

Abstract
Objective
To assess potential effects of variants in six lipid modulating genes (SORT1, HMGCR, MLXIPL, FADS2, APOE and MAFB) on early development of dyslipidemia independent of the degree of obesity in children, we investigated their association with total (TC), low density lipoprotein (LDL-C), high density lipoprotein (HDL-C) cholesterol and triglyceride (TG) levels in 594 children. Furthermore, we evaluated the expression profile of the candidate genes during human adipocyte differentiation.
Results
Expression of selected genes increased 101 to >104 fold during human adipocyte differentiation, suggesting a potential link with adipogenesis. In genetic association studies adjusted for age, BMI SDS and sex, we identified significant associations for rs599839 near SORT1 with TC and LDL-C and for rs4420638 near APOE with TC and LDL-C. We performed Bayesian modelling of the combined lipid phenotype of HDL-C, LDL-C and TG to identify potentially causal polygenic effects on this multi-dimensional phenotype and considering obesity, age and sex as a-priori modulating factors. This analysis confirmed that rs599839 and rs4420638 affect LDL-C.
Conclusion
We show that lipid modulating genes are dynamically regulated during adipogenesis and that variants near SORT1 and APOE influence lipid levels independent of obesity in children. Bayesian modelling suggests causal effects of these variants.
Introduction
Alterations in blood lipid phenotypes culminating in dyslipidemia are important risk factors for the development of cardiovascular disease [1]. Elevated blood low-density lipoprotein cholesterol (LDL-C) and triglycerides (TG) are strongly related to the likelihood of existing or future coronary heart disease [2, 3], whereas elevated blood high-density lipoprotein cholesterol (HDL-C) has a protective effect [4]. The most common cause of dyslipidemia is obesity [5]. However, a relevant proportion of patients with elevated blood lipid levels does not show an abnormal BMI [6].
Meta-analyses of genome-wide association (GWA) studies revealed many genetic loci influencing blood lipid levels underlying the polygenic cause of dyslipidemia and thereby identified suspected as well as unsuspected new candidate genes [7–10]. However, these meta-analyses concern adult cohorts. So far, there are only very few data on selected genes associated with altered blood lipid phenotypes in children and adolescents [11–14].
Investigation of childhood cohorts has several advantages though. They are much less biased by chronic disease and treatments but already show considerable heterogeneity regarding blood lipid levels. Also, considering future prediction of developing dyslipidemia, it is important to assess whether associations between genetic variants and blood lipid phenotypes observed in adults are already evident in children and adolescents [15]. Due to the lower influence of co-morbidities and other life-style related factors, we suppose that primary genetic effects are stronger in children than in adults. Thus, we hypothesize that we can detect at least some of the variants even with the lower number of individuals available for childhood cohorts.
In the present study, we aimed at assessing associations of six variants with lipid traits in a sample of mainly obese children. Selected variants are located in or near the genes SORT1 (sortilin 1), HMGCR (3-hydroxy-3-methylglutaryl-Coenzyme A reductase), MLXIPL (MLX interacting protein), FADS2 (fatty acid desaturase 2), APOE (apolipoprotein E) and MAFB (V-maf musculoaponeurotic fibrosarcoma oncogene homolog B) for which high effect-sizes regarding lipid phenotypes were reported. Going beyond classical association analysis, we additionally performed a Bayesian modelling approach to identify unconfounded relationships between genetic and non-genetic covariables and lipid phenotypes. Considering that obesity is a risk factor for dyslipidemia per se and that adipose tissue is an important tissue for lipid metabolism, we also assessed a potential relationship of the candidate gene expression for adipogenesis by studying time-series of gene-expression during human adipocyte differentiation.
Methods
Selection of Candidate Genes and Variants
Genes were selected according to evidence of genotype-phenotype-associations established in meta-analyses of adult cohorts [8–10, 16, 17]. We prioritized genetic variants by applying a score integrating (i) GWAS for lipid genes and obesity (p-value), (ii) gene expression data from adipocytes, (iii) minor allele frequency and effect size, (iv) verification in replication analyses,. Based on these criteria, we selected six variants rs6102059 (MAFB), rs4420638 (APOE), rs599839 (SORT1), rs3846663 (HMGCR), rs174570 (FADS2) and rs3812316 (MLXIPL). We excluded well-known SNPs in genes like LDL-receptor because we were interested in new candidate genes influencing blood lipid levels.
Cis-eQTL effects of SNPs in linkage disequilibrium with our variants were observed for SORT1 [18–20], HMGCR [21] and FADS2 [21–23].
Sample
A total of 683 children were recruited from the Leipzig area via our out-patient obesity clinic. We applied German reference data for the calculation of the SDS as suggested by the National German Guidelines for Pediatric Obesity [24]. Obesity was defined as BMI SDS >1.88 corresponding to the 97th percentile.
White children were phenotyped by age, sex, height, weight, pubertal state, laboratory parameters and other clinical characteristics. Assessment of pubertal stage was performed by clinical examination according to Tanner [25, 26]. Blood lipid levels (triglycerides, total cholesterol, HDL and LDL) were determined with direct enzymatic colorimetric assays by the certified laboratory at the Institute of Laboratory Medicine, Clinical Chemistry and Molecular Diagnosis at the University of Leipzig. Written informed consent was obtained from all parents and from participants ≥12 years of age. This study has been approved by the ethics committee of the University of Leipzig and has been conducted according to the principles expressed in the Declaration of Helsinki (October 2000).
We excluded children with chronic inflammatory diseases, metabolic diseases, genetic disorders and diseases that required medication influencing lipid metabolism (N = 89).
For the remaining 594 children, data on glucose metabolism and lipid phenotypes (TC, LDL-C, HDL-C, TG) were available. Anthropometric and metabolic characterisation of included samples is presented in Table 1.
Table 1. Anthropometric and metabolic characterization of study samples.
| Sex (male / female) | 277 / 317 |
| n (non-obese / obese) | 122 / 472 |
| BMI SDS | 2.39 (0.85) |
| Age (years) | 11.67 (5.23) |
| Total Cholesterol (mmol/L) | 4.08 (0.99) |
| HDL-C (mmol/L) | 1.22 (0.37) |
| LDL-C (mmol/L) | 2.46 (0.89) |
| Triglyceride (mmol/L) | 0.99 (0.70) |
Quantitative variables are presented as median (interquartile range). Obesity is defined as BMI SDS>1.88.
Gene expression analysis during human adipocyte differentiation
Gene expression profiles of selected genes were determined via qRT-PCR for human preadipocyte SGBS (Simpson-Golabi-Behmel syndrome) cells during differentiation into mature adipocytes. Adipocyte differentiation was induced as described previously [27].
RNA extraction was performed using the RNeasy MiniKit (Qiagen, Hilden, Germany) including DNase digestion according to the manufacturer’s instructions. Reverse transcription of 50 ng/μl RNA was carried out using the M-MLV Reverse Transcriptase Kit (Invitrogen, Karlsruhe, Germany) with random hexamer [p(dN)6] primers (Roche, Basel, Switzerland). Primer sequences are provided in S1 Table.
Experiments were performed in three distinct experiments each in triplicates. Target gene-expression was normalized to the averaged expression of three housekeeping genes TBP, HPRT and USF2.
DNA-Isolation and genotyping
Fasting venous EDTA blood samples were stored at -80°C. After washing with phosphate buffered saline, erythrocyte depletion by NH4-lysis, and centrifugation, we extracted DNA using QIAmp DNA Blood MiniKit (Qiagen) according to the manufacturer’s manual.
Genotyping probes and primers were obtained from Applied Biosystems (Darmstadt, Germany). Primer sequences are listed in S2 Table. We used qPCR MasterMix Kit for probe Assay and Low Rox Plus (Eurogentec, Köln, Germany) for genotyping according to the manufacturers’ manuals. Genotyping was performed on ABI Prism 7500 sequence detector (Applied Biosystems, Lincoln, USA). At least 5% of all samples were re-assessed on a different plate with concordance rate of 100%. Genotype frequencies of all SNPs were consistent with Hardy-Weinberg equilibrium. SNP characteristics are presented in S3 Table.
Classical statistical analysis
In classical analysis of genetic data, every combination of a single SNP and a single phenotype is tested for association. Prior to analysis, lipid phenotypes TC, LDL-C, HDL-C and TG were log transformed to approximate normal distribution. Continuous phenotypes TC, LDL-C, HDL-C, TG, age and BMI SDS were standardised to zero mean and unit variance before analysis in order to obtain dimensionless effect estimates which are better comparable between different predictors and studies.
SNP associations with BMI SDS were tested with a linear model assuming three different genetic models, an additive effect of both alleles, and a dominant and recessive effect of the major allele, respectively. All models were adjusted for age and sex. Similarly, lipid SNP associations were tested with a linear model and three different genetic models. All models were adjusted for age, BMI SDS and sex. Adjustment for pubertal state instead of age is also reasonable. Due to the high correlation of age and pubertal state (Spearman r = 0.84), the genetic results are essentially the same (not shown). Also, pubertal stage is assessed by two parameters (pubic hair and breast development or testicular volume), which are not necessarily coherent. Furthermore, pubertal timing differs between boys and girls. Since dyslipidemia would be more related to age as an indicator of duration of obesity and dyslipidemia it is not necessarily influenced by pubertal development per se, this was another reason to adjust for age. We, therefore, decided to use the continuous and less ambiguously measurable trait age instead of pubertal state.
Since we tested five phenotypes, six SNPs and three genetic models, it is necessary to correct for multiple testing. However, due to multiple correlations between phenotypes and effects of genetic models, it was necessary to simulate the null-distribution. In our situation, a significance level of 6.7x10-4 controls the family-wise error rate at 5% and was therefore used to correct for multiple testing in our study.
Statistical analysis was performed using R 2.10.1.
Bayesian Model Analysis
The major drawbacks of the classical analysis mentioned above are the large number of tests to be performed due to the large number of possible combinations of SNPs and phenotypes and the assumption of a specific model of genetic and non-genetic effects. To overcome these limitations, we performed Bayesian model analysis in addition to our classical association analysis. By this approach, we can estimate plausibilities of different models and corresponding effect sizes. Bayesian modelling also allows some kind of causal inference by analysing all lipid phenotypes and possible influencing factors in parallel considering their overall correlation structure. To some extent, this avoids spurious associations.
The method is well conceived with application in analysing complex genotype-phenotype associations in medical research [28–30]. Additional insights can be derived from the modelling such as probability of different genetic risk models and estimates of unconfounded effects considering all dependencies between variables of interest. It also circumvents the above mentioned issue of multiple-testing and the uncertainty regarding the model of inheritance.
Similar to the univariate analysis, transformed and standardised data were used. Lipid phenotypes were modelled with the Bayesian variable selection approach described in [29, 31] using the reversible jump interface of WinBUGS (Version 1.4.3). Since correlation of TC and LDL-C is very high (r = 0.91) we studied models of the (three-dimensional) lipid phenotype HDL-C, LDL-C and TG. We aimed to identify the most plausible sets of co-variables having a direct influence on each lipid phenotype accounting for correlations between them.
In our analysis, the set of co-variables consists of age, BMI SDS, sex and a recessive and a dominant part for each of the six SNPs defined by indicator variables “genotype” = 0 and “genotype” = 2 respectively. If both indicator variables are included, different levels of co-dominance can be expressed by corresponding effect estimates. Hence, 15 co-variables were available for selection for each of the 3 lipid phenotypes.
Each different subset of these co-variables forms a model. Prior to analysis, one assumes that all models are equally likely. We calculated Bayesian posterior probabilities representing the plausibilities of the models given our data. Details of Bayesian modelling and fitting can be found in S1 Methods.
Bayes factors [32] are used to interpret model results. Calculation of Bayes factors is explained in the S2 Methods. A usual convention is that a Bayes factor of 1 to 3.2 is judged as “not worth more than a bare mention”, a factor of 3.2 to 10 as “substantial”, a factor of 10 to 100 as “strong” and a factor greater than 100 as “decisive” evidence for a model or effect [33]. Conversely, reciprocal values represent counter-evidence for a model. Rather than deciding whether a certain covariable has an effect or not (i.e. is in the model or not), we calculate corresponding inclusion probabilities, which can be interpreted as plausibilities regarding the impact of the covariable on the phenotype considered. Effect estimates of co-variables can be determined in the Bayesian context by averaging over all models containing this co-variable (Bayesian model averaging) weighted by the plausibility of the model. Results can be considered as analogons to Beta-coefficients of classical linear regression analysis.
Results
Classical genotype-phenotype analyses for BMI SDS and lipid phenotypes
There was no significant association between BMI SDS and any of the selected SNPs indicating that the variants are not related to the degree of obesity in our data. Results for the additive model are presented in Table 2. Results for all three genetic models are given in S1 Results.
Table 2. Association of genotypes with BMI SDS and lipid phenotypes.
| Phenotype | Variant | N | Beta | SE | CI | p-value |
|---|---|---|---|---|---|---|
| BMI SDS | rs599839 | 576 | -0.087 | 0.066 | [-0.217;0.044] | 0.193 |
| BMI SDS | rs3846663 | 572 | 0.076 | 0.061 | [-0.045;0.196] | 0.219 |
| BMI SDS | rs3812316 | 564 | -0.126 | 0.095 | [-0.312;0.06] | 0.184 |
| BMI SDS | rs174570 | 578 | 0.176 | 0.09 | [-0.001;0.353] | 0.052 |
| BMI SDS | rs4420638 | 584 | -0.004 | 0.079 | [-0.16;0.152] | 0.958 |
| BMI SDS | rs6102059 | 575 | 0.062 | 0.065 | [-0.065;0.19] | 0.335 |
| TC | rs599839 | 576 | -0.257 | 0.067 | [-0.389;-0.125] | 1.50x10 -4 |
| TC | rs3846663 | 572 | 0.141 | 0.062 | [0.019;0.263] | 0.024 |
| TC | rs3812316 | 564 | -0.022 | 0.094 | [-0.207;0.162] | 0.812 |
| TC | rs174570 | 578 | -0.04 | 0.092 | [-0.22;0.14] | 0.662 |
| TC | rs4420638 | 584 | 0.336 | 0.079 | [0.181;0.491] | 2.45x10 -5 |
| TC | rs6102059 | 575 | 0.019 | 0.065 | [-0.109;0.146] | 0.775 |
| HDL-C | rs599839 | 576 | 0.077 | 0.067 | [-0.054;0.207] | 0.25 |
| HDL-C | rs3846663 | 572 | 0.098 | 0.061 | [-0.021;0.217] | 0.106 |
| HDL-C | rs3812316 | 564 | 0.129 | 0.093 | [-0.054;0.312] | 0.168 |
| HDL-C | rs174570 | 578 | -0.078 | 0.09 | [-0.254;0.098] | 0.387 |
| HDL-C | rs4420638 | 584 | -0.13 | 0.078 | [-0.283;0.024] | 0.098 |
| HDL-C | rs6102059 | 575 | 0.038 | 0.064 | [-0.087;0.164] | 0.547 |
| LDL-C | rs599839 | 576 | -0.3 | 0.067 | [-0.431;-0.168] | 8.82x10 -6 |
| LDL-C | rs3846663 | 572 | 0.12 | 0.062 | [-0.002;0.241] | 0.054 |
| LDL-C | rs3812316 | 564 | -0.043 | 0.094 | [-0.226;0.141] | 0.649 |
| LDL-C | rs174570 | 578 | 0.021 | 0.091 | [-0.158;0.2] | 0.817 |
| LDL-C | rs4420638 | 584 | 0.382 | 0.078 | [0.228;0.536] | 1.38x10 -6 |
| LDL-C | rs6102059 | 575 | 0.018 | 0.065 | [-0.109;0.145] | 0.781 |
| TG | rs599839 | 576 | -0.114 | 0.065 | [-0.241;0.014] | 0.081 |
| TG | rs3846663 | 572 | -0.006 | 0.059 | [-0.123;0.11] | 0.913 |
| TG | rs3812316 | 564 | -0.134 | 0.088 | [-0.307;0.038] | 0.127 |
| TG | rs174570 | 578 | 0.137 | 0.087 | [-0.034;0.307] | 0.116 |
| TG | rs4420638 | 584 | 0.135 | 0.076 | [-0.014;0.285] | 0.076 |
| TG | rs6102059 | 575 | 0.075 | 0.062 | [-0.046;0.197] | 0.225 |
We present numbers of cases available for the corresponding analysis (N), beta-coefficients, their standard errors (SE), 95% confidence intervals (CI) and uncorrected p-values. Since standardized values were analysed, beta-coefficients and standard errors have unit 1. BMI SDS was analysed with the additive model adjusted for age and sex. Lipid phenotypes were logarithmized and analysed with the additive model adjusted for age, sex and BMI SDS. Associations significant after correction for multiple testing (see methods section) are printed in bold.
Next, we analysed the association between genotypes and lipid phenotypes. We found significant associations with lipid phenotypes for SORT1 rs599839 with TC (p = 1.50x10-4, β = -0.257) and LDL-C (p = 8.82x10-6, β = -0.3) as well as for APOE rs4420638 with TC (p = 2.45x10-5, β = 0.336) and LDL-C (p = 1.38x10-6, β = 0.382), whereas the variants were not associated with other lipid phenotypes (Table 2). No additional associations were found when investigating alternative models of inheritance (see S1 Results).
Bayesian model analysis
We performed Bayesian modelling of the multi-phenotype of HDL-C, LDL-C and TG. TC was not included into the model due to its strong correlation with LDL-C. Analysed relations are illustrated in Fig 1. In the following, we present the most plausible models of each lipid phenotype accounting for their pairwise correlations. The corresponding WinBUGS Model is given in detail in S3 Methods. Most probable models in decreasing order of plausibility and corresponding Bayes factors are shown in Table 3. The lists are truncated when the cumulative probability of the models exceeds 95%, i.e. all other models are less plausible according to our data. Both top models of HDL-C contain no genetic factors but age and BMI SDS as co-variables. The third most probable model includes the dominant part of rs4420638 (APOE).
Fig 1. Bayesian Model.

We present the structure of the Bayesian model analysed. Black arrows represent possible impacts of considered covariables (SNPs, age, BMI SDS, sex) on the distribution means of lipid phenotypes. Grey arrows refer to the covariance between the lipids which is accounted for in the model.
Table 3. Results of Bayesian model analysis.
| Lipid | Model | Probability | Bayes factor |
|---|---|---|---|
| HDL-C | BMI SDS | 91.89 | 371265 |
| HDL-C | age, BMI SDS | 3.08 | 1041 |
| HDL-C | rs4420638dom, BMI SDS | 0.99 | 329 |
| LDL-C | rs599839rec, rs4420638rec | 53.49 | 37691 |
| LDL-C | rs599839rec, rs4420638rec, BMI SDS | 22.88 | 9720 |
| LDL-C | rs4420638rec | 7.65 | 2714 |
| LDL-C | rs4420638rec, BMI SDS | 4.62 | 1586 |
| LDL-C | rs599839rec | 2.54 | 855 |
| LDL-C | rs599839dom, rs4420638rec | 1.03 | 340 |
| LDL-C | rs599839rec, rs4420638rec, age | 0.8 | 266 |
| LDL-C | rs599839rec, rs4420638rec, rs6102059dom | 0.77 | 254 |
| LDL-C | rs599839rec, BMI SDS | 0.74 | 244 |
| LDL-C | null | 0.56 | 186 |
| TG | age, BMI SDS | 90.47 | 311171 |
| TG | rs3812316dom, age, BMI SDS | 3.66 | 1247 |
| TG | BMI SDS | 2.55 | 856 |
Possible models of HDL-C, LDL-C, TG can contain up to 15 covariables (age, sex, BMI SDS, dominant and recessive effect of six SNPs). We present most probable models, corresponding posterior probabilities and Bayes factors. Models are ranked according to their plausibility. A cumulative probability of 95% served as cut-off for model presentation.
The top models of TG contain age and BMI SDS, too. Additionally, the second best model includes the dominant part of rs3812316 (MLXIPL).
Various different models are plausible for LDL-C: The recessive parts of SNP rs599839 (SORT1) and rs4420638 (APOE) and BMI SDS contribute to the top 5 models of LDL-C. In less probable models for LDL-C, combinations of the recessive and dominant parts of rs599839 and rs4420638, age and BMI SDS occur. Further, the dominant part of rs6102059 (MAFB) is included once.
The impact of each co-variable independent of a certain model can be assessed by interpreting the inclusion probabilities for co-variables (Fig 2). In addition to the apparent and expected impact of BMI SDS, rs599839 (SORT1) and rs4420638 (APOE) have a high certainty of affecting LDL-C independent of the degree of obesity. Conversely, the following effects cannot be ruled out (i.e. no decisive evidence against the effect was found): rs4420638 (APOE) on HDL-C, rs3846663 (HMGCR) and rs6102059 (MAFB) on LDL-C, rs3812316 (MLXIPL) on TG.
Fig 2. Inclusion probabilities of covariables for each lipid phenotype.
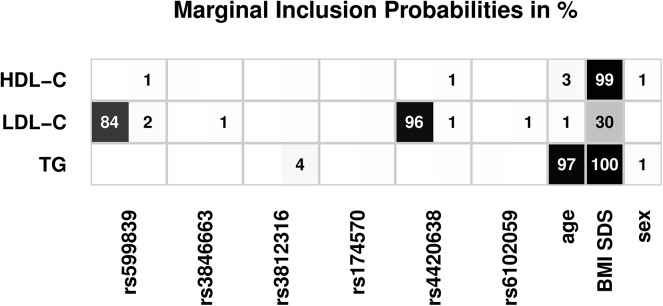
For each SNP, results are given for the recessive (first number) and dominant part (second number). Results for inclusion probabilities are rounded to integers of percentage. Effect estimates are illustrated by the shade of grey as indicated. Results rounded to zero are omitted. Results for the lipid phenotypes LDL-C, HDL-C and TG are presented. TC is omitted due to high correlation with LDL-C.
Effect estimates of co-variables with inclusion probability greater than 0.5% are listed in Table 4. Estimates and standard deviations are averaged over all models, where the respective co-variable is included (Bayesian model averaging). In comparison to classical analysis, the majority of standard deviations of estimates are smaller demonstrating higher power of the Bayesian model approach (S1 Fig).
Table 4. Inclusion probabilities of covariables and Bayesian effect sizes.
| Lipid | Variant | Probability | Estimate | SD |
|---|---|---|---|---|
| HDL-C | rs599839dom | 0.6 | 0.253 | 0.178 |
| HDL-C | rs4420638dom | 1.03 | -0.415 | 0.25 |
| HDL-C | age | 3.22 | -0.127 | 0.041 |
| HDL-C | BMI SDS | 99.34 | -0.21 | 0.041 |
| HDL-C | sex | 0.53 | -0.147 | 0.072 |
| LDL-C | rs599839rec | 84.3 | 0.32 | 0.077 |
| LDL-C | rs599839dom | 2.2 | -0.415 | 0.171 |
| LDL-C | rs3846663dom | 1.16 | 0.258 | 0.114 |
| LDL-C | rs4420638rec | 95.57 | -0.365 | 0.081 |
| LDL-C | rs4420638dom | 0.58 | 0.347 | 0.239 |
| LDL-C | rs6102059dom | 1.23 | 0.276 | 0.134 |
| LDL-C | age | 1.18 | -0.12 | 0.042 |
| LDL-C | BMI SDS | 30 | 0.146 | 0.04 |
| TG | rs3812316dom | 3.81 | -0.757 | 0.346 |
| TG | age | 97.28 | 0.172 | 0.035 |
| TG | BMI SDS | 99.98 | 0.255 | 0.044 |
| TG | sex | 1.45 | -0.166 | 0.061 |
We present probabilities for inclusion of specified covariables and resulting effect sizes and corresponding standard deviations (SD) averaged over all models containing the covariable. Only covariables with an inclusion probability greater than 0.5% are shown.
The estimated covariance of the model is shown in S2 Results. Results of the combined model of TC, HDL-C, TG are similar to those of the model of LDL-C, HDL-C, TG considered here (data not shown).
Polygenic effects for LDL-C are illustrated in S2 Fig.
Gene-expression analysis in human adipocyte precursors
We measured gene expression of SORT1, HMGCR, MLXIPL, FADS2, APOE and MAFB during differentiation of human preadipocytes into adipocytes to assess a potential physiological relevance in lipid metabolism. We observed an up-regulation of these genes by magnitudes of 10 to 104 (Fig 3).
Fig 3. mRNA expression profiles of target genes during human adipogenesis.

Fold change of gene expression for SORT1, HMGCR, MLXIPL, FADS2, APOE and MAFB mRNA during human adipocyte differentiation of SGBS cells. Data shown are averaged over 3 independent experiments, each performed in triplicates and results are given in mean±SEM. For all candidates, p<0.001 was achieved by one-way ANOVA test with Dunnet´s posthoc test.
Discussion
In this study, we aimed to assess the relevance of SNPs showing associations with lipid phenotypes from adults in a childhood sample which is less prone to confounding factors such as medication and co-morbidities and has shorter exposure to endogenous and exogenous factors.
Considering the strong impact of obesity and hence adipose tissue on circulating lipid phenotypes, we were also interested whether the candidate genes are dynamically regulated during adipogenesis. We have previously shown that genetic candidates from GWAS for obesity traits may have a functional role in human adipogenesis [27]. All selected genes from this study were expressed in adipocytes and showed considerable up-regulations during human adipocyte differentiation up to 10,000 fold. This has not been shown for these genes before. Even though this dynamic regulation during adipogenesis does not directly imply a functional relationship, this finding merits further investigation in mechanistic studies. We evaluated the dynamic regulation of candidate gene expression in SGBS preadipocytes, so far the only established model of human preadipocyte differentiation [34], which is widely applied in adipogenesis research. It has been shown that biology and molecular markers are comparable to primary human adipocyte differentiation and circumvents potential bias by patient heterogeneity due to age, risk factors, morbidities, treatment etc.
Considering the strong dependence of lipid levels on obesity, the observed regulations may affect serum lipid phenotypes and may explain SNP associations. We, therefore, verified that the six variants considered were not associated with the degree of obesity in the children prior to evaluation of associations with lipid traits.
Of the six selected SNPs located in or near the genes FADS2 (rs174570), MAFB (rs6102059), HMGCR (rs3846663), MLXIPL (rs3812316), APOE/C1/C4/C2 (rs4420638) and CELSR2/PSRC1/SORT1 (rs599839), we observed a strong impact of rs599839 (SORT1) and rs4420638 (APOE) on circulating LDL-C levels independent of the degree of obesity in conventional linear regression analyses adjusting for age, sex and BMI SDS. This was confirmed by our Bayesian model analysis suggesting causality of these two variants on LDL-C. Bayesian analysis also revealed that effects of rs4420638 (APOE) on HDL-C, rs3846663 (HMGCR) and rs6102059 (MAFB) on LDL-C as well as rs3812316 (MLXIPL) on TG cannot be ruled out. Still, these variants should be considered as candidates requiring further investigations.
The APOE-SNP rs4420638 is located on chromosome 19 in a cluster with APOC1, APOC4 and APOC2. The SNP rs599839 is located on chromosome 1, close to the genes CELSR2, PSRC1 and SORT1. Multiple other studies investigated SNPs in or near the APOE and SORT1 genes. Rs4420638 and rs599839 showed replicable associations with lipid levels (mostly LDL-C) in Caucasian and non-Caucasian population cohorts and meta-analyses [7, 9, 15, 35]. The non-Caucasian cohorts displayed lower significance regarding all SNP-lipid-associations, most likely due to lower case numbers (N ranges from 2,532 to 9,328) [15]. In a small sample, Klein et al. observed effects of rs646776, a proxy of rs599839, only in males [36]. Sex-stratified analysis of our data reveals a significant effect for both sexes. Beta estimator of females is slightly lower than that for males (Females: p = 6.5x10-4, β = -0.28, Males: p = 4.5x10-3, β = -0.32).
Associations of the other variants/candidates could not be confirmed in our sample. Correlations for rs174570 (FADS2) with TC, LDL-C and HDL-C were observed in a meta-analysis of 16 European studies [10], although others did not confirm this in Hispanic adult cohorts [37]. Admittedly, our sample is considerably smaller and, thus, weak effects may have remained undetected. However, according to our Bayesian analysis, rs174570 (FADS2) is the most implausible of the considered candidates, since, in contrast to the other variants, it is dismissed for all three lipid phenotypes analysed.
For other variants, even large sample-sized and high-powered adult studies gave controversial results. While a significant association of rs6102059 (MAFB) with LDL-C was observed by some [8], this could not be replicated by others [15]. However, the cohort of supposed European ancestry consisted of self-identified European Americans. Their genetic origin was not validated, which might have blurred the associations. In our Central European sample we observed no convincing association of rs6102059 with TC and LDL-C levels but we can also not completely rule out an effect based on our Bayesian analysis.
The intronic SNP rs3846663 in HMGCR was reported to be significantly associated with LDL-C levels in a cohort of 19,840 subjects [8]. These findings were replicated in several populations (Kosrean islands inhabitants (n = 2,346) with an even larger effect size compared to Kathiresan et al [8] and Japanese [38] or Scottish [39]. Our results did not reach significance again possibly due to our limited sample size, but by trend, are in line with the above mentioned studies. This is further confirmed by our Bayesian analysis complying with a possible causal effect of rs3846663 on LDL-C but not on HDL-C or TG.
Rs3812316 (MLXIPL) was most strongly associated with TG in adults [16], although others did not find this association [40, 41]. It was suggested that the effect on TG levels must be weak if it exists at all [40]. In our study, standard linear regression analysis did not reveal any significant association. Nevertheless our results show lower triglyceride levels in homozygous SNP-carriers with rs3812316/GG genotype (CC: 1.13 mmol/l; GG: 0.69 mmol/l adjusted for age, sex and BMI SDS) in agreement with the above mentioned observations. The effects that were seen in our analysis indicate a protective function for minor allele carriers concerning triglyceride levels in children, even in the presence of obesity [16]. Again, Bayesian model analysis supports this finding since in contrast to HDL-C and LDL-C, an effect of rs3812316 to TG cannot be excluded.
A limitation of our study is the relatively small sample size since recruitment of volunteers is more challenging for childhood cohorts. Children are a population much less affected by chronic diseases or medication. Therefore, genetic studies in childhood cohorts are intriguing. Indeed, we were able to confirm the association for children for variants which are originally detected in much larger cohorts of adults comprising several thousands of individuals. Although, the power of our study is limited, we could confirm rs599839 and rs4420638 to be associated in children. Interestingly, higher effect sizes compared to adults were observed. However, one has to note that our study population is mainly obese. Therefore, replication in a population-based sample of children is required to show general validity of our associations.
Also, besides the possibility that due to the lower influence of co-morbidities and other life-style related factors, primary genetic effects may be hypothesized to be stronger in children than in adults, an alternative possibility would be the later emergence of genetic effects on phenotype. This would particularly apply to conditions where genetic predisposition is reinforced by additional (environmental) risk factors that accumulate or increase with life time (double/multiple hit theory). Such a relationship has been discussed for the manifestation of coronary artery disease in patient with genetic risk factors [42]. It also needs to be considered, that children and adolescents do not yet present with overt disease and hence do not meet the pathological endpoints (eg. myocardial infarction), which limits interpretation on genotype-phenotype associations.
For adults it is common practise to combine diverse cohorts (i.e. The Framingham Heart Study, Invecchiare in Chianti, London Life Science Population Cohort [8], The Rotterdam Study [10], Diabetes Genetics Initiative [7, 8] or The Finland-United States Investigation of NIDDM Genetics [8, 9, 16]. These cohorts differ considerably regarding the burden of chronic illness or drug-intake which might lower the chance to detect genetic associations. However, besides all the advantages of childhood cohorts, we have to acknowledge that studies in adolescent individuals might be affected by changes of lipid metabolism during puberty [43].
By our Bayesian modelling approach we propose an innovative method of analysing multi-SNP–multi-phenotype associations independently and in addition to the classical frequentist regression modelling. This type of analysis overcomes a number of limitations of classical regression analysis: First, it allows comparisons of different types of models, i.e. different modes of inheritance and inclusion of co-variables. Although it is possible in principle to include multiple SNPs and covariables in regression analysis, this usually results in a large number of possible models with no generally accepted decision rule how to select an optimal one. Second, it considers polygenic effects and the information of other phenotypes as well. Considering the correlation structure between different phenotypes can improve detection of the underlying signal [29]. To some extent, this also allows inference regarding unconfounded effects of genotypes and co-variables, which may be indicative for direct or even causal relationships. Interestingly, as discussed above, our Bayesian model results are always in line with observations in adult studies and hence support these results.
Third, the Bayesian approach can deal with missing values, i.e. single missings in either phenotypes, co-variables or SNPs [44]. For example a classical analysis of all SNPs and phenotypes in parallel would reduce the sample size from 594 to 521 in our study whereas Bayesian analysis includes all individuals resulting in higher power. Indeed, compared to the classical analysis, standard deviations of effect estimates are typically smaller, i.e. estimates are more precise [30] and may handle smaller sample sizes.
Summary
We could show for the first time in children that rs599839 (SORT1) and rs4420638 (APOE) are strongly associated with alterations in blood lipid levels independent of the presence and degree of obesity. Our integrative Bayesian model analysis provided further candidate associations requiring further investigation of the candidates. Therefore, we conclude that this novel approach can improve the detection of weaker associations in genotype-phenotype data sets.
Supporting Information
SNP genotype corresponds to the number of minor alleles (0, 1 or 2) and 3 refer to missing values. Lipid phenotype levels are provided as hdl_c (high density lipoprotein cholesterol), ldl_c (low density lipoprotein cholesterol, tc (total cholesterol) and tg (triglyceride). Accordingly, the covariables age, sex and bmi_sds are provided.
(XLSX)
(DOCX)
Effects of identified genetic risk variants on LDL-C.
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
This work was supported by grants from the German Research Council (DFG) for the Clinical Research Center “Obesity Mechanisms” CRC1052/1 C05 and the Federal Ministry of Education and Research (BMBF), Germany, FKZ: 01EO1001 (IFB AdiposityDiseases), and the European Community’s Seventh Framework Programme (FP7/2007-2013) project Beta-JUDO under grant agreement n° 279153 to A.K. The work of A.G. was supported in part by the German Federal Research Ministry (BMBF), grant PROGRESS (01KI1010I). A.G. and M.S. and AK were funded by LIFE–Leipzig Research Center for Civilization Diseases, University of Leipzig. LIFE is funded by means of the European Union, by the European Regional Development Fund (ERDF) and by means of the Free State of Saxony within the framework of the excellence initiative.
Abbreviations
- APOE
apolipoprotein E
- TC
total-cholesterol
- FADS2
fatty acid desaturase 2
- GWA
genome-wide association
- HMGCR
3-hydroxy-3-methylglutaryl-Coenzyme A reductase
- MAFB
V-maf musculoaponeurotic fibrosarcoma oncogene homolog B
- MLXIPL
MLX interacting protein
- SORT1
sortilin 1
Data Availability
All relevant data is available in the paper and its Supporting Information files.
Funding Statement
This work was supported by grants from the German Research Council (DFG) for the Clinical Research Center “Obesity Mechanisms” CRC1052/1 C05 and the Federal Ministry of Education and Research (BMBF), Germany, FKZ: 01EO1001 (IFB AdiposityDiseases), and the European Community’s Seventh Framework Programme (FP7/2007–2013) project Beta-JUDO under grant agreement n° 279153 to A.K. The work of A.G. was supported in part by the German Federal Research Ministry (BMBF), grant PROGRESS (01KI1010I). M.S. was funded by LIFE – Leipzig Research Center for Civilization Diseases, University of Leipzig. LIFE is funded by means of the European Union, by the European Regional Development Fund (ERDF) and by means of the Free State of Saxony within the framework of the excellence initiative. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Report by the Central Committee for Report by the Central Committee for Medical and Community Program of the American Heart AssociationMedical and Community Program of the American Heart Association. Dietary fat and its relation to heart attacks and strokes. JAMA. 1961;175:389–91. [PubMed] [Google Scholar]
- 2. Mendivil CO, Rimm EB, Furtado J, Chiuve SE, Sacks FM. Low-density lipoproteins containing apolipoprotein C-III and the risk of coronary heart disease. Circulation; 2011. 2011. p. 2065–72. 10.1161/CIRCULATIONAHA.111.056986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cullen P. Evidence that triglycerides are an independent coronary heart disease risk factor. AmJCardiol. 2000;86(9):943–9. [DOI] [PubMed] [Google Scholar]
- 4. Tan MH. HDL-cholesterol: the negative risk factor for coronary heart disease. AnnAcadMedSingap. 1980;9(4):491–5. [PubMed] [Google Scholar]
- 5. Rippe JM, Crossley S, Ringer R. Obesity as a chronic disease: modern medical and lifestyle management. J Am Diet Assoc. 1998;98(10 Suppl 2):S9–15. [DOI] [PubMed] [Google Scholar]
- 6. Ruderman N, Chisholm D, Pi-Sunyer X, Schneider S. The metabolically obese, normal-weight individual revisited. Diabetes; 1998. 1998. p. 699–713. [DOI] [PubMed] [Google Scholar]
- 7. Kathiresan S, Melander O, Guiducci C, Surti A, Burtt NP, Rieder MJ, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. NatGenet. 2008;40(2):189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. NatGenet. 2009;41(1):56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. NatGenet. 2008;40(2):161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aulchenko YS, Ripatti S, Lindqvist I, Boomsma D, Heid IM, Pramstaller PP, et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. NatGenet. 2009;41(1):47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Standl M, Lattka E, Stach B, Koletzko S, Bauer CP, von BA, et al. FADS1 FADS2 gene cluster, PUFA intake and blood lipids in children: results from the GINIplus and LISAplus studies. PLoSONE. 2012;7(5):e37780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Molto-Puigmarti C, Jansen E, Heinrich J, Standl M, Mensink RP, Plat J, et al. Genetic variation in FADS genes and plasma cholesterol levels in 2-year-old infants: KOALA Birth Cohort Study. PLoSONE. 2013;8(5):e61671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu P, Qin YH, Lei FY, Pei J, Hu B, Lu L. Variable frequencies of apolipoprotein E genotypes and its effect on serum lipids in the Guangxi Zhuang and Han children. IntJMolSci. 2011;12(9):5604–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Atabek ME, Ozkul Y, Eklioglu BS, Kurtoglu S, Baykara M. Association between apolipoprotein E polymorphism and subclinic atherosclerosis in patients with type 1 diabetes mellitus. JClinResPediatrEndocrinol. 2012;4(1):8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dumitrescu L, Carty CL, Taylor K, Schumacher FR, Hindorff LA, Ambite JL, et al. Genetic determinants of lipid traits in diverse populations from the population architecture using genomics and epidemiology (PAGE) study. PLoS Genet. 2011;7(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kooner JS, Chambers JC, Aguilar-Salinas CA, Hinds DA, Hyde CL, Warnes GR, et al. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. NatGenet. 2008;40(2):149–51. [DOI] [PubMed] [Google Scholar]
- 17. Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. NatGenet. 2013;45(1):25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greenawalt DM, Dobrin R, Chudin E, Hatoum IJ, Suver C, Beaulaurier J, et al. A survey of the genetics of stomach, liver, and adipose gene expression from a morbidly obese cohort. Genome research. 2011;21(7):1008–16. 10.1101/gr.112821.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Innocenti F, Cooper GM, Stanaway IB, Gamazon ER, Smith JD, Mirkov S, et al. Identification, replication, and functional fine-mapping of expression quantitative trait loci in primary human liver tissue. PLoS genetics. 2011;7(5):e1002078 10.1371/journal.pgen.1002078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schroder A, Klein K, Winter S, Schwab M, Bonin M, Zell A, et al. Genomics of ADME gene expression: mapping expression quantitative trait loci relevant for absorption, distribution, metabolism and excretion of drugs in human liver. The pharmacogenomics journal. 2013;13(1):12–20. 10.1038/tpj.2011.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nature genetics. 2013;45(10):1238–43. 10.1038/ng.2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang X, Johnson AD, Hendricks AE, Hwang SJ, Tanriverdi K, Ganesh SK, et al. Genetic associations with expression for genes implicated in GWAS studies for atherosclerotic cardiovascular disease and blood phenotypes. Human molecular genetics. 2014;23(3):782–95. 10.1093/hmg/ddt461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zeller T, Wild P, Szymczak S, Rotival M, Schillert A, Castagne R, et al. Genetics and beyond—the transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5(5):e10693 10.1371/journal.pone.0010693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kromeyer-Hauschild K, Wabitsch M, Geller F, Ziegler A, Geiß HC, Hesse V, et al. Perzentilen für den Body Mass Index für das Kindes- und Jugendalter unter Heranziehung verschiedener deutscher Stichproben. (Centiles for body mass index for children and adolescents derived from distinct independent German cohorts). Monatsschr Kinderheilkd. 2001;149:807–18. [Google Scholar]
- 25. Marshall WA, Tanner JM. Variations in pattern of pubertal changes in girls. Arch Dis Child. 1969;44(235):291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marshall WA, Tanner JM. Variations in the pattern of pubertal changes in boys. Arch Dis Child. 1970;45(239):13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bernhard F, Landgraf K, Klöting N, Berthold A, Büttner P, Friebe D, et al. Functional relevance of genes implicated by obesity genome-wide association study signals for human adipocyte biology. Diabetologia. 2013;56(2):311–22. 10.1007/s00125-012-2773-0 [DOI] [PubMed] [Google Scholar]
- 28. Quintana MA, Schumacher FR, Casey G, Bernstein JL, Li L, Conti DV. Incorporating prior biologic information for high-dimensional rare variant association studies. Human heredity. 2012;74(3–4):184–95. 10.1159/000346021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lunn DJ, Whittaker JC, Best N. A Bayesian toolkit for genetic association studies. GenetEpidemiol. 2006;30(3):231–47. [DOI] [PubMed] [Google Scholar]
- 30. Conti DV, Cortessis V, Molitor J, Thomas DC. Bayesian modeling of complex metabolic pathways. Human heredity. 2003;56(1–3):83–93. [DOI] [PubMed] [Google Scholar]
- 31. Lunn DJ, Best N, Whittaker JC. Generic reversible jump MCMC using graphical models. Stat Comput. 2009;19:395–408. [Google Scholar]
- 32. Kass RE, Raftery AE. Bayes Factors. Journal of the American Statistical Association. 1995;90(430):773–95. [Google Scholar]
- 33. Jeffreys H. Theory of Probability. 3rd edition ed. Oxford, U.K.: Oxford University Press; 1961 1961. [Google Scholar]
- 34. Wabitsch M, Brenner RE, Melzner I, Braun M, Moller P, Heinze E, et al. Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation. IntJ ObesRelatMetabDisord. 2001;25(1):8–15. [DOI] [PubMed] [Google Scholar]
- 35. Ken-Dror G, Talmud PJ, Humphries SE, Drenos F. APOE/C1/C4/C2 gene cluster genotypes, haplotypes and lipid levels in prospective coronary heart disease risk among UK healthy men. MolMed. 2010;16(9–10):389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Klein MS, Connors KE, Shearer J, Vogel HJ, Hittel DS. Metabolomics reveals the sex-specific effects of the SORT1 low-density lipoprotein cholesterol locus in healthy young adults. Journal of proteome research. 2014;13(11):5063–70. 10.1021/pr500659r [DOI] [PubMed] [Google Scholar]
- 37. Lu Y, Feskens EJ, Dollé ME, Imholz S, Verschuren WM, Müller M, et al. Dietary n-3 and n-6 polyunsaturated fatty acid intake interacts with FADS1 genetic variation to affect total and HDL-cholesterol concentrations in the Doetinchem Cohort Study. The American Journal of Clinical Nutrition. 2010;92(1):258–65. 10.3945/ajcn.2009.29130 [DOI] [PubMed] [Google Scholar]
- 38. Hiura Y, Tabara Y, Kokubo Y, Okamura T, Goto Y, Nonogi H, et al. Association of the functional variant in the 3-hydroxy-3-methylglutaryl-coenzyme a reductase gene with low-density lipoprotein-cholesterol in Japanese. Circulation journal: official journal of the Japanese Circulation Society. 2010;74(3):518–22. [DOI] [PubMed] [Google Scholar]
- 39. Donnelly LA, Doney AS, Dannfald J, Whitley AL, Lang CC, Morris AD, et al. A paucimorphic variant in the HMG-CoA reductase gene is associated with lipid-lowering response to statin treatment in diabetes: a GoDARTS study. Pharmacogenetics and genomics. 2008;18(12):1021–6. 10.1097/FPC.0b013e3283106071 [DOI] [PubMed] [Google Scholar]
- 40. Vrablik M, Ceska R, Adamkova V, Peasey A, Pikhart H, Kubinova R, et al. MLXIPL variant in individuals with low and high triglyceridemia in white population in Central Europe. Human genetics. 2008;124(5):553–5. 10.1007/s00439-008-0577-6 [DOI] [PubMed] [Google Scholar]
- 41. Polgár N, Járomi L, Csöngei V, Maász A, Sipeky C, Sáfrány E, et al. Triglyceride level modifying functional variants of GALTN2 and MLXIPL in patients with ischaemic stroke. European journal of neurology: the official journal of the European Federation of Neurological Societies. 2010;17(8):1033–9. [DOI] [PubMed] [Google Scholar]
- 42. Munir MS, Wang Z, Alahdab F, Steffen MW, Erwin PJ, Kullo IJ, et al. The association of 9p21-3 locus with coronary atherosclerosis: a systematic review and meta-analysis. BMC Med Genet. 2014;15:66 10.1186/1471-2350-15-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moran A, Jacobs DR Jr, Steinberger J, Steffen LM, Pankow JS, Hong CP, et al. Changes in insulin resistance and cardiovascular risk during adolescence: establishment of differential risk in males and females. Circulation; 2008. 2008. p. 2361–8. 10.1161/CIRCULATIONAHA.107.704569 [DOI] [PubMed] [Google Scholar]
- 44.Lunn DJ, Osorio C, Whittaker JC. A multivariate probit model for inferring missing haplotype/genotype data. Technical report EPH-2005-02. 2005;Department of Epidemiology and Punblic Health, Imperial College London, UK.(21).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SNP genotype corresponds to the number of minor alleles (0, 1 or 2) and 3 refer to missing values. Lipid phenotype levels are provided as hdl_c (high density lipoprotein cholesterol), ldl_c (low density lipoprotein cholesterol, tc (total cholesterol) and tg (triglyceride). Accordingly, the covariables age, sex and bmi_sds are provided.
(XLSX)
(DOCX)
Effects of identified genetic risk variants on LDL-C.
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data is available in the paper and its Supporting Information files.