

Abstract
Purpose of review
Hematopoietic stem cells (HSCs) can self-renew and also give rise to the entire repertoire of hematopoietic cells. During acute infectious and inflammatory stresses, the hematopoietic system can quickly adapt to demand by increasing output of innate immune cells many-fold, often at the expense of lymphopoiesis and erythropoiesis. We review recent advances in understanding the regulation of stress-induced hematopoiesis with a specific focus on the direct effects of inflammatory signaling on hematopoietic stem and progenitor cells (HSPCs).
Recent findings
Recent studies have highlighted several areas of exciting new developments in the field, including the complex interaction and crosstalk within HSPCs and between bone marrow mesenchymal stem cells and endothelial cells needed to achieve regulated myelopoiesis, identification of increased number of inflammatory and infectious molecules with direct effects on HSPCs, the critical role of inflammatory signaling on embryonic specification of HSCs, and the ability of cytokines to instruct lineage choice at the HSPC level.
Summary
These exciting new findings will shape our fundamental understanding of how inflammatory signaling regulates hematopoiesis in health and disease, and facilitate the development of potential interventions to treat hematologic diseases associated with altered inflammatory signaling.
Keywords: inflammation, infection, hematopoiesis, hematopoietic stem cells, cytokines, toll-like receptors
Introduction
During infection and inflammation that is not limited to a local site, a stress-induced hematopoietic response is often elicited that is characterized by increased output of cells of the myeloid lineage from the bone marrow hematopoietic stem and progenitor cells (HSPCs) and mobilization of bone marrow neutrophils and HSPCs to the peripheral tissues. Similarly, hematopoiesis is shifted towards erythrocyte production during stresses like hemorrhage and anemia. The remarkable adaptability of the hematopoietic system to meet the demand of various stressed situations is critical for survival. Myeloid cells are mostly post-mitotic and need to be produced from bone marrow HSPCs when they are consumed during a fight against infection. How is the increased production of myeloid cells achieved in response to an infection? In general, committed myeloid progenitor cells are induced to proliferate and mature as a rapid immediate response, a process termed “emergency granulopoiesis”. This process is covered in a recent comprehensive review [1*]. However, studies in the recent decade have convincingly shown that uncommitted HSPCs are actively involved in stress-induced hematopoiesis.
HSPCs may respond to an infection via five general mechanisms: (i) respond to the depletion of downstream neutrophils. Although an intuitively attractive hypothesis, how HSPCs can sense and “numerate” peripheral neutrophils remains unclear. There is some evidence that a “neutrostat” system exists to sense the phagocytosis of apoptotic neutrophils consumed during an infection and thus signal the bone marrow to replenish the neutrophil pool accordingly [2-3]; (ii) respond to inflammatory cytokines produced by various hematopoietic and non-hematopoietic cells during an infection. There is clear evidence that HSPCs, including long term (LT)-HSCs, have the ability to directly respond to cytokines, including thrombopoietin (TPO), IL-3, stem cell factors (SCF), Flt3 ligand, interferons, TNFα, TGFβ, IL-6, G-CSF and M-CSF; (iii) respond to pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) directly through toll-like receptors (TLRs). The evidence to support this mechanism is also strong; (iv) respond to paracrine signals from the stem cell niche. Stem cell niche cells, including but not limited to osteoblasts, perivascular stromal cells and mesenchymal stem cells, and endothelial cells are affected in various ways during an infection, which can influence proliferation, differentiation, and mobilization of HSPCs; (v) theoretically, pathogens can affect HSPC activity by infecting them. However, we are not aware of any convincing evidence to support this. In this review, we will focus our discussion of stress-induced hematopoiesis to mechanisms ii and iii—the direct effects of PAMPs, DAMPs and cytokines on HSPCs, including LT-HSCs and short term (ST)-hematopoietic stem cells and multipotent progenitor cells (MPPs). The important effects of infectious and inflammatory signaling on the bone marrow niche have been the subjects of several recent excellent reviews [4-5**] and will not be discussed here. We will also provide recent evidence on the pathologic consequences of chronic inflammation on the hematopoietic system, and how we may utilize our knowledge in this area to treat human hematologic malignancies.
Regulation of stress-induced hematopoiesis by TLR signaling
TLRs, belonging to a group of pattern recognition receptors, bind to conserved pathogen-associated molecular patterns (PAMPs), e.g. lipopolysaccharide (LPS) of the bacteria cell wall and single-stranded RNA (ssRNA) of viruses, and signal through either MyD88-dependent or TRIF-dependent pathways, leading to activation of the NF-κB and/or the IRF family of transcription factors [6]. Nagai et al. first demonstrated that HSPCs (defined as Flk2- LSK cells) expressed TLR2 and TLR4 receptors; furthermore, stimulation of HSPCs through either TLR2 or TLR4 drove myeloid differentiation in vitro in a MyD88-dependent manner [7]. Soon after, Massberg et al. showed that both bone marrow HPSCs and egressed HSPCs from peripheral lymph differentiated into myeloid cells after implantation under the kidney capsule when stimulated with LPS in vivo [8]. Since then, many studies have confirmed these initial findings [9, 10, 11*, 12-15]. Furthermore, when highly purified LT-HSCs (LSK CD150+CD48-) were stimulated directly with LPS in vitro, NF-κB activation was readily detected using a GFP-reporter mouse, demonstrating a functional TLR-4- NF-κB axis in LT-HSCs [11*]. The consequences of TLR stimulation on HSPCs are many. In vitro activation of TLRs augments myeloid differentiation [7, 11*]. In vivo stimulation in mice generally results in increased HSC proliferation, decreased quiescence, skewed myeloid differentiation and decreased long-term repopulating ability [8-10, 13-18]. TLR stimulation of human HSPCs also induces preferentially myeloid differentiation [19-20]. While TLR4 and TLR2 are among the most commonly studied TLRs in the setting of infection, other TLRs may mediate similarly robust myelopoiesis when challenged with a real pathogen. This is highlighted in one study that subjected various genetic knockout mice to Staphylococcus aureus infection or polymicrobial peritonitis and found that stress-induced hematopoiesis did not depend on any single TLRs, cytokines or interferons, suggesting that a tremendous redundancy has evolved in the mammalian hematopoietic system to sense and respond to bacterial infection [21].
Recent studies by us and others have shed additional insight into the complex crosstalk between hematopoietic and nonhematopoietic cells that corroboratively achieve efficient pathogen detection and subsequent upregulation of myelopoiesis (Figure 1A). Taking advantage of a microfluidic single cell proteomics platform, we found that a large subset of ST-HSCs and MPPs produced a surprisingly wide range of hematopoietic growth factors and cytokines in response to direct LPS and Pam3CSK4 stimulation [11*]. This was regulated by the TLR-NF-κB axis, as tuning up or down the strength of NF-κB activity could change the amounts of cytokines produced. Interestingly, the quantity and breadth of cytokines produced by HSPCs trumped those produced by mature myeloid and lymphoid cells by many-fold. More importantly, instead of a vestigial feature, HSPC-produced cytokines, especially IL-6, promoted myeloid differentiation in an autocrine or paracrine manner. This was demonstrated in vitro and during neutropenic in vivo conditions. The short distance autocrine and paracrine communication within HSPCs is likely also functionally important in the bone marrow stem cell niche or extramedullary sites where circulating HSPCs reside. These HSPC-initiated hematopoietic centers may be able to generate a wide range of hematopoietic responses tailored to particular pathogens or other stress signals. Future studies will be needed to address the functional significance of HSPC-produced cytokines in a physiological bone marrow niche in the absence of neutropenia or in an extramedullary site containing egressed HSPCs. In addition to HSPCs as a direct translator of pathogen signals into self-directing myelopoietic cytokine signals, bone marrow mesenchymal stem cells (MSCs) were shown to be involved in promoting myelopoiesis during a viral infection. A recent study nicely demonstrated that in response to antigen stimulation or acute viral infection, cytotoxic T cells released IFN-γ, acting on MSCs to produce IL-6. Similarly, IL-6 acted on bone marrow HSPCs to promote myeloid differentiation [22*]. Given the known effect of IFN-γ on HSCs [15], it will be interesting to separate the direct and indirect effects of IFN-γ on HSCs and the stem cell niche, respectively, during a viral infection.
Figure 1.
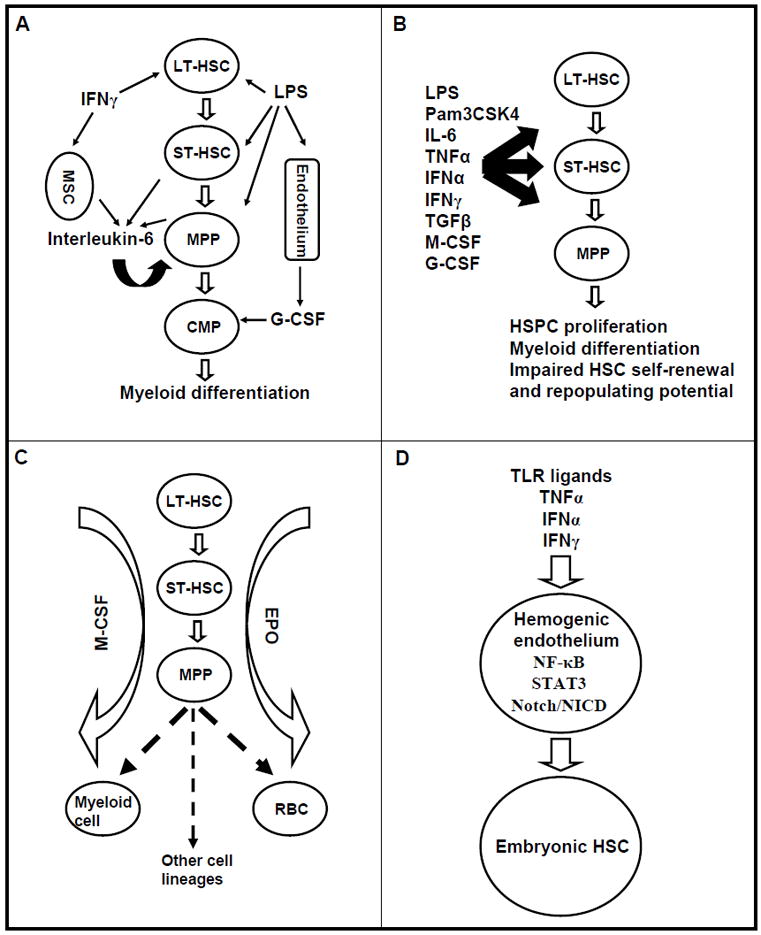
Emerging concepts in the regulation of hematopoiesis by inflammatory signaling. A. Complex paracrine and autocrine signaling within HSPCs and between bone marrow stromal cells. IL-6 is produced by ST-HSCs and MPPs upon LPS stimulation and by MSCs upon IFNγ stimulation; G-CSF is produced by endothelial cells upon LPS stimulation; IL-6, IFNγ and G-CSF stimulate myelopoiesis via paracrine and/or autocrine signaling. B. An expanding list of PAMPs, DAMPs and cytokines with direct effects on HSPCs. C. Cytokines such as M-CSF and EPO can instruct lineage choice of uncommitted HSPCs by promoting lineage-specific transcriptional factor while suppressing the alternative cell fates. D. Inflammatory molecules, including TLR agonists, TNFα, IFNα and IFNγ, promote embryonic HSC cell fate specification from hemogenic endothelium through NF-κB, Stat3 and Notch pathway.
Furthermore, endothelial cells have been shown to be an important sensor of systemic LPS or E. coli challenge, leading to G-CSF production in a MyD88-dependent but TRIF-independent manner [23*]. In these models, endothelium-produced G-CSF was shown to be the primary stimulator of emergency granulopoiesis. It is interesting to note that, in contrast to LPS injection, E. coli challenge stimulated emergency granulopoiesis, albeit at a reduced level, when MyD88 was deleted in endothelial cells. This suggests that cells other than endothelial cells and cytokines other than G-CSF can function to promote granulopoiesis in a more complex infectious model. In fact, G-CSF has been shown to be dispensable for emergency granulopoiesis in systemic fungal infection [24]. G-CSF signaling regulates granulopoiesis at multiple stages, from uncommitted HSPCs to neutrophils. However, the predominant effect of G-CSF is on lineage-committed granulocytic precursor cells, at least for steady state granulopoiesis [25-26]. It will be interesting to determine whether G-CSF acts primarily on committed myeloid progenitor cells, bone marrow niche cells, or directly on HSPCs to promote myeloid differentiation during stress conditions.
Mechanistically, the TLR-MyD88/TRIF-NF-κB axis is among the most commonly implicated pathway in regulating stress-induced hematopoiesis. Interestingly, a recent study showed that LPS induced granyzme B expression in LT-HSCs through the TLR4-TRIF-p65 NF-κB axis and granzyme B caused cell-autonomous apoptosis in HSCs [27*]. On the other hand, LPS induced cytokine production in ST-HSCs and MPPs through the TLR-p50 NF-κB pathway, leading to cytokine production and autocrine/paracrine signaling [11*]. Suffice it to say that stress-induced hematopoiesis is a highly complex and dynamic process that involves crosstalk between HSPCs, bone marrow stromal cells, and nonhematopoietic tissues to sense a pathogenic organism and convert the signal of an infection into the signal for myeloid differentiation.
Regulation of stress-induced hematopoiesis by cytokine signaling
In addition to TLR ligands, cytokines produced during stressed conditions can directly act on HSPCs to regulate hematopoiesis. SCF, TPO, Flt3 ligand, IL-3 and IL-6 are among some of the first identified cytokines to regulate HSPC proliferation, self-renewal and survival [28-33]. For example, we and other groups recently showed that IL-6 can be produced by bone marrow HSPCs during TLR stimulation and by MSCs during IFNγ stimulation and can act on neighboring HSPCs, in particularly MPPs, to stimulate myelopoiesis [11*, 22*, 34]. Recent studies have also expanded the list of cytokines and PAMPs with direct effects on HSPCs (Figure 1B) [12, 15, 35, 36]. Both IFNα and IFNγ were shown to directly stimulate quiescent HSCs to proliferate [37-40]. More recent evidence suggested that the effects of interferons on HSCs may be complex. While the finding that IFNγ exposure has a negative impact on HSC’s long-term repopulating potential is generally agreed upon, whether IFNγ stimulates or inhibits HSC proliferation has been debated [41, 42]. A recent study showed that IFNα-driven HSC proliferation was only transient and HSCs rapidly returned to a quiescent state with chronic exposure, a protective mechanism from chronic infection-driven HSC exhaustion [43*]. Effects of TNFα on HSCs are also dose- and context-dependent. In general, it appears that activation of TNF signaling in HSCs inhibits proliferation and results in decreased long-term repopulating potential [44-46]. However, studies using aged mice or different doses of TNFα showed that activation of TNFα signaling in HSCs was protective against apoptosis and thus enhanced hematopoietic engraftment in a transplant setting [47, 48]. TGFβ1 appeared to have differential regulatory effects on different subsets of HSCs, stimulating myeloid-biased HSCs to proliferate while inhibiting lymphoid-biased HSCs [49]. Mechanistically, TGFβ1 signaling in HSCs was regulated in part by transcription intermediary factor 1γ (Tif1γ) and RNA binding protein Musashi 2 (Msi-2) [50, 51].
Recent years have also seen a renewed debate over whether cytokines can instruct lineage choice of uncommitted HSPCs versus simply promote preferential proliferation and survival of stochastically committed progenitor cells [52]. Mossadegh-Keller et al. took advantage of time-lapse single cell imaging and single cell gene expression analysis to show that M-CSF, but not G-CSF or GM-CSF, directly induced PU.1 expression in LT-HSCs and instructed LT-HSCs towards myeloid lineage in the absence of proliferative or survival advantage, thus providing a strong case that cytokines can instruct lineage choice in uncommitted HSCs [53**]. Furthermore, a high level of erythropoietin (EPO) was also shown to instruct HSCs and MPPs towards the erythroid lineage at the expense of myeloid and lymphoid cells by suppressing non-erythroid transcriptional programs in uncommitted stem and progenitor cells [54*]. These studies underscore the ability of hematopoietic cytokines released under different stress conditions to actively instruct uncommitted HSPCs to differentiate into a particular cell lineage on demand (Figure 1C).
Regulation of embryonic hematopoietic stem cell specification by inflammatory signaling
Another important concept has emerged recently inflammatory signaling plays a vital role in embryonic HSC specification. Several studies have shown that inflammatory pathways, including TNFα, IFNγ, IFNα and TLR signaling, serve a surprisingly important role in the emergence of early embryonic HSPCs in mice and zebrafish (Figure 1D) [55*, 56*, 57*, 58*]. Sawamiphak et al. found that overexpressing IFNγ increased the number of emerging HSCs from the hemogenic endothelium in zebrafish, while knocking down IFNγ or its receptor had the opposite effect; the authors went to show that IFNγ likely acted downstream of Notch signaling and blood flow in an cell-autonomous manner through STAT3 to drive HSC emergence [58*]. In a parallel study, Espin-Palazon et al. examined a different inflammatory pathway involving TNFα in both zebrafish and mice and found that primitive neutrophils were the source of TNFα which acted through TNFR2 to activate again Notch signaling as well as NF-κB to promote the emergence of HSCs from hemogenic endothelium [57*]. Similar findings were reported almost concurrently in two additional studies. Li et al. demonstrated that both IFNγ and IFNα played important roles in HSPC formation in both mice and zebrafish and the source of interferons was also from primitive myeloid population [56*], while He et al. showed that inflammatory signaling involving the TLR-NF-κB-Notch axis was an important regulator of hemogenic endothelium-derived HSPC development [55*]. These studies are intriguing and have opened a new area of investigation in HSC development in the context of inflammatory signaling. Clearly, a conserved set of molecules and signaling pathways involving interferons, TNFα, TLRs and NF-κB that are traditionally linked to inflammation and infection is also critical in the early embryonic emergence of HSCs. From a more philosophical perspective, how did inflammatory molecules evolve to regulate both embryonic HSC development and stressed hematopoiesis during an infection? It is interesting that mice with genetic deletion in many of these pathways are not only viable but have surprisingly few defects in the hematopoietic stem cell compartment, suggesting that there may be substantial redundancy and/or compensatory mechanism in the signaling pathways for hematopoietic stem cell specification. Perhaps stress is required to reveal the HSC defects in these mice with genetic deletions.
Conclusions and future directions
The remarkable ability of HSPCs to adapt to various stresses, such as infection and inflammation, is critical for an organism’s survival. In the acute setting, pathogen-associated molecules and inflammatory cytokines generally induce HSPCs to proliferate and differentiate towards the myeloid lineage, serving as a protective immune response against pathogens. However, chronic uncontrolled stimulation may be detrimental to HSC’s self-renewal and long-term multi-lineage differentiation ability. Recent studies have increasingly recognized an active role of HSPCs at the heart of a well-orchestrated hematopoietic response during stressed conditions through the discovery of a highly complex cell-cell signaling network in which HSPCs participate and an expanding cast of inflammatory and infectious molecules that HSPCs react with. Several major areas of research have not been addressed. Firstly, viral, bacterial of fungal pathogens often activate multiple TLRs and stimulate production of numerous cytokines. A systems biology approach may be needed to understand how HSPCs integrate different combinations of TLR and cytokine signals into a unique hematopoietic response. Secondly, the recent discovery that basal activation of inflammatory signaling in the absence of infection promotes embryonic HSC development opens up many areas of research. Can we identify the core inflammatory signaling to aid ex vivo differentiation of HSCs from embryonic stem cells or induced pluripotent stem cells, one of the major hurdles in regenerative medicine? Is basal activation of TLR or cytokine signaling also important for steady state hematopoiesis? If so, what are the endogenous sources of TLR ligands and inflammatory cytokines? Lastly, how can we apply the knowledge of physiological stress-induced hematopoiesis to human hematologic diseases? One important question is whether dysregulation of stress-induced hematopoiesis contributes to bone marrow failure and hematopoietic malignancies in humans. Interestingly, several large scale epidemiological studies have shown that chronic immune stimulation from previous infections and autoimmune diseases increases the risk of developing myeloproliferative neoplasms (MPNs), bone marrow fibrosis, acute myelogenous leukemia (AML) and myelodysplastic syndrome (MDS) later on [59-61]. Using a genetic mouse model with deficiency in microRNA-146a, we have provided direct experimental evidence that chronic low-grade inflammation in genetic susceptible hosts was sufficient to drive HSC exhaustion, leading to bone marrow failure and myeloid malignancies in mice. Furthermore, downregulating the p50 subunit of NF-κB or IL-6 significantly dampened chronic inflammation-driven HSC exhaustion and oncogenesis, providing a potential strategy for intervention [62]. A strategy to target IRAK1, a kinase linking TLR and cytokine stimulation to NF-κB, has been tested in a pre-clinical model of MDS and shown to be effective in eliminating MDS cells while sparing normal human HSPCs [63]. One other area of clinical application lies in the utilization of cytokines to activate dormant leukemic stem cells for targeted molecular therapies. This has been tried in CML and polycythemia vera by combining interferon therapy with Bcr-Abl and JAK2 inhibitors, respectively [64-65]. In fact, combination of imatinib and IFNα was more effective in achieving a molecular response in CML patients in a Phase III clinical trial; however the mechanism underlying the improved efficacy was not studied [66-67]. Controlling chronic inflammation and targeting inflammatory signaling that is aberrantly activated in hematologic malignancies will be a promising chemopreventive and therapeutic strategy; on the other hand, waking up leukemic stem cells from dormancy with PAMPs or cytokines may finally make them vulnerable to therapeutic targeting.
Key points.
Bone marrow HSPCs can be activated by TLR ligands while MSCs can be activated by IFNγ to release IL-6 in the stem cell niche to promote myelopoiesis in a paracrine manner.
Numerous cytokines, PAMPs and DAMPs can act directly on HSPCs to stimulate hematopoiesis, leading to increased HSPC proliferation and myeloid differentiation.
Cytokines, including M-CSF and EPO, have the ability to instruct lineage choice at the HSPC level during stressed conditions.
Inflammatory signaling traditionally associated with infection and immune activation is also critical for embryonic HSC fate specification.
Long-term consequences of uncontrolled chronic inflammatory stress include HSC exhaustion and myeloid oncogenic transformation.
Acknowledgments
None.
Financial support and sponsorship
This work was supported by an R01 research grant from NIAID (D.B.) and a F30 NRSA fellowship from NHLBI (J.L.Z.) of National Institutes of Health.
Footnotes
Conflicts of interest
No conflicts of interest to declare.
References
- 1 *.Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol. 2014;14:302–314. doi: 10.1038/nri3660. A comprehensive review of emergency granulopoiesis with a focus on neutrophil production from myeloid progenitor cells during a systemic infection. [DOI] [PubMed] [Google Scholar]
- 2.Stark MA, Huo Y, Burcin TL, et al. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005;22:285–294. doi: 10.1016/j.immuni.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 3.Bugl S, Wirths S, Radsak MP, et al. Steady-state neutrophil homeostasis is dependent on TLR4/TRIF signaling. Blood. 2013;121:723–733. doi: 10.1182/blood-2012-05-429589. [DOI] [PubMed] [Google Scholar]
- 4 **.Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med. 2014;20:833–846. doi: 10.1038/nm.3647. An excellent comprehensive review on various cell types of bone marrow niche and the key niche signals in HSC maintenance during stress and aging. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5 **.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–334. doi: 10.1038/nature12984. An excellent up-to-date review on bone marrow stem cell niche with a particular focus on the distinct bone marrow architecture, osteoblasts and perivascular niche cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 7.Nagai Y, Garrett KP, Ohta S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24:801–812. doi: 10.1016/j.immuni.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Massberg S, Schaerli P, Knezevic-Maramica I, et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007;131:994–1008. doi: 10.1016/j.cell.2007.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esplin BL, Shimazu T, Welner RS, et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol. 2011;186:5367–5375. doi: 10.4049/jimmunol.1003438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Y, Ling F, Wang HC, Sun XH. Chronic TLR signaling impairs the long-term repopulating potential of hematopoietic stem cells of wild type but not Id1 deficient mice. PLoS One. 2013;8:e55552. doi: 10.1371/journal.pone.0055552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11 *.Zhao JL, Ma C, O’Connell RM, et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell. 2014;14:445–459. doi: 10.1016/j.stem.2014.01.007. The first report to demonstrate that hematopoietic stem and progenitor cells can respond to TLR stimulation by producing cytokines to regulate myeloid differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood. 2012;119:2991–3002. doi: 10.1182/blood-2011-12-380113. [DOI] [PubMed] [Google Scholar]
- 13.Megias J, Yanez A, Moriano S, et al. Direct Toll-like receptor-mediated stimulation of hematopoietic stem and progenitor cells occurs in vivo and promotes differentiation toward macrophages. Stem Cells. 2012;30:1486–1495. doi: 10.1002/stem.1110. [DOI] [PubMed] [Google Scholar]
- 14.Takizawa H, Regoes RR, Boddupalli CS, et al. Dynamic variation in cycling of hematopoietic stem cells in steady state and inflammation. J Exp Med. 2011;208:273–284. doi: 10.1084/jem.20101643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.King KY, Goodell MA. Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat Rev Immunol. 2011;11:685–692. doi: 10.1038/nri3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen C, Liu Y, Zheng P. Mammalian target of rapamycin activation underlies HSC defects in autoimmune disease and inflammation in mice. J Clin Invest. 2010;120:4091–4101. doi: 10.1172/JCI43873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Granick JL, Falahee PC, Dahmubed D, et al. Staphylococcus aureus recognition by hematopoietic stem and progenitor cells via TLR2/MyD88/PGE2 stimulates granulopoiesis in wounds. Blood. 2013;122:1770–1778. doi: 10.1182/blood-2012-11-466268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Megias J, Maneu V, Salvador P, et al. Candida albicans stimulates in vivo differentiation of haematopoietic stem and progenitor cells towards macrophages by a TLR2-dependent signalling. Cell Microbiol. 2013;15:1143–1153. doi: 10.1111/cmi.12104. [DOI] [PubMed] [Google Scholar]
- 19.De Luca K, Frances-Duvert V, Asensio MJ, et al. The TLR1/2 agonist PAM(3)CSK(4) instructs commitment of human hematopoietic stem cells to a myeloid cell fate. Leukemia. 2009;23:2063–2074. doi: 10.1038/leu.2009.155. [DOI] [PubMed] [Google Scholar]
- 20.Sioud M, Floisand Y, Forfang L, Lund-Johansen F. Signaling through toll-like receptor 7/8 induces the differentiation of human bone marrow CD34+ progenitor cells along the myeloid lineage. J Mol Biol. 2006;364:945–954. doi: 10.1016/j.jmb.2006.09.054. [DOI] [PubMed] [Google Scholar]
- 21.Scumpia PO, Kelly-Scumpia KM, Delano MJ, et al. Cutting edge: bacterial infection induces hematopoietic stem and progenitor cell expansion in the absence of TLR signaling. J Immunol. 2010;184:2247–2251. doi: 10.4049/jimmunol.0903652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22 *.Schurch CM, Riether C, Ochsenbein AF. Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell. 2014;14:460–472. doi: 10.1016/j.stem.2014.01.002. A nice study that highlights the paracrine signaling in the bone marrow niche involving bone marrow mesenchymal stem cells activating hematopoietic stem and progenitor cells during a viral infection. [DOI] [PubMed] [Google Scholar]
- 23 *.Boettcher S, Gerosa RC, Radpour R, et al. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood. 2014;124:1393–1403. doi: 10.1182/blood-2014-04-570762. Demonstrates an important role of endothelial cells as a sensor of systemic LPS stimulation and activator of emergency granulopoiesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basu S, Hodgson G, Zhang HH, et al. “Emergency” granulopoiesis in G-CSF-deficient mice in response to Candida albicans infection. Blood. 2000;95:3725–3733. [PubMed] [Google Scholar]
- 25.Schuettpelz LG, Borgerding JN, Christopher MJ, et al. G-CSF regulates hematopoietic stem cell activity, in part, through activation of Toll-like receptor signaling. Leukemia. 2014;28:1851–1860. doi: 10.1038/leu.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richards MK, Liu F, Iwasaki H, et al. Pivotal role of granulocyte colony-stimulating factor in the development of progenitors in the common myeloid pathway. Blood. 2003;102:3562–3568. doi: 10.1182/blood-2003-02-0593. [DOI] [PubMed] [Google Scholar]
- 27 *.Carnevalli LS, Scognamiglio R, Cabezas-Wallscheid N, et al. Improved HSC reconstitution and protection from inflammatory stress and chemotherapy in mice lacking granzyme B. J Exp Med. 2014;211:769–779. doi: 10.1084/jem.20131072. Demonstrates a novel mechanism that LPS and chemotherapy regulates HSC activity through induction of granzyme B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernad A, Kopf M, Kulbacki R, et al. Interleukin-6 is required in vivo for the regulation of stem cells and committed progenitors of the hematopoietic system. Immunity. 1994;1:725–731. doi: 10.1016/s1074-7613(94)80014-6. [DOI] [PubMed] [Google Scholar]
- 29.Petzer AL, Zandstra PW, Piret JM, Eaves CJ. Differential cytokine effects on primitive (CD34+CD38-) human hematopoietic cells: novel responses to Flt3-ligand and thrombopoietin. J Exp Med. 1996;183:2551–2558. doi: 10.1084/jem.183.6.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sitnicka E, Lin N, Priestley GV, et al. The effect of thrombopoietin on the proliferation and differentiation of murine hematopoietic stem cells. Blood. 1996;87:4998–5005. [PubMed] [Google Scholar]
- 31.Ueda T, Tsuji K, Yoshino H, et al. Expansion of human NOD/SCID-repopulating cells by stem cell factor, Flk2/Flt3 ligand, thrombopoietin, IL-6, and soluble IL-6 receptor. J Clin Invest. 2000;105:1013–1021. doi: 10.1172/JCI8583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang CC, Lodish HF. Cytokines regulating hematopoietic stem cell function. Curr Opin Hematol. 2008;15:307–311. doi: 10.1097/MOH.0b013e3283007db5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilbert LA, Hemann MT. Context-specific roles for paracrine IL-6 in lymphomagenesis. Genes Dev. 2012;26:1758–1768. doi: 10.1101/gad.197590.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reynaud D, Pietras E, Barry-Holson K, et al. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell. 2011;20:661–673. doi: 10.1016/j.ccr.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borghesi L. Hematopoiesis in steady-state versus stress: self-renewal, lineage fate choice, and the conversion of danger signals into cytokine signals in hematopoietic stem cells. J Immunol. 2014;193:2053–2058. doi: 10.4049/jimmunol.1400936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mirantes C, Passegue E, Pietras EM. Pro-inflammatory cytokines: emerging players regulating HSC function in normal and diseased hematopoiesis. Exp Cell Res. 2014;329:248–254. doi: 10.1016/j.yexcr.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Essers MA, Offner S, Blanco-Bose WE, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 38.Sato T, Onai N, Yoshihara H, et al. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med. 2009;15:696–700. doi: 10.1038/nm.1973. [DOI] [PubMed] [Google Scholar]
- 39.Baldridge MT, King KY, Boles NC, et al. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang L, Dybedal I, Bryder D, et al. IFN-gamma negatively modulates self-renewal of repopulating human hemopoietic stem cells. J Immunol. 2005;174:752–757. doi: 10.4049/jimmunol.174.2.752. [DOI] [PubMed] [Google Scholar]
- 41.de Bruin AM, Demirel O, Hooibrink B, et al. Interferon-gamma impairs proliferation of hematopoietic stem cells in mice. Blood. 2013;121:3578–3585. doi: 10.1182/blood-2012-05-432906. [DOI] [PubMed] [Google Scholar]
- 42.de Bruin AM, Voermans C, Nolte MA. Impact of interferon-gamma on hematopoiesis. Blood. 2014;124:2479–2486. doi: 10.1182/blood-2014-04-568451. [DOI] [PubMed] [Google Scholar]
- 43 *.Pietras EM, Lakshminarasimhan R, Techner JM, et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med. 2014;211:245–262. doi: 10.1084/jem.20131043. Demonstrates an intrinsic protective mechanism to prevent HSC exhaustion during chronic interferon stimulation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dybedal I, Bryder D, Fossum A, et al. Tumor necrosis factor (TNF)-mediated activation of the p55 TNF receptor negatively regulates maintenance of cycling reconstituting human hematopoietic stem cells. Blood. 2001;98:1782–1791. doi: 10.1182/blood.v98.6.1782. [DOI] [PubMed] [Google Scholar]
- 45.Pronk CJ, Veiby OP, Bryder D, Jacobsen SE. Tumor necrosis factor restricts hematopoietic stem cell activity in mice: involvement of two distinct receptors. J Exp Med. 2011;208:1563–1570. doi: 10.1084/jem.20110752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Harada A, Bluethmann H, et al. Tumor necrosis factor (TNF) is a physiologic regulator of hematopoietic progenitor cells: increase of early hematopoietic progenitor cells in TNF receptor p55-deficient mice in vivo and potent inhibition of progenitor cell proliferation by TNF alpha in vitro. Blood. 1995;86:2930–2937. [PubMed] [Google Scholar]
- 47.Rebel VI, Hartnett S, Hill GR, et al. Essential role for the p55 tumor necrosis factor receptor in regulating hematopoiesis at a stem cell level. J Exp Med. 1999;190:1493–1504. doi: 10.1084/jem.190.10.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rezzoug F, Huang Y, Tanner MK, et al. TNF-alpha is critical to facilitate hemopoietic stem cell engraftment and function. J Immunol. 2008;180:49–57. doi: 10.4049/jimmunol.180.1.49. [DOI] [PubMed] [Google Scholar]
- 49.Challen GA, Boles NC, Chambers SM, Goodell MA. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell. 2010;6:265–278. doi: 10.1016/j.stem.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park SM, Deering RP, Lu Y, et al. Musashi-2 controls cell fate, lineage bias, and TGF-beta signaling in HSCs. J Exp Med. 2014;211:71–87. doi: 10.1084/jem.20130736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quere R, Saint-Paul L, Carmignac V, et al. Tif1gamma regulates the TGF-beta1 receptor and promotes physiological aging of hematopoietic stem cells. Proc Natl Acad Sci U S A. 2014;111:10592–10597. doi: 10.1073/pnas.1405546111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Endele M, Etzrodt M, Schroeder T. Instruction of hematopoietic lineage choice by cytokine signaling. Exp Cell Res. 2014;329:207–213. doi: 10.1016/j.yexcr.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 53 **.Mossadegh-Keller N, Sarrazin S, Kandalla PK, et al. M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature. 2013;497:239–243. doi: 10.1038/nature12026. The first report to provide strong evidence for M-CSF to instruct lineage choice of hematopoietic stem cell in a physiologically relevant condition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54 *.Grover A, Mancini E, Moore S, et al. Erythropoietin guides multipotent hematopoietic progenitor cells toward an erythroid fate. J Exp Med. 2014;211:181–188. doi: 10.1084/jem.20131189. The first report to support an instructive role of erythropoietin to promote erythroid differentiation from uncommitted hematopoietic stem and progenitor cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55 *.He Q, Zhang C, Wang L, et al. Inflammatory signaling regulates hematopoietic stem and progenitor cell emergence in vertebrates. Blood. 2014 doi: 10.1182/blood-2014-09-601542. Among the first reports to demonstrate a role of TLR-NF-κB in the emergence of embryonic hematopoietic stem and progenitor cells. [DOI] [PubMed] [Google Scholar]
- 56 *.Li Y, Esain V, Teng L, et al. Inflammatory signaling regulates embryonic hematopoietic stem and progenitor cell production. Genes Dev. 2014;28:2597–2612. doi: 10.1101/gad.253302.114. One of the first reports to support a conserved role of interferon alpha and gamma in embryonic hematopoietic stem cell development in the absence of infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57 *.Espin-Palazon R, Stachura DL, Campbell CA, et al. Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell. 2014;159:1070–1085. doi: 10.1016/j.cell.2014.10.031. The first report to support a conserved role of TNF signaling in embryonic hematopoietic stem cell development in the absence of infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58 *.Sawamiphak S, Kontarakis Z, Stainier DY. Interferon gamma signaling positively regulates hematopoietic stem cell emergence. Dev Cell. 2014;31:640–653. doi: 10.1016/j.devcel.2014.11.007. One of the first reports to support a conserved role of interferon gamma in embryonic hematopoietic stem cell development in the absence of infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anderson LA, Pfeiffer RM, Landgren O, et al. Risks of myeloid malignancies in patients with autoimmune conditions. Br J Cancer. 2009;100:822–828. doi: 10.1038/sj.bjc.6604935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kristinsson SY, Bjorkholm M, Hultcrantz M, et al. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. J Clin Oncol. 2011;29:2897–2903. doi: 10.1200/JCO.2011.34.8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hasselbalch HC. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood. 2012;119:3219–3225. doi: 10.1182/blood-2011-11-394775. [DOI] [PubMed] [Google Scholar]
- 62.Zhao JL, Rao DS, O’Connell RM, et al. MicroRNA-146a acts as a guardian of the quality and longevity of hematopoietic stem cells in mice. Elife. 2013;2:e00537. doi: 10.7554/eLife.00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rhyasen GW, Bolanos L, Fang J, et al. Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer Cell. 2013;24:90–104. doi: 10.1016/j.ccr.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mullally A, Bruedigam C, Poveromo L, et al. Depletion of Jak2V617F myeloproliferative neoplasm-propagating stem cells by interferon-alpha in a murine model of polycythemia vera. Blood. 2013;121:3692–3702. doi: 10.1182/blood-2012-05-432989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schurch C, Riether C, Amrein MA, Ochsenbein AF. Cytotoxic T cells induce proliferation of chronic myeloid leukemia stem cells by secreting interferon-gamma. J Exp Med. 2013;210:605–621. doi: 10.1084/jem.20121229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Preudhomme C, Guilhot J, Nicolini FE, et al. Imatinib plus peginterferon alfa-2a in chronic myeloid leukemia. N Engl J Med. 2010;363:2511–2521. doi: 10.1056/NEJMoa1004095. [DOI] [PubMed] [Google Scholar]
- 67.Simonsson B, Gedde-Dahl T, Markevarn B, et al. Combination of pegylated IFN-alpha2b with imatinib increases molecular response rates in patients with low- or intermediate-risk chronic myeloid leukemia. Blood. 2011;118:3228–3235. doi: 10.1182/blood-2011-02-336685. [DOI] [PubMed] [Google Scholar]