

Abstract
Current in vitro models for tumor growth and metastasis are poor facsimiles of in vivo cancer physiology and thus, are not optimal for anti-cancer drug development. Three dimensional (3D) tissue organoid systems, which utilize human cells in a tailored microenvironment, have the potential to recapitulate in vivo conditions and address the drawbacks of current tissue culture dish 2D models. In this study, we created liver-based cell organoids in a rotating wall vessel bioreactor. The organoids were further inoculated with colon carcinoma cells in order to create liver-tumor organoids for in vitro modeling of liver metastasis. Immunofluorescent staining revealed notable phenotypic differences between tumor cells in 2D and inside the organoids. In 2D they displayed an epithelial phenotype, and only after transition to the organoids did the cells present with a mesenchymal phenotype. The cell surface marker expression results suggested that WNT pathway might be involved in the phenotypic changes observed between cells in 2D and organoid conditions, and may lead to changes in cell proliferation. Manipulating the WNT pathway with an agonist and antagonist showed significant changes in sensitivity to the anti-proliferative drug 5-fluoruracil. Collectively, the results show the potential of in vitro 3D liver-tumor organoids to serve as a model for metastasis growth and for testing the response of tumor cells to current and newly discovered drugs.
Keywords: Tumor models, Metastasis, Organoids, Drug screening, Tissue engineering
INTRODUCTION
Metastasis from the intestinal-colonic environment to the liver is a common occurrence in patients with colon carcinoma, leading to the 2nd highest number of cancer-related deaths in the U.S. This progression is a function of several phenomena, including (1) epithelial- to-mesenchymal transition (EMT), (2) dissociation of the tumorigenic cells from the host organ (colon) (3) intravasation into vascular circulation and (4) invasion into the 3D environment of the target tissue (liver).33,47 The cellular pathways and environmental influences linked to these phenomena represent important areas of exploration for understanding metastasis and identifying potential treatment targets. In metastatic colon carcinoma, death is not usually caused by the primary tumor, but by cancer metastases to other organs. Approximately 20% of patients have distant metastatic disease at the time of presentation, with the first site of hematogenous dissemination usually to the liver. Patients with hepatic metastatic disease from colorectal cancer have a poor prognosis, with 5-year survival rates below 5%.56
Although years of cancer research have been devoted to studying EMT, research has been limited with respect to the subsequent stages of dissociation, invasion, and engraftment of metastases due to the inability to model metastasis in a controlled environment. Animal models allow only limited manipulation and study of these mechanisms, and are not necessarily predictive of results in humans.51 On the other hand, in vitro methods, such as traditional 2D cultures, fail to recapitulate the 3D microenvironment of in vivo tissues. Drug diffusion kinetics vary dramatically, drug doses effective in 2D are often ineffective when scaled to patients, and cell–cell/cell– matrix interactions and other phenotype aspects are inaccurately modeled.1,11,17 These limitations result in “top level” drug candidates reaching clinical trials and failing because they have not been tested in accurate human-based models. Recently developed 3D in vitro human metastasis models comprised of tumor cells, host tissue cells, and extracellular matrix (ECM) biomaterial- based 3D microenvironments represent a better solution.
Three-dimensional tissue constructs can be created by employing a variety of biofabrication techniques and biomaterials. Our laboratory has extensive experience working with a hydrogel comprised of thiolated hyaluronic acid (HA) and gelatin, crosslinked through disulfide bonding of thiols or polyethylene glycol diacrylate (PEGDA) linkages, which was originally developed by the Prestwich group.39 These and other HA-derivatives have been extensively implemented in regenerative medicine applications in recent years,8 and examples include wound healing of corneal lacerations,26 embryonic stem cell expansion,15 soft tissue engineering in vitro and in vivo,12,21,39,40 tumor xenograft models,22,35,38 bioprinting of cellularized tubular structures,43,44 and primary hepatocyte liver constructs.45
We previously developed microcarrier beads using the HA materials described above, which were used to form tissue organoids under simulated microgravity culture in rotational wall vessel (RWV) bioreactors.42 The RWV is an in vitro optimized suspension culture system in which cells are grown in a physiological low fluid-shear environment in 3D.28,31,37 Rotation of the bioreactor results in a state commonly referred to as simulated microgravity,50 in which the dynamic culture conditions of the RWV induce cells to aggregate based on natural cellular affinities, form 3D structures and acquire properties of highly differentiated cells.3,29,50 To date, more than 50 RWV-derived tissue models have been engineered, including liver, neuronal tissue, cardiac muscle, cartilage, adipose tissue, and epithelial tissues of the lung, bladder, small intestine, colon, placenta and vagina.9,10,13,16,18,20,27,30,36,46,54,55 Cells cultured in the RWV are able to recapitulate in vivo 3D spatial organization and polarity, cellular differentiation, multi-cellular complexity, and functionality. The ease with which these organoids could be formed suggests that they may be adopted as a versatile system for creating large quantities of 3D tumor models for drug screening.
Our overarching hypothesis is that the 3D environment surrounding tumor cells has an integral role in influencing their metastatic properties. Specifically, we believe 3D in vitro tissue constructs provide the appropriate tissue microenvironments to better mimic the environments in vivo inside of which cancer resides and progresses. Here we demonstrate the use of liver-based cell organoids formed within RWV simulated microgravity bioreactors that serve as host tissues in which colon carcinoma progression can be monitored, a more accurate metastatic phenotype can be achieved, and drug susceptibility and resistance can be manipulated for drug screening.
MATERIALS AND METHODS
2D Culture of HCT-116 Colon Carcinoma Cells
Human colon carcinoma cells [HCT-116, transfected previously with red fluorescent protein (RFP)] and human hepatoma cells (HepG2) were expanded in 2D on tissue culture plastic using 15 cm tissue-treated dishes until 90% confluence with Dulbecco’s Minimum Essential Medium (DMEM, Sigma, St. Louis, MO), containing 10% fetal bovine serum (FBS, Hyclone, Logan, UT). Cells were detached from the substrate with Trypsin/EDTA (Hyclone) and resuspended in media before use in further studies.
Preparation of Hyaluronic Acid-Coated Microcarriers
HA hydrogel-coated microcarrier beads were prepared as previously described.42 Briefly, crosslinked dextran Sephadex G-50 beads (GE Healthcare Biosciences, Uppsala, Sweden) were coated with a formulation of Extracel (ESI-BIO, Alameda, CA) without the PEGDA crosslinker. Specifically, the thiol-functionalized hyaluronic acid and thiol-functionalized gelatin were allowed to crosslink by disulfide bond formation. The hydrogel solution was prepared by dissolving Glycosil and Gelin-S (the HA and gelatin components, respectively) in water to make 2% w/v solutions, and then mixed in equal volumes. Sephadex beads (0.5 g) were added to a round bottom flask and the pressure was reduced in the flask using a vacuum. A valve was opened to allow 10 mL of the ungelled Extracel solution to flow into the flask. The reduced pressure environment allowed the aqueous hydrogel solution to be taken up into the pores of the beads. Excess liquid was removed, and the hydrogel solution was allowed to crosslink overnight in air by disulfide bond formation. The coated beads were then transferred to 50 mL centrifuge tubes, frozen, and lyophilized to yield the HA-coated microcarriers (HAMs).
Cell Seeding and Rotating Wall Vessel Bioreactor Culture of Liver-Based Organoids Containing HCT-116 Tumor Foci
Prior to cell culture, HAMs were sterilized in Ca2+ and Mg2+-free PBS (HyClone, Logan, UT) by autoclaving at 115 °C for 15 min. The sterilized beads were then stored at 4 °C. Cell culture medium comprised of the DMEM described above with the addition of amphotericin B was used for all bioreactor culture studies. In preparation for seeding on HAMs, cells were initially cultured as described above. Trypsin– EDTA was used to free the cells from their substrate. Cells and HAMs in medium were added to a 50 mL RWV bioreactor (Synthecon, Houston, TX) to reach a final density of 80,000 cells/5 mg beads/mL medium with a ratio of 10:1 HEPG2 cells:HCT-116 cells. Rotation of the RWV bioreactors was started immediately and continued for 14 days. Medium was first changed on day 5 of culture in order to allow the cells to bind to the beads, after which medium was changed every 2nd day. The RWV bioreactors were set to initially rotate at 18 rpm; the rate of rotation was manually increased throughout culture to keep the clusters in “freefall” as they increased in size. Aliquots were removed on days 5, 8, 11, and 14 days post-seeding. Aliquots were used to assess growth by cell proliferation through size quantification of light microscopy images (n of 10 or higher) and MTS proliferation assays (n of 4) quantified at 490 nm on a plate reader. In addition, composite images were taken in which the organoids were also imaged with epi-fluorescence at 594 nm to analyze the progression of the RFP-labled HCT-116 cells within the host liver organoids.
For initial 2D characterization, cells were seeded into 24-well plates as HCT-116 cells only, HEPG2 cells only, and a 1:1 coculture at a density of 20,000 cells per well in order to determine their relative proliferation rates. The plates were transferred to an incubator and cell proliferation was determined using MTS (Promega, Madison, WI) mitochondrial metabolism assays at day 1, day 4, and day 7 of culture. Media was changed on day 4.
Immunocytochemistry Characterization of HCT-116 Cells and HEPG2-HCT-116 Cocultures in 2D
To assess a selected panel of markers, HCT-116 cells were seeded into 4-chamber chamber slides at a density of 20,000 cells per chamber in DMEM. Additionally, HEPG2 cells and HCT-116 cells were seeded together at the same cell density in 10:1 ratio, mimicking the ratio in 3D. After 5 days, the cells were fixed with 4% PFA for 15 min and washed with PBS. Slides were stored in PBS at 4 °C prior to immunocytochemical staining. HCT-116 cells were immunostained with antibodies against ZO-1 (a tight junction marker), β-catenin (a protein associated with E-cadherin-mediated cell–cell adhesions), and N-cadherin (associated with mesenchymal cells or metastatic cancer cells after an epithelial-to-mesenchymal transition49). All incubations were carried out at room temperature. Antigen retrieval was performed with proteinase K (Dako, Carpinteria, CA) incubations for 5 min, followed by permeablization in 0.1% Triton-X in PBS for 5 min. Nonspecific blocking was performed with Dako Antibody Protein-Block, Serum-Free (Dako) for 30 min. Next, samples were incubated with primary antibodies for 1 h: ZO-1 (raised in rabbit, cat. # 61-7300, Invitrogen, Grand Island, NY), β-catenin (raised in rabbit, cat. # 71-2700, Invitrogen), and N-cadherin (raised in mouse, 610921, BD Biosciences, San Jose, CA). Each primary antibody was prepared in Dako Antibody Diluent at a 1:200 dilution. After incubation, samples were washed with 1× PBS three times. Samples were then incubated for 1 h with anti-rabbit or anti-mouse Alexa Fluor 488 secondary antibodies (Invitrogen) as appropriate in Dako Antibody Diluent (1:200 dilution). Cells were counterstained with DAPI for 5 min, and washed three times with 1× PBS prior to fluorescent imaging. Samples were imaged by epifluorescence using excitation band filters with central excitation wavelengths 380, 488, and 594 nm with a Leica DM 4000B upright microscope (Leica Microsystems, Buffalo Grove, IL). Negative controls were performed in parallel with the primary antibody incubations and included incubation with blocking solution in place of the primary antibody. No immunoreactivity was observed in the negative control sections using the same imaging conditions used for the experimental samples.
Immunohistochemical Characterization of Liver-Tumor Organoids
Aliquots of organoids removed from the RWV on day 14 were also assessed by immunohistochemistry (IHC) for specific epithelial, mesenchymal, and metastatic markers. Samples were processed for IHC in the same manner as normal animal tissues, but with a shorter fixation period. Briefly, organoids were fixed with 4% paraformaldehyde for 1 h, dehydrated with graded ethanol washes followed by xylene, embedded in paraffin, and sectioned at 5 μm.
For IHC, all incubations were carried out at room temperature unless otherwise stated. Slides were warmed at 60 °C for 1 h to increase bonding to the slides. Antigen retrieval was performed on all slides and achieved with incubation in Proteinase K (Dako, Carpinteria, CA) for 5 min. Sections were permeabilized by incubation in 0.05% Triton-X for 5 min. Nonspecific antibody binding was blocked by incubation in Protein Block Solution (Abcam) for 15 min. Sections were incubated for 60 min in a humidified chamber with the primary ZO-1 (raised in rabbit, cat. # 61-7300, Invitrogen), β-catenin (raised in rabbit, cat. # 71-2700, Invitrogen), E-cadherin (raised in mouse, cat. # 610181, BD Biosciences), vinculin (raised in mouse, cat. # V9264, Sigma Aldrich), N-cadherin (raised in mouse, 610921, BD Biosciences), and matrix metallo-proteinase` 9 (MMP9, raised in rat, cat. # ab38898, Abcam), all at 1:200 dilutions in antibody diluent (Abcam).
Following primary incubation, slides were washed three times in PBS for 5 min. Samples were then incubated for 1 h with anti-rabbit, anti-mouse, or antirat Alexa Fluor 488 secondary antibodies (Invitrogen) as appropriate in antibody diluent (1:200 dilution). Cells were counterstained with DAPI for 5 min, and washed three times with 1× PBS prior to fluorescent imaging. Negative controls were performed in parallel with the primary antibody incubations and included incubation with blocking solution in place of the primary antibody. No immunoreactivity was observed in the negative control sections. Samples were imaged with fluorescence at 488, 594, and 380 nm with a Leica DM 4000B upright microscope.
WNT pathway modulation of proliferation
Next we employed 2 different drugs, BIO (6-bromoindirubin- 3′-oxime, Stemgent, Cambridge, MA) a WNT signaling agonist, and XAV939 (3,5,7,8-tetrahydro- 2-[4-(trifluoromethyl)phenyl]-4H-thiopyrano[ 4,3-d]pyrimidin-4-one, Sigma Aldrich), a WNT signaling antagonist to manipulate the WNT/β-catenin pathway of the HCT-116 cells and evaluated their effects on 5-FU susceptibility and proliferation. We prepared and cultured several parallel batches of liver-tumor organoids in RWVs, using the methods described above. Beginning on day 5—the first media change after the cultures were established—and on days 8, 11, and 14 we removed aliquots of organoids for analysis of proliferation by MTS and we administered or replenished drug-free DMEM, DMEM containing 5 μM BIO, or DMEM containing 3.3 μM XAV939.
On day 14, additional aliquots were fixed IHC analysis of proliferative markers PCNA (proliferating cell nuclear antigen) and KI67. These organoids were fixed, processed and sectioned as described above. BIO and XAV939-treated organoid sections were stained for PCNA and KI67 to assess the incidence of co-localization of proliferative markers with the WNT-modulated RFP-labeled HCT-116 cells. IHC staining was performed as described above using primary PCNA (raised in rabbit, cat. # 07-2162, Millipore) or KI67 (raised in rabbit, cat. # ab15580, Abcam) antibodies, and secondary anti-rabbit Alexa Fluor 488 antibodies, all at 1:200 dilutions in antibody diluent. Cells were counterstained with DAPI for 5 min, and washed three times with 1X PBS prior to fluorescent imaging at 488, 594, and 380 nm.
5-FU Drug Administration and WNT Modulation
Organoids were prepared and cultured in the RWV as described above. On day 14, aliquots of organoids were removed from culture and transferred to nonadherent agarose-coated 24-well plates for 5-FU treatment at 0, 1, 10, and 100 mM concentrations in cell culture media. Following incubation for 24 h organoids were fixed, processed and sectioned as described above. Organoid sections were stained with caspase 3, an apoptotic marker, to assess the incidence of cell death and the efficiency of 5-FU to target the rapidly proliferating HCT-116 cells rather than the HepG2 host tissue. IHC staining was performed as described above using primary caspase 3 antibodies (raised in rabbit, cat. # ab18847, Abcam) and secondary anti-rabbit Alexa Fluor 488 antibodies, both at 1:100 dilutions in antibody diluent. Cells were counterstained with DAPI for 5 min, and washed three times with 1× PBS prior to epifluorescent imaging at 488, 594, and 380 nm, as described above. Correlation between caspase 3-positive cells and RFP-positive cells was determined quantitatively by cell counting blind to the experimental groups.
Lastly, aliquots of organoids treated with drug-free DMEM, DMEM containing 5 μM BIO, or DMEM containing 3.3 μM XAV939 from day 5 onward, were removed from culture and transferred to non-adherent agarose-coated 24-well plates for 5-FU treatment at 0, 1, 10, and 100 mM concentrations in cell culture media as described above. Following incubation for 24 h, relative mitochondrial metabolic levels were assessed by MTS.
Statistical Analysis
All quantitative results are presented as mean ± standard deviation (SD). Experiments were performed in triplicate or greater. Values were compared using Student’s t test (2-tailed) with two sample unequal variance, and p<0.05 or less was considered statistically significant.
RESULTS
Liver-Tumor Organoid Formation and Progression of Metastatic Tumor Foci
For this particular model, HepG2 hepatoma cells were chosen to act as a liver mimetic, since they have the ability to secrete measurable levels of albumin and urea, as well as metabolize some drugs through cytochrome p450 activity,24 and we have cultured them successfully as spheroids in RWV cultures previously. 41 HCT-116 is a human metastatic colon carcinoma cell line used expensively to study metastasis in vitro and in vivo.2,5,7 HCT-116 cells were engineered to express RFP in order to facilitate tracking of HCT-116 foci within the organoids. Three-dimensional cultures were initiated by combining HepG2 hepatoma cells and HCT-116 metastatic colon carcinoma cells in a 10:1 ration together with hyaluronic acid and gelatin-based microcarriers inside RWV bioreactors in a manner described previously.42 These microcarriers provide a scaffolding surface for cell attachment, thus facilitating suspended culture of adherent cells in the RWV bioreactors.
Organoid formation and growth was documented on days 5, 8, 11, and 14 using a light microscope to observe aliquots removed from the RWV. Figure 1a shows evidence of organoids and clusters of organoids that have formed around some of the microcarriers. Over the 14-day time course, organoids and clusters grew in size, increasing in diameter over time, with statistically significant differences in size from one time point to the next (Fig. 1b, p<0.05). Additionally, MTS assays were used to measure relative mitochondrial metabolism at each time point, revealing an increase in metabolism that eventually stabilized after day 11 (Fig. 1c). The metabolic rates measured on day 8 and day 11 were both significantly increased compared to day 5 (p<0.05). Although not all microcarriers generated organoids, the unseeded microcarriers did not impede subsequent experiments or forms of analysis because they had no cell associated activities. Metabolic activity significantly increased (p<0.05) from day 5 and day 8 to day 11. These data suggest that the increase in organoid size over time is likely due to both cell proliferation as well as fusion between individual organoids into irregular-shaped clusters of organoids.
FIGURE 1.

Liver-tumor organoid model total growth and metabolism over time. (a) Liver-tumor organoids form within 5 days in the RWV bioreactor and increase in size over the 14-day period, as demonstrated by light microscopy images. (b) Average organoid size increases steadily over time, and (c) metabolic activity of the organoids, as measured by MTS assays, increases until leveling off at day 11. Significance: *p <0.05; **p <0.01. D) RFP-labeled HCT-116 colon carcinoma cells proliferate over time within the organoids, resulting in tumor foci that increase in size from day 5, through days 8, 11, and 14. Scale bars—250 μm.
Aliquots of organoids removed from the RWV at the same time points (days 5, 8, 11, and 14), and images were prepared by merging light microscopy-captured organoid images together with fluorescent images highlighting only the RFP-labeled HCT-116 portions of the cultures (Fig. 1d). On day 5, the tumor foci occupy a relatively unsubstantial percentage of the overall organoid volumes. However, the tumor foci rapidly expanded in size; eventually occupying a significant portion of the organoids, mirroring the distinct growth rates of the 2 cell types observed in 2D culture (not shown). These dynamic changes in size of the organoids, apparent size of the RFP-positive tumors within the organoids, and organoid metabolic rates suggest that these organoids are viable at time of imaging, which may allow future use of this model as a tool for real-time tumor tracking in vitro.
HCT-116 Colon Carcinoma Cells Show Different Patterns of Cell Surface Marker Expression in 2D and 3D Culture Conditions
To assess epithelial vs. mesenchymal phenotype in 2D, we stained HCT-116 cultures and HEPG2-HCT-116 cocultures with antibodies against ZO-1, β-catenin, and N-cadherin antibody. In HCT-116 only cultures, cells displayed clear staining of membrane-bound ZO-1 (Fig. 2a), a tight junction marker, as well as membrane-bound β-catenin (Fig. 2b), a protein associated with E-cadherin-mediated cell–cell adhesions. Notably, these proteins are generally localized to the cell membrane to facilitate cell–cell tight junctions and E-cadherin binding in cells presenting with epithelial phenotype.14 Despite the epithelial origin of HCT-116 cells, these observations were surprising because these cells are widely considered as having undergone epithelial-to-mesenchymal transition in order to acquire more invasive and metastatic properties. In addition, we observed a lack of staining for a mesenchymal marker, N-cadherin (Fig. 2c), in HCT- 116 cells, further indicating that the cells appear to take on an epithelial phenotype rather than a mesenchymal phenotype in the 2D environment. To provide a direct comparison against the 3D organoids, containing HEPG2 and HCT-116 cells, 2D cocultures were stained for the same markers described above, showing the same phenotypes. Figures 2d and 2e show both cell types (HCT-116 cells are RFP-positive are indicated by red fluorescence, while HEPG2 cells are indicated by lack of red fluorescence) supporting the cell membrane-associated ZO-1 and β-catenin staining indicating evidence of epithelial phenotype. Furthermore, cocultures stained for N-cadherin were negative for the mesenchymal marker regardless of cell type (Fig. 2f).
FIGURE 2.

Expression of cell surface markers and MMP in HCT-116 cells in 2D tissue culture dishes and inside the 3D organoids. HCT-116 cells in tissue culture dishes (a–c) and inside the liver-tumor organoids (D-I) were immunostained with antibodies against different cell surface markers and MMP. In chamber slides, HCT-116 cells in 2D presented membrane-bound ZO-1 (a) and β-catenin (b). Conversely, HCT-116 cells fail to express N-cadherin (c). HEPG2-HCT-116 cocultures in 2D present the same staining of membrane-bound ZO-1 (d) and β-catenin (e), and fail to express N-cadherin (f) regardless of which cell type. Inside the 3D liver organoids, HCT-116 cells lose membrane-bound expression of ZO-1 (g), β-catenin (h), E-cadherin (i), and vinculin (j). Additionally, HCT-116 cells express N-cadherin (k) and matrix metallo-proteinase 9 (l). For panels g–l, top images show antibody-specific stain in green, RFP-labeled HCT-116 in red, and DAPI-stained nuclei in blue, while lower images show only the antibody-specific stain in green for improved visualization. Dotted white lines highlight regions containing concentrated presence of HCT-116 cells. Scale bars: (a–c) 100 μm; (d–i) 50 μm.
Next, we examined cell surface marker expression in HCT-116 cells in the 3D liver-tumor model. Despite the relatively small size of the organoids (400–600 μm in diameter on day 14), the organoids maintained a robust structure that facilitated the use of normal tissue processing methods such as dehydration, paraffin embedding, and sectioning by microtome.
We observed a remarkable difference in the phenotype of HCT-116 cells cultured as tumor foci within the organoids, compared to the HCT-116 cells cultured in tissue culture dishes 2D conditions. In these foci, ZO-1, β-catenin, E-cadherin, and vinculin, which are all involved in cell–cell interactions, shift location away from the cell membrane interfaces (Figs. 2g–2j). The shift in cell–cell binding protein localization further support the possible EMT transition of HCT-116 cells within the 3D liver organoids.14,25 Importantly, the shift of β-catenin staining from the membrane into the cytoplasm and nucleus is common in EMT when the WNT/β-catenin pathway is activated, resulting in β-catenin traveling to the nucleus and acting as a transcription factor that can induce an invasive phenotype. 4,6,34,48
In addition, we did not detect N-cadherin, a marker typically associated with mesenchymal phenotypes,49 in HCT-116 cells in tissue culture dishes 2D conditions. In contrast, N-cadherin staining was observed in the tumor foci regions of the live-tumor organoids, co-localizing withHCT-116 cells (Fig. 2k). An additional aspect of an invasive phenotype is the expression of matrix metallo-proteinases to enzymatically degrade ECM components and facilitate cell migration.19,57 Immunostaining for MMP9 showed co-localization with RFP-labeled HCT116 cell tumor foci inside the organoids (Fig. 2l). HepG2 cells within the liver-tumor organoids expressed the epithelial markers ZO-1, β-catenin, E-Cadherin, and vinculin, with the strongest visible signal at the cell membrane. Importantly, the HepG2 regions of the liver-tumor organoid didn’t show N-cadherin and MMP 9 expression. Taken together these results support the hypothesis that the 3D liver organoid environmental support an invasive HCT-116 phenotype.
WNT Pathway Manipulation Changes HCT-116 Cell Response to Chemotherapy
The previous results (Figs. 1 and 2) suggested involvement of the WNT pathway in HCT-116 proliferation. To test this hypothesis, liver-tumor organoids were initiated in the RWV system, and on day 5 one group (control) was administered standard media only (identical to previous conditions), a 2nd group was administered media containing the WNT pathway agonist, BIO (5 μM), and a 3rd group was administered media containing the WNT pathway antagonist, XAV939 (3.3 μM). Aliquots of organoids form the three groups were removed at 4 time points during the 14-day time-course and cell proliferation was measured using an MTS assay. Organoids in control media showed normal proliferation, whereas XAV939-treated organoids proliferated to a lesser extent (Fig. 3a). In contrast, BIO-treated organoids did not proliferate over the 14-day time course (Fig. 3a). A similar proliferation assay was performed 9in HCT-116 cells cultured in tissue culture dishes 2D conditions, showing virtually no difference in control and XAV939-treated cells; whereas, BIO-treated cells did not proliferate (Fig. 3b). To further validate these results, we fixed organoids from the BIO and XAV939-treated groups on day 14 and stained them for markers of cell proliferation, PCNA and KI67. In BIO-treated organoids, we observed very few PCNA or KI67 staining of the liver-tumor organoids (Figs. 3c and 3d, respectively), suggesting that BIO treatment inhibited both HCT-116 and HepG2 cell proliferation. Conversely, in XAV939- treated organoids, we observed co-localization of PCNA and KI67 staining with RFP-labeled HCT-116 cells (Figs. 3e and 3f, respectively), indicating that the majority of proliferating cells are HCT-116.
FIGURE 3.
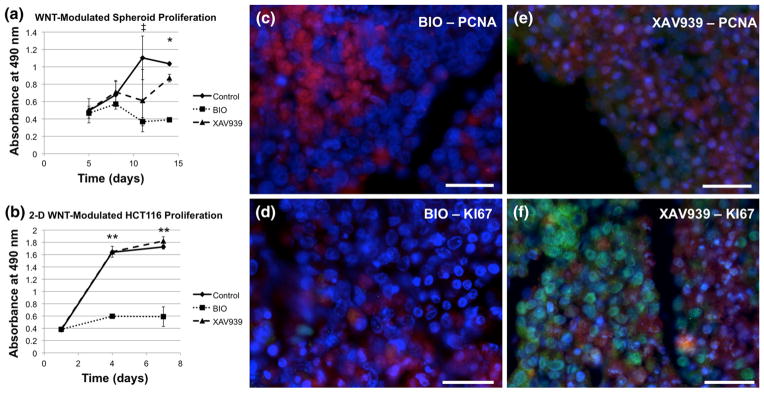
Manipulation of the WNT pathway modulates HCT-116 proliferation. Liver-tumor organoids were incubated with WNT agonist BIO and WNT antagonist XAV939, as described in the text. The effects of BIO (a, c, d) and XAV939 (a, d, f) on organoid growth over a 14 day period was measured by metabolic activity (a) and staining for proliferating cell nuclear antigen (PCNA, c, e)) and KI67 (d, f). The effects of BIO and XAV939 on HCT-116 proliferation in tissue culture dishes were measured by metabolic activity (b). Green—indicated stain; Red—RFP-labeled HCT-116 cells; Blue—DAPI. Scale bars 50 μm. Significance: *p<0.05 between all three groups (Control, BIO, and XAV939); ‡p <0.01 between BIO and Control; **p <0.01 BIO vs. Control, BIO vs. XAV939.
The results described above (Fig. 3) suggested that the WNT pathway plays a role in modulating the proliferative activity of HCT-116 cells. We next tested the effects of WNT pathway manipulation on the response of the liver-tumor organoid to a cytotoxic drug, 5-FU. This drug is commonly used to treat colon carcinoma; it disrupts the synthesis of the pyrimidine thymidine and thus induces apoptosis in fast growing cells such as those found in cancer.23,53 Specifically, we were interested in the effects of 5-FU on the rapidly proliferating HCT-116 tumor foci vs. the more slowly dividing HepG2 cells inside the organoids, in order to test the liver-tumor organoid system as a model for drug candidate screening. Organoids cultured for 14 days in DMEM-only (control) were transferred to non-adherent well plates and incubated in the presence of four 5-FU concentrations (0, 1, 10, and 100 mM) in cell culture media for 24 h. Subsequently, the organoids were collected and stained for caspase 3, a marker of apoptosis. The results showed clearly that 1) caspase 3 staining generally increased in correlation with the increase in 5-FU concentrations (Figs. 4a–4d), yet the correlation between caspase 3 staining and RFP-labeled HCT-116 was not as completely clear. Correlative data was formed using cell counts of RFP-positive cells and caspase 3-positive cells. Figure 4e shows an increase in the percentage of RFP-positive cells that also stained positive for caspase 3, indicating an increase in apoptosis of HCT-116 cells as the 5-FU concentration increased. In general, these percentages were all significantly different when compared in pairs (p<0.05), with the exception of 0 mM vs. 1 mM, and 10 mM vs 100 mM, which were not statistically significant from one another. Next, we calculated the percentage of cells that stained positive for caspase 3 that also were RFP-positive. This data would determine how accurate 5-FU was at targeting tumor cells vs. the HepG2 population (Fig. 4f). Interestingly, we observed a high percentage of correlation (near 100%) in the lower concentrations (1 and 10 mM), indicating good specificity. However, at 100 mM 5-FU, we observed approximately 50% accuracy, significantly lower than the other three groups (p<0.05), indicating that the effects of 5-FU “spilled over” into the HepG2 population, likely due to the toxic nature of the drug at such a high concentration.
FIGURE 4.

Response of liver-tumor organoids to 5-FU treatment is modified by WNT pathway manipulation. (a–d) Liver-tumor organoids were incubated with increasing concentrations of 5-FU, as indicated, harvested and stained with anti-caspase 3 antibodies. Green—caspase 3; Red—RFP-labeled HCT-116 cells; Blue—DAPI. Scale bars—50 μm. e–f) Quantification of RFP and caspase 3 correlation after 5-FU treatment. (e) Percentage of RFP-positive cells that are also caspase 3-positive. Significance: All pairs statistically significant at p<0.05, except comparisons noted by ‡. (f) Percentage of caspase 3-positive cells that are also RFP-positive. Significance: #p <0.05. (g) Liver-tumor organoids were first treated with BIO or XAV939 and subsequently with increasing concentrations of 5-FU for 24-h. Organoid growth was measured by metabolic activity. Significance: *p <0.05 between all three groups (Control, BIO, and XAV939); ***p <0.01 Control vs. XAV939, BIO vs. XAV939. (h) In 2D cultures (HCT-116-only and HEPG2-HCT-116 cocultures, 5-FU has an initial dose dependent effect on metabolic activity, but the effect appears to plateau above 10 mM 5-FU.
Next, to evaluate the effects of the WNT pathway manipulation on 5-FU sensitivity, we cultured organoids for 5 days in DMEM only (control), DMEM containing BIO, or DMEM containing XAV939, and subsequently exposed them for 24 h to increasing doses of 5-FU (0, 1, 10, and 100) (Fig. 4g). Measurements of mitochondrial metabolism in control organoids (without pre-treatment), using an MTS assay, showed a decrease in metabolism with an increase in 5-FU concentrations, as expected from the results in Figs. 4a–4d. In comparison, when performed in 2D, both with HCT-only cultures or HEPG2-HCT-116 cocultures, 5-FU elicits an initial dose dependent decrease in metabolic activity. However, at 10 mM 5-FU and above, dose dependence seems to be lost and the metabolic response plateaus (Fig. 4h). Organoids pre-treated with WNT agonist BIO showed little to no decreases in mitochondrial metabolism at increasing 5-FU concentrations. In contrast, pre-treatment with WNT antagonist XAV939 resulted in an increased sensitivity to 5-FU, compared with control organoids. These results suggest that WNT pathway inhibition, which permits HCT-116 proliferation, may cause an increased sensitivity to chemotherapeutic drugs, such as 5-FU. In contrast WNT pathway activation, which inhibits HCT-116 proliferation, may cause resistance to 5-FU in the liver-tumor organoid model.
DISCUSSION
Three-dimensional tissue models have been explored in recent years, and despite a few exceptions,52 most fail to incorporate a key component of most cancers— the host tissue in which the tumor resides or metastasizes to. In the current study, we have developed a liver-tumor organoid system, in which HepG2 hepatocyte-like cells and metastatic HCT-116 colon carcinoma cells were cultured in RWV bioreactors with hyaluronic acid and gelatin microcarrier beads to give rise to liver tissue organoids containing fluorescent colon carcinoma tumor foci. This system builds on our previous microcarrier and RWV bioreactor technology, which has successfully supported initiation and growth of cell aggregates of other tissue types.42 HCT- 116 cells cultured in traditional 2D settings appeared epithelial, and expressing tight junction markers such as the ZO-1 and β-catenin at the cell membrane. In addition, the cells did not express N-cadherin, which is commonly associated with mesenchymal cell phenotype. This epithelial phenotype is simply not an accurate representation of a metastatic tumor cell, and if employed in drug-screening applications could potentially result in outcomes that do not translate to humans. In contrast, when HCT-116 cells were grown as tumor foci inside the liver organoids, they showed a phenotype indicative of cells having undergone EMT. HCT-116 regions of the organoids showed weak expression of ZO-1, E-cadherin, and vinculin, yet showed cytoplasmic expression of β-catenin, as well as, expression of N-cadherin and MMP-9, suggesting a switch to a mesenchymal, mobile, and metastatic phenotype that is more similar to colon carcinoma metastases in vivo. These characteristics suggest that this 3D liver-tumor organoid model may serve as a better model of metastatic tumors compared with traditional 2D cell cultures.
The liver-tumor organoids showed increased growth and metabolism, indicating ongoing cell proliferation. HCT-116 tumor foci within the organoids were largely the proliferative component in the cultures. Accumulation of β-catenin in the cytoplasm and nuclei of HCT-116 cells in 3D is often associated with WNT pathway activation, and subsequently initiation of EMT and cancer cell invasiveness, motility, and proliferation. Manipulation of the WNT pathway with an agonist (BIO) inhibited cell proliferation while the antagonist (XAV939) permitted cell proliferation, indicating that the WNT pathway has a role in HCT- 116 proliferation. Indeed, activation of the Frizzled-7 receptor by the WNT pathway ligand Wnt11 has been shown to be influential in modulating HCT-116 cell proliferation and migration in vitro, and Wnt11 mRNA expression was determined to be significantly higher than baseline levels in patients with recurring colorectal tumors or inpatients that had died after surgical interventions.32
One goal of this study was to develop a system that was simple to adapt to drug screening studies. The cost of bringing a pharmaceutical product to market increases dramatically after the initial research and development when moved into human clinical trials. The tests on which early go/no-go decisions are made for progression into clinical trials are primarily based on pre-clinical animal models and 2D cell cultures, which often fail to provide accurate predictions of how drugs will behave when transitioned to humans.51 As such, there are growing needs and opportunities for in vitro models comprised of human cells that can recapitulate human tissues more accurately.1,11,17 Therefore, we performed drug screens with a range of 5-FU concentrations with the organoids, noting a dose dependent decrease in organoid metabolism and increased apoptosis inside the HCT-116 tumor foci with increasing 5-FU concentrations. Although at high 5-FU concentrations, we observed a negative impact on the surrounding HepG2 organoid tissue. Since the mechanism of action of 5-FU is through DNA replication interruption, we hypothesized that manipulation of WNT signaling and subsequent changes in proliferative activity would influence the efficacy of 5-FU treatments. We were able to demonstrate that (1) WNT pathway activation via BIO effectively induced resistance to 5-FU, and conversely, (2) WNT pathway inhibition via XAV939 induced increased drug sensitivity to 5-FU. These results may have potential implications for development of more effective anticancer treatments. Inhibition of the WNT pathway can increase cell proliferation and increase tumor cell sensitivity to anti-proliferative drugs such as 5-FU. On the other hand, activating the WNT pathway can produce a tumor model of drug resistance, thus creating a testing platform with which new therapeutics can be screened.
With an ever increasing need for improved testing models for more efficient drug development and screening, the liver-tumor organoid system presented here has the potential to provide researchers with a set of tools that can streamline the pharmaceutical pipeline. An additional area of exploration that will become important after initial drug compound screening is understanding the effects of potential drug candidates on the normal host tissue surrounding the tumor foci. Many chemotherapeutic agents are toxic to healthy tissues and the incorporation of host tissue in our liver-tumor organoid system allows study of these concerns, as we have begun to demonstrate in the drug toxicity experiments. We also envision adaptation of this in vitro metastasis model to other tissue types, which will allow extensive investigation of many cancer varieties and exploration of the complex mechanisms at play during metastasis, and ultimately providing a powerful platform for use in future drug screening, toxicology studies, and personalized medicine.
Acknowledgments
This work was supported by the Golfers Against Cancer and Wake Forest University Baptist Medical Center Comprehensive Cancer Center.
ABBREVIATIONS
- 5-FU
5-Fluorouracil
- BIO
6-Bromoindirubin-3′-oxime
- ECM
Extracellular matrix
- EMT
Epithelial-to-mesenchymal transition
- DMEM
Dulbecco’s Minimum Essential Medium
- FBS
Fetal bovine serum
- HA
Hyaluronic acid
- IHC
Immunohistochemistry
- PEGDA
Polyethylene glycol diacrylate
- RFP
Red fluorescent protein RWV, Rotating wall vessel
- XAV93
3,5,7,8-tetrahydro-2-[4-(trifluoromethyl) phenyl]-4H-thiopyrano [4,3-d]pyrimidin-4-one
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts of interest to disclose.
References
- 1.Achilli TM, Meyer J, Morgan JR. Advances in the formation, use and understanding of multi-cellular spheroids. Expert Opin Biol Ther. 2012;12:1347–1360. doi: 10.1517/14712598.2012.707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Attoub S, Rivat C, Rodrigues S, Van Bocxlaer S, Bedin M, Bruyneel E, Louvet C, Kornprobst M, Andre T, Mareel M, Mester J, Gespach C. The c-kit tyrosine kinase inhibitor STI571 for colorectal cancer therapy. Cancer Res. 2002;62:4879–4883. [PubMed] [Google Scholar]
- 3.Barrila J, Radtke A, Crabbé A, Sarker S, Herbst-Kralovetz MM, Ott CM, Nickerson CA. Organotypic 3D cell culture models: using the rotating wall vessel to study host-pathogen interactions. Nat Rev Microbiol. 2010;8:791–801. doi: 10.1038/nrmicro2423. [DOI] [PubMed] [Google Scholar]
- 4.Behrens J. Control of beta-catenin signaling in tumor development. Ann NY Acad Sci. 2000;910:21–33. doi: 10.1111/j.1749-6632.2000.tb06698.x. (discussion 33–25) [DOI] [PubMed] [Google Scholar]
- 5.Botchkina IL, Rowehl RA, Rivadeneira DE, Karpeh MS, Jr, Crawford H, Dufour A, Ju J, Wang Y, Leyfman Y, Botchkina GI. Phenotypic subpopulations of metastatic colon cancer stem cells: genomic analysis. Cancer Genom Proteom. 2009;6:19–29. [PubMed] [Google Scholar]
- 6.Brantjes H, Barker N, van Es J, Clevers H. TCF: Lady Justice casting the final verdict on the outcome of Wnt signalling. Biol Chem. 2002;383:255–261. doi: 10.1515/BC.2002.027. [DOI] [PubMed] [Google Scholar]
- 7.Brattain MG, Fine WD, Khaled FM, Thompson J, Brattain DE. Heterogeneity of malignant cells from a human colonic carcinoma. Cancer Res. 1981;41:1751–1756. [PubMed] [Google Scholar]
- 8.Burdick JA, Prestwich GD. Hyaluronic acid hydrogels for biomedical applications. Adv Mater. 2011;23:H41– H56. doi: 10.1002/adma.201003963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bursac N, Loo Y, Leong K, Tung L. Novel anisotropic engineered cardiac tissues: studies of electrical propagation. Biochem Biophys Res Commun. 2007;361:847– 853. doi: 10.1016/j.bbrc.2007.07.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carterson AJ, Honer zu Bentrup K, Ott CM, Clarke MS, Pierson DL, Vanderburg CR, Buchanan KL, Nickerson CA, Schurr MJ. A549 lung epithelial cells grown as three-dimensional aggregates: alternative tissue culture model for Pseudomonas aeruginosa pathogenesis. Infect Immun. 2005;73:1129–1140. doi: 10.1128/IAI.73.2.1129-1140.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drewitz M, Helbling M, Fried N, Bieri M, Moritz W, Lichtenberg J, Kelm JM. Towards automated production and drug sensitivity testing using scaffold-free spherical tumor microtissues. Biotechnol J. 2011;6:1488–1496. doi: 10.1002/biot.201100290. [DOI] [PubMed] [Google Scholar]
- 12.Erickson IE, Huang AH, Chung C, Li RT, Burdick JA, Mauck RL. Differential maturation and structure-function relationships in mesenchymal stem celland chondrocyte-seeded hydrogels. Tissue Eng Part A. 2009;15:1041–1052. doi: 10.1089/ten.tea.2008.0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frye CA, Patrick CW. Three-dimensional adipose tissue model using low shear bioreactors. Vitro Cell Dev Biol Anim. 2006;42:109–114. doi: 10.1290/0509055.1. [DOI] [PubMed] [Google Scholar]
- 14.Gavert N, Ben-Ze’ev A. Coordinating changes in cell adhesion and phenotype during EMT-like processes in cancer. F1000 Biol Rep. 2010;2:86. doi: 10.3410/B2-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerecht S, Burdick JA, Ferreira LS, Townsend SA, Langer R, Vunjak-Novakovic G. Hyaluronic acid hydrogel for controlled self-renewal and differentiation of human embryonic stem cells. Proc Natl Acad Sci USA. 2007;104:11298–11303. doi: 10.1073/pnas.0703723104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hjelm BE, Berta AN, Nickerson CA, Arntzen CJ, Herbst-Kralovetz MM. Development and characterization of a three-dimensional organotypic human vaginal epithelial cell model. Biol Reprod. 2010;82:617–627. doi: 10.1095/biolreprod.109.080408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho WJ, Pham EA, Kim JW, Ng CW, Kim JH, Kamei DT, Wu BM. Incorporation of multicellular spheroids into 3D polymeric scaffolds provides an improved tumor model for screening anticancer drugs. Cancer Sci. 2010;101:2637–2643. doi: 10.1111/j.1349-7006.2010.01723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Honer zu Bentrup K, Ramamurthy R, Ott CM, Emami K, Nelman-Gonzalez M, Wilson JW, Richter EG, Goodwin TJ, Alexander JS, Pierson DL, Pellis N, Buchanan KL, Nickerson CA. Three-dimensional organotypic models of human colonic epithelium to study the early stages of enteric salmonellosis. Microbes Infect. 2006;8:1813–1825. doi: 10.1016/j.micinf.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 19.Karakiulakis G, Papanikolaou C, Jankovic SM, Aletras A, Papakonstantinou E, Vretou E, Mirtsou-Fidani V. Increased type IV collagen-degrading activity in metastases originating from primary tumors of the human colon. Invasion Metastasis. 1997;17:158–168. [PubMed] [Google Scholar]
- 20.LaMarca HL, Ott CM, Honer Zu Bentrup K, Leblanc CL, Pierson DL, Nelson AB, Scandurro AB, Whitley GS, Nickerson CA, Morris CA. Threedimensional growth of extravillous cytotrophoblasts promotes differentiation and invasion. Placenta. 2005;26:709–720. doi: 10.1016/j.placenta.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Leach JB, Bivens KA, Collins CN, Schmidt CE. Development of photocrosslinkable hyaluronic acidpolyethylene glycol-peptide composite hydrogels for soft tissue engineering. J Biomed Mater Res A. 2004;70:74–82. doi: 10.1002/jbm.a.30063. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Shu XZ, Prestwich GD. Tumor engineering: orthotopic cancer models in mice using cell-loaded, injectable, cross-linked hyaluronan-derived hydrogels. Tissue Eng. 2007;13:1091–1101. doi: 10.1089/ten.2006.0297. [DOI] [PubMed] [Google Scholar]
- 23.Macedo FI, Makarawo T. Colorectal hepatic metastasis: Evolving therapies. World J Hepatol. 2014;6:453– 463. doi: 10.4254/wjh.v6.i7.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.May JE, Xu J, Morse HR, Avent ND, Donaldson C. Toxicity testing: the search for an in vitro alternative to animal testing. Br J Biomed Sci. 2009;66:160–165. doi: 10.1080/09674845.2009.11730265. [DOI] [PubMed] [Google Scholar]
- 25.McGrail DJ, Mezencev R, Kieu QM, McDonald JF, Dawson MR. SNAIL-induced epithelial-to-mesenchymal transition produces concerted biophysical changes from altered cytoskeletal gene expression. FASEB J. 2014;29:1–10. doi: 10.1096/fj.14-257345. [DOI] [PubMed] [Google Scholar]
- 26.Miki D, Dastgheib K, Kim T, Pfister-Serres A, Smeds KA, Inoue M, Hatchell DL, Grinstaff MW. A photopolymerized sealant for corneal lacerations. Cornea. 2002;21:393–399. doi: 10.1097/00003226-200205000-00012. [DOI] [PubMed] [Google Scholar]
- 27.Myers TA, Nickerson CA, Kaushal D, Ott CM, Honer zu Bentrup K, Nelman-Gonzalez M, Ramamurthy R, Pierson DL, Philipp MT. Closing the phenotypic gap between transformed neuronal cell lines in culture and untransformed neurons. J Neurosci Methods. 2008;174:31–41. doi: 10.1016/j.jneumeth.2008.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Navran S. The application of low shear modeled microgravity to 3D cell biology and tissue engineering. Biotechnol Annu Rev. 2008;14:275–296. doi: 10.1016/S1387-2656(08)00011-2. [DOI] [PubMed] [Google Scholar]
- 29.Nickerson CA, Ott CM. A new dimension in modeling infectious disease. ASM News. 2004;70:169–175. [Google Scholar]
- 30.Nickerson CA, Goodwin TJ, Terlonge J, Ott CM, Buchanan KL, Uicker WC, Emami K, LeBlanc CL, Ramamurthy R, Clarke MS, Vanderburg CR, Hammond T, Pierson DL. Three-dimensional tissue assemblies: novel models for the study of Salmonella enterica serovar Typhimurium pathogenesis. Infect Immun. 2001;69:7106–7120. doi: 10.1128/IAI.69.11.7106-7120.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nickerson CA, Richter EG, Ott CM. Studying host-pathogen interactions in 3D: organotypic models for infectious disease and drug development. J Neuroimmune Pharmacol. 2007;2:26–31. doi: 10.1007/s11481-006-9047-x. [DOI] [PubMed] [Google Scholar]
- 32.Nishioka M, Ueno K, Hazama S, Okada T, Sakai K, Suehiro Y, Okayama N, Hirata H, Oka M, Imai K, Dahiya R, Hinoda Y. Possible involvement of Wnt11 in colorectal cancer progression. Mol Carcinog. 2013;52:207–217. doi: 10.1002/mc.21845. [DOI] [PubMed] [Google Scholar]
- 33.Ogunwobi OO, Liu C. Therapeutic and prognostic importance of epithelial-mesenchymal transition in liver cancers: insights from experimental models. Crit Rev Oncol Hematol. 2012;83:319–328. doi: 10.1016/j.critrevonc.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 34.Orsulic S, Huber O, Aberle H, Arnold S, Kemler R. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J Cell Sci. 1999;112(Pt 8):1237–1245. doi: 10.1242/jcs.112.8.1237. [DOI] [PubMed] [Google Scholar]
- 35.Prestwich GD. Evaluating drug efficacy and toxicology in three dimensions: using synthetic extracellular matrices in drug discovery. Acc Chem Res. 2008;41:139–148. doi: 10.1021/ar7000827. [DOI] [PubMed] [Google Scholar]
- 36.Sainz B, Jr, TenCate V, Uprichard SL. Threedimensional Huh7 cell culture system for the study of Hepatitis C virus infection. Virol J. 2009;6:103. doi: 10.1186/1743-422X-6-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwarz RP, Goodwin TJ, Wolf DA. Cell culture for three-dimensional modeling in rotating-wall vessels: an application of simulated microgravity. J Tissue Cult Methods. 1992;14:51–57. doi: 10.1007/BF01404744. [DOI] [PubMed] [Google Scholar]
- 38.Serban MA, Scott A, Prestwich GD. Use of hyaluronan-derived hydrogels for three-dimensional cell culture and tumor xenografts. Curr Protoc Cell Biol. 2008;Chapter 10(Unit 10):14. doi: 10.1002/0471143030.cb1014s40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shu XZ, Liu Y, Palumbo FS, Luo Y, Prestwich GD. In situ crosslinkable hyaluronan hydrogels for tissue engineering. Biomaterials. 2004;25:1339–1348. doi: 10.1016/j.biomaterials.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 40.Shu XZ, Ahmad S, Liu Y, Prestwich GD. Synthesis and evaluation of injectable, in situ crosslinkable synthetic extracellular matrices for tissue engineering. J Biomed Mater Res A. 2006;79:902–912. doi: 10.1002/jbm.a.30831. [DOI] [PubMed] [Google Scholar]
- 41.Skardal A. PhD Dissertation. Department of Bioengineering, University of Utah; 2010. Hyaluronan and gelatin biomaterials for bioprinting engineered tissues; p. 208. [Google Scholar]
- 42.Skardal A, Sarker SF, Crabbe A, Nickerson CA, Prestwich GD. The generation of 3D tissue models based on hyaluronan hydrogel-coated microcarriers within a rotating wall vessel bioreactor. Biomaterials. 2010;31:8426–8435. doi: 10.1016/j.biomaterials.2010.07.047. [DOI] [PubMed] [Google Scholar]
- 43.Skardal A, Zhang J, McCoard L, Oottamasathien S, Prestwich GD. Dynamically crosslinked gold nanoparticle—hyaluronan hydrogels. Adv Mater. 2010;22:4736– 4740. doi: 10.1002/adma.201001436. [DOI] [PubMed] [Google Scholar]
- 44.Skardal A, Zhang J, Prestwich GD. Bioprinting vessel-like constructs using hyaluronan hydrogels crosslinked with tetrahedral polyethylene glycol tetracrylates. Biomaterials. 2010;31:6173–6181. doi: 10.1016/j.biomaterials.2010.04.045. [DOI] [PubMed] [Google Scholar]
- 45.Skardal A, Smith L, Bharadwaj S, Atala A, Soker S, Zhang Y. Tissue specific synthetic ECM hydrogels for 3D in vitro maintenance of hepatocyte function. Biomaterials. 2012;33:4565–4575. doi: 10.1016/j.biomaterials.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith YC, Grande KK, Rasmussen SB, O’Brien AD. Novel three-dimensional organoid model for evaluation of the interaction of uropathogenic Escherichia coli with terminally differentiated human urothelial cells. Infect Immun. 2006;74:750–757. doi: 10.1128/IAI.74.1.750-757.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steinert G, Scholch S, Koch M, Weitz J. Biology and significance of circulating and disseminated tumour cells in colorectal cancer. Langenbecks Arch Surg. 2012;397:535– 542. doi: 10.1007/s00423-012-0917-9. [DOI] [PubMed] [Google Scholar]
- 48.Strillacci A, Valerii MC, Sansone P, Caggiano C, Sgromo A, Vittori L, Fiorentino M, Poggioli G, Rizzello F, Campieri M, Spisni E. Loss of miR-101 expression promotes Wnt/beta-catenin signalling pathway activation and malignancy in colon cancer cells. J Pathol. 2013;229:379– 389. doi: 10.1002/path.4097. [DOI] [PubMed] [Google Scholar]
- 49.Tania M, Khan MA, Fu J. Epithelial to mesenchymal transition inducing transcription factors and metastatic cancer. Tumour Biol. 2014;35:7335–7342. doi: 10.1007/s13277-014-2163-y. [DOI] [PubMed] [Google Scholar]
- 50.Unsworth BR, Lelkes PI. Growing tissues in microgravity. Nat Med. 1998;4:901–907. doi: 10.1038/nm0898-901. [DOI] [PubMed] [Google Scholar]
- 51.Vandamme TF. Use of rodents as models of human diseases. J Pharm Bioallied Sci. 2014;6:2–9. doi: 10.4103/0975-7406.124301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wheeler SE, Clark AM, Taylor DP, Young CL, Pillai VC, Stolz DB, Venkataramanan R, Lauffenburger D, Griffith L, Wells A. Spontaneous dormancy of metastatic breast cancer cells in an all human liver micro-physiologic system. Br J Cancer. 2014;111(12):2342–2350. doi: 10.1038/bjc.2014.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wilson PM, Danenberg PV, Johnston PG, Lenz HJ, Ladner RD. Standing the test of time: targeting thymidylate biosynthesis in cancer therapy. Nat Rev Clin Oncol. 2014;11:282–298. doi: 10.1038/nrclinonc.2014.51. [DOI] [PubMed] [Google Scholar]
- 54.Yoffe B, Darlington GJ, Soriano HE, Krishnan B, Risin D, Pellis NR, Khaoustov VI. Cultures of human liver cells in simulated microgravity environment. Adv Space Res. 1999;24:829–836. doi: 10.1016/s0273-1177(99)00079-4. [DOI] [PubMed] [Google Scholar]
- 55.Yoshioka T, Mishima H, Ohyabu Y, Sakai S, Akaogi H, Ishii T, Kojima H, Tanaka J, Ochiai N, Uemura T. Repair of large osteochondral defects with allogeneic cartilaginous aggregates formed from bone marrow-derived cells using RWV bioreactor. J Orthop Res. 2007;25:1291–1298. doi: 10.1002/jor.20426. [DOI] [PubMed] [Google Scholar]
- 56.Young M, Ordonez L, Clarke AR. What are the best routes to effectively model human colorectal cancer? Mol Oncol. 2013;7:178–189. doi: 10.1016/j.molonc.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zaman MH, Trapani LM, Sieminski AL, Mackellar D, Gong H, Kamm RD, Wells A, Lauffenburger DA, Matsudaira P. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc Natl Acad Sci USA. 2006;103:10889–10894. doi: 10.1073/pnas.0604460103. [DOI] [PMC free article] [PubMed] [Google Scholar]