

Abstract
Regulation of 5-aminolevulinate synthase (ALAS) is at the origin of balanced heme production in mammals. Mutations in the C-terminal region of human erythroid-specific ALAS (hALAS2) are associated with X-linked protoporphyria (XLPP), a disease characterized by extreme photosensitivity, with elevated blood concentrations of free protoporphyrin IX and zinc protoporphyrin. To investigate the molecular basis for this disease, recombinant hALAS2 and variants of the enzyme harboring the gain-of-function XLPP mutations were constructed, purified, and analyzed kinetically, spectroscopically and thermodynamically. Enhanced activities of the XLPP variants resulted from accelerations in the rate at which the product 5-aminolevulinate (ALA) was released from the enzyme. Circular dichroism spectroscopy revealed that the XLPP mutations altered the microenvironment of the pyridoxal 5’-phosphate cofactor, which underwent further and specific alterations upon succinyl-CoA binding. Transient kinetic analyses of the variant-catalyzed reactions and protein fluorescence quenching upon ALA binding to the XLPP variants demonstrated that the protein conformational transition step associated with product release was predominantly affected. Of relevance, XLPP could also be modeled in cell culture. We propose that 1) the XLPP mutations destabilize the succinyl-CoA-induced hALAS2 closed conformation and thus accelerate ALA release, 2) the extended C-terminus of wild-type mammalian ALAS2 provides a regulatory role that allows for allosteric modulation of activity, thereby controlling the rate of erythroid heme biosynthesis, and 3) this control is disrupted in XLPP, resulting in porphyrin accumulation.
Keywords: Heme biosynthesis, 5-aminolevulinate, 5-aminolevulinate synthase, porphyria, X-linked erythropoietic protoporphyria, porphyrin, tetrapyrrole, pyridoxal 5’-phosphate
5-Aminolevulinate synthase [succinyl-CoA:glycine C-succinyltransferase (decarboxylating); ALAS; EC 2.3.1.37] is a homodimeric pyridoxal 5’-phosphate (PLP)-dependent enzyme that catalyzes the first and rate-limiting step of heme formation in vertebrates.1-4 The reaction involves the condensation of glycine and succinyl-CoA to produce 5-aminolevulinate (ALA), CoA, and carbon dioxide.1,4 Animal genomes encode two highly conserved, but differentially expressed, ALAS genes, a housekeeping gene (ALAS1)5,6 and an erythroid-specific gene (ALAS2).6-8 Eukaryotic mature ALAS,5,6 regardless of the isoform, is considerably larger than proteobacterial ALAS9 due to a longer, and non-identical, N-terminal sequence, and a highly conserved 30-40 amino acid C-terminal extension of unknown function.2 The N-terminal region, designated as region 22,10 and encoded by exons 3 and 4 of the human ALAS2 (hALAS2) gene,10 is not required for activity11 and contains an heme-regulatory motif.12 Binding of heme to the corresponding heme-regulatory motif and two identical motifs in the mitochondrial targeting sequence prevents the import of the precursor form of ALAS 1 into the mitochondrion.12,13
Genetic lesions in hALAS2 can result in two very different blood disorders, X-linked sideroblastic anemia (XLSA; MIM 300751)10,14 and X-linked protoporphyria (XLPP; MIM 300752).15-20 A variety of point mutations in exons 4-11 of the hALAS2 gene, which encode the highly conserved catalytic core region of the enzyme,10 are associated with XLSA, an inherited blood disorder characterized by heme-deficient, iron-overloaded red cells.10,14 The molecular basis for XLSA is thus a partial loss of hALAS2 activity, due to reduced catalytic function and/or protein stability.10,14,21 Unlike the XLSA mutations, those causing XLPP enhance ALAS2 activity,16,17,20 hence their designation as ‘gain-of-function’ mutations,16,20 and lead to protoporphyrin IX and zinc-protoporphyrin IX accumulation.19,20 In fact, patients suffering from XLPP had traditionally been diagnosed as having erythropoietic protoporphyria (EPP; MIM 177000), which is symptomatically similar but results from a deficiency of ferrochelatase (EC 4.99.1.1), the enzyme responsible for converting protoporphyrin IX and ferrous iron into heme.22 Approximately 5-10% of patients presenting EPP symptoms do not have a ferrochelatase deficiency.15,20 Most of these patients instead have XLPP, with defective and more active forms of hALAS2,16,17,20,23 and while they share the phenotypic hallmark of elevated protoporphyrin IX levels with EPP patients, they differ in that they also accumulate zinc protoporphyrin in their erythrocytes.19,23,24
In contrast to XLSA,10,14,16,25 all known XLPP mutations reside in a single exon, exon 11, encoding the far-C-terminus of hALAS2, and give rise to truncated or extended variants of the enzyme.10,14,15,17,20 The 33 C-terminal amino acids are highly conserved and yet have diverged from the ALAS1 isoform, suggesting that these residues endow the enzyme with an erythroid-specific function and/or regulation.20 Additionally, the C-terminal amino acids are conserved in higher eukaryotes but absent in prokaryotes, and deleting the final 33 amino acids of recombinant hALAS2 results in an increase in the catalytic activity.25 Kadirvel et al.25 investigated the “ex vivo” and “in vitro” catalytic activity and stability of hALAS2 variants with C-terminal mutations reflecting those identified in XLSA patients and concluded that the C-terminal region of hALAS2 “acts as an intrinsic modifier for its catalytic activity and protein stability”. Of relevance, the XLSA mutations in this C-terminal region prevented the binding of ALAS to the succinyl-CoA synthetase (SCS) ADP-forming β subunit (SUCLA2 or SCS-βA).26 Subsequent analyses of the increased catalytic activities of hALAS2 variants with XLPP mutations in relation to those of engineered hALAS2 variants and wild-type enzyme led to the definition of the hALAS2 gain-of-function domain as an approximately 45 C-terminal amino acid region.16,17 Significantly, an XLPP variant with the gain-of-function domain truncated could not bind either SUCLA2 or SCS.16
Located about 20 N-terminal amino acids from the predicted “gain-of-function” domain is a mobile active site loop, that when mutated yields ALAS variants significantly more active than wild-type ALAS and with reactions not limited by product release27 and thus contrasting with the wild-type enzyme-catalyzed reaction. In murine ALAS2, a surprisingly diverse array of single and multiple mutations, located within the corresponding loop of residues that reversibly cover the active site during the catalytic cycle, led to hyperactive variants of the enzyme.27 The basis for the hyperactivity in these in vitro generated enzymes was postulated to be an enhancement in the conformational mobility of the active site loop, which controls the overall rate of reaction by slowly moving and opening to allow release of the product from the active site cleft.27 Importantly, Rhodobacter capsulatus ALAS, which lacks the extended C-terminus homologous to the XLPP mutations site in hALAS2, is the only ALAS for which a crystallographic structure has been solved.9 Thus, any tertiary-quaternary structural or functional relationship between the extended C-terminus and the active site loop is currently unknown.
In order to determine the molecular mechanism underlying the gain of ALAS2 function observed in XLPP, we have refined and expanded on the role of the C-terminal region of hALAS2, as advanced by Kadirvel et al.25, by charactering the structural and kinetic properties of purified recombinant hALAS2 and XLPP hALAS2 variants and comparing them to the ex vivo ALAS activity and porphyrin accumulation in hALAS2- and XLPP-expressing mammalian cells. Our data indicate that the XLPP variants possess enhanced ALAS activity and ALA dissociation rates, as well as distinct structural properties from those of wild-type hALAS. We suggest that the primary molecular basis of XLPP is ALAS2 C-terminal mutations conferring augmented enzyme activity due to stabilization of a protein conformational state that facilitates product release. Further, we propose that the extended C- terminus of mammalian ALAS2 fulfills a regulatory role that allows for modulation of activity in vivo and subsequently erythroid heme biosynthesis.
EXPERIMENTAL PROCEDURES
Materials
Aprotinin, pepstatin, leupeptin, phenylmethylsulfonyl fluoride (PMSF), tricine, ampicillin, DEAE-Sephacel, Ultrogel Aca-44, β-mercaptoethanol, pyridoxal 5’-phosphate, succinyl-CoA, α-ketoglutarate dehydrogenase, HEPES-free acid, MOPS, thiamine pyrophosphate and NAD+ were obtained from Sigma-Aldrich Chemical Company. Glucose, glycerol, acetic acid, methanol, glycine, disodium ethylenediamine tetraacetic acid dihydrate, tricine, ammonium sulfate and potassium hydroxide were purchased from Fisher Scientific. Centricon concentrators were from Millipore. SDS-PAGE reagents and Phusion DNA Polymerase were acquired from Thermo Scientific. 5-Aminolevulinic acid (ALA) hydrochloride was purchased from Acros Organics. Bmtl, Sall, BlpI, and BamHI restriction enzymes were obtained from New England BioLabs, Inc. Oligonucleotides were synthesized by Integrated DNA Technologies. T4 DNA ligase and ligase buffer were obtained from Thermo Scientific Fermentas and bicinchoninic acid protein determination kits were purchased from Thermo Scientific Pierce.
Plasmids for expression in bacterial and mammalian cells
The following mutations: c. 1699-1700 AT deletion (delAT, which results in a frame-shift mutation leading to a truncated hALAS2 protein with a glutamate (E567) as the C-terminal amino acid,20 c.1705-1708 AGTG deletion (delAGTG, which results in the E569G mutation and a 23 amino acid, non-related hALAS2 C-terminus,20 and Q548X, where X stands for a stop codon, (Figure 1) were introduced into the cDNA of hALAS2 using the QuikChange method (Stratagene), and confirmed by DNA sequencing. The mutated ALAS2 DNA-encoding fragments were digested with Sall and BamHI and subcloned in the bacterial expression plasmid for murine ALAS2, pGF23,28 replacing the mouse wild-type region. The resulting plasmids for wild-type hALAS2, delAT, delAGTG, and Q548X were designated pSD1, pSD2, pSD3 and pQ548X, respectively, and used for production of the recombinant proteins in Escherichia coli and their subsequent purification. In addition, the mutated sequences were used in the construction of plasmids for expression of the XLPP variants in mammalian cells. Toward this end, initially, the wild-type precursor hALAS2 cDNA (GenBank: X56352.1) was individually subcloned into the multiple cloning site of the pIRES2-ZsGreenl vector (purchased from Clontech Laboratories, Inc. Mountain View, CA) using the Bmtl and BamHI resfriction sites. The digested hALAS2-encoding fragment was ligated into the digested and purified pIRES2-ZsGreenl vector using T4 DNA ligase in ligase buffer at 16°C. Electrocompetent BL21(DE3) cells were transformed by electroporation with the ligated plasmid DNA and selected by spreading the transformed cells on LB agar medium containing 10 μg/mL kanamycin sulfate. Plasmid DNA was purified from a single colony using a QiaPrep Spin Miniprep kit (Qiagen Inc.), and the sequence of the cloned DNA was verified by Genewiz, Inc. in New Brunswick, NJ. The resulting plasmid was named pEF27. For the mammalian expression plasmids encoding the precursor XLPP variants, the cDNAs encoding the mutated hALAS2 C-termini were retrieved from pSD2, pSD3 and pQ548X and subcloned into pEF27 using the BlpI and BamHI restriction sites. The resulting plasmids were named pEF28, pEF29 and pEF30, encoding delAT, delAGTG and Q548X, respectively.
Figure 1.
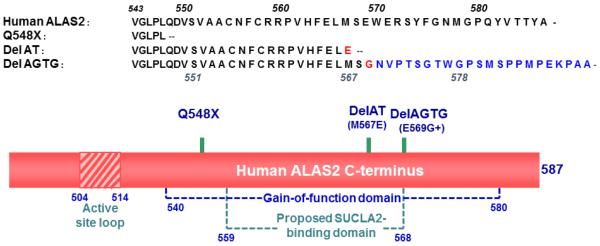
Schematic representation of the hALAS2 C-terminal domain and changes introduced with the XLPP mutations. The hALAS2 mutations observed in XLPP patients yield either truncated ALAS2 proteins or variants of ALAS2 with a different C-terminus (blue font).15-17,22,24 The gain-of-function16,17 and proposed SUCLA2-binding16 domains and active site loop are illustrated. hALAS2 has 587 amino acids and a C-terminus 33 amino acid longer than R. capsulatus ALAS.
Plasmids for protein overproduction in bacterial cells and protein purification
Plasmids pSD1, pSD2 and pSD3 were used for overproduction of wild-type hALAS2, delAT and delAGTG in E. coli BL21(DE3) cells and purification of the proteins used in the intrinsic protein fluorescence quenching experiments. Because all of the other experiments required larger amounts of protein fluorescence, we had to resort to the purification of His-tagged wild-type hALAS2 and XLPP variants.
Plasmids for purification of histidine-tagged proteins
In order to purify hALAS2 and the XLPP variant proteins using nickel affinity chromatography, we added the sequence for six histidine codons to the cDNA sequence encoding the mature hALAS2 N-terminus. The mature wild-type hALAS2 was amplified from pSD1 using the oligonucleotides hALAS22 (5’- CGT GTC GAC GAT GCA CCA TCA CCA CCA TCA CGG GAA GAG CAA GAT TGT GCA GAA G-3’) and r-hALAS23 (5’-AAG TGG TAA AGA TGA AGC CTG CAG CAT- 3’) as forward and reverse primers, respectively. The Sall site and the codons for the six histidines are indicated in italics and bold, respectively, in the sequence for the hALAS22 primer (above). The r-hALAS23 reverse primer was designed to anneal to the DNA coding strand, 531 bp downstream of the BlpI site. The generated PCR product (1040bp) was digested with SalI and BlpI to yield a 505bp-DNA fragment that was subsequently subcloned into the previously digested pSD1, pSD2, pSD3, and pQ548X vectors using T4 DNA ligase in ligase buffer (Thermo Scientific Fermentas) at 16 °C. Electrocompetent BL21(DE3) cells were transformed by electroporation with the ligated plasmids and selected by spreading the transformed cells on LB agar medium containing 50 μg/mL ampicillin. Plasmid DNA was purified from a single colony using a QiaPrep Spin Miniprep kit (Qiagen Inc.). The N-terminal ALAS- and histidineencoding sequences were verified by DNA sequencing using the primer r-hALAS17 (5’- CTG GTC ATA ACT GAA GAC-3 ’), which anneals complementarily to the DNA coding strand and 208 bp downstream of the initiation and histidine codons introduced in the sequences for the mature hALAS2 and XLPP variants. The resulting plasmids, pEF40, pEF41, pEF42 and pEF43, were used for the expression of histidine-tagged wild-type hALAS2, delAT, delAGTG and Q548X, respectively.
Cell culture
K562 cells were maintained in RPMI-1640 culture medium (Mediatech, Inc.), with 10% FBS (Thermo Scientific™ HyClone™), gentamicin (Mediatech, Inc., 50 μg/mL), penicillin (Mediatech, Inc., 60 μg/mL) and streptomycin (Mediatech, Inc., 100 μg/mL) at 37°C in a humidified incubator with 5% CO2. HeLa cells were maintained in DMEM with 4.5 g/L glucose, L-glutamine, and sodium pyruvate (Mediatech, Inc.), with 10% FBS, gentamicin (50 μg/mL), penicillin (60 μg/mL) and streptomycin (100 μg/mL) at 37°C in a humidified incubator with 5% CO2.
Transient transfection of HeLa cells
On the day prior to transfection, HeLa cells were trypsinized and counted. Approximately 2 × 104 cells were seeded into each well of a 24-well plate in 0.5 mL of DMEM. Cell density was ~30-50% confluent on the day of transfection. For each transfection, 250 ng of DNA was diluted into 100 μL of DMEM without serum. One μL of Lipofectamine™ LTX was added into the diluted DNA solution, mixed gently and incubated for 30-45 minutes at room temperature to form DNA-Lipofectamine™ LTX complexes. The DNA-Lipofectamine™ LTX complexes were added dropwise to each well containing cells and mixed gently by manually rocking the plate back and forth for a few seconds. After 4 hours of incubation with the DNA-Lipofectamine LTX complexes, the medium was aspirated out of each well and fresh DMEM with 10% FBS, gentamicin (50 μg/mL), penicillin (60 μg/mL) and streptomycin (100 μg/mL) was added to each well of cells. Cells were incubated at 37°C in a CO2 incubator for 24 hours post-transfection before assaying. For cells treated with glycine, glycine dissolved in DMEM (1M) was added to the culture medium 4 hours post-transfection.
Transient transfection of K562 human erythroleukemia cells
K562 cells were transfected with Lipofectamine™ LTX and PLUS™ Reagent, purchased from Invitrogen (San Jose, CA), according to the supplier’s optimized protocol for K562 cells. On the day of transfection, a hemocytometer and trypan blue staining were used to count the cells and determine culture density and viability. In a 6-well plate, K562 cells (5 × 105 cells per well) were seeded in a 6-well plate at a volume of 2 mL of RPMI-1640 growth medium with 10% FBS 30 minutes prior to transfection. For each transfection, 2.5 μg of DNA was added into 500 μL ofRPMI medium without serum. 2.5 μL of PLUS™ reagent (at a 1:1 ratio to DNA) was then added directly to the diluted DNA. After gentle mixing and a 10 minute incubation at room temperature, 10 μL of Lipofectamine™ LTX was added into the diluted DNA solution, mixed gently and incubated for 35 minutes at room temperature to form DNA-Lipofectamine™ LTX complexes. The DNA-Lipofectamine™ LTX complexes were added dropwise to each well containing cells and mixed gently by manually rocking the plate back and forth. Cells were incubated at 37°C in a CO2 incubator for 24 hours post-transfection before assaying. For cells treated with glycine, glycine dissolved in RPMI (1 M) was added to the culture medium 4 hours post-transfection.
Preparation of cells for FACS and quantitation of PPIX
HeLa and K562 cells were washed, scraped and resuspended in PBS (80 mM disodium hydrogen orthophosphate, 20 mM sodium dihydrogen orthophosphate, 100 mM sodium chloride, pH 7.5) before pipetting into BD Falcon tubes with cell strainer caps. Preparation of cells for FACS was done under very low light conditions (1-2 Lux as measured by a Pyle PLMT68 light meter) in order to minimize phototoxicity caused by PPIX accumulation. FACS analyses were performed using a BD LSR II Analyzer (Becton, Dickinson, and Company) and FACSDiva Version 6.1.3 software. ZsGreenl emission was measured between 515 nm and 545 nm (530/30BP filter) when cells were excited using the 488 nm laser. In order to eliminate any background red fluorescence, the 633 nm-red laser was blocked during the collection of the PPIX emission data. PPIX emission was determined in the 619 nm and 641 nm range (630/22BP filter) when cells were excited with the 405 nm laser. Forward-scatter (FSC) versus side-scatter (SSC) dot plots were used to gate the whole cells and thus remove the contribution of the cell debris from the population being examined. A minimum of 10,000 of the gated whole cells were then depicted in dot plots of SSC versus ZsGreenl fluorescence, and the “green-fluorescent population” gate was defined based on untransfected HeLa cells as negative controls. Dot plots of SSC versus PPIX fluorescence were used to define the PPIX-accumulating cells for both the “green-fluorescent” and the “non-green fluorescent” populations. The PPIX gating was based on the negative control for PPIX, the pIRES2-ZsGreenl vector-expressing cells. Normalized PPIX fluorescence values were obtained by dividing the mean PPIX fluorescence by the mean ZsGreenl fluorescence for each cell population.
Protein purification
The XLPP variants (delAT and delAGTG) and wild-type hALAS2 were purified from E. coli BL21 bacterial cells harboring pSD2, pSD3 and pSD1, respectively, as previously described by Ferreira and Dailey28 with some modifications. Briefly, BL21 cells harboring the expression plasmids were grown in a low phosphate medium containing 100 μg/mL ampicillin at 37 °C for 16 hours. After centrifugation the bacterial pellet was suspended in buffer A (20mM potassium phosphate, lmM EDTA, 5 mM β-mercaptoethanol, 10% glycerol, 1 mg/ml protamine sulfate and 20 μM PLP, at pH 8.0). The cells were then lysed using a French press cell, the wild-type and variant enzymes were precipitated with 45 % ammonium sulfate. Following the 45% ammonium sulfate precipitation, the pellet was dissolved in buffer A containing 5 % DMSO and loaded onto an Ultrogel Aca-44 gel filtration column equilibrated with buffer A. Following elution, the sample was loaded onto a DEAE-Sephacel column equilibrated with buffer A. The resin was sequentially washed with buffer A, buffer A containing 20 mM KCl, and buffer A containing 50 mM KCI. hALAS2 (or XLPP variant) was then eluted with buffer A containing 100 mM KCl.
Purification of His-tagged wild-type hALAS2 and His-tagged XLPP variants
BL21(DE3) cells harboring the expression plasmids for His-tagged hALAS2 were grown in two 125 mL-flasks with 50 mL of LB medium containing 100 μg/mL ampicillin at 37°C for 8 hours. In each of eight 2 L-flasks, 10 mL of bacterial culture was added to 1 L MOPS medium containing 100 μg/mL ampicillin. The bacterial cultures were grown for 16 hours at 37 °C. The cells were then harvested by centrifugation and stored at −20 °C until the day of purification.
All purification procedures were carried out at 4°C as described previously29 with minor changes. For proteins purified for activity assays, the cell pellet was resuspended in 30-40 mL of resuspension buffer (20 mM tricine, pH 8.0 containing 5 mM β-mercaptoethanol, 20 μM PLP, 10% glycerol, and 0.2% Triton X-100). The following protease inhibitors were also added in all buffers just before usage: aprotinin, leupeptin, and pepstatin at 1 μg/mL, and PMSF at a final concentration of 10 μg/mL. Following resuspension, the cells were homogenized and lysed by passing the cells twice through a French press at 10,000 psi. Cell debris was removed by ultracentrifugation in a swinging bucket rotor at 104,000 × g for 1 hour. The supernatant containing the protein was then loaded onto an affinity chromatography column (1.5 × 10cm) packed with HisPur™ Ni-NTA resin (Thermo Fisher Scientific Inc.) previously equilibrated with an “equilibration buffer” (20 mM tricine, pH 8, containing 20 μM PLP, and 10% glycerol,). The resin was subsequently washed with 100 mL of buffer A (20 mM tricine, pH 8, containing 5 mM β-mercaptoethanol, 20 μM PLP, and 10% glycerol), 50 mL of buffer A, pH 8, with 0.5 M NaCl, 50 mL of a 25 mM imidazole buffer (i.e., 25 mM imidazole, pH 7.2, containing 5 mM β-mercaptoethanol, 20 μM PLP, and 10% glycerol), and finally 50 mL of a 50 mM imidazole buffer (i.e., 50 mM imidazole, pH 7.2, containing 5 mM β-mercaptoethanol, 20 μM PLP, and 10% glycerol). The protein (either hALAS2 or XLPP variants) was eluted with 50 mL of a 100 mM imidazole buffer (i.e., 100 mM imidazole, pH 7.5, containing 5 mM β-mercaptoethanol, 20 μM PLP, and 10% glycerol). The eluted protein was immediately concentrated in protein concentrators with a 20,000 molecular weight cutoff (Thermo Scientific™ Pierce™). During concentration, the buffer was gradually exchanged to 20 mM tricine buffer, pH 8, containing 20 μM PLP, and 10% glycerol.
For the purification of proteins used in structural studies, glycerol and free PLP were eliminated from the buffers. Briefly, the cell pellet was resuspended in 30-40 mL of 20mM sodium phosphate, pH 8.0, containing 300 mM sodium chloride, 20 μM PLP, and 0.2% Triton X-100. The supernatant containing the protein was then loaded onto an affinity chromatography column (1.5 × 10cm) packed with HisPur™ Ni-NTA resin previously equilibrated with a phosphate equilibration buffer (20 mM sodium phosphate, pH 7.4, 300 mM sodium chloride, and 10 mM imidazole). The resin was first washed with phosphate equilibration buffer, followed by washes with increasing imidazole concentrations of 25 mM and 50 mM. The protein was eluted with 50 mL of a 150 mM imidazole buffer (i.e., 20 mM sodium phosphate, pH 7.4, containing 300 mM sodium chloride, 150 mM imidazole). The buffer of the eluted protein was gradually exchanged to 20 mM phosphate buffer, pH 8, and the protein concentrated.
Protein purify and concentration
Purity of the protein was assessed by SDS-PAGE. Protein concentration was determined using the bicinchoninic acid assay30 with bovine serum albumin as the standard. Protein concentrations are reported on the basis of subunit molecular masses of 57.4 kDa, 55.0 kDa, 57.4 kDa, and 52.7 kDa, for the wild-type hALAS2, delAT, delAGTG, and Q548X variants, respectively, calculated using the amino acid sequences and ProtPram.31
Spectrophotometric determination of ALAS activity
ALAS activity of wild-type hALAS2 and XLPP and variants was determined at 30 and 37 °C using an established coupled enzyme assay.32
Transient kinetics
Reactions of glycine saturated wild-type hALAS2 or XLPP variants with succinyl-CoA were monitored under single turnover conditions and recorded using a KinTek stopped-flow spectrophotometer, model SF-2001 (KinTek Corp, Austin, TX) at 20 °C. Experiments were conducted in a buffer system composed of 20 mM HEPES, pH 8.0, containing 10% (by volume) glycerol. Reactant concentrations are reported in the pertinent figure legends. Time courses for the formation and decay of a spectroscopically observable quinonoid intermediate, corresponding to the decarboxylation and protonation steps of Scheme 1, were recorded at 510 nm. Single-turnover time course data were interpreted using a two-step kinetic mechanism as described by reaction 1 and were fit to a two-exponential equation (Eq. 1) using software provided by KinTek Corp.
Scheme 1.
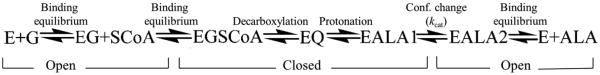
ALAS catalytic reaction cycle. E, enzyme; G, glycine; SCoA, succinyl-coenzyme A; EGSCoA, Michaelis complex of enzyme with substrates; EQ, enzyme-bound quinonoid intermediate; EALAl, product-bound enzyme in closed conformation; EALA2, product-bound enzyme in open conformation; ALA, 5-aminolevulinate. The chemical nature of the reaction steps are noted above the arrows, while the enzyme conformation is noted below.
| Reaction (1) |
| Eq. (1) |
where At is the absorbance at time t, a is the amplitude of each phase, k is the observed rate constant for each phase and c is the final absorbance.
Intrinsic protein fluorescence quenching stopped-flow spectroscopy
The pre-steady state kinetics of the product binding reaction of wild-type hALAS2 and XLPP variants (delAGTG and delAT) were examined by measuring changes in the intrinsic protein fluorescence using model RSM-1000 stopped-flow spectrophotometer (OLIS, Inc.) as previously described.33,34 The enzymes (10 μM) and ALA were maintained at 30 °C in 20 mM HEPES, pH 7 .5 containing 20 % glycerol, in separate syringes prior to mixing in the reaction chamber. Intrinsic protein fluorescence changes associated with progress towards equilibration with ALA were evaluated at several different concentrations of ALA. The excitation wavelength was 280 nm, and a 320 nm cutoff filter was placed over the detection photomultiplier to ensure any stray light was omitted from the collected data. Each data set was fit to equation 2, using the global fitting software provided with the instrument.
| Eq. (2) |
In equation 2 Fobs(t) is the observed fluorescence (in arbitrary units) at time t, A is a pre-exponential factor, kobs is the observed pseudo-first order rate constant for the rate at which the reaction approaches equilibrium, and A0 is the initial fluorescence background (offset). The observed rate constants were then plotted as a function of ALA concentration and the data were fit to equation 3 for an offset hyperbola.
| Eq. (3) |
In equation 3, kobs is the best fit to the observed pseudo-first order rate constant data acquired from Equation 2, kon is the resolved or true rate constant for ALA association with the enzyme, KD is the ALA binding constant, and koff is resolved rate constant for ALA dissociation from the enzyme.
Determination of the thermal stability of hALAS2 wild-type and variants
The thermal stability of the wild-type hALAS2 and XLPP variants was characterized by heating the enzyme at the desired temperature (20-100 °C) for three minutes in a thermocycler, cooling to 37°C, and then immediately measuring activity at 37°C as described above. The assays for each enzyme and at each temperature were performed in triplicate.
Acrylamide Quenching of Intrinsic Protein Fluorescence
Fluorescence quenching of a 2 μM protein (wild-type hALAS2 or XLPP variant) in 20 mM phosphate buffer, pH 8.0 was followed upon addition of acrylamide using a Shimadzu RE-5301 PC spectrofluorophotometer. Aliquots of a 2 M acrylamide solution, made just prior to running the experiments, were added to the protein samples to yield 100 μM increments, and the changes in fluorescence emission intensity of the tryptophan residues (λem = 330 nm) were measured by collecting the emission spectra after each addition and excitation at 295 nm. Fluorescence values were corrected for the dilution factors caused by the addition of the acrylamide aliquots. Controls were run by collecting the fluorescence emission (λex = 295 nm; λem = 330 nm) spectra for each sample containing all components except enzyme immediately prior to collecting the spectra of the corresponding samples with enzyme. These blank control spectra were subtracted from the spectra of samples containing enzyme. To obtain dynamic quenching constants for each hALAS2 variant, the fluorescence emission intensities at 330 nm were plotted against the acrylamide concentration and the data were analyzed according to the Stem-Volmer relationship and fit to Equation 4.
| Eq. (4) |
In equation 4, Fo is the fluorescence intensity without quencher, F is the fluorescence intensity with quencher, kq is the quencher rate coefficient, τ0 is the lifetime of the emissive state of tryptophan without quencher present, Q is the concentration of the quencher acrylamide, and Ksv is the dynamic quenching constant of acrylamide for the tryptophans of hALAS2 or XLPP variant.
Circular dichroism (CD) spectroscopy
CD spectra were recorded using a JASCO J-815 spectrometer continuously purged with nitrogen. For far-ultraviolet (UV) CD, the proteins were at 0.1 mg/mL in 20 mM sodium phosphate, pH 8.0 in a quartz cuvette with a 1.0 μm path length. Spectra covered a 260-190 nm range and were collected at 25°C and a scan speed of 20 nm/minute with a 0.1 nm step size and 1.0 nm bandwidth. Four spectra were accumulated and averaged for each sample. For near-UV and visible CD, the proteins were prepared at 1.0 mg/mL in 20 mM sodium phosphate, pH 8.0, in a cuvette with a 1.0 cm path length. Spectra were recorded at 25°C, over the wavelength range 500-260 nm and at a scan speed of 20 nm/minute. Three spectra were accumulated and averaged for each sample.
RESULTS
Transient expression of wild-type hALAS2 and XLPP variants and PPIX accumulation in mammalian cells
To determine whether recombinant XLPP variants could produce greater amounts and buildup of PPIX in mammalian cells than the wild-type enzyme, we evaluated the extent of PPIX accumulation in HeLa and K562 cells transiently transfected with wild-type hALAS2 and XLPP variants expression plasmids. In HeLa cells, when compared to expression of wild-type hALAS2, delAT, delAGTG, and Q548X increased PPIX accumulation by 2.9-fold (p<0.05), 3.6-fold (p<0.01), and 3.7-fold (p<0.01), respectively (Figure 2). In K562 cells, delAT, delAGTG, and Q548X increased PPIX build-up by 1.6-fold (p<0.05), 1.6-fold (p<0.05), and 2.1-fold (p<0.01), respectively. Expression of delAT, delAGTG, or Q548X resulted in significantly increased PPIX accumulation in mammalian cells in comparison to expression of wild-type hALAS2, also indicating that the XLPP variants are stable in the cellular environment.
Figure 2.

Expression of XLPP variants in mammalian cells results in accumulation of PPIX. HeLa cells are indicated by vertical-striped bars, while K562 cells are represented by checkered bars. The graph bars correspond to the results with wild-type hALAS2 (blue), delAT (green), delAGTG (pink), and Q548X (purple). Mean PPIX fluorescence values are representative of three separate experiments ± standard deviation (*p<0.05 and **p<0.01, Student’s t-test) [a.u., arbitrary units].
PPIX accumulation in HeLa cells expressing XLPP variants and grown in glycine-supplemented culture medium
Since the XLPP variants have variable affinities for glycine,17 we set out to examine if glycine supplementation of the culture medium enhanced PPIX accumulation in mammalian cells expressing XLPP variants. Similar to mALAS2,35 the Michaelis-Menten constant (Km) of hALAS2 for glycine (15 ± 3 mM)17 is higher than its intracellular concentration of approximately 2.5 mM,36 and thus we reasoned that supplementation of the cell culture medium with glycine could cause an increase in the synthesis of ALA, and subsequently, PPIX. In HeLa cells expressing wild-type hALAS2, supplementation of the medium with 100 mM glycine increased the PPIX by 2-fold (p<0.05) and in cells expressing delAGTG, PPIX increased by 1.5-fold (p<0.01) (Figure 3). However, the effect of glycine supplementation on cells expressing delAT and Q548X did not reach statistical significance (p>0.05). These data paralleled the values for the steady-state kinetic parameters determined at 30 °C17 and 37°C (data not shown) that indicate that delAGTG has a significantly increased affinity for the glycine substrate.
Figure 3.

Glycine supplementation ofthe culture medium significantly increases PPIX accumulation in HeLa cells expressing delAGTG. Plus (+) signs indicate samples in which the culture medium was supplemented with 100 glycine, and minus (−) signs indicate samples in which no glycine was added. The graph bars correspond to the results with wild-type hALAS2 (blue), delAT (green), delAGTG (pink), and Q548X (purple). Mean PPIX fluorescence values are representative of three separate experiments ± standard deviation (*p<0.05 and **p<0.01, Student’s t-test) [a.u., arbitrary units].
Protein purification
In general, the hALAS2 constructs, and particularly the variants with increased enzymatic activity, did not overexpress to the high levels observed with murine ALAS2;28,37-39 the yield of purified protein was typically on the order of 1-2mg/L of culture or 8-40mg/L, depending whether the recombinant proteins were devoid of or had an N-terminal His-tag, respectively. In general, the proteins were purified to 95% homogeneity (data not shown). Absorption spectra of wild-type hALAS2 and XLPP variants had maxima at 400 and 330 nm (data not shown), with a greater A400/A330 ratio for the variant enzymes. The absorbance maximum at 400 nm can arise from either a strained aldimine bond between the PLP cofactor and an active site lysine or an unprotonated aldimine nitrogen. While the absorption spectrum of murine ALAS2 displayed two absorbance maxima, at 330 and ~420 nm, which we assigned to two different tautomeric species of the internal aldimine, a substituted aldamine and a ketoenamine Species, respectively,40 the absorbance maximum at 400 nm suggests that the cofactor environment in the human enzyme differs slightly from that of the mouse enzyme. In all cases, the A420/A330 ratio was increased upon reaction with glycine, consistent with the formation of an aldimine linkage between the PLP cofactor and glycine.
Transient kinetics
Previously, we followed the temporal evolution of a quinonoid reaction intermediate, an ascribed species with an absorbance maximum at 510 as our spectroscopic “porthole” for studying the kinetic mechanism and reaction pathway of murine ALAS2.27,41-43 Similarly, we used stopped-flow spectroscopy to examine the hALAS2- and the XLPP variants-catalyzed reactions by mixing the specific enzyme-glycine complex with succinyl-CoA and monitoring the change in absorbance at 510 nm. The formation and decay of the quinonoid reaction intermediate were followed under single-turnover conditions, with the enzyme present in higher concentration than the succinyl-CoA substrate. The time courses for the reactions ofhALAS2 and XLPP variants (delAT, delAGTG and Q548X) were best described by a two-step sequential mechanism represented by reaction (1). In Figure 4, the data points for the time courses at 510 nm were overlaid with the best fit lines of the spectral data at the same wavelength using equation (1). Both the reaction for wild-type hALAS2 and those for the XLPP variants comprised a single kinetic step associated with quinonoid intermediate formation followed by one decay step (Figure 4). This single kinetic decay step differs from the two steps observed for the decay of the quinonoid intermediate in the murine ALAS2 reaction,27,42 which have been assigned to protonation of the covalently linked ALA-quinonoid intermediate and opening of the active site loop to allow release of ALA and regenerate the holoenzyme.27,33,42 Most probably the weaker absorbance signal at 510 nm for the hALAS2-derived enzyme reactions rendered the spectral separation of the two potential kinetic steps impossible. Nevertheless, the comparison of time courses for the reactions of hALAS2, the delAT, delAGTG and Q548X clearly indicate that the XLPP variants catalyze the formation and decay of the quinonoid intermediate at significantly faster rates than the wild-type human enzyme (Figure 4) The observed rates for the two steps range from 9.8 ± 0.4 s−1 to 14.3 ± 0.6 s−1 and from 1.37 ± 0.04 s−1 to 1.97 ± 0.05 s−1 for the reactions of the XLPP variants, whereas those for the wild-type hALAS2-catalyzed reaction were 6.6 ± 0.2 s−1 and 0. 70 ± 0.02 s−1 (Table 1 ).
Figure 4.
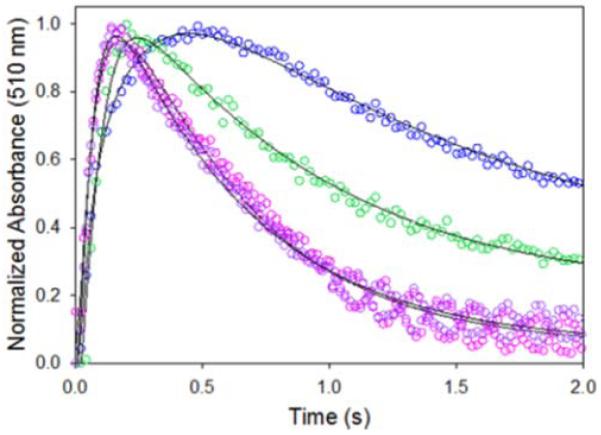
Time courses for the single-turnover reactions of wild-type hALAS2 and XLPP variants. Shown are the traces for wild-type hALAS2 (blue), delAT (green), delAGTG (magenta) and Q548X (purple), each saturated with glycine, and reacted with succinyl-CoA at a molar enzyme-glycine complex:succinyl-CoA ratio of at least 10: 1. To be directly compared, the data were normalized to the quinonoid intermediate signal (Abssionm). The solid black lines overlaying the time course data points were obtained after fitting the pertinent data to equation (1). The conditions in the observation chamber following mixing were 50 pM wild-type hALAS-2 (120 pM delAT, 34 pM delAGTG or 64 pM Q548X), 100 glycine, succinyl-CoA (3 - 12 pM range), 20 pM PLP, 10% glycerol and 20 tricine, pH 8.6. The temperature was 20 °C.
Table 1.
Observed rates of quinonoid intermediate formation and decay under the single turnover reaction conditions described in Figure 3
| Parameter | Wild-type hALA2 (s−1) |
DelAT (s−1) |
DelAGTG (s−1) |
Q548X (s−1) |
|---|---|---|---|---|
| Q f | 6.6 ± 0.2 | 9.8 ± 0.4 | 11.1 ± 0.5 | 14.3 ± 0.6 |
| Q d | 0.70 ± 0.02 | 1.37 ± 0.04 | 1.97 ± 0.05 | 1.93 ± 0.07 |
Intrinsic protein fluorescence quenching and product binding
In these experiments the rate of ALA release (koJJ) is typically observed to be coincident with the catalytic rate constant (kcat) determined in separate steady-state kinetic experiments, supporting the postulate that opening of the active site loop to allow ALA dissociation is the limiting factor in ALA production by the ALAS2 enzyme.33,34 The resolved ALA off rate constants for the XLPP variants were faster than that of the wild-type hALAS2, as seen by the increases in the y-intercept values for these variants in Figure 5. Specifically, the rate constant of ALA dissociation from wild-type hALAS2 was 0.02 ± 0.01 s−1 (kon = 0.37 ± 0.02 s−1; KD = 2.1 ± 0.4 mM), for delAGTG it was increased 8-fold to 0.16 ± 0.01 s−1 (kon = 0.34 ± 0.04 s−1; KD = 5 ± 2 mM), and for delAT it was increased 7-fold to 0.14 ± 0.02 s−1 (kon = 0.32 ± 0.05 s−1; KD = 5 ±1 mM). The rate constant of ALA binding (kon) was unaffected, while the dissociation constant for an initial collision complex (KD) was slightly increased in the XLPP variants. The elevated KD values are also consistent with an enhanced rate constant of product release.
Figure 5.
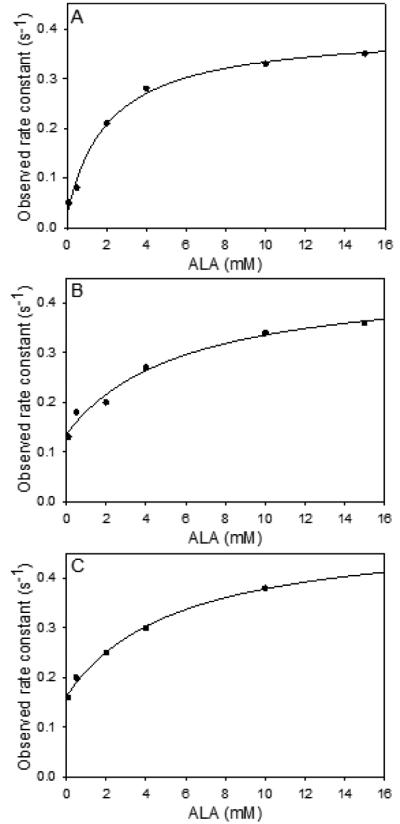
Protein fluorescence changes associated with reaction of ALA with XLPP variants indicate an increase in the rate constant of product release (corresponding to the y-axis intercept as defined in Equation 3). Observed reaction rates for (A) wild-type hALAS2 (10 pM), (B) delAT (10 pM), and (C) delAGTG (10 pM) with ALA were determined by fitting the data to Equation 3.
Thermal stability of wild-type hALAS2 and XLPP variants
Thermal stability experiments were conducted to examine the possibility that the C-terminal mutations present in the XLPP and variants might affect the structural integrity of the enzymes. Enzymatic activity was plotted as a function of temperature as shown in Figure 6. Up to 45°C, the decrease in activity was more pronounced for the XLPP variants, especially delAT, than for the wild-type enzyme. However, at temperatures above 50°C, wild-type hALAS2 lost all measurable activity, whereas the delAT and delAGTG variants retained 7% and 11 % of their initial activity, respectively, at 70°C. The calculated thermal transition temperature T1/2, which is defined as the temperature needed to achieve a 50 % activity loss, was 48.6°C, 52.5°C, 49.9°C, and 51.4°C for wild-type hALAS2, delAT, delAGTG, and Q548X, respectively. Thus, the XLPP variants were observed to be more thermostable than wild-type hALAS2. The melting temperatures (Tm) of wild-type hALAS2 and delAT were also determined using differential scanning calorimetry, yielding a Tm value ~7 °C lower for wild-type hALAS2 (data not shown).
Figure 6.
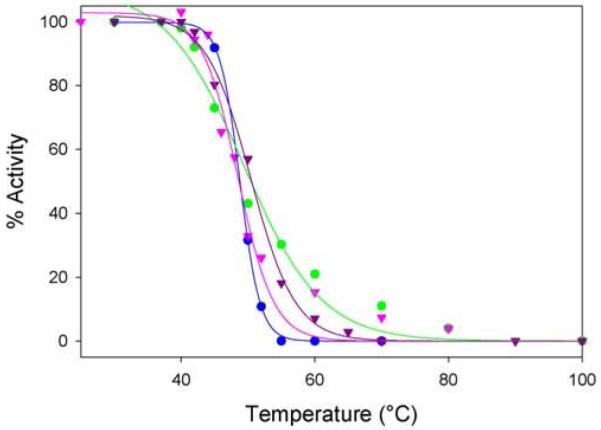
Thermal stability profiles ofwild-type hALAS2 and XLPP variants indicate that the variants are thermally stabilized. The plotted data are averages of three independent experiments and are fit to equation y = a/(1 +e(−(x-x0)/b). The observed thermal transition temperature (T1/2) values were 48.6°C, 49.9°C, 51.4°C and 52.5°C for wild-type hALAS2 (blue), delAGTG (pink), Q548X (purple) and delAT (green), respectively.
Effect of succinyl-CoA binding on the secondary structure of wild-type hALAS2 and XLPP variants
Far-UV circular dichroism (CD) spectra (260-190 nm) of wild-type hALAS2 and the XLPP variants in the absence and presence of 50 μM succinyl-CoA indicated that the wild-type enzyme did not undergo significant structural reorganization upon substrate binding, as depicted in Figure 7A. However, the integrity of the secondary structure was significantly modified with the binding of 50 μM succinyl-CoA to the XLPP variants (Figure 7B-D). Specifically, the 210 nm-elliptical minimum was blue-shifted to 208 nm and its associated intensity decreased; the intensity of the 222 nm-minimum was also reduced (Figure 7B-D).
Figure 7.

Effect of succinyl-CoA binding on circular dichroism spectra, far-UV region, of wild-type hALAS2 and XLPP variants. The spectra for (A) wild-type hALAS2, (B) delAT (C) delAGTG and (D) Q548X were recorded for the holoenzymes in the absence (solid lines) or presence of 50 μM succinyl-CoA (dashed lines). The concentrations of enzyme were 0.1 mg/mL in 20 mM phosphate buffer, pH 8.0.
Tertiary structure and acrylamide quenching of wild-type hALAS2 and XLPP variants
As near UV-CD spectra of proteins reflect their tertiary structure through the distinct spatial arrangements of aromatic amino acids,44,45 potential differences in the tertiary structures and PLP-binding sites among wild-type hALAS2 and XLPP variants were evaluated using CD spectroscopy in the near-UV (310-260 nm) and visible (310-500 nm) regions (Figure 8A). A decreased ellipticity in the near-UV signal associated with the delAT variant indicated that the truncation introduced with the delAT mutation affects the integrity of the tertiary structure of hALAS2 (Figure 8A). On the other hand, the near-UV CD spectroscopic features of delAGTG and Q548X were enhanced when compared to those of wild-type hALAS2 (Figure 8A). Possible structural differences between hALAS2 and the XLPP variants were also analyzed by assessing tryptophan exposure and quantifying the concentration of acrylamide required to quench the tryptophan emission in these enzymes. Figure 8B illustrates the quenching profiles obtained for hALAS2 and XLPP variants with respect to acrylamide concentration. The slopes of the curves for the data fit to the Stem-Volmer equation (Eq. 4) yielded the dynamic quenching constant (Ksv) values of 1.4 M−1, 3.0 M−1, 1.9 M−1, and 2.5 M−1 for wild-type hALAS2, delAT, delAGTG, and Q548X, respectively. The increased Ksv values observed with the XLPP variants could imply that, under the described experimental conditions, the variants were more dynamic than hALAS2, which elicited their tryptophans to be repositioned in an environment more accessible to the quencher.
Figure 8.

XLPP-introduced structural changes in hALAS2 and acrylamide quenching of intrinsic protein fluorescence. (A) Circular dichroism spectra, near-UV region, of wild-type hALAS2 and XLPP variants. The concentrations of enzyme were 1.0 mg/mL in 20 mM phosphate buffer, pH 8.0. Wild-type hALAS2 (blue), delAT (green), delAGTG (magenta) and Q548X (purple). (B) Stem–Volmer plots of wild-type hALAS2 and XLPP variants fluorescence quenching by acrylamide. Enzyme (either wild-type hALAS2 or XLPP variant) concentration was 2 pM in 20 mM phosphate buffer, pH 8.0. Wild-type hALAS2 (blue), delAT (green), delAGTG (magenta) and Q548X (purple).
Effect of succinyl-CoA binding on the PLP-binding site of wild-type hALAS2 and XLPP variants
CD spectra recorded in the visible region (310-500 nm) for wild-type hALAS2 and delAT variant revealed a positive Cotton effect, which is maximal at 430 nm (Figure 9A, B), whereas the CD spectra for the delAGTG and Q548X variants showed two positive dichroic bands centered at 330 and 430 nm (Figure 9C, D). However, the ratio of the molar ellipticities of the delAGTG variant for the 430 and 330 nm bands is 1.8, and that of the Q548X variant is less than 1.0 (Figure 9C, D). Cofactor-induced CD spectra of PLP-dependent enzymes originates from interactions between the cofactor and the apoenzyme, specifically from π to π* transitions of the PLP cofactor in the protonated Schiff base.46,47 The distinct molar ellipticities at 330 and 430 nm among wild-type hALAS2 and XLPP variants suggest that the PLP cofactor is bound somewhat differently in the active sites of these enzymes, possibly with changes in the orientation of the cofactor. Addition of succinyl-CoA to the wild-type hALAS2 and delAT holoenzymes reduced their molar ellipticity band at 430 nm by 55% and 20%, respectively (Figure 9A, B). When succinyl-CoA was added to either the delAGTG or the Q548X holoenzyme, not only was the intensity of the dichroic bands at 430 and 330 nm reduced by approximately 20% to 30%, but also the λmax value of the positive dichroic band at 330 nm shifted to 350 nm (Figure 9C, D). These observations indicate that binding of succinyl-CoA to the delAGTG or the Q548X induces distinct changes to the chiral environment of the PLP cofactor.
Figure 9.

Effect of succinyl-CoA binding on circular dichroism spectra, visible region, of wild-type hALAS2 and XLPP variants. The spectra for (A) wild-type hALAS2, (B) delAT, (C) delAGTG and (D) Q548X were recorded for the holoenzymes (solid lines) or holoenzymes in the presence of 50 μM succinyl CoA (dashed lines). The concentrations of enzyme were 1.0 mg/mL in 20 mM phosphate buffer, pH 8.0.
DISCUSSION
Loss- and gain-of-function mutations in the ALAS2 gene are associated with specific erythroid disorders.10,15,19 ALAS2 loss-of-function mutations, by hindering activity,7,21,48-50 structure7,21,48 stability,49,50 PLP cofactor binding,21,49 protein-protein interactions12,26 and/or mitochondrial localization51 can cause XLSA with the consequent systemic iron overload and decreased heme and hemoglobin production.10,14 Loss of function of hALAS2 and iron overload, due to a weakened binding of the cofactor accompanied with lowered protein stability, were also recently reported in females with X-linked macrocytic dyserythropoietic anemias.52 Conversely, ALAS2 gain-of-function mutations and the consequent accumulation of erythroid protoporphyrin and Zn-protoporphyrin are at the root of XLPP.15,20,24 ALAS2 mutations (e.g., c.1757 A > T in exon 11) can also modulate gene function in congenital erythropoietic porphyria.53 Here, we report the examination of the enzymological and structural properties of XLPP variants in relation to those of recombinant hALAS2.
The murine enzyme has provided the most explored model to understand the mechanistic enzymology of mammalian ALAS2.27,29,33,34,37,38,41-43,54-57 Significantly, a variety of murine ALAS2 point and multiple mutations resulted in enzymes with enhanced catalytic efficiencies.27,39 ALAS2 catalyzes the rate-determining step of porphyrin biosynthesis, so mutations leading to heightened enzyme activity could explain the elevated concentrations of porphyrins observed in XLPP. This stimulated us17 and others16 to investigate hALAS2 and the XLPP variants in order to determine whether the variants could have enhanced catalytic properties relative to the wild-type enzyme. Using steady-state kinetics, we demonstrated that the catalytic rates and specificity constants (for glycine and succinyl-CoA) of the XLPP variants were significantly greater than those of wild-type hALAS2, whereas the Km values for the 2 substrates remained within the same order of magnitude.17 Now, by utilizing the microscopic parameters obtained from the transient kinetics for the reactions catalyzed by the disorder-associated variants under single turnover conditions, we can pin down the reason for the enhancement in variant activity over that of the wild-type hALAS2 to the increased rates for the formation and decay of the ALA-quinonoid intermediate (designated “EQ” in Scheme 1). More specifically, we demonstrate that the protein conformational transition step governing ALA product release is mainly affected in the XLPP variant-catalyzed reactions.
The rate-limiting step in the murine ALAS2-calyzed reaction was proposed to be determined by a conformational change of the enzyme accompanying ALA release.27,42 This also appears to be the case with the hALAS2 and XLPP variants, as further evidenced here with studies involving intrinsic protein fluorescence quenching upon product (ALA) binding to both wild-type hALAS2 and XLPP variants (Figure 5). In fact, the calculated rates for the dissociation of ALA from the enzymes (koff) (Figure 5) were similar to the kcat values determined under the same experimental conditions.17 The increased ALA koff rates and kcat values for the XLPP variants probably arise from reduced stability of the closed conformation, affording a more rapid return to the open conformation and product release. From prior transient kinetic analyses of the murine ALAS2 reaction, we proposed a model for catalysis by ALAS in which succinyl-CoA triggers interconversion between an open conformation and a closed conformation, where catalysis take places, and turnover is defined by reversion to the open conformation, concomitant with release of ALA from the enzyme.41 Significantly, this kinetically-derived model4,42,58 is compatible with the domain movement inferred from the crystal structure of the R. capsulatus ALAS holoenzyme in relation to that of the succinyl-CoA-bound holoenzyme.9
Binding of succinyl-CoA to the XLPP variants clearly impacted the secondary and tertiary structures of these proteins in a distinct fashion when compared to wild-type ALAS2, with the variants undergoing structural reorganization upon substrate binding (Figures 7-9). Albeit, as assessed by near-UV CD spectroscopy, the different XLPP mutations altered the tertiary structure of hALAS2 to different extents (Figure 8A), the repositioning of tryptophans or overall protein conformation changes were more profound in the delAT variant (as judged by both near UV CD spectroscopy (Figure 8A) and fluorescence quenching measurements with acrylamide) (Figure 8B)). Acrylamide quenches the intrinsic fluorescence of delAT more strongly than hALAS2 and delAGTG and Q548X variants. Since diffusion of acrylamide, a polar but uncharged quencher, into the interior of proteins can be facilitated by small fluctuations in the polypeptide conformation,59 the value for the dynamic quench constant, Ksv, is an indicator of protein conformational flexibility. Thus, and in particular, the difference between the Ksv value associated with the delAT and that with hALAS2 indicates that the truncation in delAT increased the access of the quencher to the protein fluorophores (tryptophan) and leads us to suggest that delAT possesses the greatest conformational flexibility among the three XLPP variants. Overall, we propose that while the overall flexibility of hALAS2 increases with the introduction of the XLPP mutations, it is mainly the disruption of the conformational equilibrium caused by the introduction of the XLPP mutations that leads to the enhancement of product (ALA) release observed in the XLPP variants-catalyzed reactions.
Major effects on the hALAS2PLP cofactor microenvironment were also induced by the XLPP mutations, as determined using CD spectroscopy (Figure 9). The visible region CD spectra of the delAGTG and Q548X variants deviate substantially from those of wild-type hALAS2 and the delAT variant. While the CD spectra for wild-type hALAS2 and the delAT variant had a single predominant maximum at 430 nm, those for the delAGTG and Q548X variants showed an additional maximum at 330 nm; the ratio of the mean residual ellipticity at 430 nm to the one at 330 nm was greater than 1.0 for delAGTG and lower than 1.0 for the Q548X variant. The ellipticities at these two wavelengths stem from Cotton effects associated with the ketoenamine (430 nm) and substituted aldamine (330 nm) tautomers of the internal aldimine between the PLP cofactor and an active site lysine, and they reflect the microenvironment around this linkage.33,56 Previously, we assigned the ~410 nm-absorption species of murine ALAS2 to a ketoenamine species and the 330 nm-absorption species to a substituted aldamine.40 Briefly, upon excitation at 331 nm, the 330 nm-absorption species emitted fluorescence with a maximum at 385 nm, but not at 510 nm, indicating that this species is the substituted aldamine, rather than the enolimine form of the Schiff base. This fluorescence emission at 385 nm (λexc ~ 330 nm), associated with a substituted aldamine, results from the lack of double bonds conjugated with the pyridinium ring.40,60 However, the nature of the nucleophile involved in the formation of the adduct in ALAS has yet to be identified.40 Furthermore, and as observed with murine ALAS2,29,55,56 since PLP is not a chiral molecule, the positive dichroic bands with maxima at ~330 and 420 nm in the CD spectra of wild-type hALAS2 and XLPP variants indicate an equilibrium between two populations of internal aldimine species with different chiral active site environments.
Succinyl-CoA binding to the XLPP variants induced a decrease in the ellipticity at 430 nm less extensive than in the case of wild-type hALAS2, although the positive dichroic band at 330 nm in the spectra for the delAGTG and Q548X variants decreased in mean residual ellipticity and shifted to 350 nm (Figure 9). These findings indicate that the orientation of the internal aldimine in the XLPP variants differs from that observed in the wild-type enzyme. Conceivably, succinyl-CoA binding promoted conformational rearrangements, and subtle structural differences among the wild-type and XLPP proteins prompted stabilization of distinct conformational subsets and alterations in the tautomeric equilibrium of the internal aldimine. Similar to a previous interpretation we offered for murine ALAS2,33 it is possible that the weakened 430 nm dichroic band in the CD spectrum of wild-type hALAS2 resulted from partial conversion of the internal aldimine to free PLP aldehyde bound at the active site. In fact, this conversion, along with the transition to a closed conformation, was observed in three of the four R. capsulatus crystal structure succinyl-CoA-bound active sites.9 Thus, the lower reduction in the 430 nm-ellipticity for the XLPP variants than for hALAS2 suggests that, upon succinyl-CoA binding, the variants preserved the internal aldimine and did not assume a closed conformation to the same extent as in the wild-type enzyme. Moreover, the over 3-fold enhancement in kcat values with minimal effect on the Km value for succinyl-CoA17 is also consistent with destabilization of the closed conformation.
While Bishop et al.16 reported that no significant change to the Km value for succinyl-CoA was introduced with the XLPP mutations, these authors also stated that hALAS216,26 and XLPP variants16 showed positive cooperativity with succinyl-CoA binding and determined Hill coefficients of ~2.0. Our steady-state kinetic analyses of both purified hALAS2 and XLPP variants indicate that the enzymes follow Michaelis-Menten kinetics and succinyl-CoA is not an allosteric regulator of ALAS activity.17 Possible reasons for the discrepancies between the two groups include: 1) use of a discontinuous assay,16 which made it impossible to measure the critical initial reaction rate, instead of a continuous assay17,32 to determine ALAS activity, 2) linear regression (Lineweaver-Burk plot) of the steady-state kinetic data,16 with the inherent error mainly introduced from the reciprocal of the measured velocity at low substrate concentrations, rather than non-linear regression data analysis,17 and 3) degree of purification of the assayed enzymes. In contrast to our method purification, which produced hALAS2 and XLPP proteins with at least a 95% level of homogeneity, two equally predominant proteins, assigned to the full-length protein and a derived proteolytic product of 49.5 kDa,16 were present in the each of the enzyme samples used in the previously reported ALAS activity assays.16 Although the investigators stated that the specific activity of each enzyme remained constant irrespective of the ratio of the two protein forms,16 the possibility that the proteolytic product interfered in the ALAS activity determinations-cannot be ruled out. Nevertheless, both our findings and those of Bishop et al. on the greater specific activities16,17 and specificity constants17 of the XLPP variants relative to those of the wild-type enzyme are consistent with the porphyrin accumulation observed in XLPP patients. Moreover, the porphyrin overproduction associated with the enhanced ALAS activity of the XLPP variants could be recapitulated in human cervical carcinoma (HeLa) cells and human erythroleukemic (K562) cells expressing the genes for these proteins (Figure. 2 and 3). These results also emphasize the idea that the extended C-terminus of hALAS2 is a key control point for porphyrin biosynthesis and contains one or more elements that are inhibitory, possibly destabilizing, and most importantly, necessary for maintaining an optimal level of enzymatic activity.
The mechanism whereby the extended C-terminus of hALAS2 inhibits catalytic efficiency is unknown and multiple possibilities can be proposed to provide frameworks for future experiments. One possibility comes from studies with the mouse enzyme, the activity of which is controlled by the conformational flexibility of an active site loop.27,42 Although the tertiary structural details are unknown, it is possible that the C-terminal region of the human enzyme stabilizes a closed active site loop conformation, thereby slowing the rate of ALA production. In this model the XLPP mutations disrupt this stabilization, allowing product to be produced more rapidly. The data reported in Figure 5 support this possibility, as the rates of protein fluorescence changes associated with ALA release (koff; the y-intercept) are accelerated in the XLPP variants-catalyzed reactions.
Mutations leading to enhanced activity suggest the intriguing possibility that the C-terminal region of hALAS2 may have an important regulatory role in porphyrin production (Figure 6). The inhibitory effect of this region might be alleviated in a regulatory fashion, thereby facilitating a rapid response to changing cellular requirements for protoporphyrin IX. In this model the C-terminal region is envisioned as a control point for allosteric-type interactions, allowing activity to be directly controlled, and this control is lost as a result of XLPP-associated mutations. It is noteworthy that a conserved cysteine-X-X-cysteine motif, consistent with a metal ion- or porphyrin-binding site, is present in animal ALAS2 sequences very near the C-terminus. This motif is not directly altered in the XLPP variants, but the mutations might have some proximity effects on effector binding at this site. It seems unlikely that this highly conspicuous motif is conserved in a key heme biosynthetic enzyme by coincidence.
Yet another possibility is that the ALAS2 C-terminal region is important for protein-protein interactions that could regulate enzyme activity. Namely, the hALAS2-SUCLA2 interaction26,61 could not only be critical for the direct supply of succinyl-CoA to ALAS2 but also could provide a regulatory (allosteric) mechanism for controlling the rate of ALA and, likely, porphyrin syntheses specific to the erythroblast. Potentially, this specific interaction with ALAS2 would allosterically promote the active site loop to acquire a more restrained, ordered structure and ALAS2 to adopt the “closed conformation” conducive to the chemical events in ALAS catalysis. If the hALAS2-SUCLA2 interaction were hindered, as has been shown in vitro with XLPP mutations,16 then hALAS2 would be in a more open conformation, and the succinyl-CoA substrate access to, and product release from, hALAS2 would be facilitated. In contrast, a hindered hALAS2-SUCLA2 interaction that would keep hALAS2 in a more “closed conformation” would restrict succinyl-CoA accessibility to, and ALA release from, hALAS2. Another possible, and not necessarily exclusive, role for the SUCLA2 binding would be to protect hALAS2 from proteolysis. This overall model supports the idea of a dynamic complex between hALAS2 and SUCLA2, which is consistent with a reported weak interaction between the two proteins.62 Finally, the formation of the hALAS2-SUCLA2 complex might allow SUCLA2 to overcome its high Km value for succinate,63 and, in the erythroblast, the hALAS2-SUCLA2 interaction might be the checkpoint for succinyl-CoA, a substrate both for ALAS (heme biosynthesis) and SCS (TCA cycle).
The XLPP variants examined here are also thermally stabilized relative to the wild-type enzyme, but this property cannot be extrapolated to stability in vivo. The possibility that the C-terminus controls enzyme degradation and the elimination of this property in the variants has not been discounted. However, it was previously reported that hALAS2 is targeted for proteolysis via hydroxylation of proline-520 leading to ubiquination.64
In conclusion, these studies represent the first structural and mechanistic examination of the XLPP variants of hALAS2. We provide evidence that XLPP can be modeled in cell culture, and this model may aid in testing for direct action of potential therapeutics on hALAS2. We demonstrate that XLPP-associated hALAS2 mutations result in aberrant enzymes with enhanced catalytic activities, which are likely due to favoring the destabilization of the enzyme closed conformation and facilitated product release. We propose 1) that the extended C-terminus of wild-type mammalian ALAS2 fulfills a regulatory role that allows for modulation of activity, thereby controlling the rate of erythroid heme biosynthesis, and 2) that this control is disrupted in XLPP, resulting in porphyrin accumulation. Moreover, the demonstration of enhanced hALAS2 variants also provides new information about the regulation of substrate supply for heme synthesis during erythroid differentiation. Certainly, the implications can go beyond the biochemical and physiological metabolism of blood and be directly relevant to photodynamic therapy of cancer, based on intracellular protoporphyrin IX accumulation, and pharmacology in evaluation of potential ALAS inhibitors.
Acknowledgments
This work was supported by grants from the American Heart Association (#10GRNT4300073) and the National Institutes of Health (#GM080270) to G.C.F and by Grant Public Health and Consumer Protection Directorate (DG SANCO), PHEA programme from the European Commission, Brussels, Belgium: “European Porphyria Network” and the Laboratory of excellence, GR-Ex, Paris, France. The labex GR-Ex, reference ANR-11-LABX-0051 is funded by the program “Investissements d’avenir” of the French National Research Agency, reference ANR-11-IDEX-0005-02 to H.P. and L. G.
ABBREVIATIONS AND TEXTUAL FOOTNOTES
- ALA
5-aminolevulinic acid
- ALAS2
erythroid specific isoform of 5-aminolevulinate synthase
- CD
circular dichroism
- DMEM
Dulbecco's modification of Eagles medium
- EPP
erythropoietic protoporphyria
- FACS
fluorescence-activated cell sorting
- hALAS2
erythroid specific isoform of human 5-aminolevulinate synthase
- HeLa
human cervical carcinoma cells
- K562
human erythroleukemic cells
- PLP
pyridoxal 5’-phosphate
- SCS
succinyl-CoA synthetase
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SSC
side-scatter
- SUCLA2
ADP-forming β subunit of succinyl-CoA synthetase
- XLPP
X-linked protoporphyria
- XLSA
X-linked sideroblastic anemia
REFERENCES
- 1.Akhtar M, Abboud MM, Barnard G, Jordan P, Zaman Z. Mechanism and stereochemistry of enzymic reactions involved in porphyrin biosynthesis, Philosophical transactions of the Royal Society of London. Series B, Biological sciences. 1976;273:117–136. doi: 10.1098/rstb.1976.0005. [DOI] [PubMed] [Google Scholar]
- 2.Fratz EJ, Stojanvoski BM, Ferreira GC. Toward Heme: 5-Aminolevulinate Synthase and Initiation of Porphyrin Synthesis. In: Ferreira GC, Kadish KM, Smith KM, Guilard R, editors. Handbook of Porphyrin Science. World Scientific Publishing Co. Pte. Ltd.; Singapore: 2013. pp. 3–68. [Google Scholar]
- 3.Hunter GA, Ferreira GC. 5-Aminolevulinate synthase: catalysis of the first step of heme biosynthesis. Cell. Mol. Biol. (Noisy-le-grand) 2009;55:102–110. [PMC free article] [PubMed] [Google Scholar]
- 4.Hunter GA, Ferreira GC. Molecular enzymology of 5-aminolevulinate synthase, the gatekeeper ofheme biosynthesis. Biochim. Biophys. Acta. 2011;1814:1467–1473. doi: 10.1016/j.bbapap.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bishop DF. Two different genes encode δ-aminolevulinate synthase in humans: nucleotide sequences of cDNAs for the housekeeping and erythroid genes. Nucleic Acids Res. 1990;18:7187–7188. doi: 10.1093/nar/18.23.7187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riddle RD, Yamamoto M, Engel JD. Expression of 5-aminolevulinate synthase in avian cells: separate genes encode erythroid-specific and nonspecific isozymes. Proc. Natl. Acad. Sci. (U.S.A.) 1989;86:792–796. doi: 10.1073/pnas.86.3.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox TC, Bawden MJ, Abraham NG, Bottomley SS, May BK, Baker E, Chen LZ, Sutherland GR. Erythroid 5-aminolevulinate synthase is located on the X chromosome. Am. J. Hum. Genet. 1990;46:107–111. [PMC free article] [PubMed] [Google Scholar]
- 8.Yamamoto M, Yew NS, Federspiel M, Dodgson JB, Hayashi N, Engel JD. Isolation of recombinant cDNAs encoding chicken erythroid 5-aminolevulinate synthase. Proc. Natl. Acad. Sci. (U.S.A.) 1985;82:3702–3706. doi: 10.1073/pnas.82.11.3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Astner I, Schulze JO, van den Heuvel J, Jahn D, Schubert WD, Heinz DW. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. EMBO J. 2005;24:3166–3177. doi: 10.1038/sj.emboj.7600792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bottomley SS, Fleming MD. Sideroblastic Anemias: Molecular Basis, Pathophysiology, and Clinical Aspects. In: Ferreira GC, Kadish KM, Smith KM, Guilard R, editors. Handbook of Porphyrin Science. World Scientific Publishing Co. Pte. Ltd.; Singapore: 2013. pp. 43–87. [Google Scholar]
- 11.Munakata H, Yamagami T, Nagai T, Yamamoto M, Hayashi N. Purification and structure of rat erythroid-Specific 5-aminolevulinate synthase. J. Biochem. 1993;114:103–111. doi: 10.1093/oxfordjournals.jbchem.a124123. [DOI] [PubMed] [Google Scholar]
- 12.Furuyama K, Yamamoto M. Differential Regulation of 5-Aminolevulinate Synthase Isozymes in Vertebrates, In Handbook of Porphyrin Science. In: Ferreira GC, Kadish KM, Smith KM, Guilard R, editors. World Scientific Publishing Co. Pte. Ltd.; Singapore: 2013. pp. 1–39. [Google Scholar]
- 13.Dailey TA, Woodruff JH, Dailey HA. Examination of mitochondrial protein targeting of haem synthetic enzymes: in vivo identification of three functional haem-responsive motifs in 5-aminolaevulinate synthase. Biochem. J. 2005;386:381–386. doi: 10.1042/BJ20040570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bottomley SS. Sideroblastic Anemias. In: Greer JP, Arber DA, Glader B, List AF, Means RT, Paraskevas F, Rodgers GM, editors. Wintrobe's Clinical Hematology. 13th Lippincott Williams & Wilkins, a Wolters Kluwer business; Philadelphia: 2014. pp. 643–661. [Google Scholar]
- 15.Balwani M, Doheny D, Bishop DF, Nazarenko I, Yasuda M, Dailey HA, Anderson KE, Bissell DM, Bloomer J, Bonkovsky HL, Phillips JD, Liu L, Desnick RJ. Loss-of-Function Ferrochelatase and Gain-of-Function Erythroid-Specific 5-Aminolevulinate Synthase Mutations Causing Erythropoietic Protoporphyria and X-Linked Protoporphyria in North American Patients Reveal Novel Mutations and a High Prevalence of X-Linked Protoporphyria. Mol. Med. 2013;19:26–35. doi: 10.2119/molmed.2012.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bishop DF, Tchaikovskii Vassili, Nazarenko Irina, Desnick Robert J. Molecular Expression and Characterization of Erythroid-Specific 5-Aminolevulinate Synthase Gain-of-Function Mutations Causing X-Linked Protoporphyria. Mol. Med. 2013;19:18–25. doi: 10.2119/molmed.2013.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ducamp S, Schneider-Yin X, de Rooij F, Clayton J, Fratz EJ, Rudd A, Ostapowicz G, Varigos G, Lefebvre T, Deybach J-C, Gouya L, Wilson P, Ferreira GC, Minder EI, Puy H. Molecular and functional analysis of the C-terminal region of human erythroid-specific 5-aminolevulinic synthase associated with X-linked dominant protoporphyria (XLDPP) Hum. Mol. Genet. 2013;22:1280–1288. doi: 10.1093/hmg/dds531. [DOI] [PubMed] [Google Scholar]
- 18.Livideanu CB, Ducamp S, Lamant L, Gouya L, Rauzy OB, Deybach JC, Paul C, Puy H, Marguery MC. Late-Onset X-Linked Dominant Protoporphyria: An Etiology of Photosensitivity in the Elderly. J. Invest. Dermatol. 2013;133:1688–1690. doi: 10.1038/jid.2012.467. [DOI] [PubMed] [Google Scholar]
- 19.Schneider-Yin X, Minder EI. Erythropoietic Protopoorphyria and X-linked Dominant Protoporphyria. In: Ferreira GC, Kadish KM, Smith KM, Guilard R, editors. Handbook of Porphyrin Science. World Scientific Publishing Co. Pte. Ltd.; Singapore: 2013. pp. 299–328. [Google Scholar]
- 20.Whatley SD, Ducamp S, Gouya L, Grandchamp B, Beaumont C, Badminton MN, Elder GH, Holme SA, Anstey AV, Parker M, Corrigall AV, Meissner PN, Hift RJ, Marsden JT, Ma Y, Mieli-Vergani G, Deybach JC, Puy H. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am. J. Hum. Genet. 2008;83:408–414. doi: 10.1016/j.ajhg.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ducamp S, Kannengiesser C, Touati M, Garpon L, Guerci-Bresler A, Guichard JF, Vermylen C, Dochir J, Poirel HA, Fouyssac F, Mansuy L, Leroux G, Tertian G, Girot R, Heimpel H, Matthes T, Talbi N, Deybach J-C, Beaumont C, Puy H, Grandchamp B. Sideroblastic anemia: molecular analysis of the ALAS2 gene in a series of29 probands and functional studies of 10 missense mutations. Hum. Mut. 2011;32:590–597. doi: 10.1002/humu.21455. [DOI] [PubMed] [Google Scholar]
- 22.Lecha M, Puy H, Deybach JC. Erythropoietic protoporphyria. Orphanet J. Rare Dis. 2009;4:19. doi: 10.1186/1750-1172-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zoubida K, Goya L, Deybach J-C, Puy H. Heme Biosynthesis and Pathophysiology of Porphyrias. In: Ferreira GC, Kadish KM, Smith KM, Guilard R, editors. Handbook of Porphyrin Science. World Scientific Publishing Co. Pte. Ltd.; Singapore: 2013. pp. 89–118. [Google Scholar]
- 24.Puy H, Gouya L, Deybach JC. Porphyrias. Lancet. 2010;375:924–937. doi: 10.1016/S0140-6736(09)61925-5. [DOI] [PubMed] [Google Scholar]
- 25.Kadirvel S, Furuyama K, Harigae H, Kaneko K, Tamai Y, Ishida Y, Shibahara S. The carboxyl-terminal region of erythroid-specific 5-aminolevulinate synthase acts as an intrinsic modifier for its catalytic activity and protein stability. Exp. Hematol. 2012;40:477–486. doi: 10.1016/j.exphem.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 26.Bishop DF, Tchaikovskii V, Hoffbrand AV, Fraser ME, Margolis S. X-linked Sideroblastic Anemia Due to Carboxyl-terminal ALAS2 Mutations That Cause Loss of Binding to the B-Subunit of Succinyl-CoA Synthetase (SUCLA2) J. Biol. Chem. 2012;287:2894328955. doi: 10.1074/jbc.M111.306423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lendrihas T, Hunter GA, Ferreira GC. Targeting the active site gate to yield hyperactive variants of 5-aminolevulinate synthase. J. Biol. Chem. 2010;285:13704–13711. doi: 10.1074/jbc.M109.074237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferreira GC, Dailey HA. Expression of mammalian 5-aminolevulinate synthase in Escherichia coli. Overproduction, purification, and characterization. J. Biol. Chem. 1993;268:584590. [PubMed] [Google Scholar]
- 29.Gong J, Kay CJ, Barber MJ, Ferreira GC. Mutations at a glycine loop in aminolevulinate synthase affect pyridoxal phosphate cofactor binding and catalysis. Biochemistry. 1996;35:14109–14117. doi: 10.1021/bi961296h. [DOI] [PubMed] [Google Scholar]
- 30.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 31.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analyis Tools on the ExPASy Server. In: Walker JM, editor. The Proteomics Protocols Handbook. Humana Press; 2005. pp. 571–607. [Google Scholar]
- 32.Hunter GA, Ferreira GC. A continuous spectrophotometric assay for 5-aminolevulinate synthase that utilizes substrate cycling. Anal. Biochem. 1995;226:221–224. doi: 10.1006/abio.1995.1217. [DOI] [PubMed] [Google Scholar]
- 33.Lendrihas T, Hunter GA, Ferreira GC. Serine 254 enhances an induced fit mechanism in murine 5-aminolevulinate synthase. J. Biol Chem. 2010;285:3351–3359. doi: 10.1074/jbc.M109.066548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lendrihas T, Zhang J, Hunter GA, Ferreira GC. Arg-85 and Thr-430 in murine 5-aminolevulinate synthase coordinate acyl-CoA-binding and contribute to substrate specificity. Protein Sci. 2009;18:1847–1859. doi: 10.1002/pro.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fratz EJ, Hunter GA, Ferreira GC. Expression of murine 5-aminolevulinate synthase variants causes protoporphyrin IX accumulation and light-induced mammalian cell death. PLoS One. 2014;9:e93078. doi: 10.1371/journal.pone.0093078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryan WL, Carver MJ. Free amino acids ofhuman foetal and adult liver. Nature. 1966;212:292–293. doi: 10.1038/212292a0. [DOI] [PubMed] [Google Scholar]
- 37.Gong J, Hunter GA, Ferreira GC. Aspartate-279 in aminolevulinate synthase affects enzyme catalysis through enhancing the function of the pyridoxal 5'-phosphate cofactor. Biochemistry. 1998;37:3509–3517. doi: 10.1021/bi9719298. [DOI] [PubMed] [Google Scholar]
- 38.Hunter GA, Ferreira GC. Lysine-313 of 5-aminolevulinate synthase acts as a general base during formation of the quinonoid reaction intermediates. Biochemistry. 1999;38:37113718. doi: 10.1021/bi995076q. [DOI] [PubMed] [Google Scholar]
- 39.Tan D, Harrison T, Hunter GA, Ferreira GC. Role of arginine 439 in substrate binding of 5-aminolevulinate synthase. Biochemistry. 1998;37:1478–1484. doi: 10.1021/bi971928f. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, Cheltsov AV, Ferreira GC. Conversion of 5-aminolevulinate synthase into a more active enzyme by linking the two subunits: Spectroscopic and kinetic properties. Protein Sci. 2005;14:1190–1200. doi: 10.1110/ps.041258305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hunter GA, Ferreira GC. Pre-steady-state reaction of 5-aminolevulinate synthase. Evidence for a rate-determining product release. J. Biol. Chem. 1999;274:12222–12228. doi: 10.1074/jbc.274.18.12222. [DOI] [PubMed] [Google Scholar]
- 42.Hunter GA, Zhang J, Ferreira GC. Transient kinetic studies support refinements to the chemical and kinetic mechanisms of aminolevulinate synthase. J. Biol. Chem. 2007;282:2302523035. doi: 10.1074/jbc.M609330200. [DOI] [PubMed] [Google Scholar]
- 43.Stojanovski BM, Hunter GA, Jahn M, Jahn D, Ferreira GC. Unstable Reaction Intermediates and Hysteresis during the Catalytic Cycle of 5-Aminolevulinate Synthase: Implications from Using Pseudo and Alternate Substrates and a Promiscuous Enzyme Variant. J. Biol. Chem. 2014;289:22915–22925. doi: 10.1074/jbc.M114.574731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gasymov OK, Abduragimov AR, Glasgow BJ. Probing Tertiary Structure of Proteins Using Single Trp Mutations with Circular Dichroism at Low Temperature. J. Phys. Chem. B. 2014;118:986–995. doi: 10.1021/jp4120145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim. Biophys. Acta - Proteins and Proteomics. 2005;1751:119–139. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 46.Daum S, Tai C-H, Cook PF. Characterization of the S272A,D Site-Directed Mutations of 0-Acetylserine Sulfhydrylase: Involvement of the Pyridine Ring in the α,β-Elimination Reaction†. Biochemistry. 2003;42:106–113. doi: 10.1021/bi0268044. [DOI] [PubMed] [Google Scholar]
- 47.Schnackerz KD, Tai C-H, Potsch RKW, Cook PF. Substitution of Pyridoxal 5′-Phosphate ind-Serine Dehydratase from Escherichia coli by Cofactor Analogues Provides Information on Cofactor Binding and Catalysis. J. Biol. Chem. 1999;274:36935–36943. doi: 10.1074/jbc.274.52.36935. [DOI] [PubMed] [Google Scholar]
- 48.Cotter PD, Baumann M, Bishop DF. Enzymatic defect in "X-linked" sideroblastic anemia: molecular evidence for erythroid delta-aminolevulinate synthase deficiency. Proc.. Natl. Acad. Sci. (US.A.) 1992;89:4028–4032. doi: 10.1073/pnas.89.9.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cotter PD, May A, Fitzsimons EJ, Houston T, Woodcock BE, al-Sabah AI, Wong L, Bishop DF. Late-onset X-linked sideroblastic anemia. Missense mutations in the erythroid delta-aminolevulinate synthase (ALAS2) gene in two pyridoxine-responsive patients initially diagnosed with acquired refractory anemia and ringed sideroblasts [see comments] J Clin Invest. 1995;96:2090–2096. doi: 10.1172/JCI118258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cotter PD, Rucknagel DL, Bishop DF. X-linked sideroblastic anemia: identification of the mutation in the erythroid-specific delta-aminolevulinate synthase gene (ALAS2) in the original family described by Cooley. Blood. 1994;84:3915–3924. [PubMed] [Google Scholar]
- 51.Furuyama K, Fujita H, Nagai T, Yomogida K, Munakata H, Kondo M, Kimura A, Kuramoto A, Hayashi N, Yamamoto M. Pyridoxine refractory X-linked sideroblastic anemia caused by a point mutation in the erythroid 5-aminolevulinate synthase gene. Blood. 1997;90:822–830. [PubMed] [Google Scholar]
- 52.Sankaran VG, Ulirsch JC, Tchaikovskii V, Ludwig LS, Wakabayashi A, Kadirvel S, Lindsley RC, Bejar R, Shi J, Lovitch SB, Bishop DF, Steensma DP. X-linked macrocytic dyserythropoietic anemia in females with an ALAS2 mutation. J. Clin. Invest. 2015;125:0–0. doi: 10.1172/JCI78619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.To-Figueras J, Ducamp S, Clayton J, Badenas C, Delaby C, Ged C, Lyoumi S, Gouya L, de Vemeuil H, Beaumont C, Ferreira GC, Deybach J-C, Herrero C, Puy H. ALAS2 acts as a modifier gene in patients with congenital erythropoietic porphyria. Blood. 2011;118:1443–1451. doi: 10.1182/blood-2011-03-342873. [DOI] [PubMed] [Google Scholar]
- 54.Cheltsov AV, Barber MJ, Ferreira GC. Circular permutation of 5-aminolevulinate synthase. Mapping the polypeptide chain to its function. J. Biol. Chem. 2001;276:19141–19149. doi: 10.1074/jbc.M100329200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ferreira GC, Vajapey U, Hafez O, Hunter GA, Barber MJ. Aminolevulinate synthase: lysine 313 is not essential for binding the pyridoxal phosphate cofactor but is essential for catalysis. Protein Sci. 1995;4:1001–1006. doi: 10.1002/pro.5560040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stojanovski BM, Breydo L, Hunter GA, Uversky VN, Ferreira GC. Catalytically active alkaline molten globular enzyme: Effect of pH and temperature on the structural integrity of 5-aminolevulinate synthase. Biochi. Biophys. Acta (BBA) - Proteins and Proteomics. 2014;1844:2145–2154. doi: 10.1016/j.bbapap.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Turbeville TD, Zhang J, Hunter GA, Ferreira GC. Histidine 282 in 5-Aminolevulinate Synthase Affects Substrate Binding and Catalysis. Biochemistry. 2007;46:59725981. doi: 10.1021/bi062053k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J, Ferreira GC. Transient state kinetic investigation of 5-aminolevulinate synthase reaction mechanism. J. Biol. Chem. 2002;277:44660–44669. doi: 10.1074/jbc.M203584200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Szkudlarek A, Sulkowska A, Macia̧zek-Jurczyk M, Chudzik M, Rownicka-Zubik J. Effects of non-enzymatic glycation in human serum albumin. Specfroscopic analysis. Spectrochim. Acta Part A: Mol. Biomol. Spect. 2015 doi: 10.1016/j.saa.2015.01.120. [DOI] [PubMed] [Google Scholar]
- 60.Ikushiro H, Hayashi H, Kawata Y, Kagamiyama H. Analysis of the pH- and ligand-induced spectral transitions of tryptophanase: activation of the coenzyme at the early steps of the catalytic cycle. Biochemistry. 1998;37:3043–3052. doi: 10.1021/bi971995+. [DOI] [PubMed] [Google Scholar]
- 61.Furuyama K, Sassa S. Interaction between succinyl CoA synthetase and the heme-biosynthetic enzyme ALAS-E is disrupted in sideroblastic anemia. J. Clin. Investig. 2000;105:757764. doi: 10.1172/JCI6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cox TC, Sadlon TJ, Schwarz QP, Matthews CS, Wise PD, Cox LL, Bottomley SS, May BK. The major splice variant of human 5-aminolevulinate synthase-2 contributes significantly to erythroid heme biosynthesis. Int. J. Biochem. Cell. Biol. 2004;36:281–295. doi: 10.1016/s1357-2725(03)00246-2. [DOI] [PubMed] [Google Scholar]
- 63.Johnson JD, Muhonen WW, Lambeth DO. Characterization of the ATP- and GTP-specific Succinyl-CoA Synthetases in Pigeon: The enzymes incorporate the same a-subunit. J. Biol. Chem. 1998;273:27573–27579. doi: 10.1074/jbc.273.42.27573. [DOI] [PubMed] [Google Scholar]
- 64.Abu-Farha M, Niles J, Willmore WG. Erythroid-specific 5-aminolevulinate synthase protein is stabilized by low oxygen and proteasomal inhibition. Biochem. Cell Biol. 2005;83:620–630. doi: 10.1139/o05-045. [DOI] [PubMed] [Google Scholar]