

Abstract
Myotonic dystrophy (DM) is the most common form of adult onset muscular dystrophy and is caused by expansion of short nucleotide repeats that, in turn, produce toxic RNA aggregates within cells. DM is multisystemic, and the heart is primary site of pathology. DM patients exhibit cardiac conduction disorders including atrial fibrillation, atrio-ventricular heart block and ventricular arrhythmias. DM patients are also at risk for cardiomyopathy and congestive heart failure. Myotonic dystrophy is also characterized by myotonia, muscle weakness, and profound fatigue. The management of these symptoms requires input from the cardiologist and a team approach to minimize the debilitating aspects of the disorder and optimize cardiac function.
Keywords: Myotonic dystrophy, Cardiac genetics, Cardiac conduction abnormalities, Cardiomyopathy, RNA gain-of-function
INTRODUCTION
Muscular dystrophy is a genetically diverse group of disorders that can affect both children and adults. Many forms of muscular dystrophy have significant cardiac manifestations. The most common cardiac consequences in muscular dystrophy are cardiomyopathy and arrhythmias. Correct identification of the genetic subtype of muscular dystrophy is critical for proper care since it determines the degree to which a cardiologist should be involved in the care of the patient. Cardiomyopathy and arrhythmias often develop later in the course of disease so cardiac management of the neuromuscular patient often falls within the purview of the adult cardiologist. However, since early treatment can be effective at slowing the progress of disease, prepathologic treatment and surveillance for arrhythmias may fall under the pediatric cardiologist. The myotonic dystrophies represent a subset of inherited muscular dystrophy disorders. Myotonic dystrophy, or dystrophia myotonica (DM), is a multisystem disease, and the cardiovascular defects may be disabling and life-threatening. DM is dominantly inherited, so it is not uncommon that multiple family members of different generations are afflicted with the disorder. Cardiac involvement is prevalent in DM and accounts for approximately one-third of the deaths in this population (1, 2). Herein, we will discuss recommendations for screening and risk stratification of cardiac complications in the DM population.
THE MYOTONIC DYSTROPHIES
Brief Overview
Myotonic dystrophy not only affects cardiac and skeletal muscle, but also manifests in the endocrine, ocular, gastrointestinal and nervous systems. Myotonia, or delay in relaxation of muscle contraction, is a hallmark feature of DM. Myotonia can be elicited on physical exam as grip myotonia or with percussion. In addition to myotonia, patients develop progressive muscle weakness and wasting. Involvement of the respiratory musculature often develops as a late stage of disease and is life threatening (3). Endocrine abnormalities include hypogonadism and insulin resistance. Cataracts are common and typically are characterized by multicolored lens opacities referred to as “Christmas tree cataracts”. Gastrointestinal problems typically include dysphagia, reflux, hypomotility, cholestasis and incontinence reflecting both smooth and striated muscle involvement. Hypo-gammaglobulinemia may also be present but it remains in debate whether this is clinically meaningful. Central nervous system effects are broad and may include cognitive decline, impaired visual-spatial coordination and executive functioning. Hypersomnolence and fatigue are among the most debilitating complaints in DM, and are thought to arise from CNS effects, although sleep apnea-type hypoventilation from respiratory muscle weakness or from extrinsic compression due to pharyngeal weakness may also contribute. Notably DM patients are often treated with CNS stimulants such as methylphenidate and modafinil, and these agents may increase the risk of cardiac dysrhythmias (4).
Epidemiology
There are two defined genetic subtypes DM1 and DM2. DM1 is more common, approximately 1 in 8000, making it the most common adult form of muscular dystrophy. DM2 is less common, approximately 1 in 20,000. DM1 was first described by Hans Steinert in 1909, and thus is referred to as Steinert’s disease. DM patients who did not harbor the DM1 molecular signature were defined as DM2. Clinically, DM2 may be described as PROMM (proximal myotonic myopathy). DM1 is further divided into three subtypes based on age of onset and severity. The congenital subtype is the most severe with onset often at birth or in the first year of life. Congenital DM1 most commonly occurs when a child is born to a mother with DM1 (maternal transmission). Congenital DM1 babies have muscular weakness and hypotonia, developmental delay, difficulty feeding, and respiratory failure. Classical DM can be divided into child and adult forms. Clinical features of the adult form appear between the second and fourth decades of life and progress slowly. A mild adult-onset form exists, also called minimal DM, and is characterized by symptoms occurring in the fifth decade, very mild manifestations and a normal life expectancy. Overall, in DM mortality is increased to roughly seven times that of the general population and typically occurs in the fourth to fifth decade of life. The two most common causes of death in myotonic dystrophy are respiratory and cardiovascular disease.
Genetic Mechanisms in DM
Myotonic dystrophy is inherited in an autosomal dominant pattern. DM1 and DM2 each arise from expansion of a nucleotide repeat sequence; DM1 develops from expansion of a trinucleotide sequence on chromosome 19 and DM2 is associated with expansion of a tetranucleotide repeat sequence on chromosome 3. In DM1, the CTG expansion falls within the 3′-untranslated region of the myotonic dystrophy protein kinase gene (DMPK) and the 5′ end of the Six5 gene (Figure 1). Normally, there are between 4 and 37 CTG repeats, and expansion beyond 50 repeats is considered pathogenic. With congenital DM, the number of CTG repeats may be 800–1000. Individuals with between 37 and 50 repeats are considered as “pre-mutation.” Notably, the CTG expansion length may expand with subsequent generations, accounting for increasing severity of disease in each subsequent generation, a process referred to as genetic anticipation. During oogenesis, an expanded CTG may become greatly expanded and result in congenital DM. Thus, any woman with DM1 should received genetic counseling when considering pregnancy. DM2 shares a similar pathology in that a CCTG repeat is expanded within intron 1 of the zinc finger protein 9 (ZNF9) gene; the repeat expansions in DM2 commonly number in the thousands.
Figure 1.

A. DM1 is caused by an expansion in a CTG repeat on chromosome 19. Expansion to more than 100 copies is pathogenic. B. A CCTG repeat is expanded on chromosome 3 in DM2. C. Transcription of the repeated DNA sequences leads to RNA that forms hairpin structures that in turn bind splicing factors within the nucleus. D. Sequestration of the splicing factors CUBGP and MBNL lead to abnormal splicing of target genes. Abnormal splicing of the chloride channel gene (CLCN1) leads to myotonia. Abnormal splicing of TNNT2 contributes to cardiomyopathy.
Although there is a general correlation with repeat expansion length in DM1 and clinical severity, such correlation is imprecise. Generally, the mild form is associated with a range of 50–80 repeats, the classical form with 100–1000 repeats, and the congenital form with more than 1000 repeats. In DM1, CTG repeat expansion length and cardiac complications have been correlated. A longitudinal study of 50 DM1 patients (mean age 34 years) without clear cardiac involvement were followed for nearly five years (5). There was an inverse relationship between the CTG repeat length and age of onset, but not the overall frequency, of major ECG abnormalities (5). Another study of 42 patients with DM1 (mean age 27.5 years), found that increased repeat expansion length correlated with earlier age of onset of cardiac defects, as well as the frequency of conduction defects, specifically complete left bundle branch block and abnormal late potentials (6). A large study of 342 DM1 (mean age 42.5 years) patients found a direct correlation between CTG length and arrhythmias as well as with PR and QRS prolongation (7). Thus genetic information is helpful in the risk stratification of DM1 patients, although genetic information is not the entire determinant of risk.
Less is known about the cardiac risk in DM2 because the genetic underpinnings of DM2 were only more recently discovered and DM2 is more rare. A comparison of the cardiac defects between DM1 and DM2 patients suggested that cardiac abnormalities were more frequent in DM1 but were still present in DM2 (8). This study compared 76 sex matched DM1 patients with 38 DM2 patients (mean age 57 years), and found more PR prolongation and AV conduction block in DM1 compared to DM2, (8). DM2 patients remained at risk for left ventricular dysfunction and arrhythmias including sudden death. Importantly, even in this small sample, there was a correlation between the length of tetranucleotide repeat and cardiac defects DM2. However, the relationship between nucleotide expansion length and phenotype is not linear.
Early work suggested that the pathological consequences in DM1 arose from disrupting the DMPK gene. However, in a series of elegant studies, it has been shown that RNA expression of the repeat expansion is pathogenic itself. This “gain-of-function toxic RNA” mechanism is thought to mediate disease by forming aggregated double stranded RNA within nuclei. The expanded RNA accumulates as double stranded structures in the nucleus and sequesters splicing regulators, rendering them unable to facilitate normal splicing of genes. Two of the most well-studied splice regulators in DM are muscleblind protein (MBNL) and CUG-binding protein 1 (CUGBP1). Although many genes are abnormally spliced, several are thought to render the features of DM. CLCN1 encode the chloride channel and abnormal splicing of CLCN1 is thought to account for myotonia. Cardiac dysfunction is partially explained by perturbed splicing and expression of troponin T (cTnT or TNNT2). Following from this toxic RNA model are several important concepts. First, it has been shown in animal models that reversing the expression of the toxic RNA is associated with improvement of clinical manifestation, including cardiac features such as cardiomyopathy and conduction system disease (9). Second, this work now focuses therapy development on small molecules or other mechanisms that interrupt double stranded RNA (10). Lastly, the mis-spliced genes in DM1 point to the pathways that mediate disease and therefore are those to target for therapy development.
Clinical Presentation
The most common presentation of DM1 is muscle weakness and myotonia, predominantly in the distal muscles with concomitant facial and neck muscle involvement. Patients often have a typical appearance characterized by long, narrow face and atrophy of the temporal muscles as well as muscles of mastication. Upon questioning, DM1 patient will describe myotonia and often note symptoms dating back some number of years. DM may have cardiac manifestations as the primary presentation. Shown in Figure 2 is a surface EKG from a 22 year old male who presented at the age of 15 with atrial flutter that was successfully ablated. He remained without further episodes of atrial dysrhythmias for more than 5 years when a diagnosis of DM1 was made after developing myotonia affecting his hands. Atrial flutter and fibrillation in such patients may be exercised induced (11) and was the case in this patient. Similarly, in DM2, the first manifestation may be cardiac arrhythmias. Shown in Figure 3 is a surface EKG from a 54 year old female with DM2 who presented with syncope and demonstrated ventricular tachycardia on Holter monitoring. Her mother was diagnosed with PROMM and congestive heart failure in her 80’s. The cardiologist evaluating isolated atrial flutter/fibrillation or ventricular tachycardia, especially in young patients, should consider DM in the differential diagnosis.
Figure 2.

Shown is an EKG from a 22 year old male with DM1 immediately post exercise showing exercise-induced atrial fibrillation.
Figure 3.
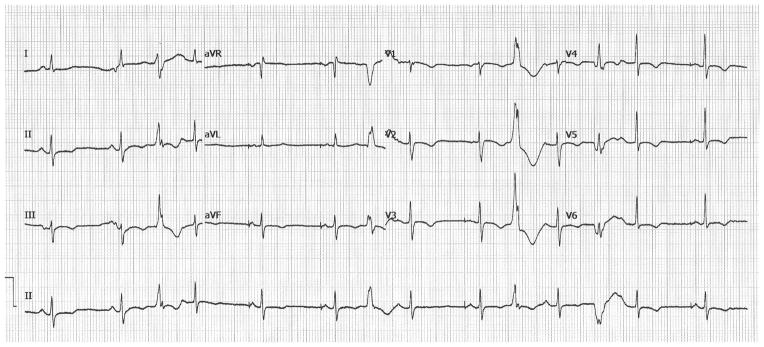
Shown is an EKG from a 54 year old female with DM2 who experienced syncope and had ventricular tachycardia on noninvasive monitoring. An ICD was placed.
Diagnosis
The gold standard for diagnosis is genetic testing performed on blood leukocytes. DM1 testing is performed first followed by DM2 testing if clinical suspicion warrants. With the increase of massively parallel sequencing now being used for whole genome analysis, it should be appreciated that repeat expansions will not be detected by the emerging DNA sequencing technology. Genetic testing for DM1 and DM2 is performed as a specific analysis of the repeat expansion region and therefore must be ordered as a specific genetic test. Genetic counseling is recommended. Prenatal diagnosis of DM1 or DM2 can be made using materials from chorionic villous biopsy or amniotic cell culture. Preimplantation genetic diagnosis is also available as a mechanism to use genetic information and avoid transmission of the CTG allele. Women with DM who plan to have children should undergo genetic testing and counseling about congenital DM. Genetic DNA testing has largely replaced previously used modalities including muscle biopsy and electromyography (EMG). Muscle biopsy may reveal variation in the size of muscle fibers, as well as atrophy and fibrosis. They may demonstrate ring fibers in which myofibril bundles are arranged perpendicular to each other rather than in parallel. It is also common to see a dramatic increase in the number of internalized nuclei in myofibers. These findings are limited by the degree of skeletal muscle involvement and sampling error. Similarly, EMG may be nondiagnostic, but is useful when it portrays the characteristic features of myotonic dystrophy. For both DM1 and DM2, EMG findings include bursts of potentials that wax and wane in intensity, giving it the “dive-bomber” or “motorcycle” pattern. Other testing modalities, such as serum muscle enzymes and neuroimaging reveal nonspecific and diagnostically insignificant findings.
CARDIAC MANAGEMENT
Myotonic dystrophy can be associated with a range of cardiovascular defects. The most common are conduction disturbances, arrhythmias, and cardiomyopathy. DM1 patients may also develop the more common age-related cardiovascular complications such as coronary artery disease and valvulopathies. Thus, the age of the DM patient will determine the likelihood of these complications.
Conduction Disturbances
Conduction disturbances are prevalent in myotonic dystrophy, affecting roughly 30–75% of DM1 (2). Although less well studied, DM2 patients also have an increased incidence of prolonged PR interval and evidence of heart block (8). It is estimated that 65% of DM1 patients have an abnormal EKG. Conduction abnormalities are a result of myocyte hypertrophy, fibrosis, focal fatty infiltration and also lymphocytic infiltration, which can occur anywhere along the conduction system including the His-Purkinje system (12). Prolongation of the PR segment occurs in roughly 20–40% of patients and QRS widening occurs in 5–25% of patients. Such depolarization abnormalities can also lead to the presence of Q waves on the EKG, when a history of myocardial infarction is lacking. Late potentials may also be evident. These are the result of delayed myocardial activation of the His-Purkinje system, rather than propagation of action potentials through focal islands of fibrosis. Late potentials are considered predictors of ventricular arrhythmias.
Arrhythmias
Both atrial and ventricular arrhythmias can occur with myotonic dystrophy. Atrial fibrillation, atrial flutter and atrial tachycardia are the most common, affecting approximately one quarter of DM patients (2). Ventricular arrhythmias are less common, but understandably raise more concern due to their life-threatening potential. Monomorphic ventricular tachycardia, polymorphic ventricular tachycardia and ventricular fibrillation may occur. There are several mechanisms that can lead to ventricular arrhythmias, including fibro-fatty degeneration of myocardium or the fascicles serving as a catalyst for re-entry within the ventricular wall or fascicular re-entry, respectively.
Myotonic dystrophy patients are predisposed to bundle branch re-entry ventricular tachycardia (BBRVT) because a diseased conduction system is essential to this mechanism (13). This results from a macro-re-entry between the left and right fascicles. Generally, BBRVT makes up only 6% of monomorphic ventricular tachycardias. Despite its relative rarity, however, morbidity is significant with 75% of patients that have inducible BBRVT presenting either with syncope or sudden cardiac death. A prolonged His-ventricular (HV) interval contributes to BBRVT and since this is the case for many patients with myotonic dystrophy, this dysrhythmia can occur in DM and should not be missed since this disorder can be successfully cured with radiofrequency ablation.
Cardiomyopathy
Patients with myotonic dystrophy are also prone to developing structural cardiomyopathy (8, 14). When applied, imaging modalities often identify evidence of structural heart disease before symptoms develop. One large registry of over 400 DM patients found some form of structural heart disease in roughly 20% patients, but only 2% of the patients had overt symptoms of heart failure (14). Radionuclide imaging and echocardiography, and in particular tissue Doppler imaging (TDI) may reveal diastolic dysfunction early in the disease course, and later may show systolic dysfunction in either ventricle. In addition, left ventricular hypertrophy, left ventricular dilatation, and left atrial dilatation may occur. As with other forms of muscular dystrophy, regional wall motion abnormalities may be present arising from non-ischemic fibrosis. In some patients with a normal resting ejection fraction (EF), stress testing may reveal failure of the EF to augment with exercise.
Other Forms of Cardiac Involvement
While structural heart disease and cardiac arrhythmias are the most common cardiovascular defects associated with myotonic dystrophy, other abnormalities have been identified. Mitral valve prolapse has been identified in up to 13–40% of patients (6, 14). Myocardial myotonia, or impaired relaxation of cardiac muscle, is the cardiac equivalent of what occurs in the skeletal muscle of these patients. It can be identified by tissue Doppler imaging as diastolic dysfunction as noted above.
Routine Surveillance and Noninvasive Testing
In DM, surveillance for arrhythmias and prophylactic treatment have been debated. While it is evident that risk of arrhythmia, including sudden cardiac death, is increased in the DM population, a clear consensus for management has not been established because of variability in presentation and course. DM patients with clinically unrecognized muscle involvement have presented initially with atrial tachyarrhythmias or even overt heart failure syndromes, and this observation is consistent with what we have observed in practice. In clinically recognized DM, where muscle symptoms are evident, sudden death is increased (2). Managing these patients should focus on risk stratification and prevention of fatal arrhythmias and sudden cardiac death (1–3, 15). Although debated, we favor the strategy of a surface EKG at least yearly and echocardiography yearly.
Symptoms such as presyncope, lightheadedness, and palpitations should prompt more formal testing. This testing may include continuous or event monitoring or an invasive electrophysiological study. Frank syncope, especially if the clinical circumstances support a cardiac cause, should merit a thorough electrophysiological evaluation with consideration of device implantation for the management of tachy and/or bradyarrhythmis. The choice of device should be influenced by the presence of conduction system defects on EKG or intracardiac measurements. An ongoing clinical trial is assessing the value of invasive monitoring in DM patients (16).
Pharmacotherapy
Medical therapy for these patients should include standard cardiomyopathy treatment, ACE inhibitor or angiotensin receptor blockade and β adrenergic blockade. These agents are thought to reverse remodeling. In Duchenne Muscular Dystrophy, prophylactic treatment with ACE inhibitors has been shown to be effective in slowing the onset of cardiomyopathy and is further associated with mortality benefit (17). Such pretreatment has not been assessed in the DM population. The longer course and lower incidence of cardiomyopathy in Duchenne Muscular Dystrophy compared to DM may not support pretreatment with cardiomyopathy agents as it does in Duchenne Muscular Dystrophy.
Medical therapy for atrial arrhythmias much be approached cautiously since DM patients are prone to bradyarrhythmias and conduction system block at the AV node. Thus, agents that reduce conduction across the AV node should be associated with close monitoring. Anticoagulation of DM patients with atrial arrhythmias must also take into account skeletal muscle symptoms and gait instability that may render patients prone to falling. For young DM patients with atrial fibrillation and normal ventricular dysfunction, anti-coagulation with warfarin may not be indicated according to the general guidelines.
The cardiologist managing the DM patient must serve as consultant to the neurologist who is often providing primary management. The neurologist will often advance treatment with carbamazipine or other agents to reduce myotonia symptoms. More recent trials have evaluated mexiletine for myotonia (18). More vexing is the use of CNS stimulants such as methylphenidate or modafinil will be pursued to treat the chronic fatigue and hypersomnolence that is most debilitating in DM. Although these agents may increase risk of cardiac arrhythmias, this risk must be weighed against potential quality of life improvement. Recent clinical trials have evaluated the safety of insulin like growth factor 1 (IGF1) to improve muscle strength (19). In principle, such an agent is not anticipated to have cardiac toxicity since IGF1 may be appropriate for the treatment of cardiomyopathy.
Device Therapy
The indications for device use, including pacemakers and defibrillators, have been clarified, in part by larger longitudinal studies of DM1 patients (2). The surface EKG is helpful in determining who benefits from device implantation since those with a PR interval longer than 240 msec and a QRS wider than 120 are at higher risk (20). Heart rate variation has also been shown to be helpful (21). For patients with myotonic dystrophy and prolonged HV intervals, pacemakers protect from bradyarrhythmias and heart block but also allow for the diagnosis of potentially life-threatening tachyarrhythmias via device interrogation. This may prompt necessary prophylaxis with defibrillator therapy and/or more aggressive evaluation for structural heart disease when there is a lack of symptoms. The 2008 ACC/AHA/HRS guidelines have been updated regarding pacemaker implantation in patients with neuromuscular diseases (22). It is now considered a class IIA indication for patients with prolonged HV intervals >100msec and a class IIB indication for any degree of AV block regardless of symptoms if there is enough concern for unpredictable progression of disease. A clinical algorithm is summarized in Figure 4.
Figure 4.

Proposed algorithm for cardiovascular management in DM. PM, pacemaker; ICD, implantable cardioverter defibrillator; WMA, wall motion abnormality; LGE, late gadolinium enhancement; BB, beta blocker; ACEI, angiotensin converting enzyme inhibitor; ARB, angiotensin receptor blocker; CAD, coronary artery disease.
Future Strategies
Current understanding of the molecular pathology in DM emphasizes that it is the collection of double stranded RNA aggregates that is the primary mediator of pathology. Small molecule approaches have identified approaches to reduce formation for these aggregates. In addition to this strategy, there are key pathways that can be targeted in DM. Wang and colleagues found that when they administered a PKC inhibitor to mice with transgenically induced DM, previously documented conduction defects and decreased contractility improved (23).
Key points.
Myotonic Dystrophy (DM) pathogenesis
Arising from nucleotide expansion and expression of toxic RNA
Disrupts splicing of multiple genes that affect heart function
Dominant inheritance
Disease course is more severe and age of onset is earlier with each subsequent generation
Women with DM can give birth to children with congenital DM
Cardiac arrhythmias in DM
Atrial arrhythmias are common and may be exercise induced
PR prolongation and QRS widening portend higher risk of arrhythmias
Risks of both brady and tachyarrhythmias are present
Sudden cardiac death risk is increased
Accompanying neuromuscular symptoms are associated with increased cardiac risk
Medical treatment
DM patients may be treated with CNS stimulants which may increase arrhythmia risks
Mexiletine is useful for the treatment of myotonia
LV dysfunction can be treated with ACE inhibitors/ARBs and beta blockers
Diabetes and other endocrinopathies may be present as part of DM
Acknowledgments
Funding: EMM is supported by the Doris Duke Charitable Foundation, the NIH and the Muscular Dystrophy Association.
Abbreviations
- BBRVT
bundle branch re-entry ventricular tachycardia
- DM
dystrophia myotonica
- EKG
electrocardiogram
- EMG
electromyelogram
- PROMM
proximal myotonic myopathy
Footnotes
Competing Interests: None
The authors have no competing interests to declare.
References
- 1.Mathieu J, Allard P, Potvin L, Prevost C, Begin P. A 10-year study of mortality in a cohort of patients with myotonic dystrophy. Neurology. 1999 May 12;52(8):1658–62. doi: 10.1212/wnl.52.8.1658. [DOI] [PubMed] [Google Scholar]
- 2**.Groh WJ, Groh MR, Saha C, Kincaid JC, Simmons Z, Ciafaloni E, et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med. 2008 Jun 19;358(25):2688–97. doi: 10.1056/NEJMoa062800. Large study of DM1 patients that describes risks for developing sudden cardiac death. [DOI] [PubMed] [Google Scholar]
- 3*.Pelargonio G, Dello Russo A, Sanna T, De Martino G, Bellocci F. Myotonic dystrophy and the heart. Heart. 2002 Dec;88(6):665–70. doi: 10.1136/heart.88.6.665. Comprehesive review of DM1 and cardiac complications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacDonald JR, Hill JD, Tarnopolsky MA. Modafinil reduces excessive somnolence and enhances mood in patients with myotonic dystrophy. Neurology. 2002 Dec 24;59(12):1876–80. doi: 10.1212/01.wnl.0000037481.08283.51. [DOI] [PubMed] [Google Scholar]
- 5.Antonini G, Giubilei F, Mammarella A, Amicucci P, Fiorelli M, Gragnani F, et al. Natural history of cardiac involvement in myotonic dystrophy: correlation with CTG repeats. Neurology. 2000 Oct 24;55(8):1207–9. doi: 10.1212/wnl.55.8.1207. [DOI] [PubMed] [Google Scholar]
- 6.Melacini P, Villanova C, Menegazzo E, Novelli G, Danieli G, Rizzoli G, et al. Correlation between cardiac involvement and CTG trinucleotide repeat length in myotonic dystrophy. J Am Coll Cardiol. 1995 Jan;25(1):239–45. doi: 10.1016/0735-1097(94)00351-p. [DOI] [PubMed] [Google Scholar]
- 7.Groh WJ, Lowe MR, Zipes DP. Severity of cardiac conduction involvement and arrhythmias in myotonic dystrophy type 1 correlates with age and CTG repeat length. J Cardiovasc Electrophysiol. 2002 May;13(5):444–8. doi: 10.1046/j.1540-8167.2002.00444.x. [DOI] [PubMed] [Google Scholar]
- 8**.Wahbi K, Meune C, Becane HM, Laforet P, Bassez G, Lazarus A, et al. Left ventricular dysfunction and cardiac arrhythmias are frequent in type 2 myotonic dystrophy: a case control study. Neuromuscul Disord. 2009 Jul;19(7):468–72. doi: 10.1016/j.nmd.2009.04.012. Describe cardiovascular complications with DM2. [DOI] [PubMed] [Google Scholar]
- 9.Koshelev M, Sarma S, Price RE, Wehrens XH, Cooper TA. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum Mol Genet. 2010 Mar 15;19(6):1066–75. doi: 10.1093/hmg/ddp570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10**.Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, et al. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science. 2009 Jul 17;325(5938):336–9. doi: 10.1126/science.1173110. Animal model of DM1 shows the reversible nature of the cardiovascular complications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11*.Bassez G, Lazarus A, Desguerre I, Varin J, Laforet P, Becane HM, et al. Severe cardiac arrhythmias in young patients with myotonic dystrophy type 1. Neurology. 2004 Nov 23;63(10):1939–41. doi: 10.1212/01.wnl.0000144343.91136.cf. Describes the pattern of arrhythmias in young patients with DM1 including exercise induced atrial arrhythmias. [DOI] [PubMed] [Google Scholar]
- 12.Nguyen HH, Wolfe JT, 3rd, Holmes DR, Jr, Edwards WD. Pathology of the cardiac conduction system in myotonic dystrophy: a study of 12 cases. J Am Coll Cardiol. 1988 Mar;11(3):662–71. doi: 10.1016/0735-1097(88)91547-1. [DOI] [PubMed] [Google Scholar]
- 13.Merino JL, Carmona JR, Fernandez-Lozano I, Peinado R, Basterra N, Sobrino JA. Mechanisms of sustained ventricular tachycardia in myotonic dystrophy: implications for catheter ablation. Circulation. 1998 Aug 11;98(6):541–6. doi: 10.1161/01.cir.98.6.541. [DOI] [PubMed] [Google Scholar]
- 14.Bhakta D, Lowe MR, Groh WJ. Prevalence of structural cardiac abnormalities in patients with myotonic dystrophy type I. Am Heart J. 2004 Feb;147(2):224–7. doi: 10.1016/j.ahj.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Morner S, Lindqvist P, Mellberg C, Olofsson BO, Backman C, Henein M, et al. Profound cardiac conduction delay predicts mortality in myotonic dystrophy type 1. J Intern Med. 2010 Jul;268(1):59–65. doi: 10.1111/j.1365-2796.2010.02213.x. [DOI] [PubMed] [Google Scholar]
- 16.Dello Russo A, Mangiola F, Della Bella P, Nigro G, Melacini P, Bongiorni MG, et al. Risk of arrhythmias in myotonic dystrophy: trial design of the RAMYD study. J Cardiovasc Med (Hagerstown) 2009 Jan;10(1):51–8. doi: 10.2459/jcm.0b013e328319bd2c. [DOI] [PubMed] [Google Scholar]
- 17**.Duboc D, Meune C, Pierre B, Wahbi K, Eymard B, Toutain A, et al. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. Am Heart J. 2007 Sep;154(3):596–602. doi: 10.1016/j.ahj.2007.05.014. Demonstrates the value of prophylactic treatment with ACE inhibitors in another form of muscular dystrophy, Duchenne Muscular Dystrophy. [DOI] [PubMed] [Google Scholar]
- 18*.Logigian EL, Martens WB, Moxley RTt, McDermott MP, Dilek N, Wiegner AW, et al. Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. Neurology. 2010 May 4;74(18):1441–8. doi: 10.1212/WNL.0b013e3181dc1a3a. Describes the use of mexiletine in DM1 for the treatment of myotonia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heatwole CR, Eichinger KJ, Friedman DI, Hilbert JE, Jackson CE, Logigian EL, et al. Open-Label Trial of Recombinant Human Insulin-like Growth Factor 1/Recombinant Human Insulin-like Growth Factor Binding Protein 3 in Myotonic Dystrophy Type 1. Arch Neurol. 2010 Sep 13; doi: 10.1001/archneurol.2010.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laurent V, Pellieux S, Corcia P, Magro P, Pierre B, Fauchier L, et al. Mortality in myotonic dystrophy patients in the area of prophylactic pacing devices. Int J Cardiol. 2010 Mar 11; doi: 10.1016/j.ijcard.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 21.Casella M, Dello Russo A, Pace M, Pelargonio G, Ierardi C, Sanna T, et al. Heart rate turbulence as a noninvasive risk predictor of ventricular tachyarrhythmias in myotonic dystrophy type 1. J Cardiovasc Electrophysiol. 2006 Aug;17(8):871–6. doi: 10.1111/j.1540-8167.2006.00517.x. [DOI] [PubMed] [Google Scholar]
- 22.Epstein AE, DiMarco JP, Ellenbogen KA, Estes NA, 3rd, Freedman RA, Gettes LS, et al. ACC/AHA/HRS 2008 Guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices): developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. Circulation. 2008 May 27;117(21):e350–408. doi: 10.1161/CIRCUALTIONAHA.108.189742. [DOI] [PubMed] [Google Scholar]
- 23.Wang GS, Kuyumcu-Martinez MN, Sarma S, Mathur N, Wehrens XH, Cooper TA. PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J Clin Invest. 2009 Dec;119(12):3797–806. doi: 10.1172/JCI37976. [DOI] [PMC free article] [PubMed] [Google Scholar]