivalent right eyes and individually contrast stretched. (E, F) Eccentricity of the LDF in the inner and outer nasal retina in all patients as a function of age. Symbols connected by lines represent serial follow-up in the same eye.
The inner nasal LDF, by definition, was delimited by the fovea and the ONH and appropriately the patients' results ranged from 4.3° to 16.5° (Fig. 3E). Of the patients with serial data, 27% did not show detectable progression at the inner nasal LDF during the follow-up period (Fig. 3E). On average, the expansion rate was 0.35°/y, which was substantially lower than the superior, inferior, and temporal expansion rates. The eccentricity at the outer nasal LDF ranged from 19° to 75° (Fig. 3F). In a subset of 16 patients with detectable outer nasal LDF at two visits, there was substantial progression in each patient (Fig. 3F); the average expansion rate was 1.98°/y, which was equivalent to the maximal expansion rate of 2.0°/y measured along nonnasal meridians. Another six patients showed only an inner nasal LDF at the first visit and developed an outer nasal LDF on the second visit. Considering the evidence that outer nasal LDF can develop independently from the inner nasal LDF (Figs. 3A, 3B), the progression rate was not calculated for these patients.
Predictive Model of Spatiotemporal Disease Progression
The initial and final eccentricities of the LDFs along the superior, inferior, and temporal (Figs. 2C–E) meridians show a wide range of disease progression rates across ABCA4-RD patients studied. Progression rate tended to be higher for patients first examined at earlier ages compared with those first examined at later ages (Fig. 4A, symbols). In addition, there appeared to be a tendency for those with relatively smaller lesions at first visit (Fig. 4A, red symbols) to expand slower compared with those with relatively larger lesions (Fig. 4A, green symbols) at any given age. Along the nasal meridian, the progression rate of the outer nasal LDF also appeared to be higher for patients first examined at earlier ages. To mathematically model the progression data a formula with exponential and linear components was considered
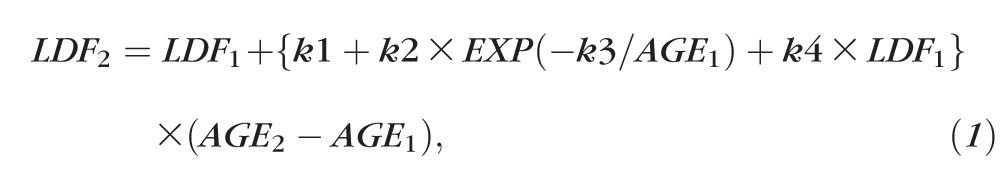 |
where LDF1 and LDF2 are the eccentricities (in degrees) of the LDF at the first and second time points, respectively, AGE1 and AGE2 (in years) are the ages at first and second time points, respectively, k1, k2, k3, and k4 are parameters. The term in curly brackets defines the rate of expansion of LDF (in °/y). All available serial data along superior, inferior, and temporal meridians (symbols in Fig. 4A) were used with a least squares fitting algorithm (Excel Solver; Microsoft Office 2013, Redmond, WA, USA) to estimate a single set of four model parameters as: k1 = −0.095 °/y, k2 = 25.25 °/y, k3 = 0.18 yrs−1, and k4 = 0.049 °/y. The use of the more limited data from the outer nasal boundary resulted in a different set of model parameters as: k1 = 0.61 °/y, k2 = 4.41 °/y, k3 = 0.06 yrs−1, and k4 = 0.008 °/y. Family of curves produced by the model (Fig. 4A, lines) appear to describe the data well.
Figure 4.

Model of the centrifugal expansion of the LDF in ABCA4-RD. (A) Measured rates of expansion of the LDF along superior, inferior, or temporal meridians as a function of age (symbols). Distinguished are the patients with the extent of the LDF at the first visit with less than 15° (red symbols) from those with greater than 15° (green symbols). Lines represent the model of disease expansion as function of age for a range of initial disease extents between 5° and 60°. (B) Comparison of the LDF eccentricity predicted from the model to that actually measured in the eyes with long-term serial data. Thin line is the linear regression fit to the data and the thicker line represents ideal match between prediction and measurement. (C) Representative predictions of the model of progression over a 3-year interval for hypothetical patients at age 10 (left two schematics) or age 30 (right two schematics), enrolling in a hypothetical clinical trial with an initial maculopathy (first and third schematics) or with extramacular disease (second and fourth schematics).
To better evaluate the accuracy of the model, the LDF at the first time point, and the ages at first and second time points from all patients were used to predict the LDF at the second time point; this prediction was directly compared to the actual measurement of the LDF at the second time point (Fig. 4B). Model predictions were well matched to the measurement (r2 = 0.94) and the intercept (1.7°) and slope (0.94) of linear regression (Fig. 4B, thinner line) were not substantially different than an ideal result zero intercept and unity slope (Fig. 4B, thicker line).
Predictive models can be particularly useful as tools for enrollment into clinical treatment trials as well as evaluation of effectiveness of a potential treatment in modifying the natural history of the underlying condition. Schematic drawings demonstrate the predicted spatiotemporal natural history of ABCA4-RD disease based on the model developed in the current study (Fig. 4C). For these schematics we assumed a 12° eccentricity difference between the outer nasal LDF and the three other meridians based on the average difference found in 33 eyes where both types of data were available. For a hypothetical 10-year-old ABCA4-RD patient with a LDF of 7° along the superior, temporal, and inferior meridians at first visit, the model would predict an ‘explosive' expansion of the LDF to 20° in 3 years (Fig. 4C, first schematic); an initial 20° eccentric LDF would be expected to expand to 34.9° in the same period (Fig. 4C, second schematic). Along the outer nasal meridian, the expansion from 19° would be to 28.6° and from 32° to 41.9°.
For a hypothetical 30-year-old ABCA4-RD patient with similar initial LDFs, predicted expansions would be much smaller. Specifically, along the superior, temporal, and inferior meridians an LDF initially at 7° would be expected to expand only to 8.1° (Fig. 4C, third schematic), whereas an initial 20° would be expected to expand to 23° over 3 years (Fig. 4C, fourth schematic). Along the outer nasal meridian, the expansion from 19° would be to 23.5° and from 32° to 36.8°. Depending on the test–retest variability of the evaluation method used, our model can be used to define enrollment criteria that would be expected to show detectable progression of LDF for a given duration of a clinical trial.
An alternative is to use the model to predict the duration for an LDF initially at 7° (typical result for a maculopathy-only stage of disease) to expand to 30° (definite extramacular disease). Along the nonnasal meridians, for a patient who is 10 years old at initial visit, the predicted duration is 7.4 years, whereas for a patient who is 30 years old at initial visit, the predicted duration is 16.4 years. Interestingly, for a 50-year-old, the predicted duration for a 7° maculopathy to expand to extramacular disease is 25.8 years. These results conform to the clinical adage of better visual prognosis for older Stargardt patients with ophthalmoscopic abnormalities limited to the macula.42–45
Yet another use of the model is to estimate the age when foveal disease started in each patient. Although the extrapolation over decades is frought with uncertainty, model results could provide estimates of severity for different genotypes. For example, among the 14 patients compound heterozygous for the common G1961E allele (Supplementary Table S1), the model would predict foveal disease in the first decade of life for three alleles (P68L;G1961E, L541P;A1038V, and T1019M), second decade of life for four alleles (R1129L, C54Y, R152Stop, and V256V) and fourth decade of life for four alleles (P1380L, R1640Q, c.5312+1 G>A, and M669fs).
Discussion
The earliest stage of disease in human ABCA4-RD is not well understood but abnormalities reported to date include changes in the RPE pigments lipofuscin and melanin, photoreceptor inner and outer segments, external limiting membrane (ELM), nuclei, and loss of light sensitivity of rods and cones.14,15,18,19,21,46–49 The end stage of human disease in ABCA4-RD, on the other hand, is well established and involves degeneration of all photoreceptors and atrophy of all RPE cells within spatially delimited retinal zones.14,16,21,50–53 Cross-sectional and longitudinal studies suggest that retinal loci transition progressively, and often slowly, from a normal structure and function to early stages of disease followed by intermediate and late stages.
In the current work, we used ultra–wide-angle coverage with NIR-RAFI to evaluate RPE melanization abnormalities in the full ABCA4-RD spectrum ranging from abnormalities limited to the macula only to disease across wide expanses of the retina. We defined the LDF as the transition from local heterogeneity of signal to homogeneity and measured its eccentricity along four cardinal meridians. We found that the eccentricity of the LDF varied continuously. We did not find supportive evidence of a bimodal distribution of eccentricities that could have been hypothesized to separate maculopathy from retina-wide disease phenotypes, such as implied previously by electrophysiological analyses.54,55 Spatiotemporal pattern of most ABCA4-RD suggests a wave of disease that centrifugally sweeps across the retina over years or decades as patients progress from maculopathy to retina-wide disease.13,45,56 Both cell-autonomous and extrinsic factors likely contribute to the natural history of disease to generate the centrifugal progression.57–61 The existence of cell-autonomous factors are supported by the initial onset of disease at the foveal/parafoveal region in all ABCA4-RD reported to date as well as the onset of disease nasal to the ONH spatially unattached from macular disease as demonstrated in the current work. Incremental expansion of the LDF from degenerate areas to neighboring healthier areas on the other hand likely involves contributions from both extrinsic as well as cell-autonomous factors. Pathophysiological bases within rod or cone photoreceptors or RPE driving specific extrinsic and cell-autonomous factors in ABCA4-RD remain mostly unknown.
Longitudinal observations of disease progression in ABCA4-RD are rare. Our studies concentrating on early RPE disease represented by the LDF and performed over an average follow-up period of approximately 6 years showed an average radial expansion rate of approximately 2°/y both along the maximally expanding nonnasal (superior or inferior or temporal) meridian as well as along the outer nasal meridian. Several other longitudinal studies of RPE structure have been reported but in each case the concentration was on the end stage of disease where investigators measured the growth of RPE atrophy within the macula and reported it in terms of areal enlargement rate.62–64 For comparison with our work, we can calculate the effective radial expansion of circular-equivalent atrophy boundaries from average areas reported and their expansion. In one study of 13 patients followed for 3 years, average radial expansion of atrophy corresponded to 0.38°/y.62 In another study of 12 patients followed for 3 years, radial expansion of the atrophy boundary corresponded to 0.53°/y (assuming a typical atrophy area of 7 mm2, which was not specified by the authors).63 In a more recent study of 67 patients, atrophy growth corresponded to radial expansion rates of 0.4 and 0.8°/y depending on different autofluoresence subtypes.64 Taking these results together, it appears that the LDF measured in the current work is expanding on average at least at twice as high a rate as compared with the expansion of the RPE atrophy reported previously. Pathophysiological basis of the implied differences in progression of early disease, as measured here, and late disease, as more commonly measured, is currently unknown.
We previously reported on the peripheral rod and cone dysfunction in ABCA4-RD measured longitudinally over 8.7 years.18 Results were consistent with two components summing to contribute to the spatiotemporal progression of the dysfunction: a central-to-peripheral gradient and a uniform retina-wide pattern. In the subset of patients showing evidence of peripheral dysfunction, the implied rate of radial expansion of early rod and cone visual dysfunction was estimated to be 3°/y,18 which is similar to the maximum LDF expansion rate of 2.6°/y in the comparable subset of 14 patients analyzed here with an initial LDF at greater than 30°. Thus, even though a direct structure–function correlation was not within the scope of the current work, expansion of the LDF appears to parallel the centrifugal progression of early rod and cone dysfunction.
Ultra–wide-angle imaging is used to visualize simultaneously the central as well as the peripheral retina.65 Scanning laser ophthalmoscope–based systems have been used to image color fundus and SW FAF imaging of the peripheral retina in AMD,66 retinitis pigmentosa,67,68 and cone–rod dystrophy.69 There was a general correspondence between peripheral autofluorescence abnormalities and visual fields.67–69 We are not aware of ultra–wide-angle imaging in Stargardt disease, or ultra–wide-angle NIR-RAFI for any retinal disease published to date. Future studies quantifying far peripheral LDF may have to first measure and then computationally account for the warping introduced by the specific combinations of lenses and imaging systems.70,71
There was an ‘explosive' expansion of the LDF in several younger patients such as P28 at age 9, P49 at age 14, and siblings P71 and P72 at ages 12 and 16, respectively. Rapid enlargement of scotoma size in younger patients with early-onset Stargardt disease have been previously reported.72 In one large study of Stargardt patients, loss of visual acuity was found to progress much faster for patients initially seen in the first 2 decades of life compared with those first evaluated at later ages.73 Patients with early-onset Stargardt disease also showed structural changes of the ELM, which was not observed in later onset disease.46 In other studies of early-onset Stargardt, RPE atrophy was noted to expand from the macula to beyond the vascular arcades.48,74 Taken together considerations of comfortable imaging and the potential for measurements involving large changes over short follow-up periods, wide-angle NIR-RAFI may be especially useful for following the progression of the early disease in early-onset forms of ABCA4-RD.
There have been many challenges in producing animal models that represent well the disease stages of ABCA4-RD observed in humans. Pigmented Abca4 knockout and L541P;A1038V knockin mice show substantial accumulation of lipofuscin in the RPE but show little, if any, retinal degeneration.61,75 Albino Abca4 knockout mice show both lipofuscin accumulation and a slow photoreceptor degeneration but similarity of photoreceptor loss in homozygote and heterozygote animals do not match the normal retinas of human heterozygote carriers.76–78 Abca4-Rdh8 double-knockout mice show hypersensitivity to acute light damage which has not been observed in human patients.59 Thus, it is currently difficult to extrapolate from preclinical experimental therapies what component and which stages of human disease may successfully be treated with different approaches. Simultaneous recording of both the leading and trailing disease fronts and their progression may be one practical strategy to follow in future clinical trials.
Supplementary Material
Acknowledgments
Supported by National Eye Institute Grant EY013203 (Bethesda, MD, USA), Foundation Fighting Blindness (Owings Mills, MD, USA), Hope for Vision (Aventura, FL, USA), Research to Prevent Blindness (New York, NY, USA), and Macula Vision Research Foundation (West Conshohocken, PA, USA).
Disclosure: A.V. Cideciyan, None; M. Swider, None; S.B. Schwartz, None; E.M. Stone, None; S.G. Jacobson, None
References
- 1. Bramall AN,, Wright AF,, Jacobson SG,, McInnes RR. The genomic biochemical, and cellular responses of the retina in inherited photoreceptor degenerations and prospects for the treatment of these disorders. Annu Rev Neurosci. 2010; 33: 441–742. [DOI] [PubMed] [Google Scholar]
- 2. Allikmets R,, Singh N,, Sun H,, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997; 15: 236–246. [DOI] [PubMed] [Google Scholar]
- 3. Martínez-Mir A,, Paloma E,, Allikmets R,, et al. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet. 1998; 18: 11–12. [DOI] [PubMed] [Google Scholar]
- 4. Cremers FP,, van de Pol DJ,, van Driel M,, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR. Hum Mol Genet. 1998; 7: 355–362. [DOI] [PubMed] [Google Scholar]
- 5. Maugeri A,, Klevering BJ,, Rohrschneider K,, et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet. 2000; 67: 960–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Briggs CE,, Rucinski D,, Rosenfeld PJ,, Hirose T,, Berson EL,, Dryja TP. Mutations in ABCR (ABCA4) in patients with Stargardt macular degeneration or cone-rod degeneration. Invest Ophthalmol Vis Sci. 2001; 42: 2229–2236. [PubMed] [Google Scholar]
- 7. Braun TA,, Mullins RF,, Wagner AH,, et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum Mol Genet. 2013; 22: 5136–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Molday LL,, Rabin AR,, Molday RS. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat Genet. 2000; 25: 257–258. [DOI] [PubMed] [Google Scholar]
- 9. Quazi F,, Lenevich S,, Molday RS. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun. 2012; 3: 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsybovsky Y,, Orban T,, Molday RS,, Taylor D,, Palczewski K. Molecular organization and ATP-induced conformational changes of ABCA4 the photoreceptor-specific ABC transporter. Structure. 2013; 21: 854–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sparrow JR,, Boulton M. RPE lipofuscin and its role in retinal pathobiology. Exp Eye Res. 2005; 80: 595–606. [DOI] [PubMed] [Google Scholar]
- 12. Maeda A,, Maeda T,, Golczak M,, et al. Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J Biol Chem. 2009; 284: 15173–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen Y,, Okano K,, Maeda T,, et al. Mechanism of all-trans-retinal toxicity with implications for Stargardt disease and age-related macular degeneration. J Biol Chem. 2012; 287: 5059–5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cideciyan AV,, Aleman TS,, Swider M,, et al. Mutations in ABCA4 result in accumulation of lipofuscin before slowing of the retinoid cycle: a reappraisal of the human disease sequence. Hum Mol Genet. 2004; 13: 525–534. [DOI] [PubMed] [Google Scholar]
- 15. Cideciyan AV,, Swider M,, Aleman TS,, et al. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Invest Ophthalmol Vis Sci. 2005; 46: 4739–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cideciyan AV,, Swider M,, Aleman TS,, et al. Reduced-illuminance autofluorescence imaging in ABCA4-associated retinal degenerations. J Opt Soc Am A Opt Image Sci Vis. 2007; 24: 1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aleman TS,, Cideciyan AV,, Windsor EA,, et al. Macular pigment and lutein supplementation in ABCA4-associated retinal degenerations. Invest Ophthalmol Vis Sci. 2007; 48: 1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cideciyan AV,, Swider M,, Aleman TS,, et al. ABCA4 disease progression and a proposed strategy for gene therapy. Hum Mol Genet. 2009; 18: 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cideciyan AV,, Swider M,, Aleman TS,, et al. Macular function in macular degenerations: repeatability of microperimetry as a potential outcome measure for ABCA4-associated retinopathy trials. Invest Ophthalmol Vis Sci. 2012; 53: 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang WC,, Cideciyan AV,, Roman AJ,, et al. Inner and outer retinal changes in retinal degenerations associated with ABCA4 mutations. Invest Ophthalmol Vis Sci. 2014; 55: 1810–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cideciyan AV,, Swider M,, Jacobson SG. Autofluorescence imaging with near-infrared excitation: normalization by reflectance to reduce signal from choroidal fluorophores. Invest Ophthalmol Vis Sci. 2015; 56: 3393–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sieving PA,, Chaudhry P,, Kondo M,, et al. Inhibition of the visual cycle in vivo by 13-cis retinoic acid protects from light damage and provides a mechanism for night blindness in isotretinoin therapy. Proc Natl Acad Sci U S A. 2001; 98: 1835–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Radu RA,, Mata NL,, Nusinowitz S,, Liu X,, Sieving PA,, Travis GH. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt's macular degeneration. Proc Natl Acad Sci U S A. 2003; 100: 4742–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maiti P,, Kong J,, Kim SR,, Sparrow JR,, Allikmets R,, Rando RR. Small molecule RPE65 antagonists limit the visual cycle and prevent lipofuscin formation. Biochemistry. 2006; 45: 852–860. [DOI] [PubMed] [Google Scholar]
- 25. Kubota R,, Boman NL,, David R,, Mallikaarjun S,, Patil S,, Safety Birch D. and effect on rod function of ACU-4429, a novel small-molecule visual cycle modulator. Retina. 2012; 32: 183–188. [DOI] [PubMed] [Google Scholar]
- 26. Dobri N,, Qin Q,, Kong J,, et al. A1120, a nonretinoid RBP4 antagonist, inhibits formation of cytotoxic bisretinoids in the animal model of enhanced retinal lipofuscinogenesis. Invest Ophthalmol Vis Sci. 2013; 54: 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kubota R,, Al-Fayoumi S,, Mallikaarjun S,, Patil S,, Bavik C,, Chandler JW. Phase 1 dose-ranging study of emixustat hydrochloride (ACU-4429), a novel visual cycle modulator, in healthy volunteers. Retina. 2014; 4: 603–609. [DOI] [PubMed] [Google Scholar]
- 28. Dugel PU,, Novack RL,, Csaky KG,, Richmond PP,, Birch DG,, Kubota R. Phase II randomized, placebo-controlled, 90-day study of emixustat hydrochloride in geographic atrophy associated with dry age-related macular degeneration. Retina. 2015; 35: 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang J,, Kiser PD,, Badiee M,, et al. Molecular pharmacodynamics of emixustat in protection against retinal degeneration. J Clin Invest. 2015; 125: 2781–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kong J,, Kim SR,, Binley K,, et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Ther. 2008; 15: 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Allocca M,, Doria M,, Petrillo M,, et al. Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J Clin Invest. 2008; 118: 1955–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Binley K,, Widdowson P,, Loader J,, et al. Transduction of photoreceptors with equine infectious anemia virus lentiviral vectors: safety and biodistribution of StarGen for Stargardt disease. Invest Ophthalmol Vis Sci. 2013; 54: 4061–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma L,, Kaufman Y,, Zhang J,, Washington I. C20-D3-vitamin A slows lipofuscin accumulation and electrophysiological retinal degeneration in a mouse model of Stargardt disease. J Biol Chem. 2011; 286: 7966–7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Charbel Issa P,, Barnard AR,, Herrmann P,, Washington I,, MacLaren RE. Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin A dimerization. Proc Natl Acad Sci U S A. 2015; 112: 8415–8520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Julien S,, Schraermeyer U. Lipofuscin can be eliminated from the retinal pigment epithelium of monkeys. Neurobiol Aging. 2012; 33: 2390–2397. [DOI] [PubMed] [Google Scholar]
- 36. Nociari MM,, Lehmann GL,, Perez Bay AE,, et al. Beta cyclodextrins bind, stabilize, and remove lipofuscin bisretinoids from retinal pigment epithelium. Proc Natl Acad Sci U S A. 2014; 111: E1402–E1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maeda A,, Golczak M,, Chen Y,, et al. Primary amines protect against retinal degeneration in mouse models of retinopathies. Nat Chem Biol. 2011; 8: 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen Y,, Palczewska G,, Mustafi D,, et al. Systems pharmacology identifies drug targets for Stargardt disease-associated retinal degeneration. J Clin Invest. 2013; 123: 5119–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sabirzhanova I,, Lopes-Pacheco M,, Rapino D,, et al. Rescuing trafficking mutants of the ATP binding cassette protein, ABCA4, with small molecule correctors as a treatment for Stargardt eye disease. J Biol Chem. 2015; 290: 19374–193755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Keilhauer CN,, Delori FC. Near-infrared autofluorescence imaging of the fundus: visualization of ocular melanin. Invest Ophthalmol Vis Sci. 2006; 47: 3556–3564. [DOI] [PubMed] [Google Scholar]
- 41. Gibbs D,, Cideciyan AV,, Jacobson SG,, Williams DS. Retinal pigment epithelium defects in humans and mice with mutations in MYO7A: imaging melanosome-specific autofluorescence. Invest Ophthalmol Vis Sci. 2009; 50: 4386–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Noble KG,, Carr RE. Stargardt's disease and fundus flavimaculatus. Arch Ophthalmol. 1979; 97: 1281–1285. [DOI] [PubMed] [Google Scholar]
- 43. Armstrong JD,, Meyer D,, Xu S,, Elfervig JL. Long-term follow-up of Stargardt's disease and fundus flavimaculatus. Ophthalmology. 1998; 105: 448–457. [DOI] [PubMed] [Google Scholar]
- 44. Oh KT,, Weleber RG,, Oh DM,, Billingslea AM,, Rosenow J,, Stone EM. Clinical phenotype as a prognostic factor in Stargardt disease. Retina. 2004; 24: 254–262. [DOI] [PubMed] [Google Scholar]
- 45. Walia S,, Fishman GA. Natural history of phenotypic changes in Stargardt macular dystrophy. Ophthalmic Genet. 2009; 30: 63–68. [DOI] [PubMed] [Google Scholar]
- 46. Lee W,, Nõupuu K,, Oll M,, et al. The external limiting membrane in early-onset Stargardt disease. Invest Ophthalmol Vis Sci. 2014; 55: 6139–6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Duncker T,, Marsiglia M,, Lee W,, et al. Correlations among near-infrared and short-wavelength autofluorescence and spectral-domain optical coherence tomography in recessive Stargardt disease. Invest Ophthalmol Vis Sci. 2014; 55: 8134–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lambertus S,, van Huet RA,, Bax NM,, et al. Early-onset Stargardt disease: phenotypic and genotypic characteristics. Ophthalmology. 2015; 122: 335–344. [DOI] [PubMed] [Google Scholar]
- 49. Greenstein VC,, Schuman AD,, Lee W,, et al. Near-infrared autofluorescence: its relationship to short-wavelength autofluorescence and optical coherence tomography in recessive Stargardt disease. Invest Ophthalmol Vis Sci. 2015; 56: 3226–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Eagle RC,, Jr, Lucier AC,, Bernardino VB,, Jr, Yanoff M. Retinal pigment epithelial abnormalities in fundus flavimaculatus: a light and electron microscopic study. Ophthalmology. 1980; 87: 1189–1200. [DOI] [PubMed] [Google Scholar]
- 51. Birnbach CD,, Järveläinen M,, Possin DE,, Milam AH. Histopathology and immunocytochemistry of the neurosensory retina in fundus flavimaculatus. Ophthalmology. 1994; 101: 1211–1219. [DOI] [PubMed] [Google Scholar]
- 52. Fishman GA,, Stone EM,, Grover S,, Derlacki DJ,, Haines HL,, Hockey RR. Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Arch Ophthalmol. 1999; 117: 504–510. [DOI] [PubMed] [Google Scholar]
- 53. Bonilha VL,, Rayborn ME,, Bell BA,, Marino MJ,, Fishman GA,, Hollyfield JG. Retinal histopathology in eyes from a patient with Stargardt disease caused by compound heterozygous ABCA4 mutations [ published online ahead of print August 21, 2015]. Ophthalmic Genet. doi:http://dx.doi.org/10.3109/13816810.2014.958861. [DOI] [PubMed]
- 54. Lois N,, Holder GE,, Bunce C,, Fitzke FW,, Bird AC. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol. 2001; 119: 359–369. [DOI] [PubMed] [Google Scholar]
- 55. Fujinami K,, Lois N,, Davidson AE,, et al. A longitudinal study of stargardt disease: clinical and electrophysiologic assessment, progression, and genotype correlations. Am J Ophthalmol. 2013; 155: 1075–1088.e13. [DOI] [PubMed] [Google Scholar]
- 56. Zahid S,, Jayasundera T,, Rhoades W,, et al. Clinical phenotypes and prognostic full-field electroretinographic findings in Stargardt disease. Am J Ophthalmol. 2013; 155: 465–473.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ripps H. Cell death in retinitis pigmentosa: gap junctions and the ‘bystander’ effect. Exp Eye Res. 2002; 74: 327–336. [DOI] [PubMed] [Google Scholar]
- 58. Cronin T,, Léveillard T,, Sahel JA. Retinal degenerations: from cell signaling to cell therapy; pre-clinical and clinical issues. Curr Gene Ther. 2007; 7: 121–129. [DOI] [PubMed] [Google Scholar]
- 59. Maeda A,, Maeda T,, Golczak M,, Palczewski K. Retinopathy in mice induced by disrupted all-trans-retinal clearance. J Biol Chem. 2008; 283: 26684–26693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cho KI,, Haque M,, Wang J,, et al. Distinct and atypical intrinsic and extrinsic cell death pathways between photoreceptor cell types upon specific ablation of Ranbp2 in cone photoreceptors. PLoS Genet. 2013; 9: e1003555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang N,, Tsybovsky Y,, Kolesnikov AV,, et al. Protein misfolding and the pathogenesis of ABCA4-associated retinal degenerations. Hum Mol Genet. 2015; 24: 3220–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen B,, Tosha C,, Gorin MB,, Nusinowitz S. Analysis of autofluorescent retinal images and measurement of atrophic lesion growth in Stargardt disease. Exp Eye Res. 2010; 91: 143–152. [DOI] [PubMed] [Google Scholar]
- 63. McBain VA,, Townend J,, Lois N. Progression of retinal pigment epithelial atrophy in stargardt disease. Am J Ophthalmol. 2012; 154: 146–154. [DOI] [PubMed] [Google Scholar]
- 64. Fujinami K,, Lois N,, Mukherjee R,, et al. A longitudinal study of Stargardt disease: quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Invest Ophthalmol Vis Sci. 2013; 54: 8181–8190. [DOI] [PubMed] [Google Scholar]
- 65. Witmer MT,, Kiss S. Wide-field imaging of the retina. Surv Ophthalmol. 2013; 58: 143–154. [DOI] [PubMed] [Google Scholar]
- 66. Reznicek L,, Wasfy T,, Stumpf C,, et al. Peripheral fundus autofluorescence is increased in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2012; 53: 2193–2198. [DOI] [PubMed] [Google Scholar]
- 67. Oishi A,, Ogino K,, Makiyama Y,, Nakagawa S,, Kurimoto M,, Yoshimura N. Wide-field fundus autofluorescence imaging of retinitis pigmentosa. Ophthalmology. 2013; 120: 1827–1834. [DOI] [PubMed] [Google Scholar]
- 68. Ogura S,, Yasukawa T,, Kato A,, et al. Wide-field fundus autofluorescence imaging to evaluate retinal function in patients with retinitis pigmentosa. Am J Ophthalmol. 2014; 158: 1093–1098. [DOI] [PubMed] [Google Scholar]
- 69. Oishi M,, Oishi A,, Ogino K,, et al. Wide-field fundus autofluorescence abnormalities and visual function in patients with cone and cone-rod dystrophies. Invest Ophthalmol Vis Sci. 2014; 55: 3572–3577. [DOI] [PubMed] [Google Scholar]
- 70. Spaide RF. Peripheral areas of nonperfusion in treated central retinal vein occlusion as imaged by wide-field fluorescein angiography. Retina. 2011; 31: 829–837. [DOI] [PubMed] [Google Scholar]
- 71. Croft DE,, van Hemert J,, Wykoff CC,, et al. Precise montaging and metric quantification of retinal surface area from ultra-widefield fundus photography and fluorescein angiography. Ophthalmic Surg Lasers Imaging Retina. 2014; 45: 312–317. [DOI] [PubMed] [Google Scholar]
- 72. Itabashi R,, Katsumi O,, Mehta MC,, Wajima R,, Tamai M,, Hirose T. Stargardt's disease/fundus flavimaculatus: psychophysical and electrophysiologic results. Graefes Arch Clin Exp Ophthalmol. 1993; 231: 555–562. [DOI] [PubMed] [Google Scholar]
- 73. Rotenstreich Y,, Fishman GA,, Anderson RJ. Visual acuity loss and clinical observations in a large series of patients with Stargardt disease. Ophthalmology. 2003; 110: 1151–1158. [DOI] [PubMed] [Google Scholar]
- 74. Fujinami K,, Zernant J,, Chana RK,, et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology. 2015; 122: 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Weng J,, Mata NL,, Azarian SM,, Tzekov RT,, Birch DG,, Travis GH. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in ABCR knockout mice. Cell. 1999; 98: 13–23. [DOI] [PubMed] [Google Scholar]
- 76. Radu RA,, Yuan Q,, Hu J,, et al. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following Vitamin A supplementation. Invest Ophthalmol Vis Sci. 2008; 49: 3821–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Charbel Issa P,, Barnard AR,, Singh MS,, et al. Fundus autofluorescence in the Abca4(−/−) mouse model of Stargardt disease—correlation with accumulation of A2E, retinal function, and histology. Invest Ophthalmol Vis Sci. 2013; 54: 5602–5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sparrow JR,, Blonska A,, Flynn E,, et al. Quantitative fundus autofluorescence in mice: correlation with HPLC quantitation of RPE lipofuscin and measurement of retina outer nuclear layer thickness. Invest Ophthalmol Vis Sci. 2013; 54: 2812–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.