

Abstract
Background
The role of microRNA-21 in isoflurane-induced cardioprotection is unknown. We addressed this issue using microRNA-21 knockout mice and explored the underlying mechanisms.
Methods
C57BL/6 and microRNA-21 knockout mice were echocardiographically examined. Mouse hearts underwent 30 min of ischemia followed by 2 h of reperfusion in vivo or ex vivo in the presence or absence of 1.0 minimum alveolar concentration of isoflurane administered before ischemia. Cardiac Akt, eNOS, and nNOS proteins were determined by Western blot. Opening of the mitochondrial permeability transition pore (mPTP) in cardiomyocytes was induced by photoexcitation-generated oxidative stress and detected by rapid dissipation of tetramethylrhodamine ethyl ester fluorescence using a confocal microscope.
Results
Genetic disruption of miR-21 gene did not alter phenotype of the left ventricle, baseline cardiac function, area at risk, and the ratios of p-Akt/Akt, p-eNOS/eNOS, and pnNOS/nNOS. Isoflurane decreased infarct size from 54±10% in control to 36±10% (P<0.05, n=8 mice/group), improved cardiac function after reperfusion, and increased the ratios of p-Akt/AKT, p-eNOS/eNOS, and p-nNOS/nNOS in C57BL/6 mice subjected to ischemia/reperfusion injury. These beneficial effects of isoflurane were lost in microRNA-21 knockout mice. There were no significant differences in time of the mPTP opening induced by photoexcitation-generated oxidative stress in cardiomyocytes isolated between C57BL/6 and microRNA-21 knockout mice. ISO significantly delayed mPTP opening in cardiomyocytes from C57BL/6 but not microRNA-21 knockout mice.
Conclusions
Isoflurane protects mouse hearts from ischemia/reperfusion injury by a microRNA-21-dependent mechanism. The Akt/NOS/mPTP pathway is involved in the microRNA-21-mediated protective effect of isoflurane.
Volatile anesthetics protect the heart against ischemia/reperfusion (I/R) injury even when administered for only a brief period of time before ischemia.1-4 This phenomenon is similar to ischemic preconditioning, termed as anesthetic preconditioning.1-4 Both basic and clinical studies have shown that anesthetic preconditioning is an effective strategy to reduce myocardial injury.1,4-7 Although the volatile anesthetic, isoflurane (ISO), potently protects the heart against I/R injury,1,2,4 the precise mechanisms have not been completely elucidated.
MicroRNAs are endogenous, noncoding single-stranded RNA molecules of approximately 22 nucleotides in length. As central regulators of gene expression, microRNAs participate in the regulation of numerous physiological and pathophysiological processes.8,9 MicroRNA-21 (miR-21) is highly expressed in cardiomyocytes, cardiac fibroblasts, vascular endothelial cells, and vascular smooth muscle cells.10-12 The physiological role of miR-21 in the cardiovascular system is incompletely known. There is evidence that miR-21 is involved in cardiac cell growth and death, regulation of cardiac fibroblast function, and vascular smooth muscle cell proliferation and apoptosis.9,13-15 However, the contribution of miR-21 to cardiovascular disease is increasingly identified, including myocardial infarction, atherosclerosis, myocardial fibrosis, cardiac hypertrophy, and heart failure.10,16-18 Recent studies indicate that miR-21 is closely associated with acute myocardial I/R injury.8,12 First, miR-21 in cardiomyocytes is up-regulated in response to hypoxia or ischemia.8 Second, down-regulation of miR-21 increases the vulnerability of myocardium to I/R injury, whereas increased expression of miR-21 enhances the tolerance of myocardium to I/R injury.17,19 Third, miR-21 inhibitor eliminates the cardioprotective effect of ischemic preconditioning.12 Recently, miR-21 is found to involve the ISO-induced protection of cardiomyocytes against hypoxia/reoxygenation injury.20 However, how miR-21 contributes to ISO-induced protection against myocardial I/R injury remains elusive. The aim of this study was to examine the role of miR-21 in ISO-induced protection against acute myocardial I/R injury using miR-21 gene knockout (KO) mice.
Materials and Methods
Animals
C57BL/6 wild-type (WT) mice and miR-21 knockout (KO) mice (weight: 26.2 ± 1.9 g; age: 9-12 weeks) were purchased from The Jackson Laboratory (Bar Harbor, ME). The animals were kept on a 12-h light-dark cycle in a temperature-controlled room. The experimental procedures were approved by the Animal Care and Use Committee of the Medical College of Wisconsin (Milwaukee, WI) and conformed to the Guide for the Care and Use of Laboratory Animals (Institute for Laboratory Animal Research, National Academy of Sciences, 8th edition, 2011).
Hemodynamic measurements
Mice were anesthetized by intraperitoneal injection of 80 mg/kg pentobarbital sodium and ventilated with room air supplemented with 100% O2 at approximately 102 breaths/min.21 The right carotid artery was cannulated with a small polyethylene tubing catheter filled with 0.9% saline containing 10 U/ml heparin, as described.22 The catheter was connected to an ADInstrument pressure transducer (MLT0380/D, ADInstruments, Colorado Springs, CO) and a Powerlab data acquisition system (ADInstruments). After a 30 min of stabilization, blood pressure was continuously recorded for 20 min. Body temperature was maintained between 36.8°C and 37.5°C throughout the experiment by using a heating pad (Model TC-1000, CWE Inc.; Ardmore, PA).
Transthoracic echocardiography
Mice were sedated by the inhalation of isoflurane (ISO, 1.50 %) and oxygen. Non-invasive transthoracic echocardiography was performed with a VisualSonics Vevo 770 High-resolution Imaging System (Toronto, Canada) equipped with a 30 MHz transducer (Scanhead RMV 707), as described previously.23 Left ventricular dimensions and ejection fraction were measured by two-dimension guided M-mode method. Pulsed Doppler waveforms recorded in the apical-4-chamber view were used for the measurements of the peak velocities of mitral E (early mitral inflow) and A (late mitral inflow) waves, E wave acceleration velocity, E wave acceleration time, E wave deceleration velocity, and E wave deceleration time, isovolumic contraction time, ejection time, and isovolumic relaxation time of LV. Myocardial performance index was calculated with the following formula: myocardial performance index = (isovolumic contraction time + isovolumic relaxation time)/ejection time.
Real-time reverse transcriptional-polymerase chain reaction
Pentobarbital-anesthetized mice were stabilized for 30 min and administered 1.0 minimum alveolar concentration (approximately 1.40% in the mouse) of ISO for 30 min via an ISO-specific vaporizer (Ohio Medical Instruments, Madison, WI). Control mice received no ISO. The heart was excised 30 min and 3 h after ISO treatment was discontinued, and the left ventricle (LV) was homogenized at 4 °C for real-time quantitative reverse transcriptional-polymerase chain reaction (qRT-PCR) analysis of miR-21.
MiR-21 extraction and qRT-PCR analysis
Total RNA from heart tissues was extracted using Qiazol reagent according to the protocol of the manufacturer (Qiagen, Valencia, CA). Chloroform was added and samples were centrifuged to facilitate phase separation. The aqueous phase was extracted and combined with ethanol in miRNeasy Mini spin columns (Qiagen). Total RNA was eluted in RNase-free water. The concentration of extracted total RNA was quantified by the Epoch spectrophotometer (Biotek, Winooski, VT). Samples were considered pure if the A260/280 ratio was between 1.9 and 2.0. One μg of total RNA from each sample was used to generate cDNA using miScript Reverse transcriptase mix, nucleics mix, and HiFlex Buffer (Qiagen). The complementary DNA product was measured in triplicate using miScript Primer Assays for miR-21 (Qiagen). qRT-PCR was conducted using the BioRad iCycler Real-Time PCR Detection System. Expression of miR-21 was normalized by expression of the housekeeping gene Rnu-6 (Qiagen). The relative gene expressions were calculated in accordance with the ΔΔCt method. Relative miRNA levels were expressed as percentages compared to non-ISO exposed controls.
PCR array of miR-21 target mRNAs
C57BL/6 mice were given 1.0 minimum alveolar concentration of ISO for 30 min via an ISO-specific vaporizer (Ohio Medical Instruments). Control mice received no ISO. The heart was excised 30 min after ISO treatment was discontinued, and the LV was homogenized at 4 °C for PCR array analysis of miR-21 target mRNAs using mouse miFinder RT2 PCR Array (SABiosciences, Valencia, CA) according to the manufacturer's instructions. These arrays investigate 84 experimentally validated and predicted targets of miR-21, including activity-dependent neuroprotector homeobox (ADNP), centrosomal protein 68kDa (CEP68), derlin 1 (DERL1), eukaryotic translation initiation factor 4A2 (EIF4A2), krev interaction trapped protein 1 (KRIT1), myristoylated alanine-rich C-kinase substrate (MARCKS), nuclear factor I/B (NFIB), phosphatase and tensin homology deleted from chromosome 10 (PTEN), Ras homolog family member B (RHOB), ribosomal protein S7 (RPS7), reticulon-4 (RTN4), sprouty 2 (SPRY2), topoisomerase I binding, arginine/serine-rich, E3 ubiquitin protein ligase (TOPORS), tropomyosin 1 (TPM1), ubiquitin-conjugating enzyme E2 D3 (UBE2D3), and Wolfram syndrome 1 (WFS1). Total RNA was extracted using the Qiagen RNeasy (Qiagen), and cDNA was prepared using the RT2 First Strand Kit (Qiagen). cDNA was diluted and combined with RT2 SYBR Green/Fluorescein Master Mix and distributed evenly across the wells of a 96-well plate in technical triplicates. The BioRad iCycler Real-Time PCR Detection System was used in qRT-PCR analysis of miR-21 target mRNAs. Samples were exposed to an initial 95°C hot-start activation step, followed by 40 cycles of 94°C denaturation and 60°C annealing/extension phases. Data collected from these experiments defined Ct values of the mRNAs present in each sample. Expression levels of the housekeeping genes β-actin and GAPDH were used as control to normalize samples. Samples without reverse transcriptase were run to confirm that no genomic DNA was present in the sample. mRNAs with Ct values greater than or equal to 35 were excluded from the study.
Myocardial I/R injury in vivo
Myocardial I/R injury was produced by occluding the left coronary artery, as previously described.12,24 MiR-21 KO and C57BL/6 mice were divided into the following 4 groups (10 mice/group): WT-I/R, miR-21 KO-I/R, WT-I/R+ISO, and miR-21 KO-I/R+ISO (Figure 1). After instrumentation was completed, all mice were stabilized for 30 min and subjected to 30 min of coronary occlusion followed by 2 h of reperfusion. ISO at 1.0 minimum alveolar concentration was administered for 30 min via an ISO-specific vaporizer (Ohio Medical Instruments) followed by a 15 min period of washout prior to coronary artery occlusion. Control mice received no ISO. Heart rate was monitored from the electrocardiogram. The infarct area was delineated by perfusing the coronary arteries with 2,3,5-triphenyltetrazolium chloride via the aortic root, and the area at risk was delineated by perfusing phthalo blue dye (Heucotech Ltd., Fairless Hill, PA) into the aortic root after tying the coronary artery at the site of previous occlusion.
Figure 1.
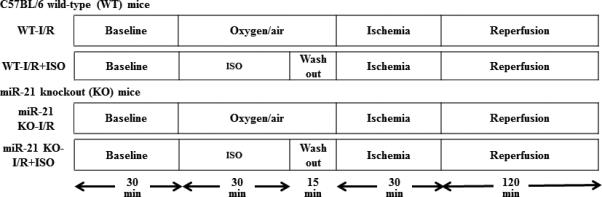
Schematic diagram depicting the experimental protocols of myocardial ischemia/reperfusion injury in vivo. ISO=isoflurane; I/R=ischemia/reperfusion.
Myocardial I/R injury ex vivo
Mouse hearts were mounted on a Langendorff apparatus and perfused retrogradely through the aorta at a constant pressure of 80 mmHg with Krebs-Henseleit buffer at 37 °C, as described.21,23 C57BL/6 and MiR-21 KO hearts were assigned to the following 6 groups (Figure 2): WT-Time C, miR-21 KO-Time C, WT-I/R, miR-21 KO-I/R, WT-I/R+ISO, and miR-21 KO-I/R+ISO. The hearts of the WT-Time C (time control, n=8 hearts) and miR-21 KO-Time C groups (n=7 hearts) were continuously perfused for 210 min with Krebs-Henseleit solution without I/R. All hearts in other 4 groups (n=10 hearts/group) were stabilized for 30 min and subjected to 30 min of no-flow global ischemia followed by 2 h of reperfusion. ISO was bubbled into Krebs-Henseleit solution using an agent-specific vaporizer (Ohio Medical Instruments) placed in the 95% oxygen/5% carbon dioxide gas mixture line. ISO concentrations in the coronary effluent were determined by gas chromatography. In the WT-I/R+ISO and miR-21 KO-I/R+ISO groups, the hearts were perfused with 2 cycles of 5-min Krebs-Henseleit solution containing 0.5 mM ISO/5-min Krebs-Henseleit solution without ISO followed by a 10-min washout prior to ischemia. Left ventricular +dP/dt (maximum rate of increase of left ventricular developed pressure) and −dP/dt (maximum rate of decrease of left ventricular developed pressure) at baseline, 20 min after ischemia, and 10, 30, 60, 90, and 120 min after reperfusion were determined.
Figure 2.
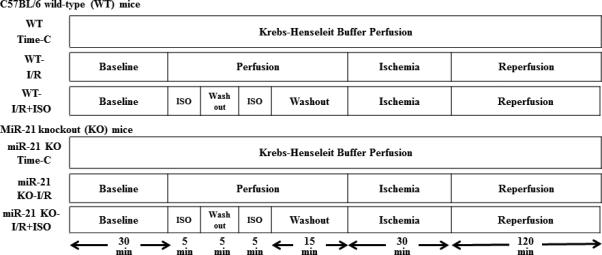
Schematic diagram depicting the experimental protocols of myocardial ischemia/reperfusion (I/R) injury in Langendorff-perfused mouse hearts. ISO=isoflurane; I/R=ischemia/reperfusion; Time-C=time control.
Measurements of mitochondrial NADH levels in Langendorff-perfused mouse hearts
NADH emits fluorescence when mitochondria are illuminated at the appropriate wavelength (for example, 350 nm, 460 nm, etc.).25 To measure NADH fluorescence in mouse hearts, Langendorff-perfused mouse hearts were placed within a light-proof Faraday cage to block incident room light, as described.26 A fiberoptic cable placed against the LV of Langendorff-prepared mouse hearts to excite and record transmyocardial fluorescence at a wavelength of 456 nm during ischemia and reperfusion. The two proximal ends of the fiberoptic cable were connected to a modified spectrophotofluorometer (Photon Technology International, London, Canada). Fluorescence (F) was excited with light at the appropriate wavelength (λ) from a xenon arc lamp at 75 W filtered through a monochromator (Delta RAM, Photon Technology International). NADH signal was recorded continuously using a Powerlab data acquisition system (ADInstruments).
Immunoblotting
Pentobarbital-anesthetized mice were subjected to myocardial I/R injury in vivo in the presence or absence of ISO, as illustrated in Figure 1. The myocardium from the area at risk of mouse hearts was harvested and homogenized in a buffer containing 20.0 mM MOPS, 2.0 mM EGTA, 5.0 mM EDTA, protease inhibitor cocktail (1:100; Calbiochem, San Diego, CA), phosphatase inhibitors cocktail (1:100; Calbiochem), 0.5% detergent (Nonidet™ P-40 detergent pH 7.4, Sigma-Aldrich, St. Louis, MO). Immunoblots were performed using standard techniques, as described.24 Briefly, tissue homogenates that contained 50 μg [for Akt or endothelial nitric oxide synthase (eNOS)] or 100 μg [for neuronal nitric oxide synthase (nNOS)] of protein were applied to 7.5% SDS-polyacrylamide gel and subjected to immunoblot analysis by incubation with a mouse anti-Akt antibody (Cell Signaling, Beverly, MA), a mouse anti-phosphorylated Akt (p-Akt) antibody (serine 473, Cell Signalling), an anti-eNOS antibody (Santa Cruz Biotechnologies, Santa Cruz, CA), an anti-phosphorylated e-NOS (p-eNOS) antibody (serine 1177, Cell Signaling), an anti-nNOS antibody (Invitrogen), or an anti-phosphorylated nNOS (p-nNOS) antibody (serine 1412, Affinity Bioreagents, Golden, CO) at 4°C. The membrane was washed and then incubated with the appropriate anti-mouse secondary antibody. Immunoreactive bands were visualized by enhanced chemiluminescence followed by densitometric analysis using image acquisition and analysis software (Image J, NIH).
Detection of opening of the mitochondrial permeability transition pore (mPTP) in cardiomyocytes
Isolation of cardiomyocytes
Cardiomyocytes were isolated from adult mice using the methods established by the Alliance for Cellular Signaling (http://www.signaling-gateway.org; protocol no. PP00000015).27 Briefly, mouse hearts were cannulated via the aorta onto a blunted 20 gauge needle and perfused for 10 min at 37 °C with perfusion buffer (in mM: 113 NaCl, 4.7 KCl, 0.6 KH2PO4, 0.6 Na2HPO4, 1.2 MgSO4-7H2O, 0.032 phenol red, 12 NaHCO3, 10.0 KHCO3, 10.0 HEPES, pH 7.4, 30 taurine, 10 2,3-butanedione monoxime, and 5.5 glucose) containing 0.25 mg/ml Liberase blendzyme I, 0.14 mg/ml trypsin, and 12.5 μM CaCl2. After perfusion, the LVs were dissected free from the atria and repeatedly passed through a plastic transfer pipette to disaggregate the cells into a single-cell suspension. Subsequently, myocytes were enriched by sedimentation in perfusion buffer containing 5% bovine calf serum while slowly exposing the cells to increasing concentrations of CaCl2 to achieve a final concentration of 1.2 mM. The final cell pellet containing calcium-tolerant myocytes was resuspended in the culture media containing Hanks’ salts, 2 mM L-glutamine, 5% bovine calf serum, 10 mM 2,3-butanedione monoxime, and 100 U/ml penicillin. After isolation, the myocytes were stored in Tyrode solution (in mM: 132 NaCl, 10 HEPES, 5 glucose, 5 KCl, 1 CaCl2, 1.2 MgCl2; adjusted to pH 7.4). Experiments were conducted at room temperature within 5 h after isolation using Tyrode solution.
Experimental protocol
Cardiomyocytes isolated from mouse hearts were assigned to the following 4 groups (12 cells/group): WT-OS (oxidative stress), miR-21 KO-OS, WT-OS+ISO, and miR-21 KO-OS+ISO. The myocytes in the WT-OS and WT-OS+ISO groups were from C57BL/6 mice, and those in the miR-21 KO-OS and miR-21 KO-OS+ISO groups from miR-21 KO mice. After 20 min of stabilization at room temperature, the myocaytes were incubated with Tyrode solution containing 0.5 mM ISO for 10 min followed by washout for 5 min or without ISO.
Detection of mPTP opening
A laser-scanning confocal microscope (Nikon Eclipse TE2000-U, Nikon Instruments Inc., Melville, NY) was used to measure opening of the mPTP in cardiomyocytes. Opening of the mPTP was induced by photoexcitation-generated oxidative stress and detected by rapid dissipation of tetramethylrhodamine ethyl ester fluorescence (TMRE), as described.28,29 All cells were loaded with 50 nM TMRE at room temperature for 20 min. After TMRE loading, the cells was washed out for 5 min, and a recording region of 30 × 30 μm was exposed to narrowly focused laser scanning. Only cells with equal initial TMRE fluorescence intensity were included in the study, and the settings of the confocal microscope were consistent to ensure equal delivery of oxidative stress. Arbitrary mPTP opening time was determined as the time of loss of average TMRE fluorescence intensity from the recorded region (excluding nucleus) by half between initial and residual fluorescence intensity. It corresponded to complete depolarization of 50% of mitochondria in the recorded region.
OS in cardiomyocytes
Cardiomyocytes were isolated from 1-2 day-old Sprague Dawley rat pups using serial digestion with collagenase and pancreatin (Sigma-Aldrich), as described.20
Experimental protocol
Cardiomyocyes were grouped (n=3 dishes/group) and received treatment as the following: (1) Control: the cells were cultured for 7 days in full supplement plating medium; (2) GFP (green fluorescence protein): the cells were cultured for 4 days and transduced with 100 MOI of GFP-labeled adenovirus for 72 h; (3) Pre-miR-21: the cells were cultured for 4 days and transduced with 100 MOI of pre-miR-21 adenovirus (Applied Biological Materials, Inc., Richmond, BC, Canada) for 72 h; (4) Anti-miR-21: the cells were transduced with 100 MOI of GFP-labeled anti-miR-21 (Applied Biological Materials, Inc.) for 72 h without H2O2 treatment. 5) H2O2: the cells were cultured for 7 days in full supplement medium and exposed to 50.0 μM H2O2 (Calbiochem, La Jolla, CA) for 4 h; (6) GFP+H2O2: the cells were cultured for 4 days, transduced with 100 MOI of GFP-labeled adenovirus for 72 h, and exposed to 50.0 μM H2O2 for 4 h; (7) Pre-miR-21+H2O2: the cells were cultured for 4 days, transduced with 100 MOI of pre-miR-21 adenovirus for 72 h, and exposed to 50.0 μM H2O2 for 4 h; (8) Anti-miR-21+H2O2: the cells were transduced with GFP-labeled anti-miR-21 for 72 h and exposed to H2O2 for 4 h. In all transduction experiments, transduction was visually confirmed through fluorescence microscropy, as well as through qRT-PCR analysis. Cell nuclei were stained with Hoechst 33342 and confirmed an approximately 70-80% transduction efficiency in cardiomyocytes.
Propidium iodide (PI) Staining
Nuclei were stained with Hoechst 33342 (Life Technologies) at a concentration of 1:800 diluted in cell culture media. PI solution (Life Technologies) at 1.0 mg/ml was diluted in media at a concentration of 1:500. Hoechst-positive nuclei were counted as a control using fluorescence microscopy and PI positive cells were taken as a percentage of the control.
Statistical analysis
Mice were randomly assigned to ISO-treated or CON groups. Western blot and qRT-PCR analyses were conducted blindly, with samples divided into randomly numbered groups. All data are expressed as mean ± S.D. Two-way repeated measures ANOVA test was used to evaluate the differences in body weight, mean arterial blood pressure, heart weight, and the ratio of heart/body weight, and echocardiographic data. Statistical analysis of heart rate, ±dP/dt, and NADH over time between groups was performed with repeated measures ANOVA followed by Bonferroni's multiple comparison. One-way ANOVA followed by Bonferroni post-hoc test was used to analyze area at risk, infarct size, the ratios of p-Akt/Akt, p-eNOS/eNOS, and pnNOS/nNOS, and the mPTP opening time. All statistical analyses were performed using GraphPad Prism 6 (GraphPad Software, Inc., La Jolla). A value of P < 0.05 (two-tailed) was considered statistically significant.
Results
Characteristics of C57BL/6 and miR-21 KO mice
Baseline characteristics of C57BL/6 and miR-21 KO mice are listed in Table 1. There were no significant differences observed in body weight, mean arterial blood pressure, heart weight, and the dimension and function of the LV between miR-21 KO and C57BL/6 mice.
Table 1.
Basic characteristics of C57BL/6 and miR-21 knockout mice
| Parameters | C57BL/6 | miR-21 knockout |
|---|---|---|
| General characteristics | ||
| Body weight, g | 25.7±1.8 | 26.7±1.9 |
| Mean arterial blood pressure, mmHg | 97±26 | 93±23 |
| Heart weight, mg | 135±13 | 129±12 |
| Heart/body weight, mg/g | 5.1±0.7 | 5.0±0.1 |
| Echocardiography parameters | ||
| Heart rate, bpm | 452±61 | 438±36 |
| Anterior wall at end diastole, mm | 0.81±0.11 | 0.84±0.11 |
| Anterior wall at end systole, mm | 1.28±0.22 | 1.32±0.13 |
| Posterior wall at end diastole, mm | 0.87±0.09 | 0.82±0.13 |
| Posterior wall at end systole, mm | 1.31±0.28 | 1.12±0.20 |
| LV end-diastolic volume, μl | 56±12 | 63±10 |
| LV end-systolic volume, μl | 18±9 | 22±5 |
| Ejection fraction, % | 70±11 | 65±6 |
| Peak E wave velocity, cm/s | 77±15 | 67±6 |
| Peak A wave velocity, cm/s | 45±13 | 48±4 |
| Peak E/A ratio | 1.79±0.38 | 1.40±0.18 |
| Isovolumic contraction time of LV, ms | 15.3±4.9 | 17.5±3.4 |
| Isovolumic relaxation time of LV, ms | 16.7±2.1 | 15.6±1.8 |
| Ejection time, ms | 44.5±6.2 | 51.0±4.6 |
| Myocardial performance index | 0.72±0.13 | 0.65±0.11 |
| Mitral E acceleration, cm/ms | 8295±2291 | 8602±1002 |
| Mitral E wave acceleration time, ms | 8.9±2.2 | 7.9±1.0 |
| Mitral E deceleration, cm/ms | 4425±1603 | 4440±954 |
| Mitral E wave deceleration time, ms | 16.5±6.6 | 17.5±2.8 |
There were no significant differences in all parameters between miR-21 knockout and C57BL/6 mice (n=10-16 mice/group).
Bpm = beats per minute; LV = left ventricle; miR-21 = microRNA-21; ms = millisecond.
Regulation of miR-21 and miR-21 target mRNAs following ISO treatment
Figure 3 shows the expression of myocardial miR-21 mRNA and miR-21 target mRNAs in C57BL/6 mice following ISO treatment. The expression of miR-21 gene was significantly increased to 294±184% and 136±21% (P < 0.05 versus the no ISO group, n=6-8 mice/group) 30 min and 3 h after ISO treatment, respectively. Among 84 miR-21 targets investigated, RHOB was significantly decreased 30 min after ISO treatment (P<0.05, n=5-7/group), and other 15 were not significantly decreased following ISO treatment (PTEN: P=0.08 between the ISO and no ISO groups, n=5-7/group) (Figure 3B).
Figure 3.
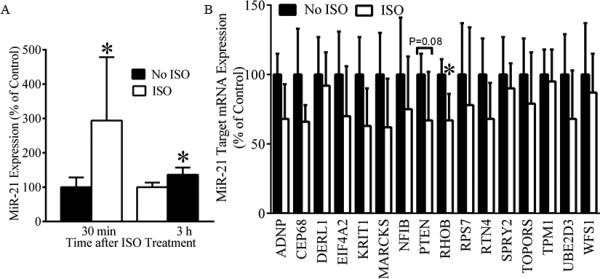
Regulation of cardiac miR-21 and miR-21 target mRNAs by isoflurane (ISO) in C57BL/6 mice. A: up-regulation of miR-21 mRNA by ISO. Pentobarbital-anesthetized mice received 1.0 minimum alveolar concentration of ISO for 30 min or oxygen/air mixture as control (no ISO). The expression of myocardial miR-21 gene was measured by real time qRT-PCR 30 min and 3 h after the treatment of ISO or oxygen/air mixture. *P< 0.05 versus no ISO (n = 6-8 mice/group). B: regulation of miR-21 target mRNAs by ISO. MiR-21 target mRNAs were investigated using mouse miFinder RT2 PCR Array 30 min after ISO or oxygen/air mixture as control (no ISO). ADNP=activity-dependent neuroprotector homeobox; CEP68=centrosomal protein 68kDa; DERL1=derlin 1; EIF4A2=eukaryotic translation initiation factor 4A2; KRIT1=krev interaction trapped protein 1; MARCKS=myristoylated alanine-rich C-kinase substrate; NFIB=nuclear factor I/B; PTEN=phosphatase and tensin homology deleted from chromosome 10; RHOB=Ras homolog family member B, RPS7=ribosomal protein S7; RTN4=reticulon-4; SPRY2=sprouty 2; TOPORS=topoisomerase I binding, arginine/serine-rich, E3 ubiquitin protein ligase; TPM1=tropomyosin 1; UBE2D3=ubiquitin-conjugating enzyme E2 D3; and WFS1=Wolfram syndrome 1. *P < 0.05 versus no ISO (n = 5-7 mice/group).
Disruption of MiR-21 gene abolished ISO-induced decreases in infarct size
Heart rate at baseline and during coronary artery occlusion and reperfusion was not different among the 4 experimental groups (Table 2). Area at risk and myocardial infarct size are shown in Figure 4. There were no significant differences (P>0.05) in area at risk between C57BL/6 and miR-21 KO mice subjected to I/R injury with or without ISO treatment. Coronary occlusion followed by reperfusion resulted in an infarct size of 54±10% and 62±11% of area at risk (n=8 mice/group) in C57BL/6 mice and miR-21 KO mice, respectively. There were no significant differences (P>0.05) in infarct size between the miR-21 KO-I/R and WT-I/R groups. Pretreatment of C57BL/6 mice subjected to with ISO significantly decreased infarct size (36±10%, P<0.05 versus CON, n=8 mice/group), however, disruption of miR-21 gene abolished ISO-induced deceases in infarct size (55±9%, P<0.05 versus the ISO group, n=8 mice/group).
Table 2.
Heart rate (bpm) during in vivo mouse experiments
| Group | Baseline | Coronary occlusion | Reperfusion |
|
|---|---|---|---|---|
| 60 min | 120 min | |||
| WT-I/R | 408±42 | 411±36 | 418±38 | 404±31 |
| miR-21 KO-I/R | 368±28 | 406±28 | 396±39 | 405±33 |
| WT-I/R+ISO | 402±34 | 419±42 | 402±32 | 399±36 |
| miR-21 KO-I/R+ISO | 360±42 | 375±44 | 394±31 | 413±24 |
There were no significant differences among groups (n=8 mice/group).
Bpm = beats per minute; I/R = ischemia/reperfusion; ISO = isoflurane; KO = knockout mice; miR-21 = microRNA-21; WT = wild-type mice.
Figure 4.
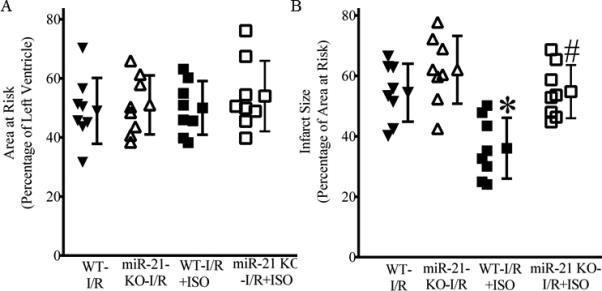
Isoflurane (ISO)-induced decreases in infarct size were blocked by disruption of miR-21 gene. A: area at risk expressed as a percentage of left ventricle area; B: myocardial infarct size expressed as a percentage of area at risk. Pentobarbital-anesthetized C57BL/6 wild-type (WT) and miR-21 knockout (KO) mice received ISO (the WT-I/R+ISO and miR-21 KOI/R+ISO groups) or air/oxygen mixture as control (the WT-I/R and miR-21 KO-I/R groups) prior to 30 min of coronary artery occlusion followed by 2 h of reperfusion (I/R). The infarct area was delineated by perfusing the coronary arteries with 2,3,5-triphenyltetrazolium chloride via the aortic root, and the area at risk was delineated by perfusing phthalo blue dye into the aortic root after tying the coronary artery at the site of previous occlusion. *P < 0.05 versus WT-I/R and miR-21 KO-I/R; #P < 0.05 versus WT-I/R+ISO (n = 8 mice/group).
MiR-21 KO abolished ISO-induced improvements in cardiac function during reperfusion
Figure 5 demonstrates time-dependent changes in cardiac function in Langendorff-perfused hearts. Baseline values of ±dP/dt were comparable among 6 groups (P>0.05). There were no significant differences in ±dP/dt values between the miR-21 KO Time-C group and the WT Time-C groups (P>0.05, n=7-8 hearts/group) throughout the experiment. Global no-flow ischemia resulted in failure of contraction and relaxation of all hearts studied. During reperfusion, the values of ±dP/dt in all 4 groups of the hearts subjected to ischemia gradually increased. At 30, 60, 90, and 120 min after reperfusion, ±dP/dt were significantly greater in the WT-I/R+ISO group (P<0.05, n=9 hearts) and depressed in the miR-21 KO-I/R group (P<0.05, n=8 hearts) compared with the WT-I/R group (n=9 hearts). Interestingly, the values of ±dP/dt were significantly greater in the WT-I/R+ISO group than the miR-21 KO-I/R+ISO group from 30 min to 2 h after reperfusion (P<0.05, n=7-9 hearts/group). There were no significant differences in the values of ±dP/dt at any time points between the miR-21 KO-I/R group and the miR-21 KO-I/R+ISO group (P>0.05).
Figure 5.
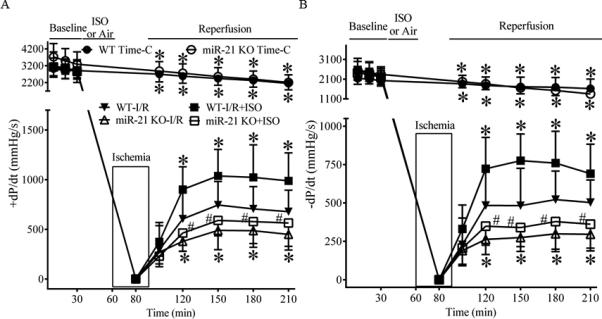
Isoflurane (ISO)-induced improvements in cardiac function were blocked by disruption of miR-21 gene in Langendorff-perfused hearts subjected to 30 min of global ischemia followed by 2 h of reperfusion. A: +dP/dt (maximum rate of increase of left ventricular developed pressure); B: −dP/dt (maximum rate of decrease of left ventricular developed pressure). In the WT (C57BL/6 heart) Time-C (time control) and miR-21 KO Time-C groups, the hearts were perfused in Langendorff apparatus for 210 min, whereas in the other groups, all hearts were stabilized for 30 min (baseline) and perfused with the buffer with or without ISO prior to 30 min of global ischemia followed by 2 h of reperfusion. ISO was administered by 2 cycles of 5 min ISO/5 min washout followed by a period of 10 min washout. I/R = ischemia/reperfusion. *P < 0.05 versus WT-I/R; #P < 0.05 versus WT-I/R+ISO (n = 7-9 hearts/group).
MiR-21 KO incresed mitochondrial NADH levels during ischemia in the presence or absence of ISO
Baseline mitochondrial NADH levels from Langendorff-perfused hearts was comparable among the 4 experimental groups (Figure 6). During ischemia, the NADH signal initially increased and peaked 5 min after ischemia followed by a gradual decline. Peak NADH fluorescece was significantly lower in the WT-I/R+ISO group than in the WT-I/R group and greater in either the miR-21 KO-I/R or miR-21 KO-I/R+ISO group than in the WT-I/R group (P<0.05, n=7-9 hearts/group). At all time points, no significant differences existed between the miR-21 KO-I/R group and the miR-21 KO-I/R+ISO group (P>0.05, n=7-9 hearts/group). During reperfusion, the NADH signal remained relatively stable in the 4 experimental groups. There were no significant differences in NADH fluorescence between groups during reperfusion.
Figure 6.
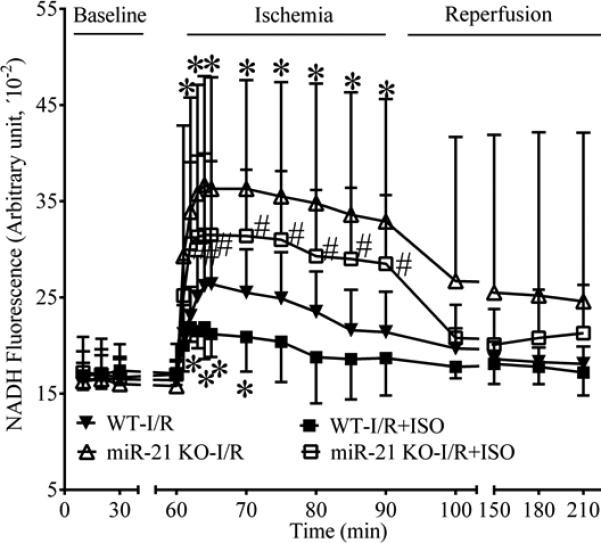
Effects of isoflurane (ISO) and miR-21 knockout (KO) on NADH fluorescence in Langendorff-perfused hearts subjected to ischemia/reperfusion (I/R) injury. All hearts were stabilized for 30 min (baseline) and perfused with the buffer with or without ISO prior to 30 min of global ischemia followed by 2 h of reperfusion, as described in Figure 5. *P < 0.05 versus WT-I/R; #P < 0.05 versus WT-I/R+ISO (n = 7-9 hearts/group).
ISO increased phosphorylated Akt, eNOS, and nNOS in C57BL/6 hearts, but not in miR-21 KO hearts
The effects of ISO on Akt and p-AKT are shown in Figure 7. There were no significant differences in the ratio of p-Akt/Akt between the miR-21 KO-I/R group and WT-I/R group (P>0.05, n=4 hearts/group). Interestingly, the ratio of p-Akt/Akt was significantly increased in the WT-I/R+ISO group (P<0.05, n=4 hearts), but not in the miR-21 KO-I/R+ISO group (P>0,05, n=4 hearts) compared with the WT-I/R group. Figure 8 demonstrates the effects of ISO on the expression of eNOS and nNOS proteins. There were no significnat differences in the ratio of either p-eNOS/eNOS or p-nNOS/nNOS between the miR-21 KO-I/R and WT-I/R groups. ISO significantly elevated the ratio of either p-eNOS/eNOS or p-nNOS/nNOS in C57BL/6 (P<0.05 versus WT-I/R, n=4 hearts/group) but not miR-21 KO hearts (P<0.05 between the miR-21 KOI/R+ISO and WT-I/R+ISO groups, n=4 hearts/group).
Figure 7.
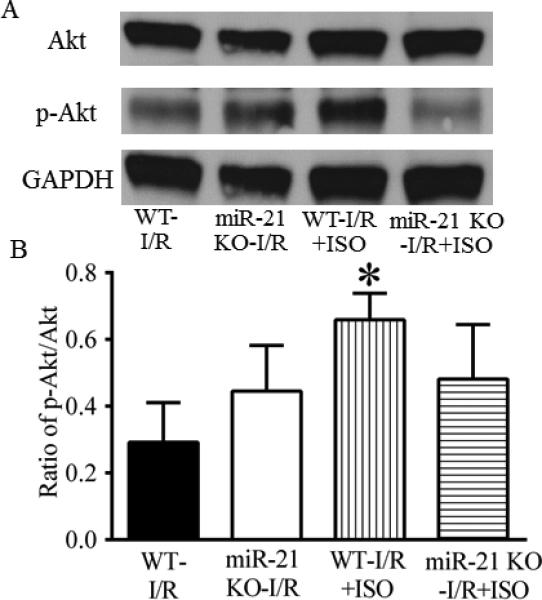
Isoflurane (ISO) increased the phosphorylation of Akt in ischemic/reperfused myocardium of C57BL/6 wild-type (WT) mice, but not in that of miR-21 KO mice. A: representative Western blot bands of Akt, phosphorylated Akt (p-Akt), and GAPDH as a housekeeping protein; B: ratio of p-Akt/total Akt. C57BL/6 and miR-21 KO mice were treated, as described in Figure 4. *P < 0.05 versus WT-I/R (n = 4 mice/group).
Figure 8.
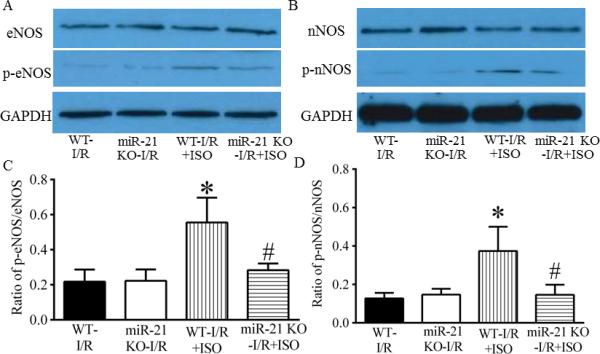
Isoflurane (ISO)-induced increases in the phosphorylation of eNOS and nNOS were abolished by disruption of miR-21 gene. A: representative Western blot bands of eNOS, phosphorylated eNOS (p-eNOS), and GAPDH as a house-keeping protein in ischemic/reperfused myocardium; B: representative Western blot bands of nNOS, phosphorylated nNOS (p-nNOS), and GAPDH as a house-keeping protein in ischemic/reperfused myocardium; C: ratio of p-eNOS/total eNOS; D: ratio of p-nNOS/total nNOS. C57BL/6 wild-type (WT) and miR-21 KO mice were treated, as described in Figure 4. *P < 0.05 versus WT-I/R; #P < 0.05 versus WTI/R+ISO (n = 4 mice/group).
MiR-21 KO blocked the ISO-induced delay in the mPTP opening
There were no significant differences in the opening time of the mPTP between the miR-21 KOOS and WT-OS groups (P>0.05, n=12 cells/group) (Figure 9). The treatment of the cardiomyocytes isolated from C57BL/6 mice with ISO delayed opening of the mPTP from 87.5±7.8 s in the WT-I/R group to 103.3±3.8 s in the WT-OS+ISO group (P<0.05 between the WT-OS+ISO and WT-OS groups, n=12 cells/group). This beneficial effect of ISO on the mPTP was eliminated by disruption of miR-21 gene (P<0.05 between the miR-21 KO-OS+ISO and WT-OS+ISO groups and P>0.05 between the miR-21 KO-OS+ISO and WT-OS groups).
Figure 9.
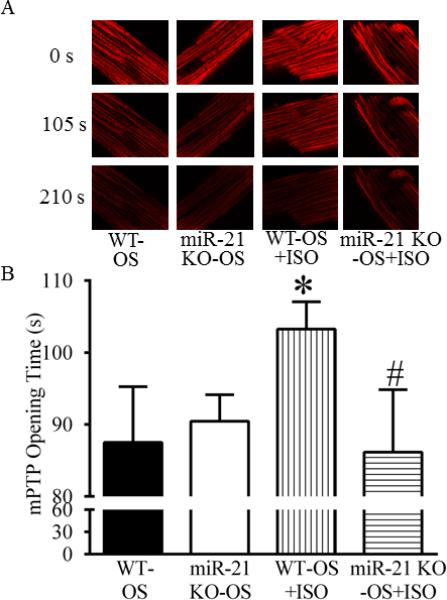
Isoflurane (ISO)-delayed opening of the mitochondrial permeability transition pore (mPTP) was blocked by disruption of miR-21 gene. A: representative confocal images of mitochondria; B: the mPTP opening time. Cardiomyocytes isolated from C57BL/6 wild-type (WT) or miR-21 KO mice were treated with Tyrode solution containing 0.5 mM ISO (WTOS+ISO and miR-21 KO-OS+ISO groups) or Tyrode solution without ISO as control (WT-OS and miR-21 KO-OS groups). Tetramethylrhodamine ethyl ester (TMRE) fluorescence image was obtained with a confocal microscope. The mPTP opening was induced by photoexcitation-generated oxidative stress (OS) and evaluated by measuring mitochondrial membrane potential. *P < 0.05 versus WT-OS; #P < 0.05 versus WT-OS+ISO (n = 12 cells/group).
Regulation of cardiomyocyte injury by MiR-21
The effects of pre-miR-21, anti-miR-21, and H2O2 on cardiomyocyte injury are shown in Figure 10. GFP and pre-miR-21 did not change PI-positive cardiomyocytes compared with the control group (P>0.05). Anti-miR-21 and oxidative stress induced by H2O2 increased PI-positive cardiomyocytes (P<0.05, n=3/group). The detrimental effect of H2O2 on cardiomyocytes was abolished by pre-miR-21 (P<0.05 between the pre-miR-21+H2O2 and H2O2 groups, n=3/group) and not significantly altered by anti-miR-21 (P>0.05).
Figure 10.
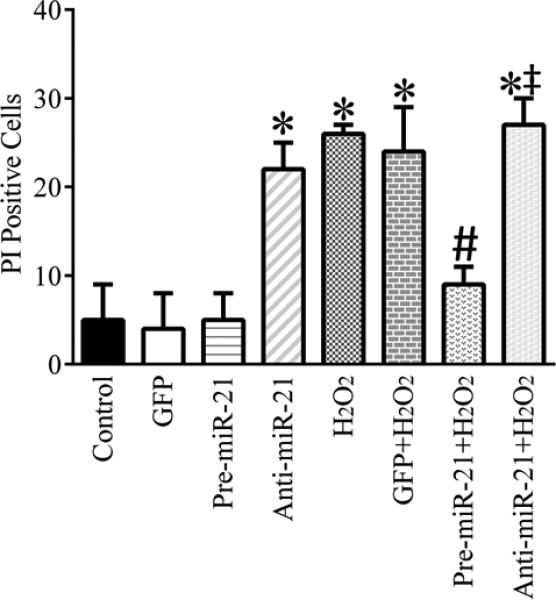
Effects of pre-miR-21, anti-miR-21, and H2O2 on cell injury in cardiomyocytes. Neonatal cardiomyocytes were transduced with green fluorescence protein (GFP)-labed adenovirus (GFP group), pre-miR-21 adenovirus (Pre-miR-21), GFP-labeled anti-miR-21 (Anti-miR-21) in the presence or absence of H2O2 and stained with Hoechst 33342 and propidium iodide (PI). Hoechst-positive nuclei were counted as a control using fluorescence microscopy and PI positive cells were taken as a percentage of the control. *P < 0.05 versus control; #P < 0.05 versus GFP+H2O2; ‡P < 0.05 versus pre-miR-21+ H2O2 (n = 3/group).
Discussion
The results of the present study demonstrate the involvement of miR-21 in ISO-induced protection of mouse hearts against acute I/R injury. In the WT mice, ISO up-regulates shortly the expression of the miR-21 gene, down-regulates the miR-21 target RHOA, decreases myocardial infarct size and the NADH levels during ischemia, improves the recovery of cardiac function after I/R injury, increases the phosphorylation of Akt, eNOS, and nNOS in ischemic/reperfused myocardium, and delays opening of the mPTP. These beneficial effects of ISO are lost in miR-21 KO mice. These results provide mechanistic evidence that the Akt/NOS/mPTP cascade is involved in the miR-21-mediated protective effect of ISO.
Our results indicate that that disruption of miR-21 gene did not alter heart rate, heart weight, mean arterial blood pressure, wall thickness of the LV, and systolic and diastolic function of the LV in mice. A previous study demonstrated that cardiomyocyte-specific overexpression of miR-21 by targeted gene transfer did not impact dimensions and function of the LV.8 Taken together, these data suggest that alterations in cardiomyocyte miR-21 do not significantly impact overall cardiovascular function in mice. However, in heart disease, such as cardiac hypertrophy and heart failure, miR-21 appears to have a disposition to be aberrantly modified in cardiac fibroblasts.10,16 Increased overexpression of miR-21 in cardiac fibroblasts contributes to the development of hypertrophy and fibrosis.10,16
In the present study, the expression of cardiac miR-21 was up-regulated following ISO treatment in C57BL/6 mice. The molecular mechanisms involved in miR-21 regulation by ISO remain elusive. Accumulating evidence indicates that pretreatment of the heart and brain with ISO increases the expression of NF-κB, a key transcription factor that regulates gene programing through positive and negative feedback mechanisms.4,27,30 Recently, a study demonstrated that NF-κB mediated the regulation of miR-21 in cardiomyocyes.14 It is reasonable to speculate that increased expression of NF-κB by ISO contributes to up-regulation of myocardial miR-21.
MiR-21 targets multiple components of many important signal pathways, including programmed cell death 4 (PDCD4), PTEN, SPRY 1 and 2, and mitochondrial apoptosis tumor-suppressive pathways.12,14,31 Initially, we used qRT-PCR to analyze the expression of PDCD4, SPRY 1 and 2, and PTEN mRNAs in C57BL/6 mouse hearts following ISO treatment. There were no significant differences in the levels of PDCD4, SPRY 1 and 2, and PTEN mRNAs between the ISO-treated and control groups (P=0.09 in PTEN between the ISO and no ISO group, n=7) (data not shown). Furthermore, we used mouse miFinder RT2 PCR Array to analyze 84 experimentally validated and predicted targets of miR-21. RHOB was inhibited following ISO treatment. Recently, we have indicated that PDCD4 is a down-stream target of miR-21 in ISO-induced protection of cardiomyocytes.20 Thus, ISO-induced up-regulation of miR-21 may alter the expression of multiple gene targets.
The pathophysiological role of miR-21 in myocardial I/R injury is cell-specific, depending on the progression of myocardial infarction.9,17 In the present study, inhibition of miR-21 with anti-miR-21 increases cell injury, and the miR-21 mimic, pre-miR-21, diminishes H2O2-induced cell injury in cultured cardiomyocytes. These data are consistent with a previous study showing that increased expression of miR-21 in cardiac myocytes reduces myocardial I/R injury in the early phase of myocardial infarction.17 However, in the late phase of myocardial infarction, miR-21 is overexpressed predominantly in cardiac fibroblasts, that is associated with fibrosis and cardiac remodeling.32 To examine the dependence of ISO-induced cardioprotection on miR-21, we used miR-21 KO mice. Myocardial infarct size was comparable between miR-21 KO mice and WT mice subjected to a 30 min coronary occlusion followed by 2 h reperfusion. Nonetheless, myocardial infarct size was significantly decreased by ISO in WT mice, but not in miR-21 KO mice. Furthermore, cardiac function during reperfusion was significantly improved by ISO in isolated C57BL/6 hearts, but not in miR-21 KO hearts. These in vivo and ex vivo data indicate that miR-21 is involved in cardioprotection against acute I/R injury by ISO.
NADH is a substrate for respiratory complex I in the electron transport chain in mitochondria. It can emit fluorescence when mitochondria are illuminated at the appropriate wavelength. Under physiological conditions, NADH levels remain stable by the balance between its generation (mitochondrial dehydrogenase) and its consumption (electron transfer). During myocardial ischemia/hypoxia, NADH fluorescence is significantly increased mainly due to dysfunction of electron transfer in mitochondria.26 During the present investigation, ischemia-induced increases in NADH signal were inhibited by ISO in WT hearts, and this finding was consistent with previous observations in guinea-pig hearts.26 Interestingly, disruption of miR-21 gene resulted in a significant increase in NADH fluorescence during ischemia compared with WT hearts. Previous studies demonstrated that changes in mitochondrial NADH fluorescence were consistent with alterations in mitochondrial dysfunction.26,28 Our results suggest that disruption of the miR-21 gene may exacerbate impairment of mitochondrial function during ischemia. While disruption of miR-21 did not impact both baseline NADH levels and cardiac function, loss of miR-21 causes disturbance of mitochondrial respiration, thereby diminishing energy production and possibly increasing superoxide generation in ischemic/reperfused myocardium.33 Alterations of mitochondrial homeostasis regulate cardiac tolerance to I/R injury and have been identified as an important component of cell death secondary to I/R.33,34 Future studies will examine the impact of miR-21 KO on the expression and activity of mitochondrial complexes I, II, III, and IV, the production of high energy phosphates (ATP, ADP, and AMP) and superoxide in ischemic/reperfused myocardium.
The phosphorylation of Akt plays a key role in ISO-induced cardioprotection against I/R injury.2,35 In the present study, ISO enhanced phosphorylated Akt in WT mouse hearts, but not in miR-21 KO hearts. These results suggest that Akt is a downstream effector of miR-21 in ISO-induced cardioprotection. Akt protects the heart against I/R injury through activation of other proteins such as GSK3β, Pim-1, and nitric oxide synthase (NOS).13,36,37 In the present study, ISO increased phosphorylation of both eNOS and nNOS, and this action was blocked by disruption of miR-21 gene. In contrast, phosphorylated GSK3β and Pim-1 were not altered by disruption of miR-21 gene (data not shown). Thus, both eNOS and nNOS appear to be the down-stream targets of Akt in the ISO-induced cardioprotection against I/R injury.
The mPTP plays a crucial role in myocardial I/R injury and protection.38,39 In the present study, we measured time of the mPTP opening in cardiomyocytes isolated from adult mice. Opening of the mPTP induced by photoexcitation-generated oxidative stress was delayed by ISO in cardiomyocytes isolated from WT mice. This confirms our previous findings in cardiomyocytes isolated from rat hearts.28,39 Interestingly, the protective effects of ISO on mitochondria were abolished in the miR-21 KO mice, suggesting that mitochondria are a downstream effector of miR-21. The signaling pathway linking miR-21 to the mPTP remains elusive. Previously, we indicate that eNOS regulates function of mPTP at early reperfusion in mice.39 Given that disruption of miR-21 gene blocked ISO-induced phosphorylation of both eNOS and nNOS, nitric oxide generated by eNOS and nNOS may be involved in modulation of the mPTP opening.40
One limitation of this study is that there are no array-based analyses of mcroRNAs in mouse hearts following the treatment of ISO. In rat hearts or neonatal rat cardiomyocytes, our array analyses of microRNAs indicate that ISO up-regulated 11 out of 87 microRNAs studied, including miR-21, microRNA-210, and microRNA-30 family.20 It is likely that multiple microRNAs are involved in the cardioprotective effect of ISO. However, miR-21 plays an important role in protection of cardiomyocytes against hypoxia/reoxygenation injury.20 Another limitation is that this study does not involve the long-term outcome of miR-21 up-regulation by ISO, since overexpression of miR-21 in fibroblasts is associated with hypertrophy and fibrosis in diseased state.10,16 However, ISO-induced increase in miR-21 mRNA lasted about 6 h after ISO exposure. It is reasonably believed that this temporary up-regulation of miR-21 is not sufficient to cause significant hypertrophy and fibrosis.
In summary, the present study demonstrates the pivotal role of miR-21 in ISO-induced cardioprotection against acute I/R injury. The findings reveal new mechanisms involved in miR-21-induced myocardial protection. As a main “cardiac” microRNA, miR-21 is up-modulated in cardiomyocytes in the early phase of myocardial infarction, which contributes to myocardial protection. However, in the late phase of myocardial infarction, miR-21 is overexpressed predominantly in fibroblasts, that causes fibrosis and cardiac remodeling. Since ISO up-regulates the expression of miR-21 in myocardium, future studies will examine the effect of ISO on cardiac miR-21 and the contribution of miR-21 modulation to cardiac fibrosis and remodeling after myocardial infarction.
Acknowledgements
We thank Drs. John A. Auchampach (Ph.D., Professor of Pharmacology and Toxicology), Tina C. Wan (Ph.D., Research Scientist), and Garrett J. Gross (Ph.D., Emeritus Professor) (all from Department of Pharmacology and Toxicology, Medical College of Wisconsin) and Dr. Xiaowen Bai (Ph.D., Assistant Professor, Department of Anesthesiology) for their equipment, and David Schwabe (B.S., Engineer), Shelley L. Baumgardt (B.S., Research Associate), and John Tessmer (B.S., Lab Manager) (all from Department of Anesthesiology, Medical College of Wisconsin) for their excellent technical assistance.
Disclosure of Funding
This work was supported, in part, by National Institutes of Health research grants P01GM 066730 (to Drs. Bosnjak, Warltier, and Liang), HL098490-03 (to Dr. Riess), and HL123227-01 (to Dr. Riess) and the Department of Veterans Affairs grant IK2 BX001278 (to Dr. Riess).
Footnotes
Disclosure of Conflict
This work was presented in part at the American Society of Anesthesiologists meeting, San Francisco, CA, October 15, 2013.
Competing Interests
The authors declare no competing interests.
References
- 1.Kersten JR, Schmeling TJ, Pagel PS, Gross GJ, Warltier DC. Isoflurane mimics ischemic preconditioning via activation of K(ATP) channels: reduction of myocardial infarct size with an acute memory phase. Anesthesiology. 1997;87:361–70. doi: 10.1097/00000542-199708000-00024. [DOI] [PubMed] [Google Scholar]
- 2.Raphael J, Rivo J, Gozal Y. Isoflurane-induced myocardial preconditioning is dependent on phosphatidylinositol-3-kinase/Akt signalling. Br J Anaesth. 2005;95:756–63. doi: 10.1093/bja/aei264. [DOI] [PubMed] [Google Scholar]
- 3.Raphael J, Abedat S, Rivo J, Meir K, Beeri R, Pugatsch T, Zuo Z, Gozal Y. Volatile anesthetic preconditioning attenuates myocardial apoptosis in rabbits after regional ischemia and reperfusion via Akt signaling and modulation of Bcl-2 family proteins. J Pharmacol Exp Ther. 2006;318:186–94. doi: 10.1124/jpet.105.100537. [DOI] [PubMed] [Google Scholar]
- 4.Lang XE, Wang X, Zhang KR, Lv JY, Jin JH, Li QS. Isoflurane preconditioning confers cardioprotection by activation of ALDH2. PLoS One. 2013;8:e52469. doi: 10.1371/journal.pone.0052469. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.De Hert SG, ten Broecke PW, Mertens E, Van Sommeren EW, De Blier IG, Stockman BA, Rodrigus IE. Sevoflurane but not propofol preserves myocardial function in coronary surgery patients. Anesthesiology. 2002;97:42–9. doi: 10.1097/00000542-200207000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Fräßdorf J, Borowski A, Ebel D, Feindt P, Hermes M, Meemann T, Weber R, Mullenheim J, Weber NC, Preckel B, Schlack W. Impact of preconditioning protocol on anesthetic-induced cardioprotection in patients having coronary artery bypass surgery. J Thorac Cardiovasc Surg. 2009;137:1436–42. doi: 10.1016/j.jtcvs.2008.04.034. [DOI] [PubMed] [Google Scholar]
- 7.Pagel PS. Myocardial protection by volatile anesthetics in patients undergoing cardiac surgery: a critical review of the laboratory and clinical evidence. J Cardiothorac Vasc Anesth. 2013;27:972–82. doi: 10.1053/j.jvca.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 8.Sayed D, He M, Hong C, Gao S, Rane S, Yang Z, Abdellatif M. MicroRNA-21 is a downstream effector of AKT that mediates its antiapoptotic effects via suppression of Fas ligand. J Biol Chem. 2010;285:20281–90. doi: 10.1074/jbc.M110.109207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Wang DZ. microRNAs in cardiovascular development. J Mol Cell Cardiol. 2012;52:949–57. doi: 10.1016/j.yjmcc.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol. 2007;170:1831–40. doi: 10.2353/ajpath.2007.061170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suarez Y, Fernandez-Hernando C, Pober JS, Sessa WC. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ Res. 2007;100:1164–73. doi: 10.1161/01.RES.0000265065.26744.17. [DOI] [PubMed] [Google Scholar]
- 12.Cheng Y, Zhang C. MicroRNA-21 in cardiovascular disease. J Cardiovasc Transl Res. 2010;3:251–5. doi: 10.1007/s12265-010-9169-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Zhao L, He X, Yang T, Yang K. MiR-21 inhibits c-Ski signaling to promote the proliferation of rat vascular smooth muscle cells. Cell Signal. 2014;26:724–9. doi: 10.1016/j.cellsig.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 14.Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A, Just A, Remke J, Zimmer K, Zeug A, Ponimaskin E, Schmiedl A, Yin X, Mayr M, Halder R, Fischer A, Engelhardt S, Wei Y, Schober A, Fiedler J, Thum T. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124:2136–46. doi: 10.1172/JCI70577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong S, Ma W, Hao B, Hu F, Yan L, Yan X, Wang Y, Chen Z, Wang Z. microRNA-21 promotes cardiac fibrosis and development of heart failure with preserved left ventricular ejection fraction by up-regulating Bcl-2. Int J Clin Exp Pathol. 2014;7:565–74. [PMC free article] [PubMed] [Google Scholar]
- 16.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–4. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 17.Dong S, Cheng Y, Yang J, Li J, Liu X, Wang X, Wang D, Krall TJ, Delphin ES, Zhang C. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J Biol Chem. 2009;284:29514–25. doi: 10.1074/jbc.M109.027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duygu B, Da Costa Martins PA. miR-21: a star player in cardiac hypertrophy. Cardiovasc Res. 2015;105:235–7. doi: 10.1093/cvr/cvv026. [DOI] [PubMed] [Google Scholar]
- 19.Toldo S, Das A, Mezzaroma E, Chau VQ, Marchetti C, Durrant D, Samidurai A, Van Tassell BW, Yin C, Ockaili RA, Vigneshwar N, Mukhopadhyay ND, Kukreja RC, Abbate A, Salloum FN. Induction of microRNA-21 with exogenous hydrogen sulfide attenuates myocardial ischemic and inflammatory injury in mice. Circ Cardiovasc Genet. 2014;7:311–20. doi: 10.1161/CIRCGENETICS.113.000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olson JM, Yan Y, Bai X, Ge ZD, Liang M, Kriegel AJ, Twaroski DM, Bosnjak ZJ. Up-regulation of microRNA-21 mediates isoflurane-induced protection of cardiomyocytes. Anesthesiology. 2015;122:795–805. doi: 10.1097/ALN.0000000000000567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ge ZD, Peart JN, Kreckler LM, Wan TC, Jacobson MA, Gross GJ, Auchampach JA. Cl-IBMECA [2-chloro-N6-(3-iodobenzyl)adenosine-5'-N-methylcarboxamide] reduces ischemia/reperfusion injury in mice by activating the A3 adenosine receptor. J Pharmacol Exp Ther. 2006;319:1200–10. doi: 10.1124/jpet.106.111351. [DOI] [PubMed] [Google Scholar]
- 22.Saegusa N, Sato T, Saito T, Tamagawa M, Komuro I, Nakaya H. Kir6.2-deficient mice are susceptible to stimulated ANP secretion: K(ATP) channel acts as a negative feedback mechanism? Cardiovasc Res. 2005;67:60–8. doi: 10.1016/j.cardiores.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 23.Small EM, Frost RJ, Olson EN. MicroRNAs add a new dimension to cardiovascular disease. Circulation. 2010;121:1022–32. doi: 10.1161/CIRCULATIONAHA.109.889048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ge ZD, Ionova IA, Vladic N, Pravdic D, Hirata N, Vasquez-Vivar J, Pratt PF, Jr., Warltier DC, Pieper GM, Kersten JR. Cardiac-specific overexpression of GTP cyclohydrolase 1 restores ischaemic preconditioning during hyperglycaemia. Cardiovasc Res. 2011;91:340–9. doi: 10.1093/cvr/cvr079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramanujam N. Fluorescence spectroscopy of neoplastic and non-neoplastic tissues. Neoplasia. 2000;2:89–117. doi: 10.1038/sj.neo.7900077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riess ML, Camara AK, Kevin LG, An J, Stowe DF. Reduced reactive O2 species formation and preserved mitochondrial NADH and [Ca2+] levels during short-term 17 °C ischemia in intact hearts. Cardiovasc Res. 2004;61:580–90. doi: 10.1016/j.cardiores.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 27.Chen CH, Chuang JH, Liu K, Chan JY. Nitric oxide triggers delayed anesthetic preconditioning-induced cardiac protection via activation of nuclear factor-kB and upregulation of inducible nitric oxide synthase. Shock. 2008;30:241–9. doi: 10.1097/SHK.0b013e318162ad19. [DOI] [PubMed] [Google Scholar]
- 28.Sedlic F, Pravdic D, Hirata N, Mio Y, Sepac A, Camara AK, Wakatsuki T, Bosnjak ZJ, Bienengraeber M. Monitoring mitochondrial electron fluxes using NAD(P)H-flavoprotein fluorometry reveals complex action of isoflurane on cardiomyocytes. Biochim Biophys Acta. 2010;1797:1749–58. doi: 10.1016/j.bbabio.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Canfield SG, Sepac A, Sedlic F, Muravyeva MY, Bai X, Bosnjak ZJ. Marked hyperglycemia attenuates anesthetic preconditioning in human-induced pluripotent stem cell-derived cardiomyocytes. Anesthesiology. 2012;117:735–44. doi: 10.1097/ALN.0b013e3182655e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ge ZD, van der Hoeven D, Maas JE, Wan TC, Auchampach JA. A3 adenosine receptor activation during reperfusion reduces infarct size through actions on bone marrow-derived cells. J Mol Cell Cardiol. 2010;49:280–6. doi: 10.1016/j.yjmcc.2010.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papagiannakopoulos T, Shapiro A, Kosik KS. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res. 2008;68:8164–72. doi: 10.1158/0008-5472.CAN-08-1305. [DOI] [PubMed] [Google Scholar]
- 32.Roy S, Khanna S, Hussain SR, Biswas S, Azad A, Rink C, Gnyawali S, Shilo S, Nuovo GJ, Sen CK. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res. 2009;82:21–9. doi: 10.1093/cvr/cvp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moens AL, Champion HC, Claeys MJ, Tavazzi B, Kaminski PM, Wolin MS, Borgonjon DJ, Van Nassauw L, Haile A, Zviman M, Bedja D, Wuyts FL, Elsaesser RS, Cos P, Gabrielson KL, Lazzarino G, Paolocci N, Timmermans JP, Vrints CJ, Kass DA. High-dose folic acid pretreatment blunts cardiac dysfunction during ischemia coupled to maintenance of high-energy phosphates and reduces postreperfusion injury. Circulation. 2008;117:1810–9. doi: 10.1161/CIRCULATIONAHA.107.725481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sack MN, Murphy E, Schulz R. The regulation and control of mitochondrial homeostasis in changing cardiac tolerance to ischemia-reperfusion injury: a focused issue. Basic Res Cardiol. 2009;104:111–2. doi: 10.1007/s00395-009-0005-7. [DOI] [PubMed] [Google Scholar]
- 35.Jamnicki-Abegg M, Weihrauch D, Pagel PS, Kersten JR, Bosnjak ZJ, Warltier DC, Bienengraeber MW. Isoflurane inhibits cardiac myocyte apoptosis during oxidative and inflammatory stress by activating Akt and enhancing Bcl-2 expression. Anesthesiology. 2005;103:1006–14. doi: 10.1097/00000542-200511000-00015. [DOI] [PubMed] [Google Scholar]
- 36.Vigneron F, Dos Santos P, Lemoine S, Bonnet M, Tariosse L, Couffinhal T, Duplaà C, Jaspard-Vinassa B. GSK-3β at the crossroads in the signalling of heart preconditioning: implication of mTOR and Wnt pathways. Cardiovasc Res. 2011;90:49–56. doi: 10.1093/cvr/cvr002. [DOI] [PubMed] [Google Scholar]
- 37.Sussman MA, Volkers M, Fischer K, Bailey B, Cottage CT, Din S, Gude N, Avitabile D, Alvarez R, Sundararaman B, Quijada P, Mason M, Konstandin MH, Malhowski A, Cheng Z, Khan M, McGregor M. Myocardial AKT: the omnipresent nexus. Physiol Rev. 2011;91:1023–70. doi: 10.1152/physrev.00024.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res. 2004;61:372–85. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 39.Ge ZD, Pravdic D, Bienengraeber M, Pratt PF, Jr., Auchampach JA, Gross GJ, Kersten JR, Warltier DC. Isoflurane postconditioning protects against reperfusion injury by preventing mitochondrial permeability transition by an endothelial nitric oxide synthase-dependent mechanism. Anesthesiology. 2010;112:73–85. doi: 10.1097/ALN.0b013e3181c4a607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heusch G, Boengler K, Schulz R. Cardioprotection: nitric oxide, protein kinases, and mitochondria. Circulation. 2008;118:1915–9. doi: 10.1161/CIRCULATIONAHA.108.805242. [DOI] [PubMed] [Google Scholar]